

Abstract
Chemotherapy is the mainstream treatment of anaplastic large cell lymphoma (ALCL). However, chemotherapy can cause severe adverse effects in patients because it is not ALCL-specific. In this study, we developed a multifunctional aptamer-nanomedicine (Apt-NMed) achieving targeted chemotherapy and gene therapy of ALCL. Apt-NMed was formulated by self-assembly of synthetic oligonucleotides containing CD30-specific aptamer and ALK-specific siRNA followed by self-loading of the chemotherapeutic drug doxorubicin (DOX). Apt-NMed exhibited a well-defined nanostructure (diameter 59 mm) and stability in human serum. Under aptamer guidance, Apt-NMed specifically bound and internalized targeted ALCL cells. Intracellular delivery of Apt-NMed triggered rapid DOX release for targeted ALCL chemotherapy and intracellular delivery of the ALK-specific siRNA induced ALK oncogene silencing, resulting in combined therapeutic effects. Animal model studies revealed that upon systemic administration, Apt-NMed specifically targeted and selectively accumulated in ALCL tumor site, but did not react with off-target tumors in the same xenograft mouse. Importantly, Apt-NMed not only induced significantly higher inhibition in ALCL tumor growth, but also caused fewer or no side effects in treated mice compared to free DOX. Moreover, Apt-NMed treatment markedly improved survival rate of treated mice, opening a new avenue for precision treatment of ALCL.
Keywords: Anaplastic large T-cell lymphoma (ALCL), anaplastic lymphoma kinase (ALK), aptamer nanostructures, CD30 receptor, cell-targeted chemotherapy, nanomedicine
Graphical Abstract
INTRODUCTION
Nanomedicines offer notable advantages compared to traditional drug delivery methods including transportation of higher drug payloads, prolonged drug circulation and improved bioavailability, synergistic effects due to simultaneous administration of multiple drugs with different activities, and improved therapeutic indices through enhanced permeability, retention, and programed design.[1–3] Recent studies showed that single-stranded DNA nanostructures provide tunable, precise, rationally designed drug delivery vehicles[4–6] that can be further functionalized with judiciously chosen ligands for cell-specific and targeted therapy[7–10].
Aptamers are a class of short, single-stranded oligonucleotides (RNA or ssDNA) with high affinity and specificity for a wide range of biological targets[11–14]. Similarly to protein antibodies, aptamers bind to their cognate targets via conformational recognition and thus, are often referred to as “chemical antibodies”. As small-sized oligonucleotide biomaterials, aptamers can be rapidly synthesized and easily conjugated to various functional agents for different clinical needs[15–17]. The aforementioned features provide aptamers with notable advantages over protein antibodies as targeting ligands to formulate nanoparticles and nanomedicines.[18–21]
Anaplastic large cell lymphoma (ALCL) is the most common T-cell lymphoma in children. Biologically, lymphoma cells express high-levels of the surface CD30 receptor, a signaling molecule regulating cell fate and a distinct biomarker for ALCL diagnosis. Genetically, lymphoma cells express the Anaplastic Lymphoma Kinase (ALK) oncogene, a key pathogenic factor for lymphoma development in the majority of children. Currently, CHOP chemotherapy, comprising cyclophosphamide, hydroxyl-doxorubicin, oncovin (Vincristine) and prednisone, is the mainstay of treatment for ALK+ ALCL.[22, 23] However, the CHOP regimen is not ALCL cell-selective or ALK gene-specific, and thus, has serious adverse side effects. Recently, FDA approved CD30 antibody drug-conjugates for cell-targeted chemotherapy treatment of relapsed/refractory ALCL[24, 25]. Also, RNA interference (RNAi) approaches[26, 27] and kinase inhibitors[28, 29] to restrain expression or activity of ALK oncogene have been investigated. Moreover, stimulation of CD30 by induced polymerization led to apoptotic death of ALK+ ALCL cells in vitro and antitumor effects in animal model.[30],[31] However, treatment with single therapeutic modalities appears to be ineffective in curing ALK+ ALCL, and also carries the risk of inducing drug-resistance of lymphoma cells.
To address the aforementioned clinical challenges, we previously reported a functionalized siRNA nanocomplex, consisting of a CD30-specific aptamer and ALK gene-specific siRNA on nanosized polyethylenimine (PEI) polymer carriers.[27] Functional analysis revealed that the siRNA-aptamer nanocomplexes preferentially bound to and were internalized by ALCL cells, whereas they did not bind to off-target cells, resulting in down-regulation of cellular ALK gene expression and growth inhibition of ALCL cells. Notwithstanding the advantages of the functionalized nanocomplex for targeted ALCL therapy, its further development for clinical use is limited due to the unknown risks of PEI as a foreign material to the human body and the short lifetime of RNA-based aptamers due to susceptibility to environmental nucleases.
To overcome the shortcomings of the previous nanocomplex,[27] in this study, we developed a multifunctional aptamer-nanomedicine (Apt-NMed) for ALCL therapy, through self-assembly of functional oligonucleotides, comprising aptamers and siRNA sequences, followed by self-loading of the therapeutic drug doxorubicin (DOX). As synthetic biodegradable oligonucleotides, the DNA-based aptamers rendered Apt-NMed both biostable and biocompatible. Functional studies demonstrated that Apt-NMed achieved both ALCL cell-targeted chemotherapy and ALK gene-specific therapy. The therapeutic efficacy of Apt-NMed was validated in a preclinical model. Our studies showed that the resulting combination therapy not only achieved higher therapeutic efficacy but also had fewer non-specific toxic side effects on normal cells/tissues.
RESULTS AND DISCUSSION
Development and characterization of aptamer-nanomedicine
Apt-NMed was simply formulated through self-assembly of functional oligonucleotide sequences and self-loading of DOX (Figure 1A). We hypothesized that the Apt-NMed would specifically bind and act on ALCL cells under CD30-specific aptamer guidance. It would then induce combined cell-targeted chemotherapy- via intracellular release of its drug payload- and cell-targeted gene therapy to achieve ALK oncogene silencing via intracellular delivery of siRNA.
Figure 1. Development and characterization of aptamer-nanomedicine.

(A) Self-assembly scheme of aptamer nanostructure carrying gene specific and cell-specific entities, and the formed Apt-NS/DOX-siRNA (Apt-NMed) inducing targeted chemotherapy and gene therapy. (B) Formation of Apt-NS/siRNA was confirmed by change in the size of ssDNA oligonucleotide mixture (ssDNA #1, ssDNA #2, ssDNA #3) on 5% PAGE gel; Lane 1, markers between 50 and 1000 bp; Lane 2, DNA oligonucleotide #1; Lane 3, DNA oligonucleotide #2; Lane 4, DNA oligonucleotide #3; Lane 5, mixture of #1+#2; Lane 6, mixture of #1+#2+#3. (C) Formed Apt-NS/siRNA produced two fragments after restriction enzyme digestion with HindIII and BamHI, indicating successful formation of Apt-NS/siRNA; Lane marked with M shows markers between 50 and 1000 bp. (D) Saturation point determination of DOX loading in Apt-NS/siRNA by mixing different Apt-NS/siRNA to DOX ratios. Changes in fluorescence were used to monitor DOX intercalation into Apt-NS/siRNA; red arrow indicates saturation point of DOX loading in Apt-NS/siRNA. Size (E) and zeta-potential (F) of Apt-NS/siRNA and Apt-NMed were examined by Zeta-sizer nano-detector.
To develop Apt-NMed, an aptamer-nanostructure carrying siRNA (Apt-NS/siRNA) was initially formulated through a self-assembly process (Figure 1A). To this end, three complementary functional single-stranded DNA (ssDNA) sequences were synthesized, including CD30-specific aptamer sequences to target ALCL cells[32] and ALK-specific siRNA for oncogene silencing.[26,27] As shown in Materials and Methods, the synthetic oligonucleotide ssDNA #1 contains two separate CD30 aptamers and two complementary arm sequences; ssDNA #2 contains three complementary arm sequences for nanostructure formation; and ssDNA/siNRA #3 contains a complementary arm with an aptamer sequence at 5′-end and a chimeric siRNA sequence at 3′-end. Notably, for biosafety purposes, the complementary arm sequences were derived from human immunoglobulin sequences. For the purpose of validation, restriction sites for two endonucleases were introduced into the arm sequences of the Apt-NS/siRNA, as described in Materials and Methods. Additional nanostructures were also prepared and used in the control experiments (Figure S1A, B and C). The Apt-NS/siRNA was formed by simply mixing equimolar amounts of three synthetic complementary oligonucleotides, and self-assembly was confirmed by polyacrylamide gel electrophoresis (PAGE). Compared to the individual ssDNA oligonucleotides, which had different mobility shifts on PAGE gel, the Apt-NS/siRNA products generated a single band, indicating nanostructure formation (Figure 1B). In addition, dynamic light scattering (DLS) analysis showed that the three assembled ssDNA sequences formed the compact particle Apt-NS/siRNA with a single peak at 50 nm, whereas the parental individual ssDNA sequences had peaks ranging from 80 to 250 nm (Figure S2). For further validation, Apt-NS/siRNA was exposed to restriction endonucleases, and the resultant products were analyzed by PAGE. As shown in Figure 1C, two digested fragments were detected post-treatment with endonucleases HindIII and BamHI. The previous findings confirmed Apt-NS/siRNA formation through self-assembly of three complementary ssDNA sequences because endonucleases only cleave dsDNA nanostructures.
Subsequently, self-loading of DOX in Apt-NS/siRNA was conducted given that DOX is a key component of the chemotherapeutic regimen for ALCL. Notably, due to its chemical properties, DOX can freely intercalate into dsDNA (GC or CG sequences) via non-covalent interactions.[33, 34] Therefore, the Apt-NS/siRNA was incubated with free DOX overnight, at different molar ratios ranging from 1:1 to 1:40 (Figure 1D). Given that fluorescence of free DOX is quenched upon intercalation into dsDNA.[35,36] self-loading of DOX into Apt-NS/siRNA was quantified by detecting fluorescence of residual free DOX using a microplate reader (Eem = 590 nm). As shown in Figure 1D, the Apt-NS/siRNA fully incorporated up to 10-fold DOX payload (mol/mol). The physical properties of the formulated Apt-NS/DOX-siRNA (Apt-NMed) were then characterized. DLS assay indicated that DOX self-loading had minimal effect on Apt-NMed size because it had a peak at 59 nm (Figure 1E). In addition, the zeta-potentials of the aptamer-nanomedicine became more positive than that of Apt-NS/siRNA (0.60 vs. 0.058) (Figure 1F), indicating that Apt-NMed is more stable than aptamer nanostructure. Furthermore, atomic force microscopy (AFM) and scanning electron microscopy (SEM) studies were performed (Figures 2A, 2B), and revealed the uniform size and shape of Apt-NMed, in accordance with the findings from measurements of the physical properties.
Figure 2. Morphology and specificity of formed Apt-NMed.

Size and morphology characterization of Apt-NMed by AFM (A) and SEM (B). (C) SEM imaging clearly displaying Apt-NMed bound to ALCL cells K299, but not to control cells U937. (D) Flow cytometry analysis of specific cell binding of Apt-NS and Apt-NS/siRNA to ALCL cells.
Apt-NMed specifically targeted ALCL cells for both chemotherapy and gene therapy
To examine cell binding, cultured cells were treated with Apt-NMed and examined by SEM. The Apt-NMed specifically bound ALCL cells (K299), but did not react with control U937 cells, which do not express surface CD30 (Figure 2C). In addition, individual nanostructures were labeled with fluorochrome Cy3 reporter and incubated with cultured cells. Flow cytometry analysis revealed that Apt-NS and Apt-NS/siRNA specifically bound to K299 cells, but not to U937 cells, with similar reaction patterns to those observed with CD30 aptamer or anti-CD30 antibody (Figure 2D). However, the control nanostructure (NS) alone showed no cell binding due to lack of conjugated aptamer.
Recent studies showed that specific cell binding of aptamers resulted in endocytosis into lysosomes and micropinocytosis into cytoplasm[37, 38]. To validate the potential mechanism of intracellular delivery, the Apt-NS/siRNA was labeled with fluorochrome Cy3 and incubated with K299 cells, in which lysosomes had been pre-stained with LysoTracker Green for tracking purposes. Confocal microscopy studies revealed intracellular delivery of Apt-NS-siRNA in the cytoplasm only (red fluorescence) and co-localization with lysosomes (merged yellow signal from red and green) as shown in Figures 3A and Figure S4. To distinguish between the intracellular delivery pathways, K299 cells were pre-exposed to an endocytosis inhibitor (Dynasore). Blockage of endocytosis eliminated lysosomal delivery of Apt-NS/siRNA, but had no effect on cytoplasmic delivery. Moreover, pre-exposure of K299 cells to both endocytosis and micropinocytosis (Cytochalasin D) inhibitors completely abolished intracellular delivery of Apt-NS/siRNA, confirming presence of two different intracellular delivery pathways, namely endocytosis to cell lysosomes and micropinocytosis to cytoplasm (Figure S4).
Figure 3. Intracellular delivery of Apt-NMed.

(A) Determination of internalization pathway of Cy3-labeled-Apt-NS/siRNA by confocal fluorescence microscopy. Upon treatment with endocytosis inhibitor Dynasore, Apt-NS/siRNA (red) internalization was partially blocked in K299 cells, compared to untreated cells. Apt-NS/siRNA uptake was completely blocked in the presence of both endocytosis and micropinocytosis inhibitors. (B) Stability of Apt-NMed was determined by incubating with 100% human serum. (C) Drug release from Apt-NMed was demonstrated by incubation in PBS buffer along with DNase I or cell lysate. PBS buffer and free DOX were used as negative and positive controls, respectively. Final fluorescence intensity was measured with microplate reader at excitation (470 nm) and emission (590 nm) wavelengths. (D) Fluorescence microscopy showed rapid DOX delivery and release into target cells at indicated time points. Flow cytometry analysis further confirmed specific cell binding of Apt-NMed (E) and DOX release (F) in target cells. FAM-labeled Apt-NMed and released DOX were detected with FITC and PE channels of flow cytometer, respectively. Data shown are mean ± SEM, n=3; ***p < 0.001.
An ideal delivery system for targeted therapy should be stable under physiological conditions and also have the ability to release drug payload in cells of interest. Therefore, the Apt-NMed was incubated in human serum and its stability was evaluated by kinetically detecting released DOX given that intercalated DOX is optically silent. Fluorescence measurements with a microplate reader revealed that Apt-NMed was stable in serum and release of free DOX was not detected post incubation for 12 h (Figure 3B). Apt-NMed was also stable in fresh plasma from human blood or under different pH values ranging from 4.0 to 7.4 (Figure S3, B). To test drug release potential, Apt-NMed was treated with DNase I or fresh ALCL cell lysates as a source of cellular nucleases. Resultant degradation of Apt-NMed led to rapid release of DOX payload, reaching the maximum within 30 min (Figure 3C). To confirm intracellular drug release capacity, cultured cells were treated with Apt-NMed and examined under the fluorescence microscope. Intracellular fluorescence, derived from free DOX that was released from the degraded Apt-NMed was detected in K299 cells 15 min post treatment, and reached the maximum at 1 h (Figure 3D). For further validation, the Apt-NMed was labeled with FITC fluorescent reporter and incubated with cultured cells. Flow cytometry analysis showed that Apt-NMed specifically bound to K299 cells and reached maximal binding in 30 min, but did not react with U937 cells (Figure 3E). Quantitative flow cytometry analysis detected intracellular DOX release in K299 cells with maximal release in 2 h, but no DOX in control U937 cells (Figure 3F). Taken together, these findings indicate that Apt-NMed specifically targeted K299 cells, was internalized in nuclease-rich lysosomes via endocytosis, and thus, released drug payload exclusively within cells of interest.
On the other hand, specific cell binding of Apt-NS/siRNA also triggered cytoplasmic delivery via micropinocytosis (Figure 3A), which has been demonstrated to allow siRNA-induced gene silencing.[39,40] To test its gene silencing potential, K299 cells were treated with Apt-NS/siRNA only (without drug payload) for 48 h, and stained with anti-ALK antibody to detect cellular ALK proteins. Flow cytometry analysis revealed that treatment with Apt-NS/siRNA significantly decreased cellular ALK protein level (Figure 4A). In the control experiment, conventional lipofectamine-mediated transfection with equimolar amount of siRNA was conducted, and resulted in a mild effect on cellular ALK protein expression, compared to untreated cells. In addition, cells were also stained with acridine orange/ethidium bromide (AO/EB) to highlight apoptotic and dead cells, and were examined by flow cytometry. As shown in Figure 4B, treatment with Apt-NS/siRNA resulted in a significantly higher (%) increase in dead and apoptotic cells than lipofectamine-mediated transfection, but had no effect on control U937 cells.
Figure 4. Specific effects of Apt-NMed on ALCL cells for chemotherapy and gene therapy in vitro.

(A) ALK gene silencing by Apt-NS/siRNA. Cultured Karpas 299 cells were treated with Apt-NS/siRNA for 48 h, followed by staining with anti-ALK antibody. Reduction of ALK expression was analyzed by flow cytometry. (B) Apt-NS/siRNA induced significant silencing of ALK gene, leading to cell apoptosis and death. (C) Fluorescence microscopy imaging of induced-apoptotic and dead cells, detected by AO/EB staining at 24 h post treatment with Apt-NS/DOX. Viable and dead cells were stained green and red, respectively. (D) Rates (%) of apoptotic and dead cells detected by AO/EB staining, at 48 h post treatment. Data shown are Mean ± SEM, n=3; ** p < 0.01, ***p < 0.001, NS, no statistically significant difference.
To evaluate therapeutic potential, cultured cells were treated with Apt-NS/DOX, carrying DOX payload at final concentration 0.25 μM, for 24 h. Cells were then collected and stained with AO/EB to highlight viable (green) and dead (red) cells. Fluorescence microscopy studies revealed that treatment with Apt-NS/DOX induced a significantly higher death rate of K299 cells than the control groups treated with equimolar amount of free DOX or without treatment (Figure 4C). Importantly, the Apt-NS/DOX had minimal effect on control U937 cells, although they showed similar sensitivity to free DOX treatment, which suggests fewer off-target effects.
To validate potential combination therapeutic effects and rule out non-specific toxicity, cultured cells were exposed to different treatments, as illustrated in Figure 4D. In particular, the cells were treated with the nanostructure core (NS), nanostructure carrying DOX only (NS/DOX), nanostructure carrying siRNA only (NS/siRNA), nanostructure carrying both DOX and siRNA (NS/DOX-siRNA), Apt-NS, Apt-NS/DOX, Apt-NS/siRNA, Apt-NMed, and an equimolar amount of free DOX (0.25 μM final concentration). Cells were collected 48 h post-treatment and stained with AO/EB to highlight apoptotic and dead cells. Flow cytometry analysis revealed that treatment with free DOX exhibited similar toxicity to both K299 and U937 cells, while treatment with NS/DOX showed no toxic effect to the cells due to lack of free DOX in the cultures (Figure 4D). However, treatment with Apt-NS/DOX specifically induced death of K299 cells (from 6.5% at baseline to 21% death rate), but had no toxicity against control U937 cells. More importantly, treatment with Apt-NMed further increased the death rate of K299 cells (up to 28%), thus showing the combined effects of targeted chemotherapy, via released DOX, and gene therapy through ALK-specific siRNA on ALK+ ALCL cells only.
Apt-NMed treatment inhibited ALCL tumor growth and improved mouse survival
For therapeutic study of ALCL, mouse model bearing ALCL and CD30 (-) control tumors, derived from K299 cells and MDA-MB-231 breast cancer cells, respectively, was established. Development of both xenograft tumors in the same mouse was monitored by bioluminescence imaging (Figure 5A), given that tumor cells stably expressed luciferase. To validate targeted delivery, Apt-NS/siRNA was conjugated to the near-infrared fluorescent reporter IRD800CW (for tracking purposes), and systemically administrated through the tail veins of tumor-bearing mice (6.7 μg/mouse). Whole body optical imaging was performed using the IVIS 200 Imaging System immediately post administration. In comparison to tumor signals recorded by bioluminescence, Apt-NS/siRNA selectively highlighted ALCL tumor, but did not react with control tumor in the same mouse (Figure 5A). Quantitative imaging analysis revealed that imaging signal at ALCL tumor site was 5-fold higher than that detected in control tumor and 8-fold higher than body background. For confirmation, tumor tissues were removed from the mice, and re-scanned ex vivo (Figure 5B). As expected, in contrast to breast cancer tumor, ALCL tumor fluoresced due to Apt-NS/siRNA, but both tumors emitted bioluminescent radiation. In addition, fresh tumor cells were collected and examined under fluorescence microscope. The ALCL tumor cells (K299) were specifically highlighted by Apt-NS/siRNA, but no signal was detected in breast cancer tumor cells MDA-MB-231 (Figure S5). Histological and immunohistochemical studies of tumor tissues demonstrated expression of CD30 in ALCL tumors (Figure 5C). To evaluate biostability, kinetic imaging was conducted at different time points, post-systemic administration of Apt-NS/siRNA. As demonstrated in Figure 5D, selective accumulation of Apt-NS/siRNA in ALCL tumor was detected immediately post administration; the signal lasted up to 8 h, indicating the capacity of Apt-NS/siRNA for drug delivery and therapeutic use.
Figure 5. In vivo targeted delivery of Apt-NS/siRNA into xenograft tumors in mouse model.

(A) Specific delivery of IRD800 labeled-Apt-NS/siRNA to ALCL xenograft tumors in mouse model detected by IVIS 200 imaging. (B) Tumors tissues were removed from mouse, and re-scanned ex vivo. (C) Tissue immunostaining with CD30 antibody to confirm that IRD800-Apt-NS/siRNA was delivered into tumors expressing CD30 antibody (magnification x400). (D) Whole body imaging of IRD800-Apt-NS/siRNA versus time to determine particle retention in tumor area.
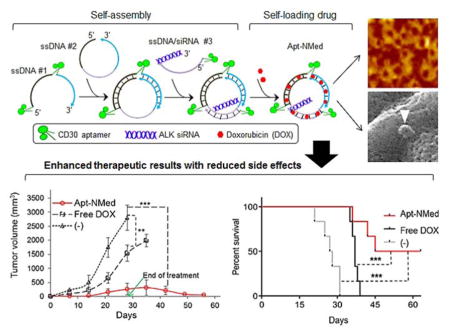
To study the therapeutic potential, xenograft mice were generated, and randomly assigned to receive different treatments (6 mice/group). As illustrated in Figure 6A, mice were systemically administered Apt-NMed (0.24 mg/mouse carrying 20 μg of DOX payload), equimolar amount of free DOX (20 μg/mouse), or vehicle alone as an untreated control (-), three times per week for a total 12 doses. Tumor growth was monitored by whole body bioluminescence, and tumor cell signals were recorded every 7 days (Figure 6B). Changes in tumor volume were then calculated and graphed. As depicted in Figure 6C, at the end point of treatment (day 28), the average tumor volume in the Apt-NMed-treated group was 10-fold lower than the untreated control group (-) and 6-fold lower than free DOX-treated group, findings which corroborate the high therapeutic efficacy of Apt-NMed.
Figure 6. Antitumor efficacy of Apt-NMed in vivo.

(A) Scheme of xenograft tumor treatment: initial treatment took place 9 days post tumor cell inoculation; serial treatment followed by IV administration every other day for four weeks (DOX: 1 mg/kg). (B) Bioluminescence imaging of tumor growth in treated and untreated groups (6 mice/group) during Apt-NMed administration. (C) Graph showing relative tumor volume change in treated and untreated groups (6 mice/group) post treatment. (D) Body weight change of tumor-bearing mice during treatment. (E) Kaplan-Meier survival curve of mice bearing ALCL tumors. Mice died naturally or were executed when tumor volume exceeded 2,000 mm3. (F) Tumor-free percentage of mice (16.7%) post-treatment. Tumor recurrence at 80 days after stopping treatment. Data shown are mean ± SEM, n=6; * p < 0.05, ***p < 0.001.
To evaluate non-specific toxic effects of treatment, the body weights of the mice were measured every 7 days and graphed. As shown in Figure 6D, treatment with Apt-NMed resulted in insignificant changes in mice body weight, suggesting minimal toxicity in mice. In contrast, treatment with free DOX induced a considerable decrease in mouse body weight with 20% loss at day 35, consistent with severe side effects of DOX. Notably, the body weight of untreated mice increased due to rapid growth of tumor masses (Figure 6D). To study survival rate, mice were observed for 65 days post first dose treatment (Figure 6E). All the mice died in 31 days in the untreated group. In contrast, treatment with free DOX slightly prolonged survival of tumor-bearing mice, and the last mouse died at day 38. Interestingly, treatment with Apt-NMed significantly improved mouse survival, as shown by the 50% survival rate at 65 days. More importantly, the tumor masses in surviving mice had regressed after completion of treatment, and these mice were tumor-free at the end point. After treatment was stopped, the tumors slowly recurred (Figures 6C and 6F).
CONCLUSIONS
In this study, we demonstrated that Apt-NMed, which can be simply formulated through self-assembly of functional oligonucleotides containing and self-loading of the chemotherapeutic drug DOX, successfully performed selective chemotherapy and gene therapy of ALCL. Under CD30-specific aptamer guidance, the multifunctional Apt-NMed specifically bound ALCL cells and performed targeted chemotherapy through intracellular delivery of DOX and specific oncogene silencing via intracellular delivery of ALK-specific siRNA. The combination of cell-selective chemotherapy and gene therapy of Apt-NMed achieved higher growth inhibition rates of ALCL tumors and significantly higher survival rates of xenograft mice as compared to single agent therapy. Importantly, treatment with Apt-NMed showed negligible toxicity in mice in contrast to treatment with an equimolar amount of free DOX, which caused severe toxicity in mice.
Notably, as a synthetic biomaterial, Apt-NMed is completely biocompatible and biodegradable, and will not cause adverse effects in the human body. The majority of materials previously conjugated to aptamers, such as inorganic and organic carriers,[41] including gold or magnetic nanomaterials,[42–45] single-walled carbon nanotubes,[46–48] mesoporous silica nanoparticles,[49–50] quantum dots, liposomes, copolymers, and protein-based nanomaterials[51–56] are foreign to the human body and may pose unknown risks for in vivo use. It is noteworthy that Apt-NMed can be synthesized by a simple method, which is highly reproducible, cost-effective, and can be easily scaled up for industrial production of this multifunctional nanomedicine. Apt-NMed can also be developed to logically combine multiple therapeutic modalities for additional or synergistic efficacy, including chemotherapy, gene therapy,[55,57–60] immunotherapy, biotherapy, kinase inhibitors, thermotherapy,[43,61] and photodynamic therapy.[44–45] The design of Apt-NMed is contingent upon several factors aimed at precision therapy, such as the nature of the disease, biology of cells/tissues of interest, chemical and physical properties of aptamers, selection of appropriate nanomaterials, and appropriate therapeutic agents/drugs. The aforementioned findings provide a solid foundation for development of new precision nanomedicines that can selectively treat ALCL and other diseases with high efficacy and minimal or no adverse effects in patients. Further preclinical and clinical studies are necessary to translate Apt-NMed from bench to bedside.
MATERIALS AND METHODS
Reagents and cell lines
Karpas299 (K299) cells expressing receptor CD30 were used at the National Cancer Institute/National Institutes of Health, in collaboration with Dr. Mark Raffeld. CD30-negative U937 and MDA-MB-231 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA). K299 and MDA-MB231 cells, stably expressing GFP and luciferase, were used for tumor formation in mouse model. Suspension cells K299 and U937 were cultured with RPMI1640 medium (Fisher Scientific, Pittsburgh, PA) with 10% FBS (Atlanta Biologicals, Lawrenceville, GA). Adhesion cells MDA-MB-231 were cultured in DMEM (Atlanta Biologicals, Lawrenceville, GA) with 10% FBS. The chemotherapeutic drug DOX was purchased from Sigma (St. Louis, MO).
Synthetic oligonucleotides and formation of aptamer nanostructure
To generate the aptamer nanostructure linked with siRNA (Apt-NS/siRNA), three single-stranded oligonucleotides containing aptamer, complementary ssDNA and/or siRNA sequences were synthesized. To ensure biosafety, synthetic complementary sequences were adapted from human IgG heavy chain (NCBI database: GenBank: X82593.1). To target cells of interest, ssDNA CD30-specific aptamer was employed as previously reported[32]. For oncogene silencing, the previously validated ALK-specific siRNA sequence[27] was directly linked to the aptamer. To validate aptamer-nanostructure formation, two endonuclease restriction sites, namely HindIII and BamHI (underlined nucleotides in sequences shown below), were introduced into the arm sequences of ssDNA. All oligonucleotides were synthesized by Integrated DNA Technologies (IDT, Coralville, IA). The scheme for self-assembly of the aptamer nanostructure is depicted in Figure 1A. In addition, Apt-NS without siRNA, and control NS or NS/siRNA without aptamer were also synthesized (Figure S1). The ssDNA sequence for aptamer nanostructure formation is displayed below:

Apt-NS/siRNA was formed by self-assembly of three single-stranded oligonucleotides via programed hybridization of complementary ssDNA sequences. Briefly, synthetic oligonucleotides were mixed in equal molar ratio (1:1:1), each at 10 μM final concentration, in TRIS EDTA buffer (Sigma Aldrich, St. Louis, MO) supplemented with 10 mM MgCl2 (Fisher Scientific, Pittsburgh, PA). Reactions were heated at 95°C for 5 min and then cooled to 4°C for programed hybridization of oligonucleotides to form Apt-NS/siRNA.
Characterization of Apt-NS/siRNA formation by native PAGE
First, the formed Apt-NS/siRNA (1μg) was separated on 5% non-denaturing PAGE, and analyzed on a Gel Doc EZ imager (Bio-Rad, Hercules, CA). The individual oligonucleotides (ssDNA #1, ssDNA #2, and ssDNA/siRNA #3), and a mixture of oligonucleotides (ssDNA #1+ssDNA #2) were treated under the same conditions as the controls (Figure 1B). For further confirmation, the annealed aptamer nanostructure (1 μg) was digested with the restriction endonucleases HindIII and/or BamHI for at least 2 h, in an incubator at 37°C, and analyzed by PAGE (Figure 1C).
Drug loading into Apt-NS/siRNA
DOX intercalation can be measured by monitoring changes in DOX fluorescence because DOX fluorescence is dramatically quenched upon intercalation into DNA. Briefly, to determine the loading ability of Apt-NS/siRNA for DOX, Apt-NS/siRNA and DOX were mixed at specific molar ratios (from 1:1 to 1:40), and kept overnight at room temperature to allow saturation of drug loading (DOX was fixed to 0.1 nmol). Fluorescence intensity was measured by microplate reader (Eex = 470 nm, Eem = 590 nm, Biotek, Winooski, VT). Saturation of DOX loading was determined based on fluorescence intensity of free DOX.
Dynamic light scattering analysis of Apt-NS/siRNA and Apt-NMed
The size and zeta-potential of the formed Apt-NS/siRNA and drug-loaded Apt-NS/siRNA (10 nM) were characterized with a Zetasizer Nano ZS ZEN3600 (Malvern Instruments, Worcestershire, UK); the scattering angle was 173°. Three sets of measurements, each comprising 15 runs with a 20 s duration for each, were performed at 25 °C. For the calculations, the viscosity η = 0.8872 mPa.s and refractive index = 1.33 were used. The autocorrelation function was evaluated by ALV-Correlation software v 3.0, using an exponential regularized fit to get the size distribution curves and Zetasizer Software 6.34 to get the z-average and PDI values.
Characterization of Apt-NMed
Samples were studied by AFM and SEM to determine the size and morphology of freshly made Apt-NMed. For AFM imaging, prior to sample loading, the sample holder mica was pre-treated with 0.1 % (v/v) APTES (3-aminopropyltriethoxy-silane) water solution for 10 min, washed with pure water, and then dried with compressed nitrogen. The formed nanostructure Apt-NMed was diluted to 10 nM in Tris buffer supplemented with 10 mM MgCl2 in 10 μl; this solution was placed on a previously modified surface, absorbed for 5 min, washed with pure water, and dried with compressed nitrogen for imaging with MultiMode M8 AFM (Bruker, Spring, TX). For high-resolution images, an SNL-10 probe (Veeco Inc., USA), with super-sharp tips (2–3 nm radius), was used. For SEM imaging, a drop of solution (5 μl, 10 nM) was spotted onto silicon pre-treated with aminopropylsilane (APS) for 5 min to allow strong adsorption. The samples were air-dried and carefully mounted on an aluminum stub using either silver paint or a double stick carbon tape. Samples were then placed in the chamber of the sputter coater, and coated with a very thin film of gold. All SEM images were taken at 10–15 kV (accelerating voltage) with electron-beam spot size 3, in the FEI Nova NanoSEM 230 (Hillsboro, OR). Chamber pressure was between 1.0 × 10−7 and 1.0 × 10−6 mbar (1 mbar = 100 Pa).
Specific cell binding of Apt-NMed to ALCL cells analyzed by SEM
Briefly, the treated cells were fixed with 2.5% glutaraldehyde at 4°C for 2 h, followed by sequential dehydration for 10 min in 20, 30, 50, 70, 90, and 100% ethanol. The fixed cells were sputter-coated with PtPd by Cressington 208 HR Sputter Coater (Cressington Scientific, Cranberry Twp, PA), and examined by SEM (Nova NanoSEM 230, FEI Co., Hillsboro, USA).
Specific cell binding of Apt-NS and Apt-NS/siRNA to ALCL cells analyzed by flow cytometry
Cy3-labeled aptamer nanostructure (Apt-NS), control Cy3-labeled control nanostructure (NS), Cy3-directly labeled CD30 aptamer (Cy3-Apt) and CD30 antibody were incubated with 1× 105 K299 and U937 cells at 100 nM for 30 min at room temperature (RT). The cells were washed once with 500 μl PBS, and re-suspended in 500 μl PBS for flow cytometry analysis. Fluorescence intensity was determined with a FACScan cytometer (LSRII, BD Biosciences, San Jose, CA) by counting 10,000 events.
Internalization pathway of Apt-NS/siRNA
To examine internalization pathway of Apt-NS/siRNA, K299 cells were incubated with both Cy3-labeled Apt-NS/siRNA (100 nM) and endocytosis inhibitor Dynasore (80 μM) or Cy3-labeled Apt-NS/siRNA along with endocytosis inhibitor Dynasore (80 μM) and micropinocytosis inhibitor Cytochalasin D (1 μM) for 2h at 37°C; Cy3-labeled Apt-NS/siRNA (100 nM) alone was used as control. Post-PBS buffer washing, the fixed cells were visualized by confocal fluorescence microscopy (Olympus, Fluo ViewTM 1000).
Stability of Apt-NMed
To detect stability of nanostructure/DOX conjugates, Apt-NMed (100 nM) was incubated at 37°C in 100 μl of 100% human serum (Atlanta Biological, Lawrenceville, GA, USA), At indicated time in Figure 3B, fluorescence intensity of the released DOX was detected by microplate reader, at the excitation and emission wavelengths 480 ± 20 and 570 ± 20 nm, respectively (Biotek, Winooski, VT).
Drug release from Apt-NMed by DNA degradation
The formed Apt-NMed (100 nM, ratio of nanostructure to DOX 1:10) was incubated at 37°C in 100 μl of PBS buffer with 2 units of DNase I (Life technologies, Grand Island, NY), or cell lysate from K299 cells for 2 h. Fluorescence from the released DOX was detected at 590 nm after excitation at 480 nm (Biotek, Winooski, VT). Cell lysate and PBS buffer alone were considered negative controls. The same amounts of free DOX were considered as controls for 100% drug release from Apt-NMed.
Intracellular delivery of Apt-NMed observed by fluorescence microscopy and flow cytometry
Apt-NMed (100 nM) was incubated with K299 and U937 cells at 37°C. Fluorescent signal from the released DOX was recorded by a fluorescence microscope (Olympus FluoViewTM 1000, Olympus America, and Center Valley, PA) at different time points (Figure 3D). Cell-selective intracellular delivery and drug release from Apt-NMed were also determined by flow cytometry (BD Biosciences, San Jose, CA). Briefly, DOX was loaded into FAM-labeled Apt-NMed to treat K299 and U937 cells. As illustrated in Figure 3D, cells were collected, and analyzed by flow cytometry at different time points. Cell binding to Apt-NMed and released DOX were detected using FITC and PE channels, respectively. The efficiency of particle delivery and drug release was determined by quantification of changes in fluorescence.
Silencing of ALK gene by Apt-NS/siRNA
To address of silencing of ALK expression by Apt-NS/siRNA, cultured Karpas 299 cells were treated with 100 nM Apt-NS containing ALK-targeted siRNA, or 100 nM siRNA for 4 h at 37°C. The previous solution was removed; cells were treated with fresh medium (10% FBS), and cultured for 24 h. Cells were collected, and stained with FITC-labeled mouse anti-human ALK antibody (BD Biosciences, San Jose, CA), and analyzed by flow cytometry to determine silencing of ALK (BD Biosciences, San Jose, CA). To determine the effect of ALK gene knockdown on cell growth and the rates of apoptotic (very weak green) and dead cells (bright red) in the treated and control groups, the collected cells were stained with AO (5μg/ml)/EB (3μg/ml), and analyzed by flow cytometry.
Demonstration of specific and enhanced effect of Apt-NS/DOX on target cells by fluorescence microscopy
25 nM Apt-NS/DOX (DOX 0.25 μM) and free DOX (0.25 μM) were incubated with K299 (CD30+) cells and U937 (CD30-) at RT for 2–4 h, respectively. The treatment solution was removed, and complete fresh medium was added to continue culturing for 24 h. The treated cells were stained with AO (5 μg/ml)/ EB (3 μg/ml), according to the manufacturer’s instructions. After washing, slides of cell smears were prepared and examined under a fluorescence microscope (Olympus FluoViewTM 1000, Olympus America, Center Valley, PA). Viable and dead cells were stained green and red, respectively.
Combined effect of Apt-NMed on ALCL cells
To determine the combined effect of Apt-NMed, K299 cells were treated with aptamer nanostructures including Apt-NS, Apt-NS/siRNA, Apt-NS/DOX, Apt-NMed, and control nanostructures including NS, NS/siRNA, NS/DOX, NS/DOX-siRNA, in RPMI 1640 medium for 2 h. After replacement with fresh medium containing 10% serum, cells were cultured for an additional 48 h, stained with AO (5 μg/ml)/ EB (3 μg/ml) for 5 min at RT, and analyzed by flow cytometry to determine the apoptosis (very weak green) and death (bright red) rates in the treated and control groups.
In vivo targeted delivery of Apt-NS/siRNA into xenograft tumors in mouse model
For imaging studies, 4–6-weeks old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NOD SCID) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Each mouse was subcutaneously inoculated with cultured CD30-expressing lymphoma cells (Karpas 299, 6×106) and CD30-negative control tumor cells (MDA-MB-231, 2×106) at the same time, but at different anatomic sites, as shown in Figure 5A. Both cells stably expressed Green fluorescence protein (GFP) and luciferase. Tumor development was monitored by bioluminescence scanning, and confirmed by physical examination until the tumor reached a diameter ≥5 mm (approximately 20–25 days post tumor cell implantation). Apt-NS/siRNA labeled with reporter IRD800CW (6.7 μg in 100 μl PBS buffer) was administered systemically via tail vein injection, and whole body imaging using the IVIS 200 Imaging System was performed immediately and up to 6 h after injection, as shown in Figure 5D. Imaging signals of regions of interest (ROIs) from lymphoma tumors, control tumors and body areas without tumor were recorded in digital format, using Xenogen software. The increase in signal intensity was calculated according to the manufacturer’s instructions as follows:
To confirm imaging findings, tumors and adjacent tissues were removed at the end-point and re-scanned as previously described. Cells extracted from tumor tissues were observed by fluorescence microscopy. In addition, tumor tissues were fixed and immunostained for CD30 expression in our pathology laboratory, following a standard protocol.
Anticancer efficacy of Apt-NMed in vivo
Mice NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NOD SCID), 4–6-weeks old, were used to develop xenograft tumor models by subcutaneously injecting 6 × 106 cultured CD30-expressing lymphoma cells (Karpas 299) (in 100 μl of PBS buffer) into mice on the back. Dorsal tumor nodules were allowed to grow to a volume of 4 mm3 before treatment initiation. Tumor-bearing mice were randomly assigned to three groups (with six mice in each group) as follows: (i) group administered with 0.25 mg Apt-NMed carrying 20 μg DOX to each mouse; (ii) group administered 20 μg free DOX to each mouse, and (iii) untreated group. Drugs were injected through tail veins every other day, and the treatment was continued for four weeks. Tumor length and width were measured with calipers weekly for each mouse. Tumor volume was calculated using the following equation: Tumor volume = Length * Width2 * 0.52. The body weight of each mouse was also measured weekly to monitor potential drug toxicity. Mice were euthanized when tumor volume exceeded 2,000 mm3.
Supplementary Material
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Tigli Aydin RS. Mini Rev Med Chem. 2015;14:1048. doi: 10.2174/1389557514666140820115052. [DOI] [PubMed] [Google Scholar]
- 2.Auffan M, Rose J, Bottero JY, Lowry GV, Jolivet JP, Wiesner MR. Nat Nanotechnol. 2009;4:634. doi: 10.1038/nnano.2009.242. [DOI] [PubMed] [Google Scholar]
- 3.Toy R, Bauer L, Hoimes C, Ghaghada KB, Karathanasis E. Adv Drug Delivery Rev. 2014;76:79. doi: 10.1016/j.addr.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen YJ, Groves B, Muscat RA, Seelig G. Nat Nanotechnol. 2015;10:748. doi: 10.1038/nnano.2015.195. [DOI] [PubMed] [Google Scholar]
- 5.Angell C, Xie S, Zhang L, Chen Y. Small. 2016;12:1117. doi: 10.1002/smll.201502167. [DOI] [PubMed] [Google Scholar]
- 6.Jiang D, England CG, Cai W. J Controlled Release. 2016;27:239. doi: 10.1016/j.jconrel.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee H, Lytton-Jean AK, Chen Y, Love KT, Park AI, Karagiannis ED, Sehgal A, Querbes W, Zurenko CS, Jayaraman M, Peng CG, Charisse K, Borodovsky A, Manoharan M, Donahoe JS, Truelove J, Nahrendorf M, Langer R, Anderson DG. Nat Nanotechnol. 2012;7:389. doi: 10.1038/nnano.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J, Wei T, Zhao J, Huang Y, Deng H, Kumar A, Wang C, Liang Z, Ma X, Liang XJ. Biomaterials. 2016;91:44. doi: 10.1016/j.biomaterials.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 9.Zhu J, Huang H, Dong S, Ge L, Zhang Y. Theranostics. 2014;4:931. doi: 10.7150/thno.9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu G, Zheng J, Song E, Donovan M, Zhang K, Liu C, Tan W. Proc Natl Acad Sci USA. 2013;110:7998. doi: 10.1073/pnas.1220817110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banerjee J, Nilsen-Hamilton M. J Mol Med. 2013;91:1333. doi: 10.1007/s00109-013-1085-2. [DOI] [PubMed] [Google Scholar]
- 12.Wandtke T, Wozniak J, Kopinski P. Viruses. 2015;7:751. doi: 10.3390/v7020751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimbres FM, Tarnok A, Ulrich H, Wrenger C. Biomed Res Int. 2013;2013:731516. doi: 10.1155/2013/731516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng B, Lin Y, Wang C, Li F, Wang Z, Zhang H, Li XF, Le XC. Anal Chim Acta. 2014;837:1. doi: 10.1016/j.aca.2014.04.055. [DOI] [PubMed] [Google Scholar]
- 15.Jo H, Ban C. Exp Mol Med. 2016;48:e230. doi: 10.1038/emm.2016.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen K, Liu B, Yu B, Zhong W, Lu Y, Zhang J, Liao J, Liu J, Pu Y, Qiu L, Zhang L, Liu H, Tan W. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;9:e1438. doi: 10.1002/wnan.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gopinath SC, Lakshmipriya T, Chen Y, Arshad MK, Kerishnan JP, Ruslinda AR, Al-Douri Y, Voon CH, Hashim U. Appl Microbiol Biotechnol. 2016;100:6955. doi: 10.1007/s00253-016-7686-2. [DOI] [PubMed] [Google Scholar]
- 18.Sun H, Tan W, Zu Y. Analyst. 2016;141:403. doi: 10.1039/c5an01995h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKeague M, Derosa MC. J Nucleic Acids. 2012;2012:748913. doi: 10.1155/2012/748913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman AD, Claypool SE, Liu R. Curr Pharm Des. 2013;19:6315. doi: 10.2174/13816128113199990375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy-Nissenbaum E, Radovic-Moreno AF, Wang AZ, Langer R, Farokhzad OC. Trends Biotechnol. 2008;26:442. doi: 10.1016/j.tibtech.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 22.Ma H, Abdul-Hay M. Int J Clin Oncol. 2017;22:18. doi: 10.1007/s10147-016-1045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher RI, Gaynor ER, Dahlberg S, Oken MM, Grogan TM, Mize EM, Glick JH, Coltman CA, Jr, Miller TP. N Engl J Med. 1993;328:1002. doi: 10.1056/NEJM199304083281404. [DOI] [PubMed] [Google Scholar]
- 24.de Goeij BE, Lambert JM. Curr Opin Immunol. 2016;40:14. doi: 10.1016/j.coi.2016.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Diefenbach CS, Leonard JP. American Society of Clinical Oncology Educational Book American Society of Clinical Oncology Meeting. 2012:162. doi: 10.14694/EdBook_AM.2012.32.83. [DOI] [PubMed] [Google Scholar]
- 26.Hsu FY, Zhao Y, Anderson WF, Johnston PB. Cancer Invest. 2007;25:240. doi: 10.1080/07357900701206372. [DOI] [PubMed] [Google Scholar]
- 27.Zhao N, Bagaria HG, Wong MS, Zu Y. J Nanobiotechnol. 2011;9:2. doi: 10.1186/1477-3155-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turturro F, Arnold MD, Frist AY, Pulford K. Clin Cancer Res. 2002;8:240. [PubMed] [Google Scholar]
- 29.Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, Xia G, Steensma R, Chopiuk G, Jiang J, Wan Y, Ding P, Liu Y, Sun F, Schultz PG, Gray NS, Warmuth M. Proc Natl Acad Sci USA. 2007;104:270. doi: 10.1073/pnas.0609412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schirrmann T, Steinwand M, Wezler X, Ten Haaf A, Tur MK, Barth S. BioDrugs. 2014;28:181. doi: 10.1007/s40259-013-0068-8. [DOI] [PubMed] [Google Scholar]
- 31.Pierce JM, Mehta A. Expert Rev Hematol. 2017;10:29. doi: 10.1080/17474086.2017.1270202. [DOI] [PubMed] [Google Scholar]
- 32.Parekh P, Kamble S, Zhao N, Zeng Z, Portier BP, Zu Y. Biomaterials. 2013;34:8909. doi: 10.1016/j.biomaterials.2013.07.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porciani D, Tedeschi L, Marchetti L, Citti L, Piazza V, Beltram F, Signore G. Mol Ther Nucleic Acids. 2015;4:e235. doi: 10.1038/mtna.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez-Arnaiz C, Busto N, Leal JM, Garcia B. J Phys Chem B. 2014;118:1288. doi: 10.1021/jp411429g. [DOI] [PubMed] [Google Scholar]
- 35.Agudelo D, Bourassa P, Berube G, Tajmir-Riahi HA. Int J Biol Macromol. 2014;66:144. doi: 10.1016/j.ijbiomac.2014.02.028. [DOI] [PubMed] [Google Scholar]
- 36.Zunino F, Di Marco A, Zaccara A, Gambetta RA. Biochim Biophys Acta. 1980;607:206. doi: 10.1016/0005-2787(80)90073-8. [DOI] [PubMed] [Google Scholar]
- 37.Zhou J, Rossi JJ. Oligonucleotides. 2011;21:1. doi: 10.1089/oli.2010.0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyer C, Hahn U, Rentmeister A. J Nucleic Acids. 2011;2011:904750. doi: 10.4061/2011/904750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hou KK, Pan H, Ratner L, Schlesinger PH, Wickline SA. ACS Nano. 2013;7:8605. doi: 10.1021/nn403311c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reyes-Reyes EM, Teng Y, Bates PJ. Cancer Res. 2010;70:8617. doi: 10.1158/0008-5472.CAN-10-0920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun H, Zu Y. Small. 2015;20:2352. doi: 10.1002/smll.201403073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao N, You J, Zeng Z, Li C, Zu Y. Small. 2013;9:3477. doi: 10.1002/smll.201202694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pala K, Serwotka A, Jelen F, Jakimowicz P, Otlewski J. Int J Nanomedicine. 2014;9:67. doi: 10.2147/IJN.S52539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, You M, Zhu G, Shukoor MI, Chen Z, Zhao Z, Altma MB, Yuan Q, Zhu Z, Chen Y, Huang CZ, Tan W. Small. 2013;9:3678. doi: 10.1002/smll.201202155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shiao YS, Chiu HH, Wu PH, Huang YF. ACS Appl Mater Interfaces. 2014;6:21832. doi: 10.1021/am5026243. [DOI] [PubMed] [Google Scholar]
- 46.Taghdisi SM, Lavaee P, Ramezani M, Abnous K. Eur J Pharm Biopharm. 2011;77:200. doi: 10.1016/j.ejpb.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 47.De Puig H, Cifuentes Rius A, Flemister D, Baxamusa SH, Hamad-Schifferli K. PLoS One. 2013;8:e68511. doi: 10.1371/journal.pone.0068511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu Z, Tang Z, Phillips JA, Yang R, Wang H, Tan W. J Am Chem Soc. 2008;130:10856. doi: 10.1021/ja802913f. [DOI] [PubMed] [Google Scholar]
- 49.He X, Zhao Y, He D, Wang K, Xu F, Tang J. Langmuir. 2012;28:12909. doi: 10.1021/la302767b. [DOI] [PubMed] [Google Scholar]
- 50.Zhu CL, Lu CH, Song XY, Yang HH, Wang XR. J Am Chem Soc. 2011;133:1278. doi: 10.1021/ja110094g. [DOI] [PubMed] [Google Scholar]
- 51.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Proc Natl Acad Sci USA. 2006;103:6315. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xing H, Tang L, Yang X, Hwang K, Wang W, Yin Q, Wong NY, Dobrucki LW, Yasui N, Katzenellenbogen JA, Helferich WG, Cheng J, Lu Y. J Mater Chem B Mater Biol Med. 2013;1:5288. doi: 10.1039/C3TB20412J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang L, Radovic-Moreno AF, Alexis F, Gu FX, Basto PA, Bagalkot V, Jon S, Langer RS, Farokhzad OC. ChemMedChem. 2007;2:1268. doi: 10.1002/cmdc.200700121. [DOI] [PubMed] [Google Scholar]
- 54.Huang F, You M, Chen T, Zhu G, Liang H, Tan W. Chem Commun. 2014;50:3103. doi: 10.1039/c3cc49003c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li L, Hou J, Liu X, Guo Y, Wu Y, Zhang L, Yang Z. Biomaterials. 2014;35:3840. doi: 10.1016/j.biomaterials.2014.01.019. [DOI] [PubMed] [Google Scholar]
- 56.Wu J, Song C, Jiang C, Shen X, Qiao Q, Hu Y. Mol Pharm. 2013;10:3555. doi: 10.1021/mp300686g. [DOI] [PubMed] [Google Scholar]
- 57.Guo S, Tschammer N, Mohammed S, Guo P. Hum Gene Ther. 2005;16:1097. doi: 10.1089/hum.2005.16.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shu D, Shu Y, Haque F, Abdelmawla S, Guo P. Nat Nanotechnol. 2011;6:658. doi: 10.1038/nnano.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haque F, Shu D, Shu Y, Shlyakhtenko LS, Rychahou PG, Evers BM, Guo P. Nano Today. 201;7:245. doi: 10.1016/j.nantod.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou J, Shu Y, Guo P, Smith DD, Rossi JJ. Methods. 2011;54:284. doi: 10.1016/j.ymeth.2010.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J, Sefah K, Altman MB, Chen T, You M, Zhao Z, Huang CZ, Tan W. Chem-Asian J. 2013;8:2417. doi: 10.1002/asia.201300375. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.