

Abstract
Increasing evidence shows that diabetes causes cardiac dysfunction. We hypothesized that a glucagon-like peptide-1 (GLP-1) analog, liraglutide, would attenuate cardiac dysfunction in diabetic rats. A total of 24 Sprague–Dawley (SD) rats were divided into two groups fed either a normal diet (normal, n=6) or a high-fat diet (HFD, n=18) for 4 weeks. Then, the HFD rats were injected with streptozotocin (STZ) to create a diabetic rat model. Diabetic rats were divided into three subgroups receiving vehicle (diabetic, n=6), a low dose of liraglutide (Llirag, 0.2 mg/kg/day, n=6), or a high dose of liraglutide (Hlirag, 0.4 mg/kg/day, n=6). Metabolic parameters, systolic blood pressure (SBP), heart rate (HR), left ventricular (LV) function, and whole genome expression of the heart were determined. Diabetic rats developed insulin resistance, increased blood lipid levels and oxidative stress, and impaired LV function, serum adiponectin, nitric oxide (NO). Liraglutide improved insulin resistance, serum adiponectin, NO, HR, and LV function and reduced blood triglyceride (TG), total cholesterol (TC) levels, and oxidative stress. Moreover, liraglutide increased heart nuclear receptor subfamily 1, group H, member 3 (Nr1h3), peroxisome proliferator activated receptor (Ppar) α (Pparα), and Srebp expression and reduced diacylglycerol O-acyltransferase 1 (Dgat) and angiopoietin-like 3 (Angptl3) expression. Liraglutide prevented cardiac dysfunction by activating the PPARα pathway to inhibit Dgat expression and oxidative stress in diabetic rats.
Keywords: cardiac function, Diabetes, glucagon-like peptide-1 analogue, PPAR
Introduction
Diabetes is a worldwide public health problem that has prevalence greater than 5.71% in adults [1]. Chronic hyperglycemia leads to a high risk of cardiovascular events [2]. Cardiovascular disease is a leading cause of morbidity and mortality worldwide. Diabetic cardiomyopathy (DCM) is defined as cardiac hypertrophy that is independent of hypertension and coronary artery disease (CAD). The three main risk factors of DCM are insulin resistance, hyperinsulinemia, and hyperglycemia. To date, some cellular and molecular defects, including impaired insulin signaling, hyperglycemia, glucotoxicity, cardiac lipotoxity, mitochondrial dysfunction, oxidative stress, endoplasmic reticulum (ER) stress, and cardiomyocyte apoptosis, have been reported as primary causes of DCM pathogenesis [3,4]. Treatments providing glycemic control and cardiovascular protection are important to improve the health of people all over the world [5].
Glucagon-like peptide-1 (GLP-1) is secreted from L-cells in the gut. In addition, to controlling blood glucose levels, GLP-1 also reduces gastric emptying and inhibits appetite. However, GLP-1 can be digested quickly by dipeptidyl peptidase-4 (DPP-4). In clinical practice, the GLP-1 receptor (GLP-1R) agonist liraglutide was effective at controlling blood glucose levels. In addition to pancreatic α and β cells, GLP-1Rs are also found in the heart. More and more clinical trials and animal experiments have shown evidence of the protective cardiac effects of liraglutide, independent of its effects on blood glucose levels. Short-term liraglutide treatment mildly improves left ventricular ejection fraction (LVEF) in ST-segment elevation myocardial infarction patients [6]. In type 1 diabetic rats, liraglutide inhibits cardiac steatosis, oxidative stress, and apoptosis through activating the activated protein kinase (AMPK)-sirtuin 1 (Sirt1) pathway [7]. However, the exact mechanism of the beneficial effects of liraglutide on cardiac tissue in diabetic rats remains to be elucidated.
Therefore, the present study aimed to investigate whether liraglutide has protective cardiac effects and its underlying mechanism in type 2 diabetic rats. We employed a global microarray analysis combined with bioinformatics to explore key genes and pathways affected by liraglutide in cardiac tissue from diabetic rats.
Materials and methods
Animal treatments and diets
Five-week-old male Sprague–Dawley (SD) rats, provided by the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China, SCXK-2014-0013), were maintained in a pathogen-free environment with a 12-h light/dark cycle and free access to food and water. Animal experiments followed the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH publication number 85-23, revised 1996) and were approved by the Animal Care Committee of the Peking Union Medical Hospital Animal Ethics Committee (Project XHDW-2015-0051, 15 February 2015). After acclimatization, the rats were randomly divided into four groups (n=6 for each group): control, diabetic, low-dose liraglutide (Llirag), and high-dose liraglutide (Hlirag) groups. The control group was fed a standard rodent diet (kcal%: 10% fat, 20% protein, and 70% carbohydrate; 3.85 kcal/g); the other three groups were fed a high-fat diet (HFD) (kcal%: 45% fat, 20% protein, and 35% carbohydrate; 4.73 kcal/g, Research Diet, New Brunswick, NJ, U.S.A.). After 4 weeks of HFD feeding, diabetes was induced in the rats by streptozotocin (STZ, 30 mg/kg) injection. Fasting blood glucose levels higher than 11.1 mmol/l were considered standard for the diabetic model. Then, 0.2 or 0.4 mg/kg/day liraglutide (i.h.) was administered to the Llirag and Hlirag groups, respectively. Control and diabetic groups were injected with the same volume of normal saline. Body weights and fasting blood glucose levels (Bayer Contour TS glucometer, Hamburg, Germany) were recorded monthly. After 12 weeks of treatment, an oral glucose tolerance test, in which 20% glucose was gavaged at a dose of 2 g/kg, was performed after 10 h of fasting. Tail vein blood glucose levels at 0, 30, 60, and 120 min were measured. At the end of the study, all rats were fasted for 10 h and anesthetized with ketamine (100 mg/kg i.p., Pharmacia and Upjohn Ltd, Crawley, U.K.), then blood samples were collected from the abdominal aorta. Finally, the rats were killed by decapitation. Cardiac tissue was quickly collected, frozen in liquid nitrogen, and stored at −80°C for gene microarray analysis.
Metabolic profile analysis
Triglyceride (TG), total cholesterol (TC), high-density lipoprotein (HDL), and low-density lipoprotein (LDL) levels were measured by an enzyme end point method (Roche Diagnostics, GmbH, Mannheim, Germany). Fasting serum adiponectin and insulin levels were determined by enzyme-linked immunosorbent assay (Millipore, Billerica, MA, U.S.A.). Homeostasis model assessment of insulin resistance (HOMA-IR) scores was calculated using the following formula: FBG (mmol/l) × fasting insulin (μIU/ml)/22.5.
Serum nitric oxide and antioxidant markers
Serum nitric oxide (NO) and GSH/GSSG levels were determined using Fluorometric Assay Kit (Cayman Chemical, Ann Arbor, MI, U.S.A.) and Thiol Green Indicator fluorometric method (Abcam, Cambridge, MA, U.S.A.), respectively.
Tail cuff systolic blood pressure and heart rate measurements
Systolic blood pressure (SBP) and heart rate (HR) were measured by tail-cuff plethysmography (BP98A, Softron, Tokyo, Japan). After prewarming at 25°C for at least 5 min, the first five cycles were used as acclimatization cycles. After that, the mean blood pressure was recorded for the next five consecutive cycles.
Left ventricle function measurement by echocardiography
Rats were anesthetized by inhaling 1% isoflurane with 99% O2. During anesthesia, left ventricle (LV) diameter was determined using a Vevo 2100 Ultrasound System (Visual Sonics, Toronto, Ontario, Canada). Fractional shortening (FS) was calculated as follow: FS% = (LVEDD – LVESD)/LVEDD × 100, where LVEDD and LVESD are the LV end-diastolic diameter and the LV end-systolic diameter, respectively [8]. Increased FS indicated better LV contractile function [9].
RNA extraction and gene microarray hybridization
Total RNA was extracted from cardiac tissue by using a mirVana™ RNA Isolation Kit (Ambion, Sao Paulo, SP, Brazil). Total RNA was transcribed into double-stranded cDNA and then synthesized into double-stranded cRNA. The second cycle cRNA was then labeled with biotin. The biotinylated cRNA was purified, fragmented, and hybridized to an Affymetrix GeneChip Rat Gene 2.0 ST whole transcript-based array (Affymetrix Technologies, Santa Clara, CA, U.S.A.). After washing and staining, the microarrays were scanned using an Affymetrix Scanner 3000 7G (Santa Clara, CA, U.S.A.).
Microarray bioinformatics analysis
Expression Console Software (version 1.4.1, Affymetrix, Santa Clara, CA, U.S.A.) was used to analyze the microarray signals. Differentially expressed genes were defined as having a fold change >1.5 and P-value <0.05 (one-way ANOVA). The raw microarray data have been submitted to the Gene Expression Omnibus (GEO) repository (GSE102194). The enrichment analysis of differentially expressed genes was performed by gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis with Database for Annotation, Visualization, and Integrated Discovery (DAVID) software (http://david.abcc.ncifcrf.gov/) [10]. The gene interaction network was drawn using String software (http://string-db.org/) [11].
Real-time PCR
Total RNA was extracted from cardiac tissue. Reverse transcription products were tested by real-time PCR. The primers are listed in Table 1. Real-time PCR was performed on an ABI Prism 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, U.S.A.). The cycling conditions were 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, and 60°C for 30 s. β-actin was used as an internal control. Samples were run in triplicate. The 2−ΔΔCt method was used to calculate the relative expression levels.
Table 1. Oligonucleotide sequences for qPCR analysis.
| Gene symbol | Genbank ID | Forward primer | Reverse primer | Product size (bp) |
|---|---|---|---|---|
| Angptl3 | NM_001025065 | AAAGGGTTTTGGGAGGCTTGA | CCCAAAAGCGCTATGGTCTC | 117 |
| Dgat1 | NM_053437 | GAACCGCTTCTTCCAAGGGA | AGAACTCCAGGCCCAGGTTA | 177 |
| Dgat2 | NM_001012345 | ACCTACCTCGGATCTCGACC | CTGATCCATGCCCCAGCC | 105 |
| Ephx2 | NM_022936 | CGTTCGACCTTGACGGAGTG | CTGGAAAGCGCCAAGTAGGA | 107 |
| Nr1h3 | NM_031627 | GAGTCATCCGAGCCTACAGC | AAGAATCCCTTGCAGCCCTC | 191 |
| Pparα | NM_013196 | ATTGGCGTTCGCAGCTGTTT | CTCGTGTGCCCTCCCTCAAG | 102 |
| Srebf1 | NM_001276707 | CCATGGACGAGCTACCCTTC | GGCATCAAATAGGCCAGGGA | 149 |
| β-actin | NM_031144 | ACTCTGTGTGGATTGGTGGC | CGCAGCTCAGTAACAGTCCG | 140 |
Abbreviations: Angptl3, angiopoietin-like 3; Dgat1, diacylglycerol O-acyltransferase 1; Dgat2, diacylglycerol O-acyltransferase 2; Ephx2, epoxide hydrolase 2; Nr1h3 (LXRα), nuclear receptor subfamily 1, group H, member 3; Pparα, peroxisome proliferator activated receptor α, Srebf1, sterol regulatory element binding transcription factor 1.
Statistical analysis
GraphPad Prism software (version 5.0, San Diego, CA, U.S.A.) was used for statistical analyses. All values are shown as the mean ± S.D. Group data were analyzed using one-way ANOVA followed by Student’s t test. P<0.05 was considered to be statistically significant.
Results
Effect of liraglutide on body weight, serum lipid profile, blood glucose, adiponectin, and serum fasting insulin levels
Liraglutide significantly reduced the body weights of diabetic rats (P<0.05, Figure 1A). In addition, liraglutide dose dependently reduced fasting blood glucose levels and blood glucose and area under the curve (AUC) values for oral glucose tolerance tests ( OGTTs, P<0.01, Figure 1B–D). The rats that underwent 12-week liraglutide treatment had lower TC (P<0.01, Figure 1E) and LDL-c levels (P<0.05, Figure 1H). Only the high dose of liraglutide reduced serum TG levels and increased serum adiponectin level in diabetic rats (P<0.01, Figure 1F,I). Compared with diabetic rats, liraglutide-treated rats had lower serum fasting insulin levels and HOMA-IR scores (P<0.01, Figure 1J,K).
Figure 1. Effect of liraglutide on metabolic indexes in diabetic rats.

(A) Body weight, (B) fasting blood glucose, (C) blood glucose in OGTT, (D) AUC in OGTT, (E) TC, (F) TG, (G) HDL, (H) LDL, (I) adiponectin, (J) fasting insulin, (K) HOMA-IR, (L) NO, (M) GSH, (N) GSSG, and (O) GSH/GSSG. Values are mean ± S.D. (n=6), *P<0.05, **P<0.01 compared with normal group; #P<0.05, ##P<0.01 compared with diabetic group; $$P<0.01 compared with Llirag group.
Effect of liraglutide on serum NO and antioxidant markers
Serum NO, GSH level, and GSH/GSSG ratio in diabetic rats were lower than control rats (P<0.01, Figure 1L,M,O). Liraglutide treatment moderated this decrease (P<0.01, Figure 1L,M,O). Serum GSSG level in diabetic group was higher than control rats (P<0.01, Figure 1N). Liraglutide reduced serum GSSG level dose independently (P<0.01, Figure 1N).
Effect of liraglutide on cardiac function
SBP, HR, LVEDD, and LVESD levels in the diabetic group were significantly increased (P<0.01, Figure 2A–D). However, diabetic rats had lower %FS values than normal control rats (P<0.01, Figure 2E). Liraglutide treatment decreased SBP, HR, LVEDD, and LVESD levels and increased % FS values (P<0.01, Figure 2A–E). These results suggest that liraglutide moderated LV dysfunction and decreased blood pressure and HRs.
Figure 2. Effect of liraglutide on cardiac function in diabetic rats.

(A) SBP, (B) HR, (C) LVEDD, (D) LVESD, and (E) FS. n=6. **P<0.01 compared with control; ##P<0.01 compared with diabetic.
Microarray data analysis of Hlirag and diabetic groups
A total of 269 differentially expressed genes were screened out from Hlirag group (fold change >1.5, P<0.05); these included 166 up-regulated genes and 105 down-regulated genes. The differentially expressed genes were enriched in 11 pathways (P<0.001, Table 2). The top five pathways were cardiac muscle contraction, non-alcoholic fatty liver disease (NAFLD), oxidative phosphorylation, metabolic pathways, metabolic pathways, and Alzheimer’s disease. The significant biological processes (BPs) in the GO categories are listed in Table 3 (P<0.01). The top ten BP terms were hydrogen ion transmembrane transport, cholesterol homeostasis, lipid homeostasis, positive regulation of TG biosynthetic process, fatty acid β-oxidation using acyl-CoA dehydrogenase, fatty acid β-oxidation, response to cAMP, very long-chain fatty acid catabolic process, circadian rhythm, and lipid metabolic process.
Table 2. The enriched KEGG pathway with differentially expressed genes (P<0.001).
| Pathway ID | Pathway name | Count | Fold enrichment | P-value |
|---|---|---|---|---|
| rno04932 | NAFLD | 14 | 6.003 | 4.39 × 10−7 |
| rno04260 | Cardiac muscle contraction | 10 | 8.905 | 1.42 × 10−6 |
| rno01100 | Metabolic pathways | 39 | 2.095 | 4.16 × 10−6 |
| rno00190 | Oxidative phosphorylation | 12 | 5.7889 | 5.79 × 10−6 |
| rno05012 | Parkinson’s disease | 12 | 5.484 | 9.74 × 10−6 |
| rno05010 | Alzheimer’s disease | 13 | 4.961 | 9.80 × 10−6 |
| rno00640 | Propanoate metabolism | 6 | 14.885 | 4.09 × 10−5 |
| rno01200 | Carbon metabolism | 10 | 5.740 | 5.27 × 10−5 |
| rno00280 | Valine, leucine, and isoleucine degradation | 7 | 8.840 | 1.21 × 10−4 |
| rno05016 | Huntington’s disease | 12 | 4.106 | 1.41× 10−4 |
| rno01130 | Biosynthesis of antibiotics | 12 | 3.806 | 2.73 × 10−4 |
Table 3. The enriched GO terms with differentially expressed genes (P<0.01).
| Term ID | Term name | Count | P-value | Fold enrichment | Catalog |
|---|---|---|---|---|---|
| GO:1902600 | Hydrogen ion transmembrane transport | 8 | 1.221 × 10−5 | 0.0193 | BP |
| GO:0042632 | Cholesterol homeostasis | 7 | 0.000124 | 0.197 | BP |
| GO:0055088 | Lipid homeostasis | 6 | 0.000148 | 0.236 | BP |
| GO:0010867 | Positive regulation of triglyceride biosynthetic process | 4 | 0.00105 | 1.669 | BP |
| GO:0033539 | Fatty acid β-oxidation using acyl-CoA dehydrogenase | 4 | 0.00148 | 2.329 | BP |
| GO:0006635 | Fatty acid β-oxidation | 5 | 0.00231 | 3.616 | BP |
| GO:0051591 | Response to cAMP | 6 | 0.00245 | 3.825 | BP |
| GO:0042760 | Very long-chain fatty acid catabolic process | 3 | 0.00296 | 4.601 | BP |
| GO:0007623 | Circadian rhythm | 7 | 0.00361 | 5.589 | BP |
| GO:0006629 | Lipid metabolic process | 6 | 0.00458 | 7.039 | BP |
| GO:0090181 | Regulation of cholesterol metabolic process | 3 | 0.00499 | 7.647 | BP |
| GO:0043401 | Steroid hormone mediated signaling pathway | 5 | 0.00538 | 8.209 | BP |
| GO:0006631 | Fatty acid metabolic process | 5 | 0.00606 | 9.213 | BP |
| GO:0019217 | Regulation of fatty acid metabolic process | 3 | 0.00619 | 9.399 | BP |
| GO:2000188 | Regulation of cholesterol homeostasis | 3 | 0.00619 | 9.399 | BP |
| GO:0030522 | Intracellular receptor signaling pathway | 4 | 0.00737 | 11.085 | BP |
| GO:0019432 | Triglyceride biosynthetic process | 3 | 0.00894 | 13.296 | BP |
| GO:0006366 | Transcription from RNA polymerase II promoter | 12 | 0.00976 | 14.419 | BP |
| GO:0005739 | Mitochondrion | 43 | 1.503 × 10−6 | 2.199 | Cellular components |
| GO:0005743 | Mitochondrial inner membrane | 13 | 0.000465 | 3.403 | Cellular components |
| GO:0005777 | Peroxisome | 8 | 0.000731 | 5.373 | Cellular components |
| GO:0005746 | Mitochondrial respiratory chain | 3 | 0.00620 | 24.584 | Cellular components |
| GO:0004129 | Cytochrome c oxidase activity | 6 | 4.237 × 10−5 | 15.081 | Molecular function |
| GO:0003700 | Transcription factor activity, sequence-specific DNA binding | 22 | 0.000415 | 2.359 | Molecular function |
| GO:0001077 | Transcriptional activator activity, RNA polymerase II core promoter proximal region sequence-specific binding | 11 | 0.00127 | 3.483 | Molecular function |
| GO:0000978 | RNA polymerase II core promoter proximal region sequence-specific DNA binding | 12 | 0.00397 | 2.789 | Molecular function |
| GO:0043565 | Sequence-specific DNA binding | 16 | 0.00479 | 2.265 | Molecular function |
| GO:0009055 | Electron carrier activity | 5 | 0.00505 | 7.181 | Molecular function |
| GO:0003707 | Steroid hormone receptor activity | 5 | 0.00505 | 7.181 | Molecular function |
| GO:0004879 | RNA polymerase II transcription factor activity, ligand-activated sequence-specific DNA binding | 4 | 0.00912 | 9.192 | Molecular function |
| GO:0046982 | Protein heterodimerization activity | 15 | 0.00946 | 2.170 | Molecular function |
| GO:0044212 | Transcription regulatory region DNA binding | 9 | 0.00950 | 3.054 | Molecular function |
All the 271 differentially expressed genes were mapped using String online software. The results showed that there were 253 interactions with a total of 256 joint edges (Figure 3). Twenty-one nodes had more than ten joint edges. These nodes involved 137 joint edges. These 21 genes are listed in Table 4. The top ten genes were citrate synthase (Cs), ubiquinol-cytochrome c reductase core protein I (Uqcrc1), acyl-CoA dehydrogenase, very long chain (Acadv1), 3-oxoacid CoA transferase 1 (Oxct1), NADH dehydrogenase (ubiquinone) flavoprotein 1 (Ndufv1), succinate-CoA ligase, alpha subunit (Suclg1), enoyl CoA hydratase, short chain, 1, mitochondrial (Echs1), acyl-CoA dehydrogenase, C2–C3 short chain (Acads), and branched chain ketoacid dehydrogenase E1, α-polypeptide (Bckdha).
Figure 3. Protein–protein interaction network in Hlirag group compared with diabetic group.

The nods stand for differentially expressed genes in Hlirag group compared with diabetic group. The lines stand for the interactions between two proteins.
Table 4. A list of genes with connective degree more than ten in the String network.
| Gene accession | Gene symbol | Gene name | Degree |
|---|---|---|---|
| NM_130755 | Cs | Citrate synthase | 20 |
| NM_001004250 | Uqcrc1 | Ubiquinol-cytochrome c reductase core protein I | 18 |
| NM_012891 | Acadvl | Acyl-CoA dehydrogenase, very long chain | 17 |
| NM_001127580 | Oxct1 | 3-Oxoacid CoA transferase 1 | 16 |
| NM_001006972 | Ndufv1 | NADH dehydrogenase (ubiquinone) flavoprotein 1 | 15 |
| NM_053752 | Suclg1 | Succinate-CoA ligase, alpha subunit | 15 |
| NM_078623 | Echs1 | Enoyl CoA hydratase, short chain, 1, mitochondrial | 14 |
| NM_022512 | Acads | Acyl-CoA dehydrogenase, C-2 to C-3 short chain | 13 |
| NM_012782 | Bckdha | Branched chain ketoacid dehydrogenase E1, α-polypeptide | 13 |
| NM_134364 | Atp5b | ATP synthase, H+ transporting, mitochondrial F1 complex, β-polypeptide | 12 |
| NM_017202 | Cox4i1 | Cytochrome c oxidase subunit IV isoform 1 | 12 |
| NM_145783 | Cox5a | Cytochrome c oxidase, subunit Va | 12 |
| NM_001107793 | Acss2 | Acyl-CoA synthetase short-chain family member 2 | 11 |
| NM_138883 | Atp5o | ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit | 11 |
| ENSRNOT00000077826 | Decr2 | 2,4-Dienoyl CoA reductase 2, peroxisomal | 11 |
| NM_001044242 | Pdhx | Pyruvate dehydrogenase complex, component X | 11 |
| NM_001108387 | Sucla2 | Succinate-CoA ligase, ADP-forming, β-subunit | 11 |
| NM_001025134 | Uqcrq | Ubiquinol-cytochrome c reductase, complex III subunit VII | 11 |
| NM_019267 | Bckdhb | Branched chain keto acid dehydrogenase E1, β-polypeptide | 10 |
| NM_012814 | Cox6a1 | Cytochrome c oxidase, subunit VIa, polypeptide 1 | 10 |
| NM_053493 | Hacl1 | 2-Hydroxyacyl-CoA lyase 1 | 10 |
Confirmation by quantitative Polymerase Chain Reaction (qPCR)
To validate the microarray results, we analyzed the mRNA expression levels of representative gene by using qPCR. As shown in Figure 4, the relative mRNA levels of LXRα, nuclear receptor subfamily 1, group H, member 3 (Nr1h3), sterol regulatory element binding transcription factor 1 (Srebf1) and peroxisome proliferator activated receptor α (PPARα) in the Hlirag group were significantly higher, whereas the mRNA levels of angiopoietin-like 3 (Angptl3), diacylglycerol O-acyltransferase 1 (Dgat1), diacylglycerol O-acyltransferase 2 (Dgat2), and epoxide hydrolase 2 (Ephx2) were lower than those in diabetic rat group (P<0.01). These outcomes were consistent with the microarray results.
Figure 4. Confirmation of five representative differentially expressed genes by qPCR.
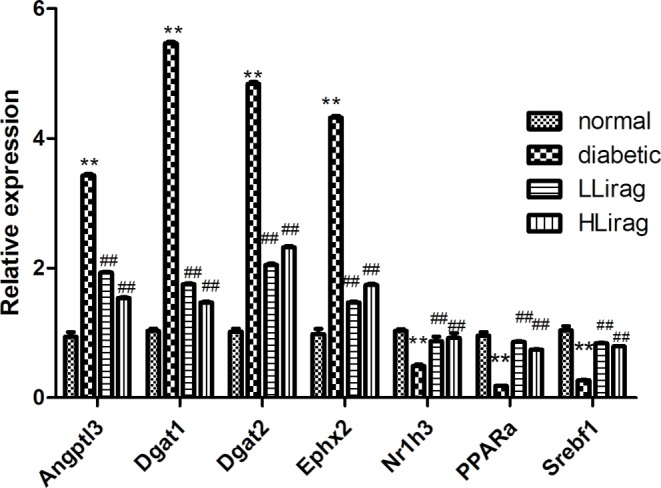
Values are mean ± S.D. (n=6), **P<0.01 compared with normal group; ##P<0.01 compared with diabetic group.
Discussion
In the present study, as expected, liraglutide reduced blood glucose levels and moderated insulin resistance in diabetic rats. Moreover, we found that liraglutide reduced the body weights in diabetic rats. Liraglutide-treated rats also had lower TC and LDL-c levels and higher adiponectin levels. Clinical trials proved that liraglutide was effective at reducing blood glucose levels and body weights [12–14]. Liraglutide at 3.0 mg/day has been approved by the U.S. Food and Drug Administration (FDA) for treating obesity since 2014. In human studies, liraglutide treatment at 3.0 mg for 56 weeks decreased insulin resistance [15]. In addition, liraglutide treatment dose dependently increased plasma adiponectin in Chinese type 2 diabetes [16].
Regarding cardiac function, we found that liraglutide reduced SBP, HR, LVEDD, and LVESD levels, increased %FS values and serum NO levels in diabetic rats. These results indicate the beneficial effects of liraglutide on cardiac function. HFD-fed mice have cardiac ceramide accumulation. One week of liraglutide treatment improved cardiac ER homeostasis and cardiac function [17]. A clinical trial revealed that liraglutide reduced LVEF in patients with heart failure (HF) [18]. Interestingly, liraglutide reduced both systolic and diastolic blood pressure in hypertensive mice through the atrial natriuretic peptide (ANP) axis [19]. In a meta-analysis of 16 randomized controlled trials, GLP-1 receptor agonists (exenatide and liraglutide) reduced systolic pressure (SBP) and diastole pressure (DBP) by 1–5 mmHg compared with other antidiabetic drugs in diabetic patients [20]. Reductions in blood pressure were not related to weight loss or hemoglobin A1c (HbA1c) improvement [21].
We found that liraglutide increased PPARα expression in the cardiac tissue of diabetic rats. PPARs have three forms: α, γ, and δ. They can bind with retinoid X receptor (RXR) to regulate energy utilization and storage [22]. Recent results implicate PPARs in the regulation of inflammation and atherosclerosis [23]. In the heart, both PPARα and PPARδ can regulate lipid metabolism. In addition to lipid metabolism, PPAR-γ also modulates glucose metabolism [24–26]. Previous studies found that PPAR-α expression is down-regulated in diabetic rat hearts [27–30]. Many studies indicate that oxidative stress increases in diabetic status [31,32] and contributes to inhibition of PPAR-α in cardiomyocytes [33]. Our data also showed that diabetic rats had lower GSH/GSSG ratio, and liraglutide treatment increased serum GSH/GSSG ratio. GSH/GSSG ratio is an important antioxidant biomarker [34].
Interestingly, in our study, liraglutide reduced Dgat1 and Dgat2 expression in the hearts of diabetic rats. DGAT has two isoforms: DGAT1 and DGAT2. They are the enzymes that catalyze the final step in the biosynthesis of TG [35]. DGAT is the target gene of PPARα. DGAT2 appears to be a key enzyme that controls TG homeostasis in vivo and regulates fatty acid storage [36]. A previous study found that DGAT1 and DGAT2 expression was increased in diabetic rat hearts [37]. Increased DGAT expression generated reactive oxidative stress, and caused myocardial damage in DM cardiomyopathy [38]. Thus, our results indicate that liraglutide reverses oxidative stress generated by diabetic status to increase PPARα expression, leads to reduce the expression of DGAT, and also finally inhibits reactive oxidative stress.
Our research found that liraglutide increased Nr1h3 (LXRα) and Srebf1 expression in diabetic rat hearts. LXRs have important role in the regulation of cholesterol and fatty acid metabolism. It forms heterodimer with RXR [39]. Srebf is directly induced by LXRs through an RXR/LXR-binding site on the Srebf gene promoter [40,41]. SREBF is a transcription factor that regulates lipogenic enzymes by binding to sterol response elements [42]. Thus, our data supports that liraglutide treatment activates cardiac LXRα and Srebf1 expression in diabetic rats.
We also found that liraglutide reduced Ephx2 expression in diabetic rat hearts. Soluble epoxide hydrolase (sEH) is an Ephx2 gene product. Arachidonic acid (AA) can be transferred to epoxyeicosatrienoic acids (EETs) by cytochrome P-450 (CYP) epoxygenases. EETs are signaling molecules that regulate blood pressure [43–47], inflammation [44,45,47,48], and glucose homeostasis [49,50]. However, EETs have a short half-life and may be metabolized by sEH into dihydroxyeicosatrienoic acids (DHETs) with relatively weak activity. Hearts in sEH null mice had improved post-ischemic recovery of Lv developed pressure (LVDP), reduced infarct size after ischemia and reperfusion [51], and reduced survival after cardiac arrest and cardiopulmonary resuscitation [52]. Therefore, sEH is a promising target for treating cardiovascular disease. sEH inhibitors have been indicated as beneficial treatments in animal models of high blood pressure [43,44,46], inflammation [44,48,53], myocardial injury [44,54–56], ischemia–reperfusion [57,58], pathological cardiac hypertrophy [44], and insulin resistance [49,50] and have been shown to protect heart structure and function [59]. Our study provided evidence that as a sEH ingibitor, liraglutide can preserve cardiac function.
We also found that liraglutide reduced Angptl3 expression in diabetic rat hearts. Lipoprotein lipase (LPL) metabolized TG into free fatty acids (FFAs). LPL overexpression is correlated with reduced plasma TG levels and decreased cardiovascular risks [60]. However, LPL null models have severe hypertriglyceridemia [60]. The angiopoietin-like protein (ANGPTL) family is a key regulator of LPL [61,62]. ANGPTL3 is an endogenous inhibitor of LPL. Rare loss-of-function variants for ANGPTL3 have been associated with decreased TG levels as well as decreased low-density lipoprotein-cholesterol (LDL-c) and high-density lipoprotein-cholesterol (HDL-c) levels in family and general population studies in humans [62–70]. In addition, these subjects demonstrate an absence of coronary atherosclerotic plaques [71]. Another human population study showed that plasma ANGPTL3 levels were increased in myocardial infarction patients [71]. Heterozygous carries of ANGPTL3 loss-of-function mutations had a 34% reduction in CAD risk [71]. Angptl3 deletion was also reported to reduce the development of atherosclerosis in apolipoprotein E (apoE)-deficient mice [72]. Recently, a human monoclonal antibody against Angptl3 in dyslipidemic mice and against ANGPTL3 in healthy human subjects with elevated levels of TGs or LDL-c significantly reduced serum TG, HDL-c, and LDL-c levels and decreased the odds of atherosclerotic cardiovascular disease [73]. Another research group showed that treating mice and human subjects with antisense oligonucleotides targeting Angptl3 messenger RNA reduced atherogenic lipoproteins and retarded the progression of atherosclerosis [74].
Conclusion
In conclusion, liraglutide prevents cardiac dysfunction by activating cardiac PPARα to inhibit Dgat expression and oxidative stress in diabetic rats (Figure 5). The present study provides a potential mechanism for the protective cardiac effects of a GLP-1 analog in a model of diabetes.
Figure 5. Liraglutide activates PPARα, which binds to RXR.
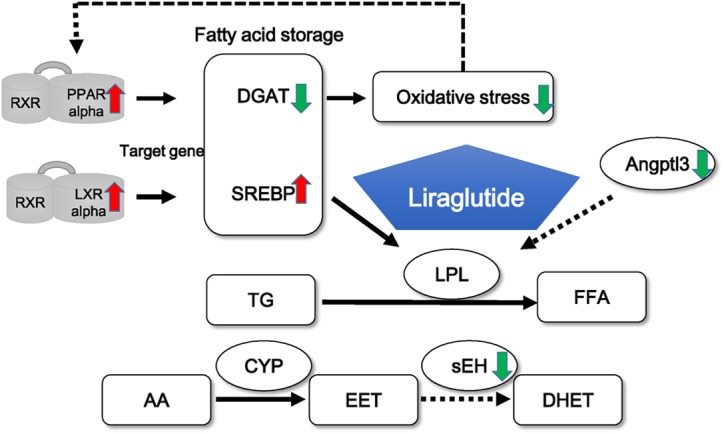
Then PPARα inhibits its target gene, DGAT to inhibit oxidative stress. Liraglutide also activates Nr1h3 and SREBP and inhibits Angptl3 to activate LPL, leading the production of FFAs. Moreover, liraglutide inhibits sEH expression to increase EET.
Acknowledgments
We thank Beijing Compass Biotechnology Company for excellent technical assistance with the microarray experiments.
Abbreviations
- ANGPTL3
angiopoietin-like 3
- AMPK
activated protein kinase
- apoE
apolipoprotein E
- AUC
area uncer the curve
- BP
biological process
- CAD
coronary artery disease
- DBP
diastole pressure
- DCM
diabetic cardiomyopathy
- DGAT
diacylglycerol O-acyltransferase
- EET
epoxyeicosatrienoic acid
- ER
endoplasmic reticulum
- FS
fractional shortening
- GLP-1
glucagon-like peptide-1
- GLP-1R
GLP-1 receptor
- GO
gene ontology
- HbA1c
hemoglobin A1c
- HDL-c
high-density lipoprotein-cholesterol
- HFD
high-fat diet
- Hlirag
high-dose liraglutide
- HOMA-IR
homeostasis model assessment of insulin resistance
- HR
heart rate
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LDL-c
low-density lipoprotein-cholesterol
- Llirag
low-dose liraglutide
- LV
left ventricular
- LVEDD
LV end-diastolic diameter
- LVEF
LV ejection fraction
- LVESD
LV end-systolic diameter
- OGTT
oral glucose tolerance test
- PPAR
peroxisome proliferator activated receptor
- qPCR
quantitative polymerase chain reaction
- RXR
retinoid X receptor
- SBP
systolic blood pressure
- sEH
soluble epoxide hydrolase
- SBP
systolic pressure
- Sirt1
sirtuin 1
- STZ
streptozotocin
- TC
total cholesterol
- TG
triglyceride
Author contribution
X.X. designed the experiments, contributed reagents and materials. Q.Z., J.Z., T.W., and X.W. conducted the experiments. M.Y, M.L., and F.P. analyzed the data. Q.Z. wrote the manuscript.
Funding
This work was supported by the National Key R&D Program of China [grant number 2017YFC1309603]; the National Key Research and Development Program of China [grant number 2016YFA0101002]; the National Natural Science Foundation of China [grant numbers 81170736, 81570715]; the National Natural Science Foundation for Young Scholars of China [grant number 81300649]; the China Scholarship Council Foundation [grant number 201308110443]; the PUMC Youth Fund [grant number 33320140022]; the Fundamental Research Funds for the Central Universities; and the Scientific Activities Foundation for Selected Returned Overseas Professionals of Human Resources and Social Security Ministry.
Competing interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Ogurtsova K., da Rocha Fernandes J.D., Huang Y., Linnenkamp U., Guariguata L., Cho N.H. et al. (2017) IDF Diabetes Atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 128, 40–50 10.1016/j.diabres.2017.03.024 [DOI] [PubMed] [Google Scholar]
- 2.Capes S.E., Hunt D., Malmberg K. and Gerstein H.C. (2000) Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet 355, 773–778 10.1016/S0140-6736(99)08415-9 [DOI] [PubMed] [Google Scholar]
- 3.Jia G., DeMarco V.G. and Sowers J.R. (2016) Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 12, 144–153 10.1038/nrendo.2015.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jia G., Whaley-Connell A. and Sowers J.R. (2017) Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia, 10.1007/s00125-017-4390-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cardillo C. (2013) Drug treatments to restore vascular function and diabesity. Ann. Pharm. Fr. 71, 27–33 10.1016/j.pharma.2012.09.001 [DOI] [PubMed] [Google Scholar]
- 6.Chen W.R., Hu S.Y., Chen Y.D., Zhang Y., Qian G., Wang J. et al. (2015) Effects of liraglutide on left ventricular function in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Am. Heart J. 170, 845–854 10.1016/j.ahj.2015.07.014 [DOI] [PubMed] [Google Scholar]
- 7.Inoue T., Inoguchi T., Sonoda N., Hendarto H., Makimura H., Sasaki S. et al. (2015) GLP-1 analog liraglutide protects against cardiac steatosis, oxidative stress and apoptosis in streptozotocin-induced diabetic rats. Atherosclerosis 240, 250–259 10.1016/j.atherosclerosis.2015.03.026 [DOI] [PubMed] [Google Scholar]
- 8.Derumeaux G., Mulder P., Richard V., Chagraoui A., Nafeh C., Bauer F. et al. (2002) Tissue doppler imaging differentiates physiological from pathological pressure-overload left ventricular hypertrophy in rats. Circulation 105, 1602–1608 10.1161/01.CIR.0000012943.91101.D7 [DOI] [PubMed] [Google Scholar]
- 9.Apaijai N., Pintana H., Chattipakorn S.C. and Chattipakorn N. (2012) Cardioprotective effects of metformin and vildagliptin in adult rats with insulin resistance induced by a high-fat diet. Endocrinology 153, 3878–3885 10.1210/en.2012-1262 [DOI] [PubMed] [Google Scholar]
- 10.Dennis G. Jr, Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C. et al. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3 10.1186/gb-2003-4-5-p3 [DOI] [PubMed] [Google Scholar]
- 11.Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J. et al. (2015) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 10.1093/nar/gku1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun F., Chai S., Li L., Yu K., Yang Z., Wu S. et al. (2015) Effects of glucagon-like peptide-1 receptor agonists on weight loss in patients with type 2 diabetes: a systematic review and network meta-analysis. J. Diabetes Res. 2015, 157201 10.1155/2015/157201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris K.B. and McCarty D.J. (2015) Efficacy and tolerability of glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes mellitus. Ther. Adv. Endocrinol. Metab. 6, 3–18 10.1177/2042018814558242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amori R.E., Lau J. and Pittas A.G. (2007) Efficacy and safety of incretin therapy in type 2 diabetes: systematic review and meta-analysis. JAMA 298, 194–206 10.1001/jama.298.2.194 [DOI] [PubMed] [Google Scholar]
- 15.Christou G.A., Katsiki N. and Kiortsis D.N. (2016) The current role of liraglutide in the pharmacotherapy of obesity. Curr. Vasc. Pharmacol. 14, 201–207 10.2174/1570161113666150615111951 [DOI] [PubMed] [Google Scholar]
- 16.Li D., Xu X., Zhang Y., Zhu J., Ye L., Lee K.O. et al. (2015) Liraglutide treatment causes upregulation of adiponectin and downregulation of resistin in Chinese type 2 diabetes. Diabetes Res. Clin. Pract. 110, 224–228 10.1016/j.diabres.2015.05.051 [DOI] [PubMed] [Google Scholar]
- 17.Noyan-Ashraf M.H., Shikatani E.A., Schuiki I., Mukovozov I., Wu J., Li R.K. et al. (2013) A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation 127, 74–85 10.1161/CIRCULATIONAHA.112.091215 [DOI] [PubMed] [Google Scholar]
- 18.Moberly S.P., Mather K.J., Berwick Z.C., Owen M.K., Goodwill A.G., Casalini E.D. et al. (2013) Impaired cardiometabolic responses to glucagon-like peptide 1 in obesity and type 2 diabetes mellitus. Basic Res. Cardiol. 108, 365 10.1007/s00395-013-0365-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim M., Platt M.J., Shibasaki T., Quaggin S.E., Backx P.H., Seino S. et al. (2013) GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat. Med. 19, 567–575 10.1038/nm.3128 [DOI] [PubMed] [Google Scholar]
- 20.Wang B., Zhong J., Lin H., Zhao Z., Yan Z., He H. et al. (2013) Blood pressure-lowering effects of GLP-1 receptor agonists exenatide and liraglutide: a meta-analysis of clinical trials. Diabetes Obes. Metab. 15, 737–749 10.1111/dom.12085 [DOI] [PubMed] [Google Scholar]
- 21.Katout M., Zhu H., Rutsky J., Shah P., Brook R.D., Zhong J. et al. (2014) Effect of GLP-1 mimetics on blood pressure and relationship to weight loss and glycemia lowering: results of a systematic meta-analysis and meta-regression. Am. J. Hypertens. 27, 130–139 10.1093/ajh/hpt196 [DOI] [PubMed] [Google Scholar]
- 22.Chen L., Jia Z. and Yang G. (2014) PPARs and metabolic syndrome. PPAR Res. 2014, 832606 10.1155/2014/832606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown J.D. and Plutzky J. (2007) Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation 115, 518–533 10.1161/CIRCULATIONAHA.104.475673 [DOI] [PubMed] [Google Scholar]
- 24.Finck B.N. (2007) The PPAR regulatory system in cardiac physiology and disease. Cardiovasc. Res. 73, 269–277 10.1016/j.cardiores.2006.08.023 [DOI] [PubMed] [Google Scholar]
- 25.Yang Q. and Li Y. (2007) Roles of PPARs on regulating myocardial energy and lipid homeostasis. J. Mol. Med. (Berl.) 85, 697–706 10.1007/s00109-007-0170-9 [DOI] [PubMed] [Google Scholar]
- 26.Madrazo J.A. and Kelly D.P. (2008) The PPAR trio: regulators of myocardial energy metabolism in health and disease. J. Mol. Cell Cardiol. 44, 968–975 10.1016/j.yjmcc.2008.03.021 [DOI] [PubMed] [Google Scholar]
- 27.Lee T.I., Kao Y.H., Chen Y.C., Pan N.H. and Chen Y.J. (2010) Oxidative stress and inflammation modulate peroxisome proliferator-activated receptors with regional discrepancy in diabetic heart. Eur. J. Clin. Invest. 40, 692–699 10.1111/j.1365-2362.2010.02318.x [DOI] [PubMed] [Google Scholar]
- 28.Depre C., Young M.E., Ying J., Ahuja H.S., Han Q., Garza N. et al. (2000) Streptozotocin-induced changes in cardiac gene expression in the absence of severe contractile dysfunction. J. Mol. Cell Cardiol. 32, 985–996 10.1006/jmcc.2000.1139 [DOI] [PubMed] [Google Scholar]
- 29.Young M.E., Patil S., Ying J., Depre C., Ahuja H.S., Shipley G.L. et al. (2001) Uncoupling protein 3 transcription is regulated by peroxisome proliferator-activated receptor (alpha) in the adult rodent heart. FASEB J. 15, 833–845 10.1096/fj.00-0351com [DOI] [PubMed] [Google Scholar]
- 30.Yu B.C., Chang C.K., Ou H.Y., Cheng K.C. and Cheng J.T. (2008) Decrease of peroxisome proliferator-activated receptor delta expression in cardiomyopathy of streptozotocin-induced diabetic rats. Cardiovasc. Res. 80, 78–87 10.1093/cvr/cvn172 [DOI] [PubMed] [Google Scholar]
- 31.Cai L., Wang Y., Zhou G., Chen T., Song Y., Li X. et al. (2006) Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J. Am. Coll. Cardiol. 48, 1688–1697 10.1016/j.jacc.2006.07.022 [DOI] [PubMed] [Google Scholar]
- 32.Ye G., Metreveli N.S., Donthi R.V., Xia S., Xu M., Carlson E.C. et al. (2004) Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 53, 1336–1343 10.2337/diabetes.53.5.1336 [DOI] [PubMed] [Google Scholar]
- 33.Lee T.I., Kao Y.H., Chen Y.C. and Chen Y.J. (2009) Proinflammatory cytokine and ligands modulate cardiac peroxisome proliferator-activated receptors. Eur. J. Clin. Invest. 39, 23–30 10.1111/j.1365-2362.2008.02062.x [DOI] [PubMed] [Google Scholar]
- 34.Sentellas S., Morales-Ibanez O., Zanuy M. and Alberti J.J. (2014) GSSG/GSH ratios in cryopreserved rat and human hepatocytes as a biomarker for drug induced oxidative stress. Toxicol. In Vitro 28, 1006–1015 10.1016/j.tiv.2014.04.017 [DOI] [PubMed] [Google Scholar]
- 35.Shi Y. and Cheng D. (2009) Beyond triglyceride synthesis: the dynamic functional roles of MGAT and DGAT enzymes in energy metabolism. Am. J. Physiol. Endocrinol. Metab. 297, E10–E18 10.1152/ajpendo.90949.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stone S.J., Myers H.M., Watkins S.M., Brown B.E., Feingold K.R., Elias P.M. et al. (2004) Lipopenia and skin barrier abnormalities in DGAT2-deficient mice. J. Biol. Chem. 279, 11767–11776 10.1074/jbc.M311000200 [DOI] [PubMed] [Google Scholar]
- 37.Lee T.I., Kao Y.H., Tsai W.C., Chung C.C., Chen Y.C. and Chen Y.J. (2016) HDAC inhibition modulates cardiac PPARs and fatty acid metabolism in diabetic cardiomyopathy. PPAR Res. 2016, 5938740 10.1155/2016/5938740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J., Xue J., Wang H., Zhang Y. and Xie M. (2011) Osthole improves alcohol-induced fatty liver in mice by reduction of hepatic oxidative stress. Phytother. Res. 25, 638–643 10.1002/ptr.3315 [DOI] [PubMed] [Google Scholar]
- 39.Joseph S.B. and Tontonoz P. (2003) LXRs: new therapeutic targets in atherosclerosis? Curr. Opin. Pharmacol. 3, 192–197 10.1016/S1471-4892(03)00009-2 [DOI] [PubMed] [Google Scholar]
- 40.Repa J.J., Liang G., Ou J., Bashmakov Y., Lobaccaro J.M., Shimomura I. et al. (2000) Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 14, 2819–2830 10.1101/gad.844900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshikawa T., Shimano H., Amemiya-Kudo M., Yahagi N., Hasty A.H., Matsuzaka T. et al. (2001) Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol. Cell. Biol. 21, 2991–3000 10.1128/MCB.21.9.2991-3000.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beltowski J. (2008) Liver X receptors (LXR) as therapeutic targets in dyslipidemia. Cardiovasc. Ther. 26, 297–316 10.1111/j.1755-5922.2008.00062.x [DOI] [PubMed] [Google Scholar]
- 43.Imig J.D. (2005) Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am. J. Physiol. Renal Physiol. 289, F496–F503 10.1152/ajprenal.00350.2004 [DOI] [PubMed] [Google Scholar]
- 44.Imig J.D. and Hammock B.D. (2009) Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 8, 794–805 10.1038/nrd2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inceoglu B., Schmelzer K.R., Morisseau C., Jinks S.L. and Hammock B.D. (2007) Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (EETs). Prostaglandins Other Lipid Mediat. 82, 42–49 10.1016/j.prostaglandins.2006.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang H., Quilley J., Doumad A.B., Zhu A.G., Falck J.R., Hammock B.D. et al. (2011) Increases in plasma trans-EETs and blood pressure reduction in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 300, H1990–H1996 10.1152/ajpheart.01267.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spector A.A., Fang X., Snyder G.D. and Weintraub N.L. (2004) Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog. Lipid Res. 43, 55–90 10.1016/S0163-7827(03)00049-3 [DOI] [PubMed] [Google Scholar]
- 48.Fleming I. (2007) DiscrEET regulators of homeostasis: epoxyeicosatrienoic acids, cytochrome P450 epoxygenases and vascular inflammation. Trends Pharmacol. Sci. 28, 448–452 10.1016/j.tips.2007.08.002 [DOI] [PubMed] [Google Scholar]
- 49.Luo P., Chang H.H., Zhou Y., Zhang S., Hwang S.H., Morisseau C. et al. (2010) Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J. Pharmacol. Exp. Ther. 334, 430–438 10.1124/jpet.110.167544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luria A., Bettaieb A., Xi Y., Shieh G.J., Liu H.C., Inoue H. et al. (2011) Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 108, 9038–9043 10.1073/pnas.1103482108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seubert J.M., Sinal C.J., Graves J., DeGraff L.M., Bradbury J.A., Lee C.R. et al. (2006) Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ. Res. 99, 442–450 10.1161/01.RES.0000237390.92932.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hutchens M.P., Nakano T., Dunlap J., Traystman R.J., Hurn P.D. and Alkayed N.J. (2008) Soluble epoxide hydrolase gene deletion reduces survival after cardiac arrest and cardiopulmonary resuscitation. Resuscitation 76, 89–94 10.1016/j.resuscitation.2007.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Campbell W.B., Gebremedhin D., Pratt P.F. and Harder D.R. (1996) Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 78, 415–423 10.1161/01.RES.78.3.415 [DOI] [PubMed] [Google Scholar]
- 54.Aboutabl M.E., Zordoky B.N., Hammock B.D. and El-Kadi A.O. (2011) Inhibition of soluble epoxide hydrolase confers cardioprotection and prevents cardiac cytochrome P450 induction by benzo(a)pyrene. J. Cardiovasc. Pharmacol. 57, 273–281 10.1097/FJC.0b013e3182055baf [DOI] [PubMed] [Google Scholar]
- 55.Gross G.J., Gauthier K.M., Moore J., Falck J.R., Hammock B.D., Campbell W.B. et al. (2008) Effects of the selective EET antagonist, 14,15-EEZE, on cardioprotection produced by exogenous or endogenous EETs in the canine heart. Am. J. Physiol. Heart Circ. Physiol. 294, H2838–H2844 10.1152/ajpheart.00186.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nithipatikom K., Moore J.M., Isbell M.A., Falck J.R. and Gross G.J. (2006) Epoxyeicosatrienoic acids in cardioprotection: ischemic versus reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 291, H537–H542 10.1152/ajpheart.00071.2006 [DOI] [PubMed] [Google Scholar]
- 57.Seubert J., Yang B., Bradbury J.A., Graves J., Degraff L.M., Gabel S. et al. (2004) Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ. Res. 95, 506–514 10.1161/01.RES.0000139436.89654.c8 [DOI] [PubMed] [Google Scholar]
- 58.Seubert J.M., Zeldin D.C., Nithipatikom K. and Gross G.J. (2007) Role of epoxyeicosatrienoic acids in protecting the myocardium following ischemia/reperfusion injury. Prostaglandins Other Lipid Mediat. 82, 50–59 10.1016/j.prostaglandins.2006.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guglielmino K., Jackson K., Harris T.R., Vu V., Dong H., Dutrow G. et al. (2012) Pharmacological inhibition of soluble epoxide hydrolase provides cardioprotection in hyperglycemic rats. Am. J. Physiol. Heart Circ. Physiol. 303, H853–H862 10.1152/ajpheart.00154.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang H. and Eckel R.H. (2009) Lipoprotein lipase: from gene to obesity. Am. J. Physiol. Endocrinol. Metab. 297, E271–E288 10.1152/ajpendo.90920.2008 [DOI] [PubMed] [Google Scholar]
- 61.Dijk W. and Kersten S. (2014) Regulation of lipoprotein lipase by Angptl4. Trends Endocrinol. Metab. 25, 146–155 10.1016/j.tem.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 62.Kersten S. (2014) Physiological regulation of lipoprotein lipase. Biochim. Biophys. Acta 1841, 919–933 10.1016/j.bbalip.2014.03.013 [DOI] [PubMed] [Google Scholar]
- 63.Romeo S., Yin W., Kozlitina J., Pennacchio L.A., Boerwinkle E., Hobbs H.H. et al. (2009) Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J. Clin. Invest. 119, 70–79, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y., McNutt M.C., Banfi S., Levin M.G., Holland W.L., Gusarova V. et al. (2015) Hepatic ANGPTL3 regulates adipose tissue energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 112, 11630–11635 10.1073/pnas.1515374112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tikka A. and Jauhiainen M. (2016) The role of ANGPTL3 in controlling lipoprotein metabolism. Endocrine 52, 187–193 10.1007/s12020-015-0838-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu J., Afroza H., Rader D.J. and Jin W. (2010) Angiopoietin-like protein 3 inhibits lipoprotein lipase activity through enhancing its cleavage by proprotein convertases. J. Biol. Chem. 285, 27561–27570 10.1074/jbc.M110.144279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shimamura M., Matsuda M., Yasumo H., Okazaki M., Fujimoto K., Kono K. et al. (2007) Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler. Thromb. Vasc. Biol. 27, 366–372 10.1161/01.ATV.0000252827.51626.89 [DOI] [PubMed] [Google Scholar]
- 68.Minicocci I., Montali A., Robciuc M.R., Quagliarini F., Censi V., Labbadia G. et al. (2012) Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. J. Clin. Endocrinol. Metab. 97, E1266–E1275 10.1210/jc.2012-1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Helgadottir A., Gretarsdottir S., Thorleifsson G., Hjartarson E., Sigurdsson A., Magnusdottir A. et al. (2016) Variants with large effects on blood lipids and the role of cholesterol and triglycerides in coronary disease. Nat. Genet. 48, 634–639 10.1038/ng.3561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Musunuru K., Pirruccello J.P., Do R., Peloso G.M., Guiducci C., Sougnez C. et al. (2010) Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N. Engl. J. Med. 363, 2220–2227 10.1056/NEJMoa1002926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stitziel N.O., Khera A.V., Wang X., Bierhals A.J., Vourakis A.C., Sperry A.E. et al. (2017) ANGPTL3 deficiency and protection against coronary artery disease. J. Am. Coll. Cardiol. 69, 2054–2063 10.1016/j.jacc.2017.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ando Y., Shimizugawa T., Takeshita S., Ono M., Shimamura M., Koishi R. et al. (2003) A decreased expression of angiopoietin-like 3 is protective against atherosclerosis in apoE-deficient mice. J. Lipid Res. 44, 1216–1223 10.1194/jlr.M300031-JLR200 [DOI] [PubMed] [Google Scholar]
- 73.Dewey F.E., Gusarova V., Dunbar R.L., O’Dushlaine C., Schurmann C., Gottesman O. et al. (2017) Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N. Engl. J. Med. 377, 211–221 10.1056/NEJMoa1612790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Graham M.J., Lee R.G., Brandt T.A., Tai L.J., Fu W., Peralta R. et al. (2017) Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N. Engl. J. Med. 377, 222–232 10.1056/NEJMoa1701329 [DOI] [PubMed] [Google Scholar]