

Abstract
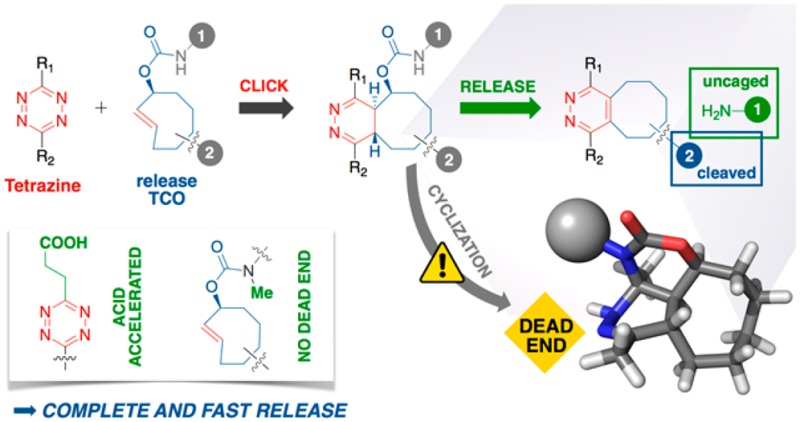
Recent developments in bond cleavage reactions have expanded the scope of bioorthogonal chemistry beyond click ligation and enabled new strategies for probe activation and therapeutic delivery. These applications, however, remain in their infancy, with further innovations needed to achieve the efficiency required for versatile and broadly useful tools in vivo. Among these chemistries, the tetrazine/trans-cyclooctene click-to-release reaction has exemplary kinetics and adaptability but achieves only partial release and is incompletely understood, which has limited its application. Investigating the mechanistic features of this reaction’s performance, we discovered profound pH sensitivity, exploited it with acid-functionalized tetrazines that both enhance and markedly accelerate release, and ultimately uncovered an unexpected dead-end isomer as the reason for poor release. Implementing facile methods to prevent formation of this dead end, we have achieved exceptional efficiency, with essentially complete release across the full scope of physiologic pH, potentiating drug-delivery strategies and expanding the dynamic range of bioorthogonal on/off control.
Introduction
Biosynthetic transformations that afford spatiotemporal control of molecular assembly and disassembly are essential and omnipresent features of the chemistry of living systems. Engineered chemical tools that can recapitulate these fundamental capabilities of ligation, cleavage, substitution, and chemical modification have only recently been envisioned, and their application for selective bioorthogonal chemistry, synthetic tools capable of activation, inactivation, and delivery in vivo, remains at an early stage. Whereas bioorthogonal ligation reactions1−7 inspired an early focus on labeling,8−11 biomolecular sensing/tracking,12−15 and molecular imaging,16−24 the current diversified effort to encompass both ligation and cleavage reactions25,26 has expanded the scope of applications in synthesis,27 diagnostics/therapeutics,25,28−30 and chemical biology31,32 tremendously (Figure 1a). Key characteristics needed for utility in click ligations still apply, with an emphasis on biocompatibility, triggering performance (i.e., the yield or efficiency of a cleavage event), and reaction kinetics.
Figure 1.

Environmental dependence of tetrazine-TCO click-to-release chemistry. (a) Bioorthogonal and catalyst-free, the tetrazine click reaction cleaves a TCO tag or linker from a captive amine, with diverse potential applications, including (i) activating a caged enzyme; (ii) releasing a drug conjugate; or (iii) inactivating a biomolecular assembly. (b) Tz/rTCO release chemistry; inert gases are the only byproduct. (c) Release probes used to quantify kinetics and yields across analytical and reaction conditions; key design features include embedded fluorophores for sensitive detection by absorbance or fluorescence and excellent water solubility. (d) Routine HPLC conditions with formic acid perturb the observed release in a time-dependent manner, relative to the true release yield observed with buffered LC conditions. (e) (i) Release yields vary as a function of reaction pH, with large differences between phosphate buffer at pH 7 and phosphate buffered saline (pH 7.4); (ii) this gap was a clue to a significant effect from buffer concentration; and (iii) further efforts to profile yields revealed a significant effect on release from even individual buffer species at matched pH. (f) pH dependence of release for 1+2 in citrate-phosphate buffer (pH 4–8), which provides a smooth composition gradient across its very broad working range, and in (*) bicarbonate-carbonate buffer at pH 9. Data are mean ± sd for two independent measurements.
In this light, tetrazine/trans-cyclooctene (Tz/TCO) click and release, key to many recent advances, stands out for its freedom from catalysts, exceptional reaction kinetics, nontoxic byproducts (N2 and CO2), and substantial chemical optimizability, enabling tuning of the reagent characteristics. Astute observations by Robillard and co-workers25 led to the initial insight that postclick tautomerization of 3-OH-functionalized TCO derivatives (release-TCO, rTCO) could drive the decarboxylative elimination of a caged amine. In this inverse electron demand Diels–Alder (IEDDA) pyridazine elimination (Figure 1b), tetrazine click functions as a bioorthogonal scissors to cleave a carbamate substituent from rTCO. This affords enormous potential for drug delivery, prodrug strategies, nanomaterials, antibody–drug conjugates, and small molecule conjugates, among other applications of in vivo release, provided that the performance of rTCO-based systems can achieve the necessary resilience and efficiency.26 Known liabilities include minimal release from bis-aryl and H-tetrazines (<5%), and variable release from aryl-alkyl and bis-alkyl tetrazines, ranging from 20% to 80% with Tz and context.25,29,31,33 While cleavage yields are relevant for potency and therapeutic index in activation-based strategies such as drug release, maximal cleavage is pivotal when near-quantitative biomolecular inactivation is desired for detoxification or conditional termination of a therapeutic function.
In the early development of this reaction, the chemical properties of minimalist prototype reagents and the concentration requirements of analytical instrumentation (i.e., LCMS, NMR) have frequently required analysis of reaction performance in the presence of significant amounts of organic solvent,25,31 which we judged worrisome, given the general environmental sensitivity of IEDDA reactions.34 To enable the design of better drug delivery and therapeutic control systems, we set out to characterize the performance of Tz-mediated click-to-release reactions under stringently defined conditions, beginning at physiologic pH, in fully aqueous buffered solution (≥98% water), with probes that were soluble and stable in this environment, and readily quantified by measuring the characteristic UV/vis absorption of the attached fluorophores (Figure 1c).
Results
Environmental Sensitivity and Analytical Artifacts
We compared click and release rates for the reaction of dimethyltetrazine (DMT, 1) and AlexaFluor350-rTCO (AF350-rTCO, 2, characteristic UV/vis absorption at 350 nm) in phosphate buffered saline (PBS, pH 7.4) as measured under the routine acidic LCMS conditions (acetonitrile–water, 0.1% formic acid) employed in prior studies25,31 with measurements made under buffered analytical LCMS conditions at pH 8.4. As is evident in Figure 1d, the apparent release rate and yield are dramatically higher in the acidic analytic environment (red). When we continued serial analyses of the same sample, however, we observed a paradoxical decline in apparent release over time, ultimately converging on the result obtained from buffered analytical conditions (blue) at equilibrium. Given the irreversibility of decarboxylative elimination of the free AF350-amine, the augmented release observed at early time points must be an artifact of the acidic analytical conditions. We therefore adopted buffered HPLC conditions for all further kinetic and end point analyses. Our subsequent investigation of the effect of reaction conditions on release yields (Figure 1e) using MayaFluor-rTCO41 (3, characteristic UV/vis absorption at 500 nm) revealed a profound environmental dependency on not only pH, but also organic solvent composition (data not shown), buffer concentration, and even the buffer species at matched pH. The exceptional working range of citrate-phosphate buffer (Cit-Phos) was required for a profile of release versus pH free of discontinuities, with release yields ranging from 95% at pH 4.2 to just 7% at pH 9 (Figure 1f).
Both the enhancement of release at higher buffer concentrations (characteristic of general acid–base catalysis) and the markedly increased release at lower pH likely reflect an impact on the rate of postclick tautomerization essential to release (Figure 2a), as per Robillard and co-workers.25 Our observation of time-dependent pH effects indicates that the ensemble of postclick intermediates is not static, but shifts from acid-sensitive to acid-insensitive species (from which release can no longer be induced) over time. The mechanistic basis of this effect in buffered/physiologic solution remains an important open question. Unfortunately, the nonreleased end-products of 1+2 (and 1+3) comprise a chromatographically inseparable mixture, including components matching the expected mass of both dihydropyridazine (e.g., A/B1/B2) and pyridazine (aromatized) derivatives, indicating a need for alternative probes and/or methods to unravel the competing pathways (vide infra).
Figure 2.

Tautomerization enhancement by acid-functionalized tetrazines (Tz-acids). (a) Proposed pathway leading to release after Tz-TCO click via 1,4-elimination from the B1 tautomer. Oxidation to the pyridazine (lower right) yields an aromatic core that ends tautomer exchange and abrogates further release; LCMS data indicate the existence of additional nonreleasing isomers of unknown structure/stereochemistry. (b) Newly synthesized Tz-acids. (c) Marked enhancement of release by Tz-acids in PBS, with complete release observed for PA2 (5). (d) Tz-acids enhance release relative to DMT across a broad pH range. Data are mean ± sd for two independent measurements. (e) The amino acid substituent lowers the pKa of Ala-MT by 0.4 pH units, contributing to decreased release relative to MPA. (f) LCMS profiles of release vs time at pH 5–7 demonstrate enhanced release at acidic pH for all four tetrazines, and biphasic release for MPA and Ala-MT, with an initial very fast phase followed by a pH-dependent phase. (g) Proposed mechanism for the tautomerization-directing effect of the propanoic acid substituent of MPA. (h) Click orientation determines fast and pH-dependent release, schematized as in (a), in which the weight of the arrow signifies the relative rate of reaction.
Acid-Enhanced Release
Given the high desirability of click-and-release tools that achieve complete release under the broadest range of relevant conditions, we sought to apply our insights to the development of new tetrazines. Enhanced release in acidic environments and at higher buffer concentration suggested a potential benefit for embedding a proton donor vicinal to the tautomerization sites. We therefore prepared acid-functionalized tetrazines (Figure 2b) starting from the corresponding carboxylic acid nitriles (for synthesis and click kinetics, see the Supporting Information). Gratifyingly, reaction of compounds 4–6 with AF350-rTCO (2) in PBS revealed improved performance by all three Tz-acids relative to DMT (Figure 2c). Further studies in Cit-Phos buffer revealed markedly enhanced release by Tz-acids across the entire pH range (Figure 2d), with full release observed for the bis-propanoic acid functionalized tetrazine (PA2, 5) from pH 4.2–8.0. A single acid substituent yielded an intermediate effect, with superior performance by MPA (4) relative to the amino acid Tz (Ala-MT, 6). The enhanced release by MPA is likely driven in part by steric factors, as well as a difference in the carboxylic acid pKa (Figure 2e).
Subsequent LCMS studies of release versus time and reaction pH proved mechanistically informative (Figure 2f). Both of the symmetric tetrazines (1 and 5) displayed single exponential kinetics, with progressively accelerated and augmented release at lower pH; PA2 release is essentially instantaneous at pH ≤ 6, too rapid to reliably quantify by LCMS. In contrast, MPA and Ala-MT release is strikingly biphasic. For MPA, the initial fast phase of release (constant at ∼50%) was much too rapid to quantify by LCMS, followed by a pH-dependent phase that exhibited a 30-fold acceleration as the pH was lowered from 7.0 to 5.0. Ala-MT exhibited a slightly lower fraction of fast release (∼40%) at a rate that was measurable at pH 7 and thereafter too fast to quantify. On this basis, we infer that (i) random click orientation is balanced for MPA and nearly so for Ala-MT; (ii) click orientation directs tautomerization via local general-acid catalysis; and (iii) ultrafast release depends on proximity between solution pH and acid pKa. A representative scheme in Figure 2g illustrates carboxylic acid-facilitated protonation of the neighboring dihydropyridazine nitrogen. Under this scenario, the click reaction proceeds in either of two orientations (Figure 2h): head-to-head click of MPA drives preferential formation of the B1 (releasing) tautomer and accounts for the observed extremely fast release; head-to-tail click, in contrast, directs predominant, although not exclusive, formation of the B2 (nonreleasing) tautomer. Accordingly, the observed pH-dependent phase of release likely corresponds to the rate of tautomer interconversion back to B1 (via A), when spontaneous (nonfacilitated) protonation leads to the formation of the other tautomer.
Measurement of Ultrafast Release Rates
Given the potential utility of ultrafast click-and-release in various applications, including imaging, we devised an optical method for real-time reporting and direct quantification of release rates. Synthesis of the black-hole-quencher (BHQ)-tetrazine conjugates 7–9 (Figure 3a) yielded analogues for DMT, Ala-MT, and MPA, respectively, with excellent water solubility and efficient quenching of fluorophores in the green spectral range. As schematized in Figure 3b, upon click reaction with 3, fluorescence dynamics allow quantification of reaction rates. To cleanly segregate the three components of this sequence for kinetic analysis, the click reaction rate must be high enough to cleanly resolve the quenching phase from the restoration of fluorescence (upon release) for accurate nonlinear fitting. Conservative estimates of the minimum release rates compatible with the LCMS data in Figure 2f implied that BHQ-Tz concentrations in the millimolar range would be necessary, prohibitively high for conventional cuvette-based fluorescence measurements; consequently, a specialized cuvette with a triangular cross section that enables surface excitation/emission geometry was required (Figure 3c).35−37
Figure 3.

Tz acids mediate ultrafast release. (a) Black hole quencher-Tz probes for kinetics. (b) FRET assay mechanism: BHQ-Tz efficiently quenches the MayaFluor emission after click reaction with 3; fluorescence is restored upon release of the MayaFluor amine, allowing direct quantification of fast release. (c) Analytical configuration for the kinetic experiments, employing a triangular cuvette due to the very high absorbance of the BHQ-Tz at the mM concentrations required to temporally resolve click and release. (d) Fluorescence intensity vs time profiles for the reaction of 3 and 7; dual exponential fits of the click and release phases are superimposed for the four independent experiments at varying reaction pH. (e) Release rates are strikingly pH dependent, increasing ∼65-fold between pH 7.4 and pH 4 in tandem with release yields (corresponding data in gray from Figure 1e). (f) Release rates for 7–9 at pH 7.4, with respective half-lives superimposed; the PA-group accelerates release by nearly 2 orders of magnitude. Data are mean ± sd for three independent measurements. (g) Release for 8 and 9 is further accelerated by lower pH; *the reported rate for PA-directed release at pH 6 is a minimum estimate, as release is too fast to fully resolve.
Fluorescence intensity versus time profiles for the reaction of 7 and 3 display the desired clean, multiphasic response, with the signal spiking upon addition of 3 before falling and then recovering with a cleanly fitted biphasic profile (Figure 3d). Plotted as a function of pH in Figure 3e, the observed rate of release tracks well with the release fraction for the reaction of 1+2 (secondary Y-axis). The corresponding data for 8 and 9 allowed quantification of the acceleration afforded by the acid-enhanced tetrazines: at physiologic pH, Ala-directed release (8+3) is 4-fold faster, while the PA-directed release (9+3) is nearly 2 orders of magnitude faster than a Me-group as the release directing substituent (Figure 3f). The comparative acceleration increases further at lower pH ranges (Figure 3g). At pH 6.0, the synergistic effects of the PA substituent (9) and acidic pH rendered release again too fast to measure, with release rates ≥ click rates even at 4 mM BHQ-Tz. This set a lower limit of 0.21 s–1, at least 600-fold higher than the release rates observed for 7 at pH 7.4, and corresponding to a t1/2 < 3.3 s. The ultrafast PA-directed release in the physiologic pH range may enable a range of applications in biomolecular switching, optical monitoring of click reactions, and/or new methods in quantitative fluorescence microscopy.
Mechanisms of Nonrelease
Having established the enhanced yield and accelerated kinetics of Tz-acid-mediated release, we returned our attention to characterizing the molecular events that govern release/nonrelease. Glycine-rTCO (10) trades the detectability and ready quantitation of the fluorophore-labeled probes (2, 3) for superior ease of analysis by NMR and the potential for chromatographic discrimination between isomers/tautomers by their UV spectra. We first adopted 10 as an NMR probe in a kinetically and pH-controlled experiment (Figure 4) to isolate the slower-releasing tautomer and thereby validate the hypothesis of orientation-controlled tautomerization (vide supra). The reaction of 10 with MPA (4) at pH 6.0 affords an isomeric mixture of fast and slow-releasing species; 15 min at this pH allows complete click and elimination of all of the fast-releasing isomer. Thereafter, we raised the pH of the mixture to slow the rate of pH-dependent release and transferred the sample to a well-aerated flask under an oxygen atmosphere; pH 7.6 proved optimal for balancing minimizing release and facilitating aromatization, the rate of which falls off rapidly with rising pH. After 72 h at 4 °C, LCMS indicated near-complete conversion to an aromatized species. Purification of this compound by reverse phase chromatography and structural assignment by NMR (Supporting Information) confirmed the head-to-tail orientation of the MPA-pyridazine (Figure 4, at right), as we had inferred from the biphasic kinetic data. Conclusively identifying the click-orientation of the fast- and slow-releasing species also supports the underlying B1/B2 tautomer hypothesis and directing effect of the PA substituent in tautomerization, that is, in facilitating protonation of the adjacent dihydropyridazine nitrogen.
Figure 4.

Elucidation of the slowly releasing tautomer. Glycine-rTCO (10) was reacted with MPA (4) under sequential controlled conditions to isolate the slowly releasing tautomer for structural characterization, verifying the head-to-tail product by NMR.
Encouraged by the utility of 10 in NMR studies, we next turned to characterization of the DMT-rTCO click byproducts. After optimization, the mixture of Gly-rTCO-DMT click products (1+10) proved more readily separated than the fluorophore-labeled rTCO reactions had been, allowing a working identification of each peak on the basis of mass, UV spectrum (no longer obscured by the fluorophore absorbance), and the kinetics of its formation/disappearance (Supporting Information). Serial chromatograms in Figure 5a(i) reveal that the initial click product (tautomer A) is replaced by an isometric species with a near-identical elution time to A. This nonreleasing dead-end is indistinguishable from A and B2 by ESI–/ESI+ spectra but has a distinct UV absorbance spectrum (Figure 5a(ii)). Although quantification of the unlabeled species is approximate given the lack of a consistent chromophore, release yields for other probes (e.g., 2, 3) under these reaction conditions indicate that the unidentified compound is the major product of the click reaction in PBS, corroborated by evaporative light scattering data (Supporting Information). Serial analyses revealed a matching time-course for the disappearance of A and the formation of this nonreleasing dead-end (Figure 5a(iii)), indicating that it is formed relatively early in the reaction sequence, potentially arising directly from A (rather than B2).
Figure 5.

Identification of an unexpected dead-end isomer. (a) (i) Serial chromatograms for the reaction of 1+10; the initial product A is replaced by a closely adjacent peak that appears immediately (t = 3 min), is best resolved at t = 60 min, and does not release. (ii) This unidentified dead end is isometric to A/B1/B2 but has a distinct absorbance spectrum; and (iii) is the major product of the reaction, arising synchronously with the disappearance of A. (b) NMR studies of the purified material revealed an unexpected tricyclic compound (11) that is broadly stable; geometry optimization using Gaussian (M06-2X/6-311+G(d,p)) allows visualization of the proximity of the carbamate nitrogen to the dihydropyridazine ring.
Scaling up the reaction, we isolated the dead-end for characterization by NMR (Supporting Information). To our intrigue, the structure revealed the unexpected intramolecular cyclization product 11 (Figure 5b). This compound is bench stable, acid stable, and extremely slow to oxidize. Formation of this tricyclic dead-end isomer accelerates at higher pH, while the tautomerization and release rates decline, countervailing trends that account for the sharp pH dependence of release yields. To verify that this compound was not unique to glycine, we repeated the experiment with the corresponding N-ε-rTCO-lysine probe, which also yielded a tricyclic dead-end corresponding to 11 (Supporting Information). The fact that an unexpected intramolecular cyclization is the basis of failed release offers an immediate and straightforward option to enhance rTCO performance (Figure 6a). Recognizing that cyclization would be impossible for a tertiary amide, we synthesized sarcosine-rTCO (12) and tested its release performance upon reaction with DMT in PBS. As desired, no tricyclic dead-end formation was observed, and release performance was accordingly significantly enhanced relative to Gly-rTCO (Figure 6b). To quantify the release yields, we turned to NMR experiments in fully deuterated PBS (pH 7.4), which revealed essentially complete release for Sar-rTCO (Figure 6c).
Figure 6.

Blocking dead-end formation with an N-Me group enables complete release. (a) Sarcosine-rTCO (12) is unable to cyclize. (b) Click with DMT confirms the absence of tricycle formation and demonstrates enhanced release. (c) Quantification of DMT-mediated release yields by NMR in deuterated phosphate buffer (equivalent to pH 7.4) reveals complete release for 12.
Cleavage of Multifunctional TCO Linkers
To confirm the generality of the benefit of Tz-acids and N-Me substitution, we extended our investigation from rTCO to a second generation multifunctionalized TCO linker, schematized in Figure 7a.29 Equipped with a carboxylic acid side chain, this second member of the 3-OH-substituted releasing TCO family allows the TCO to function as an integrated element within a bioorthogonally cleavable linker. As such, this tetrazine-cleavable TCO (cTCO) can serve as a versatile linker for multiple biochemical species (Figure 7a). We chose to explore two scenarios with distinct requirements: (i) applications in which implementation of N-substitution (N-Me) on the linking nitrogen is not feasible or not desirable, mandating tetrazine optimization; and (ii) applications in which an N-Me group can be added, blocking any potential dead-end formation and allowing free choice of the tetrazine (e.g., DMT). We prepared the corresponding AF350-tagged glycine (13) and sarcosine (14) cleavage probes and assessed their cleavage performance with DMT and PA2 (Figure 7b). At physiologic pH, cleavage of AF350-cTCO-Gly (13) with PA2 improves the yield 2.4-fold relative to DMT, reaching 95% via Tz-acid enhancement. Meanwhile, conversion to the N-Me-substituted 14 yielded complete cut at physiologic pH with DMT. In the course of these experiments, we were intrigued to note that the cleavage/release rate for the cTCO probes is significantly faster than the corresponding rTCO derivatives under matching conditions (Figure 7c). Further study will be required to delineate the mechanistic interplay of tautomerization and release that accounts for this acceleration.
Figure 7.

Expanding enhanced click-to-release to bioorthogonal cut. (a) Release TCO variants with a second handle on the TCO ring (cleavable TCO, cTCO) function as Tz-cleavable linkers, capable of bioorthogonal cut reactions with a diverse range of potential attachments. AF350-cTCO-cleavage probes 13 and 14 allow quantitation of release kinetics and the impact of N-Me substitution. (b) At pH 7.4, PA2 markedly enhances cleavage of cTCO-Gly, validating the Tz-acid enhancement for unmodified amino linkers; cTCO-Sar achieves complete release. Data are mean ± sd for two independent measurements. (c) Release kinetics are markedly faster for cTCO-Sar than for rTCO-Sar. (d) (i) Extended profiles of release vs pH reveal the global improvement in cleavage for PA2 and near-universal cleavage of 14 with DMT, independent of pH. (ii) Cleavage kinetics for AF350-cTCO-Sar + DMT as a function of pH demonstrate complete scission of the linker. (iii) LC traces for 13+DMT reveal the pH-dependent formation of a dead-end isomer. (e) Click reaction of the unlabeled cTCO-Sar (R1 = H) at pH 10 affords a single dead-end product (clicked but not cut); NMR confirmed the tricyclic dead-end 15. (f) Minimizing or eliminating dead-end formation, routes to enhanced cleavage reactions releasing (i) primary amines (Tz-acid) or (ii) N-methylated amines.
We went on to assess the impact of Tz-acid enhancement and N-Me substitution as a function of pH and were gratified to observe that the reaction of the sarcosine probe 14 and DMT achieves complete or near complete cleavage independent of pH (Figure 7d(i)). The cleavage rate was pH dependent, as for rTCO, and rapid, reaching 80% within 10 min at pH 5 and 90% within 100 min at pH 7.4, with a second phase of the reaction completing the process to achieve complete cut (Figure 7d(ii)). Meanwhile, cleavage of the glycine probe 13 recapitulates the Tz-acid effect seen with rTCO, with cleavage of 13 remaining near-complete at pH 8. At higher pH values, the degree of cleavage could not be quantified, as an unreleased byproduct coeluted with the cleaved linker, even on extended gradients. In contrast, the reaction of 13 and DMT was strongly pH dependent, with release falling off from near 100% at pH 4 to zero at pH 9. Careful chromatography enabled quantification of the transition between cleavage and a nonreleasing dead-end, isometric to the dihydropyridazine (Figure 7d(iii)). Although the AF350 tag obviates assessment of the characteristic dead-end absorbance spectrum, the formation of a nonreleasing product under basic conditions strongly suggested tricyclic dead-end formation, (i.e., the cTCO equivalent of 11). We therefore exploited the clean conversion at higher pH to verify the basis of dead-end formation for cTCO, reacting the unlabeled precursor to 13 with DMT in pH 10 buffer (Supporting Information). Isolation of the click product by reverse-phase chromatography and characterization by NMR confirmed the formation of the tricyclic dead-end 15 (Figure 7e). Prevention of intramolecular cyclization is central to the performance of both Tz-acids, which reduce it dramatically by accelerating the conversion to B1/B2, and to N-Me substitution, which blocks it completely (Figure 7f). Consolidating these observations suggests a concise format for visualization of the postclick reaction network mapped by this work, organized geometrically as a cube that places the four intermediate species in a top layer and the three final products below (Figure 8); the cube framework also suggests a simplified notation for rendering the comparative impact of present and future tools on click-to-release yields (Supporting Information).
Figure 8.

Postclick reaction network, a schematic overview of interconversion and release. The cube diagram spatially segregates intermediates (upper tier) from final products (lower tier), with annotations indicating key factors impacting release/cleavage. Oxidation of the tricyclic dead end (lower left edge) is very slow under ambient conditions, requiring weeks even under an oxygen atmosphere. Structural features intrinsic to the TCO ring system that bias tautomerization to favor formation of B1 rather than B2 are an avenue for further study.
Discussion
Bioorthogonal reactions capable of controlling molecular function in living systems face a demanding set of performance requirements, including fast reaction kinetics to allow on demand triggering at acceptably low concentrations, chemical flexibility (connecting and disconnecting a wide range of species), and excellent cleavage efficiency. Tetrazine/TCO reactions as a class have exemplary reaction kinetics, but other aspects of the click-to-release reaction had not been fully characterized under biologically relevant conditions. We learned in the course of our investigations that the release rate (after click) and the release/cleavage yield are extremely environmentally sensitive, with comparatively small changes in the reaction conditions or use of routine acidic chromatography yielding significant alterations in performance, in no small part due to the unexpected ability of the system to form the tricyclic dead-end isomer, which interrupts the expected tautomeric interconversion/release. Environmental susceptibility thus mandates cautious interpretation of click-to-release model systems and needs to be taken into account when designing experiments and probes to characterize new click-to-release tools. Conversely, deliberate modulation of experimental/environmental conditions offers promising strategies for catch and release applications or other strategies in which the slow release rate at high pH and fast release rate at mildly acidic pH (e.g., pH ≤ 6) can be exploited to control the pace of molecular disassembly.
As work to advance and apply bioorthogonal cleavage reactions continues, one key avenue of investigation will be the extension of this framework to elucidate the performance of aryl tetrazines to maximize kinetics for in vivo applications, work currently in process. Collectively, these insights advance Tz-TCO click-to-release from a reaction of uncertain mechanism and limited performance into a tool with not only the high fidelity required for efficient chemical biology probes, therapeutic activation, or drug delivery, but also the broad dynamic range necessary for applications that rely on molecular inactivation or conversion, that is, temporal control of biomolecular fusions, detoxification, or other termination of action, where residual activity needs to be minimized (Figure 1a(iii)). Finally, the discovery that Tz-acids enable ultrafast release for head-to-head click suggests further innovations in Tz and TCO design for rapid molecular activation, inactivation, and/or cleavage in a diverse range of bioorthogonal tools.
Experimental Section
Materials and Methods
Detailed synthetic methods and characterization data are presented in full in the Supporting Information, including additional presentation of 2D NMR data confirming the structural assignment for compounds 11 and 15.
LCMS Quantification of Release Yield and Rates
High performance liquid chromatography–mass spectrometry analysis (HPLC–MS, LCMS) was performed with a Waters instrument. See the Supporting Information for a full description of the instrumentation. Stock solutions of rTCO and cTCO release probes were prepared in DMSO and diluted into buffer at the reported pH, ensuring a final DMSO concentration in the sample of less than 2%, followed by addition of the tetrazine to trigger the click and release reaction. Tetrazine (11, 4–6) stock solutions were prepared in DMSO at concentrations ranging from 85 to 170 mM; Tz concentration in the analytical samples was 200 μM for all end point release experiments. For release kinetics, the concentration of 1 was kept constant at 200 μM, while the concentration of 4–6 was increased to 400 μM to ensure that the click reaction was complete within 10–20 min to allow monitoring of early time points. Chromophore peak absorbance wavelengths were selected from the diode array data, and the chromatogram was integrated.
NMR Quantification of Release Yield
A 100 mM deuterated phosphate buffer was prepared by dissolving potassium dideuterium phosphate KD2PO4 [98% D] (Cambridge Isotope Laboratories) in D2O [99.9% D] (Sigma-Aldrich) and adjusting the pH* to 7.51 by addition of sodium deuteroxide (40 wt % in D2O [99% D]). This pH* equates to a pH of 7.4, as reported by Krȩżel and Bal.38 Stock solutions of Gly-rTCO (10) and Sar-rTCO (12) were prepared in DMSO-d6 [99.9% D] at a concentration of 150 mM. 100 mM deuterated phosphate buffer was diluted to 10 mM with D2O. Ten microliters of 100 mM Gly-rTCO (10) and 100 mM Sar-rTCO (12) were added to 990 μL of 10 mM deuterated phosphate buffer containing 1 mM DMF (as internal standard), respectively, to afford a concentration of 1.5 mM of the rTCO probe for the click-to-release experiment. pH* was readjusted to pH* 7.51 = pH 7.4 by addition of sodium deuteroxide (see section B.i in the Supporting Information). 1H NMR spectra were recorded (128 scans), and the ratio of rTCO probe to DMF was calculated on the basis of the CH-signal of DMF at 7.92 ppm and the CH-signals of rTCO at ∼5.75 and ∼5.50 ppm. DMT (1) was dissolved in DMSO-d6 at a concentration of 1 M. One microliter of this solution was added to the rTCO samples to afford a concentration of 1 mM DMT (1). Immediate click reaction was observed (disappearance of pink color). Samples were stored in the dark at room temperature for 36 h before recording 1H NMR spectra (256 scans) to obtain end point values.
BHQ-Tz Experiments
The sample chamber of the PTI-QuantaMaster 400 fluorimeter was equipped with dichroic bandpass filters (OD > 5.0 for rejected wavelengths; Thorlabs) to eliminate passage of reflected/scattered light off the face of the triangular cuvette (see below). BHQ-Tz probes 7–9 were dissolved in water to prepare concentrated stock solutions. Concentrations were established by absorbance measurement of serially diluted samples in phosphate buffer pH 7.0 (50 mM). MayaFluor-rTCO (3) was diluted from the parent DMSO stock into acetonitrile at 100 μM for more rapid mixing with the aqueous samples upon addition to the fluorimeter cuvette. Into a triangular cuvette (model 24-SB-Q-10, Starna Cells) with a magnetic stir bar were added first the buffer solution and then the BHQ-Tz (7–9) to a final concentration of 1, 2, or 4 mM in 600 μL total volume. For BHQ-Tz-acids 8 and 9, the pH of the solution was checked by pH micro electrode to verify adequate buffering capacity. Data acquisition was started; after collecting a stable baseline, 6 μL of the MeCN stock solution of 3 was added via the instrument’s sample addition port (for a final concentration of MayaFluor-rTCO at 1 μM, and final MeCN concentration ≤1%), with continuous stirring and ongoing data collection. Higher concentrations of BHQ-Tz (2 and 4 mM) were used at lower pH values to accelerate the pseudo-first-order click rate for kinetic partitioning of click and release. The fluorescence traces of intensity versus time were exported to Graphpad Prism 6.0/7.0 (Graphpad Software) for double exponential nonlinear fitting of fluorescence intensity versus time.
Isolation of Slow Releasing Isomers
See the Supporting Information for full methodologic details. In brief, the rTCO or cTCO probes were diluted into buffer at the desired pH and minimal DMSO concentration (<2%) followed by the addition of the Tz to initiate the reaction. At the indicated time/conditions, the click products were isolated and purified by reverse phase chromatography for characterization.
Structural Modeling
Geometry optimization and quantum-chemical calculations at the M06-2X/6-311+G(d,p) level39 were performed using Gaussian 09.40
Acknowledgments
This work was supported by the National Institutes of Health (RO1 EB010011 J.C.T.C. and R.W.) and the Austrian Science Fund (FWF, J 3586-B23 to H.M.). We thank Jenna Klubnick, Andres Fernandez Del Castillo, Christian Hametner, Stefan Kronister, Philipp Skrinjar, and Walter Kuba for their kind support of this work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b11217.
Synthetic procedures, click kinetics, release experiments, detailed description of the isolation and identification of slowly releasing and dead-end isomers, characterization data, NMR spectra, and Cartesian coordinates of calculated optimized geometries (PDF)
Author Contributions
∥ J.C.T.C. and H.M. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Sletten E. M.; Bertozzi C. R. Angew. Chem., Int. Ed. 2009, 48 (38), 6974–6998. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Acc. Chem. Res. 2011, 44 (9), 666–676. 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson D. M.; Nazarova L. A.; Prescher J. A. ACS Chem. Biol. 2014, 9 (3), 592–605. 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- Ramil C. P.; Lin Q. Chem. Commun. 2013, 49 (94), 11007–11022. 10.1039/c3cc44272a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. J. Am. Chem. Soc. 2008, 130 (41), 13518–13519. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj R.; Fox J. M. Curr. Opin. Chem. Biol. 2013, 17 (5), 753–760. 10.1016/j.cbpa.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Trout W. S.; Liu S.; Andrade G. A.; Hudson D. A.; Scinto S. L.; Dicker K. T.; Li Y.; Lazouski N.; Rosenthal J.; Thorpe C.; Jia X.; Fox J. M. J. Am. Chem. Soc. 2016, 138 (18), 5978–5983. 10.1021/jacs.6b02168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay Craig S.; Finn M. G. Chem. Biol. 2014, 21 (9), 1075–1101. 10.1016/j.chembiol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell-Verduyn L. S.; Mirfeizi L.; Schoonen A. K.; Dierckx R. A.; Elsinga P. H.; Feringa B. L. Angew. Chem., Int. Ed. 2011, 50 (47), 11117–20. 10.1002/anie.201105547. [DOI] [PubMed] [Google Scholar]
- Lang K.; Chin J. W. Chem. Rev. 2014, 114 (9), 4764–4806. 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- Li Z.; Cai H.; Hassink M.; Blackman M. L.; Brown R. C. D.; Conti P. S.; Fox J. M. Chem. Commun. 2010, 46 (42), 8043–8045. 10.1039/c0cc03078c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haun J. B.; Devaraj N. K.; Hilderbrand S. A.; Lee H.; Weissleder R. Nat. Nanotechnol. 2010, 5 (9), 660–665. 10.1038/nnano.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K.; Weissleder R. Acc. Chem. Res. 2011, 44 (9), 816–827. 10.1021/ar200037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Cisneros B. T.; Cole C. M.; Devaraj N. K. J. Am. Chem. Soc. 2014, 136 (52), 17942–17945. 10.1021/ja510839r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Alexander S. C.; Jin S.; Devaraj N. K. J. Am. Chem. Soc. 2016, 138 (36), 11429–11432. 10.1021/jacs.6b01625. [DOI] [PubMed] [Google Scholar]
- Baskin J. M.; Prescher J. A.; Laughlin S. T.; Agard N. J.; Chang P. V.; Miller I. A.; Lo A.; Codelli J. A.; Bertozzi C. R. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (43), 16793–16797. 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K.; Weissleder R.; Hilderbrand S. A. Bioconjugate Chem. 2008, 19 (12), 2297–2299. 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughlin S. T.; Baskin J. M.; Amacher S. L.; Bertozzi C. R. Science 2008, 320 (5876), 664–667. 10.1126/science.1155106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin R.; Renart Verkerk P.; van den Bosch S. M.; Vulders R. C. M.; Verel I.; Lub J.; Robillard M. S. Angew. Chem., Int. Ed. 2010, 49 (19), 3375–3378. 10.1002/anie.200906294. [DOI] [PubMed] [Google Scholar]
- Yang J.; Šečkutė J.; Cole C. M.; Devaraj N. K. Angew. Chem., Int. Ed. 2012, 51 (30), 7476–7479. 10.1002/anie.201202122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeglis B. M.; Sevak K. K.; Reiner T.; Mohindra P.; Carlin S. D.; Zanzonico P.; Weissleder R.; Lewis J. S. J. Nucl. Med. 2013, 54 (8), 1389–96. 10.2967/jnumed.112.115840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson J. C. T.; Meimetis L. G.; Hilderbrand S. A.; Weissleder R. Angew. Chem., Int. Ed. 2013, 52 (27), 6917–6920. 10.1002/anie.201301100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk C.; Svatunek D.; Mairinger S.; Stanek J.; Filip T.; Matscheko D.; Kuntner C.; Wanek T.; Mikula H. Bioconjugate Chem. 2016, 27 (7), 1707–12. 10.1021/acs.bioconjchem.6b00234. [DOI] [PubMed] [Google Scholar]
- Vázquez A.; Dzijak R.; Dračínský M.; Rampmaier R.; Siegl S. J.; Vrabel M. Angew. Chem., Int. Ed. 2017, 56 (5), 1334–1337. 10.1002/anie.201610491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteegen R. M.; Rossin R.; ten Hoeve W.; Janssen H. M.; Robillard M. S. Angew. Chem., Int. Ed. 2013, 52 (52), 14112–14116. 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- Li J.; Chen P. R. Nat. Chem. Biol. 2016, 12 (3), 129–137. 10.1038/nchembio.2024. [DOI] [PubMed] [Google Scholar]
- Agustin E.; Asare Okai P. N.; Khan I.; Miller M. R.; Wang R.; Sheng J.; Royzen M. Chem. Commun. 2016, 52 (7), 1405–1408. 10.1039/C5CC05392G. [DOI] [PubMed] [Google Scholar]
- Matikonda S. S.; Orsi D. L.; Staudacher V.; Jenkins I. A.; Fiedler F.; Chen J.; Gamble A. B. Chem. Sci. 2015, 6 (2), 1212–1218. 10.1039/C4SC02574A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin R.; van Duijnhoven S. M. J.; ten Hoeve W.; Janssen H. M.; Kleijn L. H. J.; Hoeben F. J. M.; Versteegen R. M.; Robillard M. S. Bioconjugate Chem. 2016, 27 (7), 1697–1706. 10.1021/acs.bioconjchem.6b00231. [DOI] [PubMed] [Google Scholar]
- Mejia Oneto J. M.; Khan I.; Seebald L.; Royzen M. ACS Cent. Sci. 2016, 2 (7), 476–482. 10.1021/acscentsci.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Jia S.; Chen P. R. Nat. Chem. Biol. 2014, 10 (12), 1003–1005. 10.1038/nchembio.1656. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Li J.; Xie R.; Fan X.; Liu Y.; Zheng S.; Ge Y.; Chen P. R. ACS Cent. Sci. 2016, 2 (5), 325–331. 10.1021/acscentsci.6b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X.; Ge Y.; Lin F.; Yang Y.; Zhang G.; Ngai W. S. C.; Lin Z.; Zheng S.; Wang J.; Zhao J.; Li J.; Chen P. R. Angew. Chem., Int. Ed. 2016, 55 (45), 14046–14050. 10.1002/anie.201608009. [DOI] [PubMed] [Google Scholar]
- Wijnen J. W.; Zavarise S.; Engberts J. B. F. N.; Charton M. J. Org. Chem. 1996, 61 (6), 2001–2005. 10.1021/jo9518563. [DOI] [Google Scholar]
- Courtis A. M.; Santos S. A.; Guan Y.; Hendricks J. A.; Ghosh B.; Szantai-Kis D. M.; Reis S. A.; Shah J. V.; Mazitschek R. Bioconjugate Chem. 2014, 25, 1043–1051. 10.1021/bc400575w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz J. R.Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Singapore, 2006. [Google Scholar]
- Kwapiszewski R.; Szczudlowska J.; Kwapiszewska K.; Dybko A.; Brzozka Z. Anal. Bioanal. Chem. 2014, 406 (18), 4551–4556. 10.1007/s00216-014-7844-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco M. E.; Bruzzone L. Anal. Methods 2013, 5 (24), 6908–6910. 10.1039/c3ay41599f. [DOI] [Google Scholar]
- Krȩżel A.; Bal W. J. Inorg. Biochem. 2004, 98 (1), 161–166. 10.1016/j.jinorgbio.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Yang Y. F.; Liang Y.; Liu F.; Houk K. N. J. Am. Chem. Soc. 2016, 138 (5), 1660–1667. 10.1021/jacs.5b12054. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.