

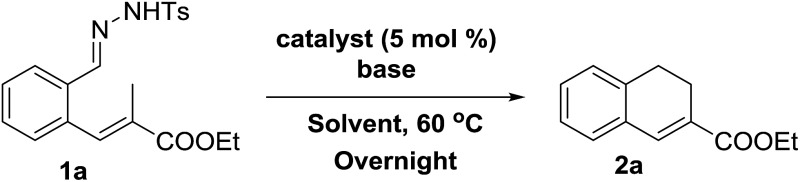
 Catalytic synthesis of substituted 1,2-dihydronaphthalenes via metalloradical activation of o-styryl N-tosyl hydrazones is presented. Substrates with an alkyl substituent on the allylic position reacted to form E-aryl-dienes rather than the expected 1,2-dihydronaphthalenes.
Catalytic synthesis of substituted 1,2-dihydronaphthalenes via metalloradical activation of o-styryl N-tosyl hydrazones is presented. Substrates with an alkyl substituent on the allylic position reacted to form E-aryl-dienes rather than the expected 1,2-dihydronaphthalenes.
Abstract
Catalytic synthesis of substituted 1,2-dihydronaphthalenes via metalloradical activation of o-styryl N-tosyl hydrazones ((E)-2-(prop-1-en-1-yl)benzene-N-tosyl hydrazones) is presented, taking advantage of the intrinsic reactivity of a cobalt(iii)–carbene radical intermediate. The method has been successfully applied to a broad range of substrates with various R1 substituents at the aromatic ring, producing the desired ring products in good to excellent isolated yields for substrates with an R2 = COOEt substituent at the vinylic position (∼70–90%). Changing the R2 moiety from an ester to other substituents has a surprisingly large influence on the (isolated) yields. This behaviour is unexpected for a radical rebound ring-closure mechanism, and points to a mechanism proceeding via ortho-quinodimethane (o-QDM) intermediates. Furthermore, substrates with an alkyl substituent on the allylic position reacted to form E-aryl-dienes in excellent yields, rather than the expected 1,2-dihydronaphthalenes. This result, combined with the outcome of supporting DFT calculations, strongly points to the release of reactive o-QDM intermediates from the metal centre in all cases, which either undergo a 6π-cyclisation step to form the 1,2-dihydronaphthalenes, or a [1,7]-hydride shift to produce the E-aryl-dienes. Trapping experiments using TEMPO confirm the involvement of cobalt(iii)–carbene radical intermediates. EPR spectroscopic spin-trapping experiments using phenyl N-tert-butylnitrone (PBN) confirm the radical nature of the catalytic reaction.
Introduction
A diverse range of transition-metal complexes have been shown to catalyse C–C coupling reactions, providing valuable alternatives to traditional methods for carbon–carbon bond formation.1 However, the use of catalytic C–C coupling reactions for the synthesis of fused ring systems remains challenging,2 in particular those producing partially unsaturated, conjugated but not fully aromatic fused 6-membered ring-systems such as 1,2-dihydronaphthalenes.3
The 1,2-dihydronaphthalene ring system is present in various natural products of therapeutic importance, including: cannabisins 48, isolated from the fruits of Cannabis sativa,4 6,7-dehydrosempervirol,5 isolated from the roots of Salvia apiana and negundin B,6 isolated from the roots of Vitex negundo. Nafoxidene is a class of biologically active dihydronaphthalene and its analogues can be prepared from 1-(4-benzyloxyphenyl)-6-methoxy-2-phenyl-3,4-dihydronaphthalene.7
Dihydronaphthalene derivatives are used as fluorescent ligands for the estrogen receptor8 and exhibit activity as Hepatitis C NS5B polymerase inhibitors.9 Recently, dihydronaphthalenes were found to be potent and selective inhibitors of aldosterone synthase (CYP11B2) for the treatment of congestive heart failure and myocardial fibrosis (Fig. 1).10 Dihydronaphthalene derivatives are also useful starting materials for the synthesis of several biologically active cyclic molecules.11 Many strategies towards these compounds have been published, of which dearomatization of the fully aromatic naphthalene substrate by organometallic reagents is perhaps one of the most useful and convenient methods.12 However, this approach requires the use of naphthalenes containing electron-withdrawing substituents (e.g. oxazolino or imino groups) as starting materials to enhance their electrophilicity, which limits the substrate scope.
Fig. 1. Representative natural products and drug molecules containing 1,2-dihydronaphthalene substructures.

Organometallic nucleophilic addition to electrophilic double bonds is another valuable approach to the dihydronaphthalene scaffold.12 Photoinitiated cyclizations to form dihydronaphthalene have been developed by Nicolaou and colleagues in the synthesis of biologically relevant tetralins.13 Suzuki14 presented a palladium(ii)-catalysed cross-coupling reaction via benzylic boronates, and Yamamoto15 reported the Cu(OTf)2-catalyzed [4 + 2]-cycloaddition reaction of alkynyl-benzenes with alkenes.
Still, several of the abovementioned synthetic protocols suffer from functional group intolerance, a necessity to use scarce and expensive (noble) metal catalysts, harsh reaction conditions, easy over-reduction/oxidation leading to loss of the C C double bond or full aromatization, and/or limitations to particular substitution patterns. Hence, the development of new, efficient, and broadly applicable catalytic routes to complement existing methods for 1,2-dihydronaphthalene synthesis certainly remains to be a worthwhile effort, especially those employing readily available and inexpensive base metal catalysts.
Cobalt(ii) porphyrin complexes and related low-spin planar cobalt(ii) complexes provide interesting opportunities in this perspective. These stable metalloradicals, with their well-defined open-shell doublet d7-electronic configuration, are known to mediate a series of uncommon radical-type ‘carbene-transfer’ reactions. Reaction of these cobalt(ii) complexes with carbene precursors such as N-tosylhydrazones or diazo compounds leads to formation of unusual cobalt(iii)–carbene radical intermediates (Scheme 1). These are best described as one-electron reduced Fischer-type carbene complexes,16 having discrete spin density at their carbene-carbon atom. As a result, these intermediates react via radical-type pathways,16–22 of interest for the development of new ring-closure protocols. Cobalt(iii)–carbene radical intermediates were recently already demonstrated to be key-intermediates in some other synthetically useful metallo-radical catalysed reactions, including (enantioselective) alkene cyclopropanation,17 C–H functionalization,18 β-ester-γ-amino ketone synthesis,19 and in the regioselective synthesis of β-lactams,20 2H-chromenes21 and 1H-indenes.22
Scheme 1. Formation of CoIII–carbene radicals (one-electron reduced Fischer-type carbenes) upon activation of a carbene precursor by planar, low spin cobalt(ii) complexes.

Herein, we report the development of a cobalt-catalysed reaction for the synthesis of substituted 1,2-dihydronapthalenes involving CoIII–carbene radical reactivity. Starting from safe to use and easy to prepare N-tosyl hydrazones as carbene precursors, this reaction gives access to a broad range of substituted products with varying functional groups. Substrates with an alkyl substituent on the allylic position reacted to form E-aryl dienes in excellent yields, rather than the expected 1,2-dihydronaphthalenes.
Mechanistic studies point to a metallo-radical pathway involving formation of a CoIII–carbene radical, followed by hydrogen atom transfer to give a benz–allylic radical. From here on, ring-closure to form the dihydronaphthalene product can follow two distinct pathways, producing the same product: (1) A direct radical-rebound step, or (2) first dissociation of an ortho-quinodimethane fragment from the catalyst,13a followed by 6π-cyclisation. Clean formation of aryl dienes when reacting substrates with an alkyl substituent on the allylic position suggests that the pathways proceeding via release of ortho-quinodimethanes are dominant. Spin- and radical-trapping experiments performed under catalytically relevant reaction conditions confirm the radical nature of the process.
Results and discussion
Initial experiments were aimed at exploring the feasibility of 1,2-dihydronaphthalene synthesis from tosyl hydrazone substrate 1a, using a cobalt(ii) metallo-radical approach. The idea behind these reactions was to activate the tosyl hydrazone moiety to generate a cobalt(iii)–carbene radical intermediate (Scheme 1), which we anticipated to undergo net (formal) carbene insertion into the allylic C–H bond of the substrate via a radical-type hydrogen atom transfer (HAT)/radical-rebound mechanism (Scheme 2).23
Scheme 2. Anticipated steps to generate dihydronaphthalenes: HAT from the benz–allylic C–H bond of a CoIII–carbene radical intermediate followed by a radical rebound ring-closure step.

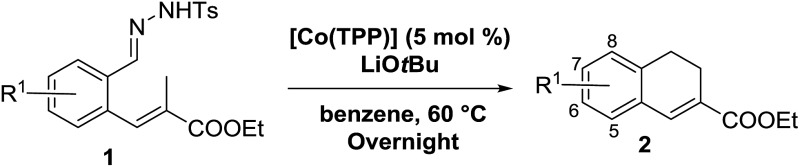
We decided to focus on the commercially available cobalt(ii)-metallo-radical catalyst [Co(TPP)]. First screening experiments indeed revealed the feasibility of this reaction, producing dihydronaphthalene 2a from substrate 1a in 81% yield (Table 1, entry 1), after which we further optimized the reaction conditions. Different bases were tested (Table 1, entries 1–5), which revealed that LiOtBu is best suited for this reaction.
Table 1. Standardization of reaction conditions.
| |||||
| Entry | Catalyst | Base | Eq. | Solvent | Yield a (%) |
| 1 | [Co(TPP)] | LiOtBu | 1.2 | PhCl | 81 |
| 2 | [Co(TPP)] | KOtBu | 1.2 | PhCl | 71 |
| 3 | [Co(TPP)] | NaOtBu | 1.2 | PhCl | 66 |
| 4 | [Co(TPP)] | K2CO3 | 1.2 | PhCl | 69 |
| 5 | [Co(TPP)] | NaOMe | 1.2 | PhCl | 67 |
| 6 | [Co(TPP)] | LiOtBu | 1.2 | PhCH3 | 95 |
| 7 | [Co(TPP)] | LiOtBu | 2 | PhCH3 | 54 |
| 8 | [Co(TPP)] | LiOtBu | 3 | PhCH3 | 0 |
| 9 | [Co(TPP)] | LiOtBu | 1.2 | THF | 84 |
| 10 | [Co(TPP)] | LiOtBu | 1.2 | CH3CN | 64 |
| 11 | [Co(TPP)] | LiO t Bu | 1.2 | Benzene | 97 (89) b |
| 12 | — | LiOtBu | 1.2 | PhCH3 | 0 |
| 13 | [Co(MeTAA)] | LiOtBu | 1.2 | Benzene | 94 |
aYields were determined by integration of the 1H NMR signals using acenaphthene as internal standard.
bIsolated yield between brackets.
Increasing the amount of base led to a large decrease in yield (Table 1, entries 6–8). Probably this is caused by decomposition or poisoning of the catalyst in the presence of excess base. Furthermore, variations in solvent revealed that in general higher yields were obtained in non-coordinating, low-polarity solvents like toluene or chlorobenzene. However, THF works rather well too (Table 1, entries 1, 6, 9–11). Finally, in the absence of a cobalt(ii) catalyst the reaction does not form the desired product 2a at all.24
We also tested the [Co(MeTAA)] catalyst for this reaction, which was shown to have a higher activity in some related carbene-transfer reactions.22,25 In this reaction, however, [Co(MeTAA)] gave similar results as [Co(TPP)] (Table 1, entry 13). Hence, because [Co(MeTAA)] is a quite air-sensitive complex, we decided to continue our investigations with the commercially available, and air and moisture stable [Co(TPP)] catalyst.
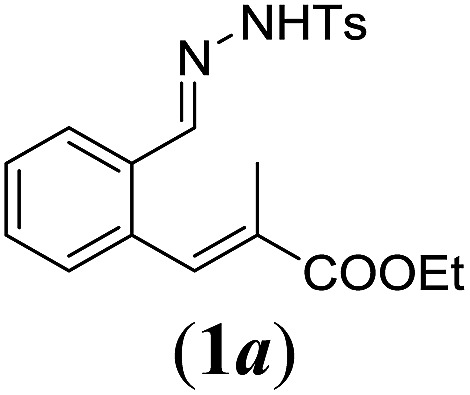
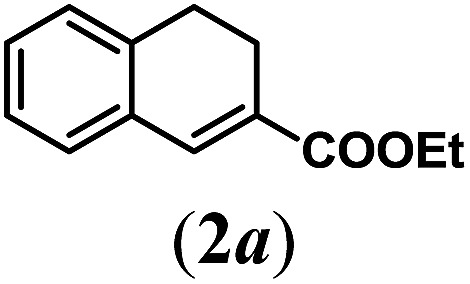
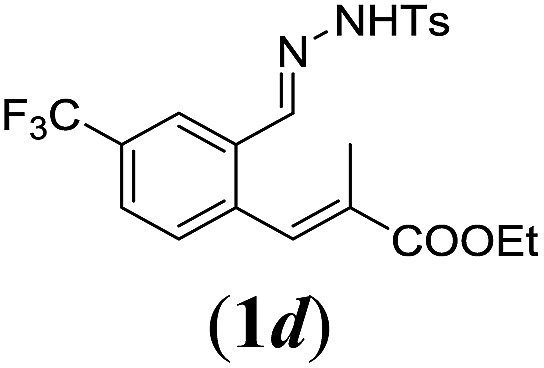
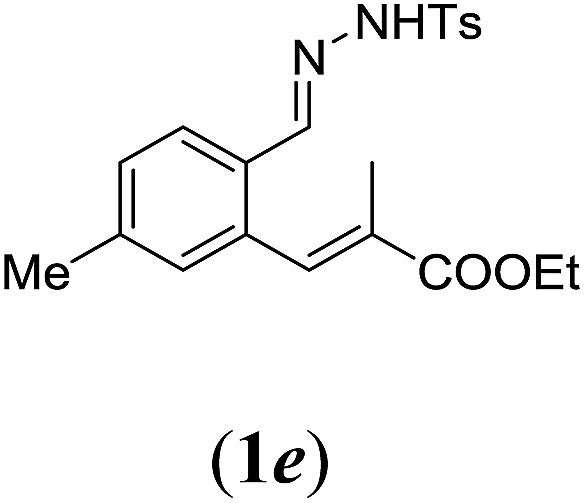
In order to investigate the scope of this new reaction, a range of substrates (1a–l; Tables 2 and 3) was synthesized with different functional groups at the aromatic ring (1a–g) and at the vinylic position (1h–l). Two general routes were used to arrive at substrates 1a–l, starting from 2-bromobenzaldehyde. The first route (Scheme 3, top) starts with protection of the aldehyde followed by converting the bromide into a new aldehyde at the ortho position, which is then converted into an alkene using an (E-selective) Horner–Wadsworth–Emmons reaction.26 Deprotection of the aldehyde and introduction of the tosylhydrazone results in the desired substrate. Alternatively, the alkene can also be introduced directly through a Heck-type reaction with 2-bromobenzaldehyde (Scheme 3, bottom).27 Substrates 1a and 1e–l were obtained as pure E-isomers. For the three substrates 1b–d mixtures of the E/Z-isomers were isolated, containing 88–94% of the major E-isomer, which were directly used in the cobalt(ii)-catalysed ring closing-reactions.
Table 2. Substrates scope varying functional groups R1 at the aromatic ring a .
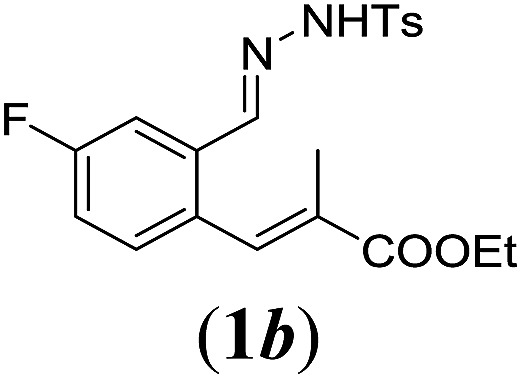
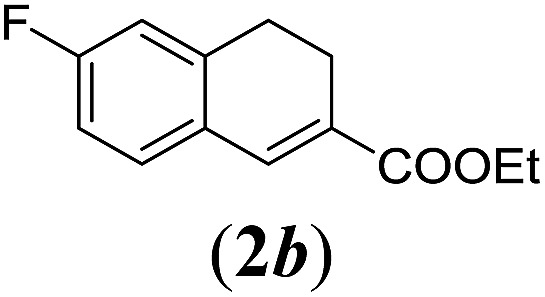
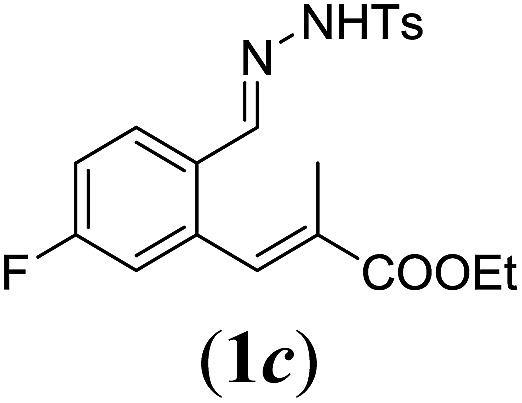
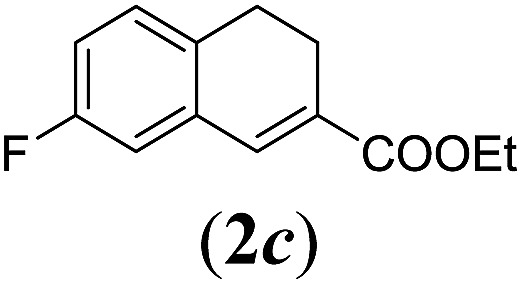
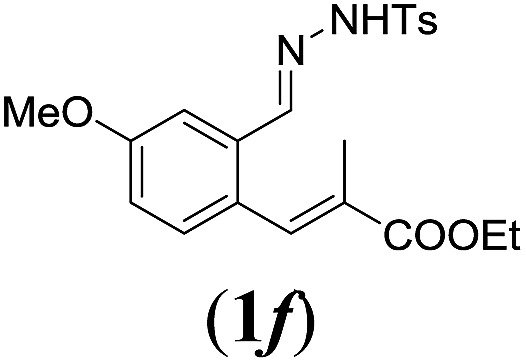
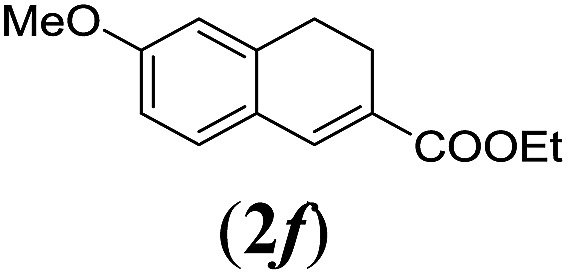
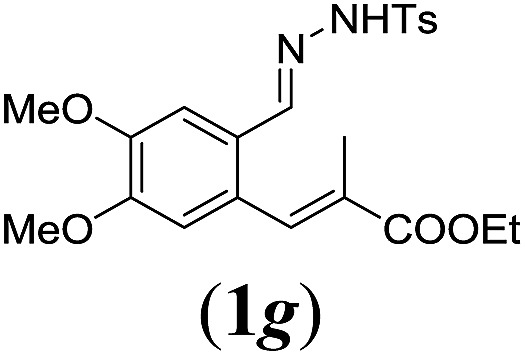
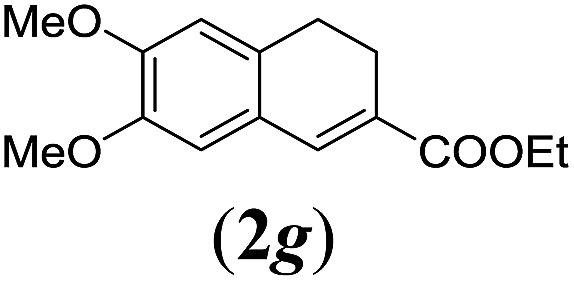
aReaction conditions: N-tosylhydrazone (1a–g) (0.1 mmol, 1.0 equiv.), LiOtBu (0.12 mmol, 1.2 equiv.), [Co(TPP)] (5 mol%), benzene (2 mL), 60 °C, overnight.
bIsolated yields after column chromatography (average of two separate experiments).
cIsolated yield of experiment performed with 1.0 mmol substrate.
dYields of these three entries based on the major E-isomer, using an E/Z-mixture containing 6–12% of the non-productive Z-isomer.
Table 3. Substrate scope varying functional groups R2 at the vinylic double bond a .
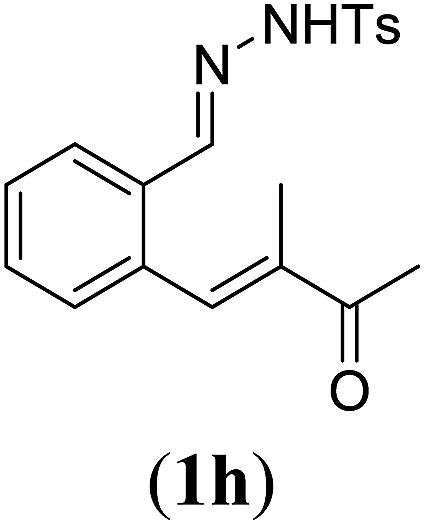
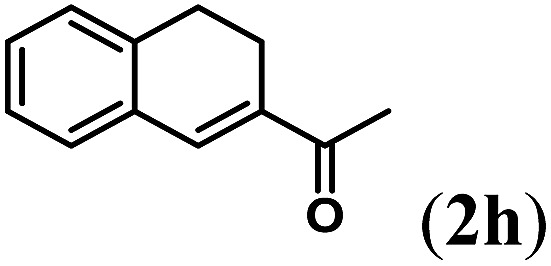
aReaction conditions: N-tosylhydrazone (1h–l) (0.1 mmol, 1.0 equiv.), LiOtBu (0.12 mmol, 1.2 equiv), [Co(TPP)] (5 mol%), benzene (2 mL), 60 °C, overnight.
bIsolated yields after column chromatography (average of two experiments).
cAlso 25% naphthalene isolated.
dCrude yield, based on integration of the 1H NMR signals using 1,3,5-trimethoxybenzene as internal standard.
Scheme 3. Substrate synthesis using either the Horner–Wadsworth–Emmons reaction (top) or a Heck-coupling approach (bottom).

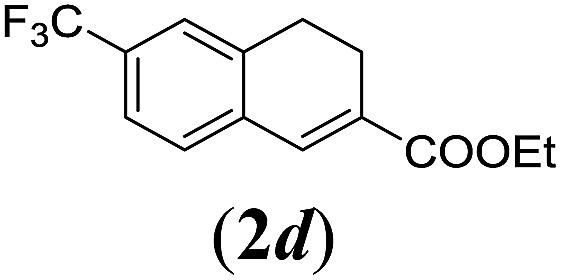
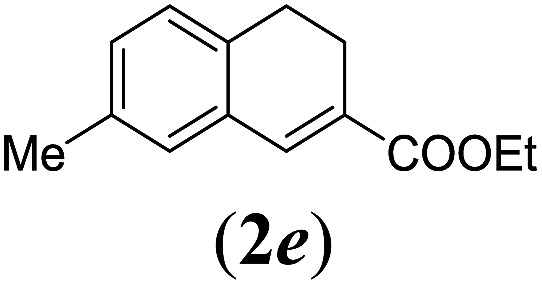
We first tested substrates 1a–g, varying in the substitution pattern of the aromatic ring (Table 2). The reaction proved to be quite tolerant to both electron donating and electron withdrawing substituents on the aromatic ring. Performing the reaction on a larger scale led to similar results (entry 2). It is clear that electron withdrawing groups at either the 6- or 7-position have a similar effect (2b–d). An electron donating methyl group at the 7-position (2e) results in a lower yield of 50%, but the even stronger donating methoxy-substituents at either the 7- or 6- and 7-position restores the yields to about 70%, comparable to the electron withdrawing groups (2f–g). Note that NMR spectra of the crude mixtures in most cases reveal higher yields than isolated, thus indicating some product loss during the purification steps using column chromatography and rotary evaporation (see for example Table 1, entry 11 and Table 3, entry 4).
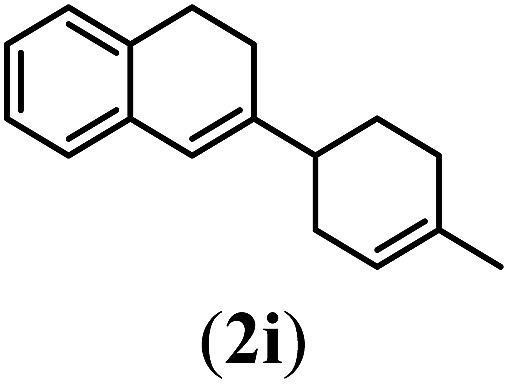
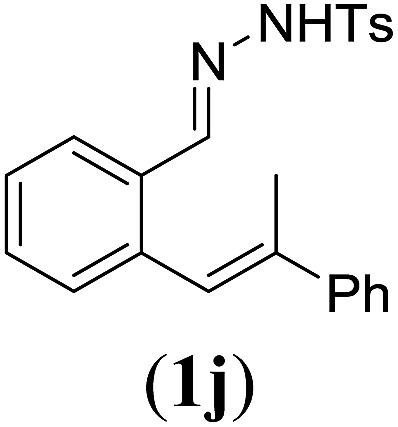
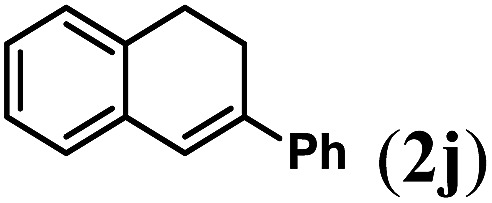
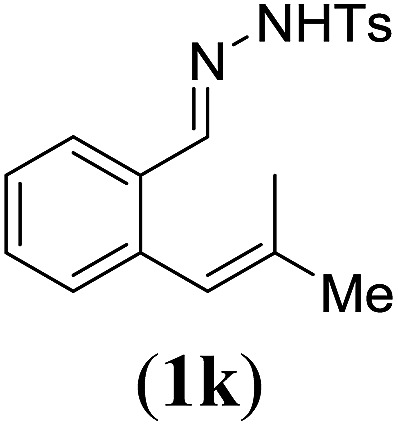
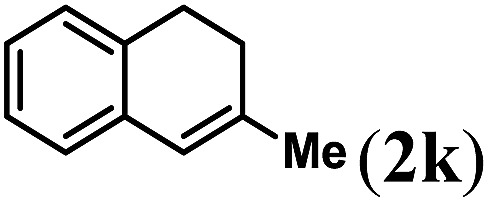
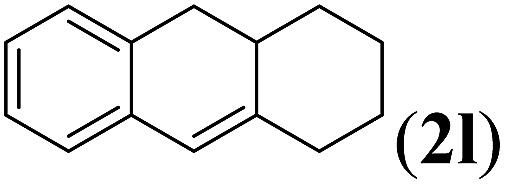
Next we tested substrates 1h–l, varying in the substitution pattern of the allylic moiety (Table 3). Surprisingly, the cobalt-catalysed ring-closing protocol proved to be rather sensitive to the nature of the R2-substituent. While ring-closure is observed for a variety of substrates with different R2-substituents at the vinylic position, the isolated yields are quite low when compared to substrates with an ester substituent at this position. When the R2-substituent is a ketone (Table 3, entry 1), azine side-product formation is indicated by 1H NMR spectroscopy, a well-known side-reaction for diazo compounds. A comparable, albeit slightly higher yield was obtained with R2 being a more bulky limonene group (Table 3, entry 2). With a phenyl-ring at the R2-position the yield increases to 50%. In this case, also 25% of the fully aromatic naphthalene was isolated as a side-product (entry 3). When the substituent is a methyl group, NMR characterization of the reaction mixture reveals a crude yield of 68%, while only 21% could be isolated. Substantial product loss during the purification steps in this case is likely caused by the volatile nature of product 2k. The isolated yield of the tricyclic compound 2l is also low (entry 5).28
Despite some purification issues (entry 4), the rather low yields of the reactions described in Table 3 as compared to those in Table 2 is surprising. Since the R2-substituent is not in conjugation with the anticipated allyl-radical intermediate, only a small influence of the R2-substituent on both the HAT and the radical-rebound steps was expected (Scheme 4a). Yet, the actually observed influence of the R2-group in the product yield is substantial, suggestive of an alternative (perhaps parallel) ring-closing mechanism involving release of a reactive ortho-quinodimethane (o-QDM) intermediate,13a followed by 6π-cyclisation to produce the 1,2-dihydronaphthalene product (Scheme 4b). Such o-QDM pathways, with the R2 substituent in direct conjugation with the double bond of the o-QDM intermediate, are expected to be influenced by the nature of the R2 substituent.
Scheme 4. ortho-Quinodimethane release followed by 6π-cyclisation (b) as an alternative to the anticipated radical rebound mechanism (a) for 1,2-dihydronaphthalene formation.

To further investigate the difference between these two mechanisms we decided to test a range of substrates containing an alkyl substituent at the allylic position (Scheme 3, Y = –CH2R3 instead of Y = H; see Table 4). This was expected to have little influence on the initially anticipated radical-rebound pathway (Scheme 4a), but for the o-QDM route (Scheme 4b) this should have a significant effect.
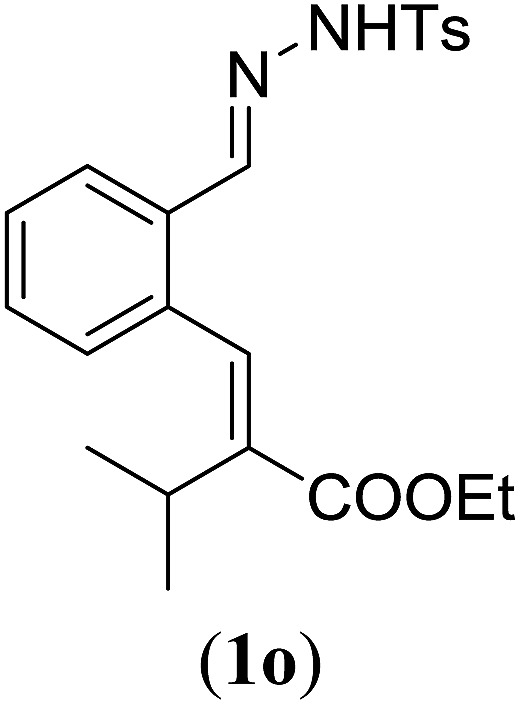
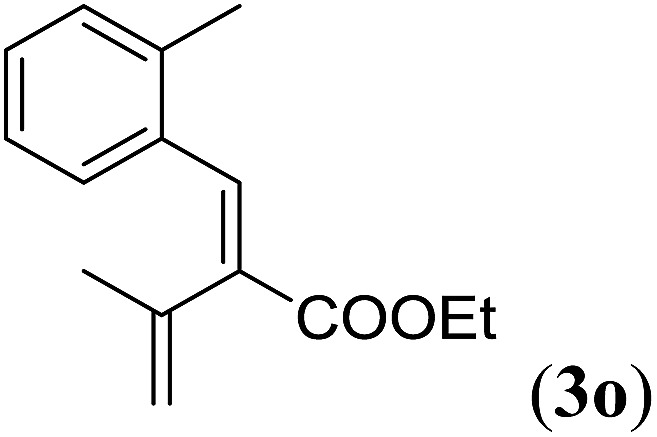
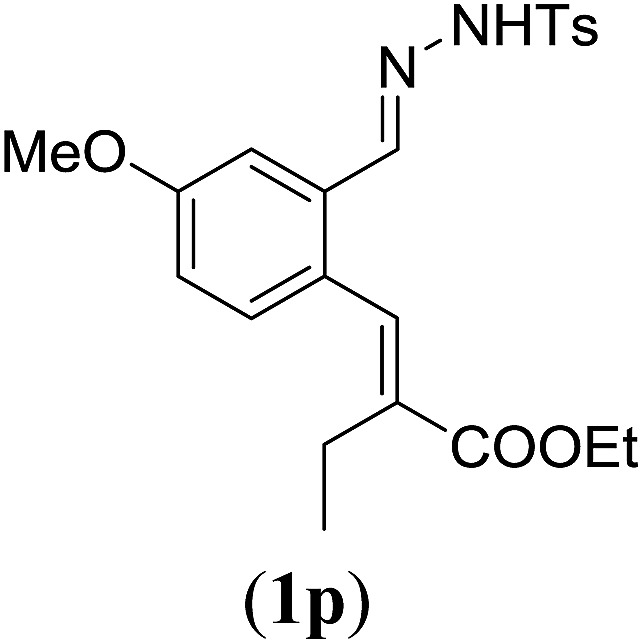
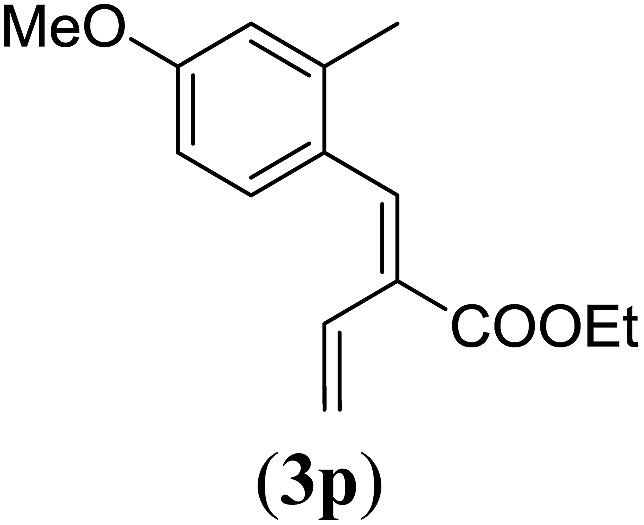
Table 4. Substrates scope varying functional groups at the allylic position a .
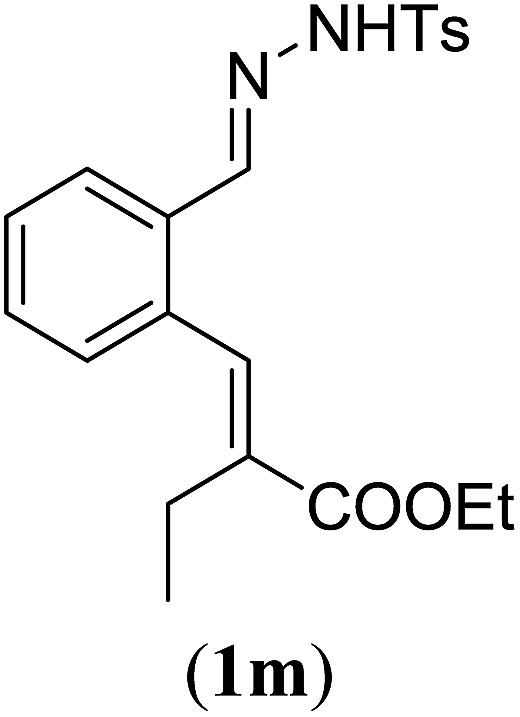
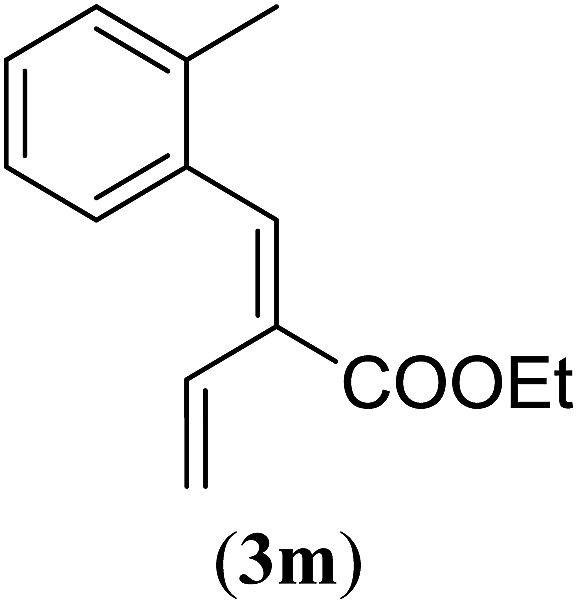
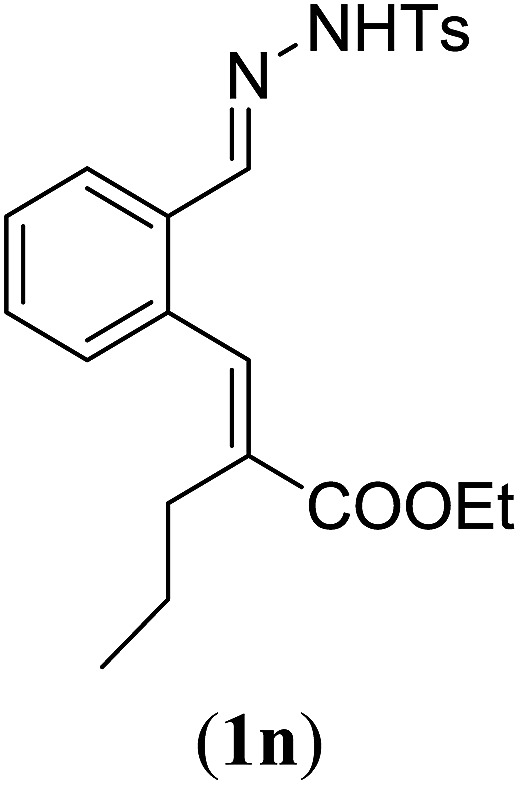
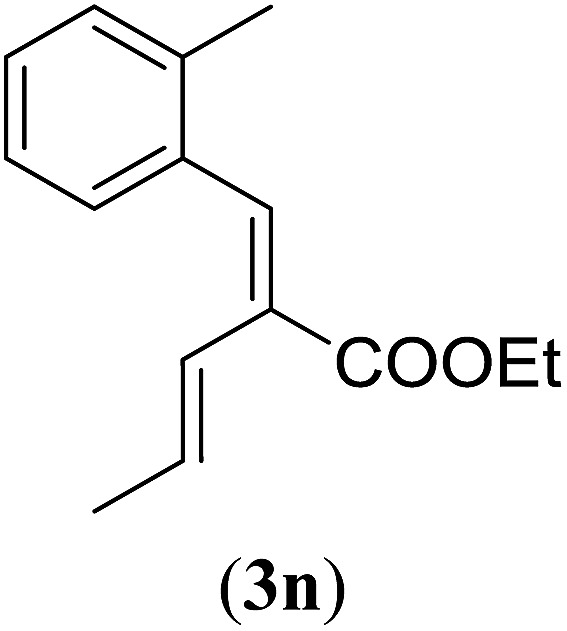
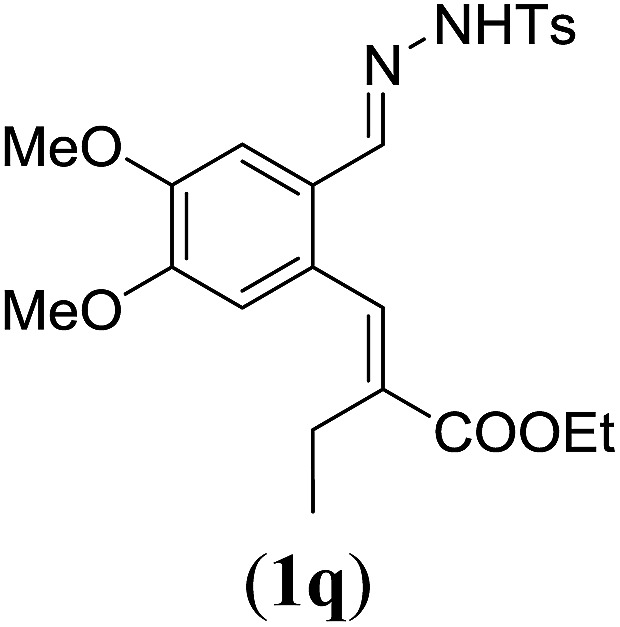
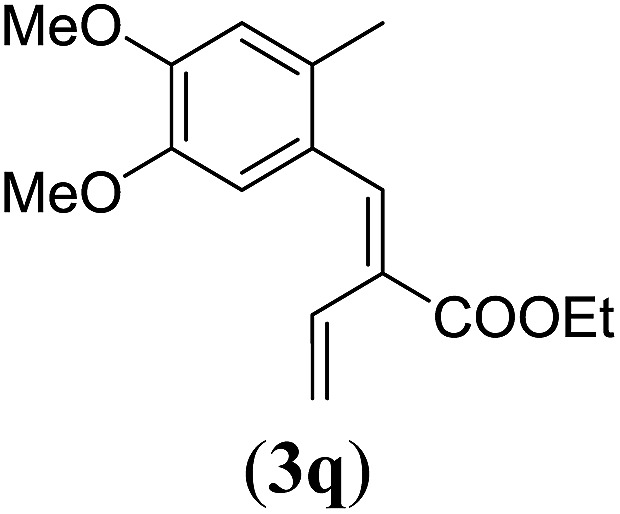
aReaction conditions: N-tosylhydrazone (1m–q) (0.1 mmol, 1.0 equiv.), LiOtBu (0.12 mmol, 1.2 equiv.), [Co(TPP)] (5 mol%), benzene (2 mL), 60 °C, overnight.
bIsolated yields after column chromatography (average of two separate experiments).
cYields of these three entries based on the major E-isomer, using an E/Z-mixture containing 11–25% of the non-productive Z-isomer.
dIsolated yield of experiment performed at 1 mmol scale.
eProduct isolated as mixture containing 10% cycloheptatriene product 4 (see ESI for details), which is formed from the non-productive Z-isomer of 1o.29
Surprisingly, for all such substrates tested (Table 4), the presence of an alkyl substituent at the allylic position (Scheme 3, Y = –CH2R3 instead of Y = H) led to a completely different outcome of the reaction. Instead of the anticipated ring-closure to form 1,2-dihydronapthalenes, the reaction led to selective formation of E-aryl dienes, which could be isolated in high yields. These products result from a net hydrogen atom transfer not only from the allylic position, but as well from the alkyl substituent. The reactions clearly point to dissociation of o-QDM intermediates, which undergo an ene-type [1,7]-hydride shift to form the observed dienes (Scheme 5).
Scheme 5. Ene-type [1,7]-hydride shift reaction of o-QDM intermediates leading to formation of the E-aryl dienes shown in Table 4.

Entry 1 of Table 4 shows the formation of benzyl diene 3m in high yield, also at a larger scale. When this substrate was extended with one carbon atom the reaction still proceeded cleanly, giving double E diene 3n in excellent isolated yield. Also double alkyl substitution (1o) leads to selective E-aryl diene formation (entry 3).29 Next we had a look at the influence of the electronic environment by substituting the aromatic ring which led to similar yields (entries 4 and 5, Table 4). Besides providing valuable mechanistic information, the reactions in Table 4 produce interesting products with various opportunities for further reactions or functionalization. Dienes are for example interesting substrates for polymerization, Diels–Alder reactions, epoxidation and [2 + 2] π cyclization to give cyclobutadienes.30
Mechanistic studies
In order to shed more light on the reaction mechanism, DFT calculations were performed. The calculated pathways for 1,2-dihydronaphthalene formation are depicted in Scheme 6. The catalytic cycle starts with coordination of the diazo compound 1a′ to the cobalt(ii) porphyrin catalyst to form intermediate B. Formation of 1a′ from the deprotonated tosylhydrazone 1a is assumed to be a non-catalysed thermal process, which is most likely rate limiting (vide infra).31 Dinitrogen loss from diazo adduct B32 over TS1 to form the cobalt(iii)–carbene radical intermediate C is a low-barrier (+10.4 kcal mol–1) and exergonic process. As observed for other carbene radical species, intermediate C has most of its spin density localized at the ‘carbene carbon atom’.16 The next step (TS2) is a HAT process from the allylic C–H bond of C,33 to produce the benz–allylic radical intermediate D.34 This is again a low-barrier (+5.6 kcal mol–1) and exergonic process. Interestingly, two competing pathways were identified for ring-closure to form the same dihydronaphthalene product from intermediate D, with very similar overall barriers. The first pathway concerns the anticipated (see also Scheme 2a) radical rebound mechanism (Scheme 6, red-coloured pathway). This process involves attack of the terminal carbon atom of the allyl-radical moiety on the anti-bonding orbital of the weak Co–C bond in intermediate D, leading to simultaneous C–C bond making and Co–C bond breaking, thus forming the desired six-membered ring product with regeneration of the catalyst in one concerted step (TS3). This elementary step has a quite low barrier (+15.2 kcal mol–1), albeit a bit higher than all preceding steps of the catalytic cycle.
Scheme 6. Computed mechanisms for [Co(por)]-catalysed 1,2-dihydronaphthalene formation.a DFT-D3 calculated (Turbomole, BP86, def2-TZVP, disp3) mechanisms for [Co(por)]-catalysed 1,2-dihydronaphthalene formation (free energies,  , in kcal mol–1). All energies, including the transition states, are reported with respect to (the toluene adduct of) species A as the reference point. A cobalt–porphyrin complex without meso-substituents was used as a simplified model of the actual [Co(TPP)] catalyst.
, in kcal mol–1). All energies, including the transition states, are reported with respect to (the toluene adduct of) species A as the reference point. A cobalt–porphyrin complex without meso-substituents was used as a simplified model of the actual [Co(TPP)] catalyst.

In view of the experimental results discussed above, bearing in mind also the weakness of the Co–C bond of D, in direct conjugation with the benz–allyl radical moiety, an alternative pathway (see also Scheme 4b) for 1,2-dihydronaphthalene formation from D was considered. This involves homolytic splitting of the Co–C bond to release the reactive ortho-quinodimethane intermediate E, followed by 6π-cyclisation (TS4) to yield the same 1,2-dihydronaphthalene product F (Scheme 6, blue-coloured pathway).13a
While formation of o-QDM intermediate E from D is endergonic, the subsequent ring-closing barrier viaTS4 is very low (+3.9 kcal mol–1). As such the pathway via the o-QDM intermediate E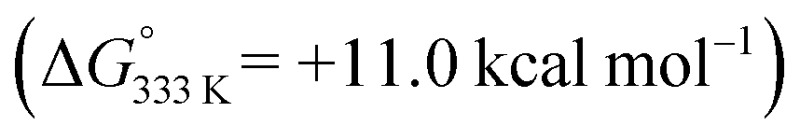 has a slightly lower, albeit very similar overall barrier for 1,2-dihydronaphthalene formation from D (+14.9 kcal mol–1) when compared to the direct radical-rebound path (+15.2 kcal mol–1). The relative TS3vs.TS4 barriers are too similar to discriminate reliably between these two pathways on the basis of DFT. However, in the presence of coordinating compounds (such as deprotonated 1a), trapping of the [Co(por)] catalyst A, after release of E, the o-QDM fragmentation pathway viaE and TS4 is likely thermodynamically favoured over the radical-rebound pathway viaTS3.
has a slightly lower, albeit very similar overall barrier for 1,2-dihydronaphthalene formation from D (+14.9 kcal mol–1) when compared to the direct radical-rebound path (+15.2 kcal mol–1). The relative TS3vs.TS4 barriers are too similar to discriminate reliably between these two pathways on the basis of DFT. However, in the presence of coordinating compounds (such as deprotonated 1a), trapping of the [Co(por)] catalyst A, after release of E, the o-QDM fragmentation pathway viaE and TS4 is likely thermodynamically favoured over the radical-rebound pathway viaTS3.
The pathway via ortho-quinodimethane E (Scheme 6, blue-coloured pathway) is of interest in view of the experimental results described in Table 3, showing that the R2-substituents have a large influence on the product yield. While this behaviour was unexpected in view of the anticipated radical-rebound mechanism (Scheme 6, red-coloured pathway), this makes perfect sense in view of a ring-closing mechanism involving o-QDM intermediate E. After all, in E the R2-moiety is in direct conjugation with the adjacent vinyl group. As such, and in line with the experimental results described in Table 3, the R2-substituents should have a direct influence on the relative stabilities of species E, thereby affecting both the thermodynamics for dissociation of E as well as the barriers for 6π-cyclisation (TS4) to form F (thus explaining competing side-product formation for some of the R2-substituents).
Furthermore, the influence of an alkyl substituent at the allylic position (Scheme 3, Y = –CH2R3 instead of Y = H) proved to be even larger, leading to formation of E-aryl dienes instead of the expected 1,2-dihydronaphthalenes, which clearly points to involvement of o-QDM intermediates in the reactions described in Table 4.
To shed more light on those reactions, we also explored the mechanism for E-aryl diene formation from allyl radical intermediate D′ (see Scheme 7). Allyl radical intermediate D′ could in principle undergo a direct radical rebound ring-closure step (TS5, Scheme 7, similar to TS3 in Scheme 6) to produce 3-methyl substituted 1,2-dihydronaphthalene product F′. This is a strongly exergonic 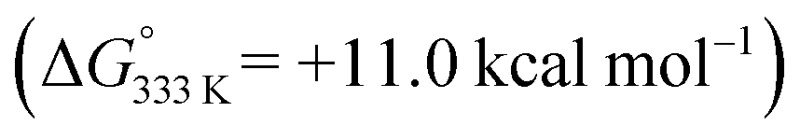 reaction step with a very accessible transition state barrier (TS5: +15.3 kcal mol–1; comparable to the TS3 barrier from D: +15.2 kcal mol–1). However, dissociation of o-QDM E′ from D′ is only slightly endergonic (+7.0 kcal mol–1), and a subsequent ene-type [1,7]-hydride shift reaction viaTS8 (Scheme 7, blue pathway) has a very low barrier (+2.0 kcal mol–1 from E′). As such, the total TS8 barrier from D′ of +9.0 kcal mol–1 to form E-aryl diene product G is much lower than the TS5 barrier for direct radical-rebound ring closure to form 1,2-dihydronaphthalene F′ (Scheme 7, top red pathway). Alternative o-QDM pathways for formation of 1,2-dihydronaphthalene F′, involving 6π-cyclisation of E′ or E′′ over TS6 or TS7, are also higher barrier processes than formation of G over TS8 according to DFT (Scheme 7, bottom red pathways). As such, the experimentally observed (Table 4) formation of E-aryl dienes for substrates containing an alkyl substituent at the allylic position instead of the thermodynamically favoured 1,2-dihydronapthalenes,35 must be a kinetically controlled process, dictated by the very low TS8 barrier.36
reaction step with a very accessible transition state barrier (TS5: +15.3 kcal mol–1; comparable to the TS3 barrier from D: +15.2 kcal mol–1). However, dissociation of o-QDM E′ from D′ is only slightly endergonic (+7.0 kcal mol–1), and a subsequent ene-type [1,7]-hydride shift reaction viaTS8 (Scheme 7, blue pathway) has a very low barrier (+2.0 kcal mol–1 from E′). As such, the total TS8 barrier from D′ of +9.0 kcal mol–1 to form E-aryl diene product G is much lower than the TS5 barrier for direct radical-rebound ring closure to form 1,2-dihydronaphthalene F′ (Scheme 7, top red pathway). Alternative o-QDM pathways for formation of 1,2-dihydronaphthalene F′, involving 6π-cyclisation of E′ or E′′ over TS6 or TS7, are also higher barrier processes than formation of G over TS8 according to DFT (Scheme 7, bottom red pathways). As such, the experimentally observed (Table 4) formation of E-aryl dienes for substrates containing an alkyl substituent at the allylic position instead of the thermodynamically favoured 1,2-dihydronapthalenes,35 must be a kinetically controlled process, dictated by the very low TS8 barrier.36
Scheme 7. Computed mechanisms for E-aryl diene formation as compared to 1,2-dihydronaphthalene formation from allyl-radical intermediate D′.a DFT-D3 calculated (Turbomole, BP86, def2-TZVP, disp3) mechanisms for [Co(por)]-catalysed 1,2-dihydronaphthalene and E-aryl diene formation from D′ (free energies,  , in kcal mol–1). All energies, including the transition states, are reported with respect to species D′ as the reference point. A cobalt–porphyrin complex without meso-substituents was used as a simplified model of the actual [Co(TPP)] catalyst.
, in kcal mol–1). All energies, including the transition states, are reported with respect to species D′ as the reference point. A cobalt–porphyrin complex without meso-substituents was used as a simplified model of the actual [Co(TPP)] catalyst.

Notably, the overall catalytic cycles involve the sequential transition of (1) a diazo compound to (2) a cobalt(ii) carbenoid (species B),32 (3) a cobalt(iii)–carbene radical (species C) and (4) a cobalt(iii)–CH2-aryl species containing a benz-allyl radical moiety (species D and D′), thus providing an intriguing sequence of single-electron steps uncommon to more traditional closed-shell organometallic catalysis. A further mechanistically interesting observation is that the benz–allyl radical moiety in species D and D′, in direct conjugation with the already quite weak CoIII–C bonds, triggers dissociation of o-QDM intermediates E and E′, which either undergo an uncatalysed ring closure (E) or ene-type [1,7]-hydride shift (E′) to form 1,2-dihydronaphtalenes (F) or E-aryldienes (G).
In an attempt to gather additional experimental mechanistic information, we performed a set of kinetic NMR studies and radical-trapping experiments. The kinetic studies were not very informative though, showing zero order kinetics in the substrate as well as the catalyst (see ESI†). This points to a rate limiting thermal conversion of the poorly soluble substrate 1b into the soluble, reactive diazo compound 1b′, and hence does not provide any information about the subsequent cobalt-catalysed steps. Radical trapping experiments proved more informative. Performing the catalytic reaction of 1b in the presence of two equivalents of 2,2,6,6-tetra-methylpiperidine-1-oxyl (TEMPO) led to formation of the (over)oxidized species 6, isolated in 59% yield (Scheme 8, top). Apparently it is possible to trap the cobalt(iii)–carbene radical intermediate C with TEMPO.37
Scheme 8. Radical scavenging experiments used to trap the CoIII–carbene radical intermediate.

In a separate EPR spin-trapping experiment, using phenyl N-tert-butylnitrone (PBN) (7) as the spin-trap, we first generated the diazo compound 1b′ at elevated temperature, cooled down the reaction mixture to room temperature, after which we added PBN and the catalyst to prepare the EPR sample (Scheme 8, bottom).
The resulting EPR spectrum is depicted in Fig. 2 (giso = 2.007, AN = 14.3 G, AH = 2.8 G). The detected proton and nitrogen hyperfine interactions of 2.8 G and 14.3 G, respectively, reveal trapping of a carbon-centred radical by the PBN spin-trap. The most likely candidate of this PBN-trapped species is intermediate C, thus forming compound 8. HR-MS spectrometry confirmed the presence of such species, with the mass of the catalyst plus carbene-substrate plus PBN. The trapping experiments are thus in agreement with the proposed metallo-radical activation steps of diazo compound 1a′ by the cobalt(ii) catalyst in order to produce carbene radical intermediate C.
Fig. 2. Isotropic X-band EPR spectrum of the PBN-trapped carbon-centred radical (T = 298 K; microwave frequency: 9.36607 GHz; power: 6.33 mW; modulation amplitude: 1.0 G).

Conclusions
Metalloradical activation of substituted o-styryl N-tosyl hydrazones provides an effective synthetic protocol for the synthesis substituted 1,2-dihydronaphthalenes and E-aryl-dienes, taking advantage of the intrinsic and controlled radical-type reactivity of a cobalt(iii)–carbene radical intermediate. The cobalt-catalysed synthesis of 1,2-dihydronaphthalene involves net intramolecular carbene insertion into the allylic C(sp3)–H bond, via HAT from the allylic C–H bond to the cobalt(iii)–carbene radical. A subsequent direct and low barrier (DFT) radical-rebound C–C bond forming step provides one viable mechanistic possibility for ring-closure to 1,2-dihydronaphthalenes. However, a competing pathway seems to prevail kinetically, involving dissociation of a reactive ortho-quinodimethane (o-QDM) intermediate from the metal, followed by concerted, pericyclic 6π-cyclisation. The ring-closure reaction has been successfully applied to a broad range of substrates with various R1 substituents at the aromatic ring, producing the desired 1,2-dihydronapthalenes in good to excellent isolated yields for substrates with an R2 = COOEt substituent at the vinylic position (∼70–90%). Changing the R2 moiety from an ester to another substituent has a surprisingly large influence on the (isolated) yields. This behaviour is unexpected for a radical rebound ring-closure mechanism, and points to a mechanism proceeding via o-QDM intermediates. Furthermore, substrates with an alkyl substituent at the allylic position (Y = CH2R3 instead of Y = H) react to form E-aryl-dienes in excellent yields, rather than the expected 1,2-dihydronaphthalenes. This result, combined with the outcome of supporting DFT calculations, strongly points to the release of reactive o-QDM intermediates from the metal centre in all cases. The latter either undergo a 6π-cyclisation reaction to form 1,2-dihydronaphthalenes, or a [1,7]-hydride shift to produce E-aryl-dienes. Both pathway are low-barrier processes according to DFT, but the [1,7]-hydride shift outcompetes the 6π-cyclisation step for the substrates with an alkyl substituent at the allylic position, thus producing E-aryl-dienes in kinetically controlled reactions rather than the expected, thermodynamically more stable 1,2-dihydronaphthalenes. Spin trapping experiments confirm the radical nature of these reactions. Follow-up studies in our laboratory currently focus on exploiting metalloradical catalysed ortho-quinodimethane formation coupled to a series of related pericyclic reactions.13a
Funding sources
Financial support from the Netherlands Organization for Scientific Research (NWO-CW VICI project 016.122.613) and the University of Amsterdam (Research Priority Area Sustainable Chemistry) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We thank Ed Zuidinga for MS measurements and Jan Meine Ernsting for NMR advice.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc03909c
References
- (a) de Meijere A. and Stang P. J., in Metal-Catalysed Cross-Coupling Reactions, Wiley-VCH, Weinheim, Germany, 2nd edn, 2004. [Google Scholar]; (b) Gladysz J. A. Chem. Rev. 2011;111:1167. doi: 10.1021/cr2000515. [DOI] [PubMed] [Google Scholar]; (c) Suzuki A. Angew. Chem., Int. Ed. 2011;50:6722. doi: 10.1002/anie.201101379. [DOI] [PubMed] [Google Scholar]; (d) Negishi E. Angew. Chem., Int. Ed. 2011;50:6738–6764. doi: 10.1002/anie.201101380. [DOI] [PubMed] [Google Scholar]
- (a) Gutiérrez-Bonet A., Juliá-Hernández F., de Luis B., Martin R. J. Am. Chem. Soc. 2016;138:6384. doi: 10.1021/jacs.6b02867. [DOI] [PubMed] [Google Scholar]; (b) Xia Y., Qu P., Liu Z., Ge R., Xiao Q., Zhang Y., Wang J. Angew. Chem., Int. Ed. 2013;52:2543. doi: 10.1002/anie.201209269. [DOI] [PubMed] [Google Scholar]
- (a) Angle S. R., Arnaiz D. O. Tetrahedron Lett. 1991;32:2327. [Google Scholar]; (b) Santi R., Bergamini F., Citterio A., Sebastiano R., Nicolini M. J. Org. Chem. 1992;57:4250. [Google Scholar]; (c) Asao N., Kasahara T., Yamamoto Y. Angew. Chem., Int. Ed. 2003;42:3504. doi: 10.1002/anie.200351390. [DOI] [PubMed] [Google Scholar]; (d) Nishizawa M., Takao H., Yadav V. K., Imagawa H., Sugihara T. Org. Lett. 2003;5:4563. doi: 10.1021/ol035622e. [DOI] [PubMed] [Google Scholar]; (e) Harrowven D. C., Tyte M. J. Tetrahedron Lett. 2002;43:5971. [Google Scholar]
- Sakakibara I., Ikeya Y., Hayashi K., Mitsuhashi H. Phytochemistry. 1992;31:3219. [Google Scholar]
- González A. G., Aguiar Z. E., Grillo T. A., Luis J. G. Phytochemistry. 1992;31:1691. [Google Scholar]
- Azhar-Ul H., Malik A., Anis I., Khan S. B., Ahmed E., Ahmed Z., Nawaz S. A., Choudhary M. I. Chem. Pharm. Bull. 2004;52:1269. doi: 10.1248/cpb.52.1269. [DOI] [PubMed] [Google Scholar]
- (a) Gennari L. Drugs Today. 2006;42:355. doi: 10.1358/dot.2006.42.6.973583. [DOI] [PubMed] [Google Scholar]; (b) Yang X., Reinhold A. R., Rosati R. L., Liu K. K. C. Org. Lett. 2000;2:4025. doi: 10.1021/ol006652+. [DOI] [PubMed] [Google Scholar]; (c) Hejtmánková L., Jirman J., Sedlák M. Res. Chem. Intermed. 2009:615. [Google Scholar]
- Scribner A. W., Haroutounian S. A., Carlson K. E., Katzenellenbogen J. A. J. Org. Chem. 1997;62:1043. [Google Scholar]
- Hutchinson D. K., Rosenberg T., Klein L. L., Bosse T. D., Larson D. P., He W., Jiang W. W., Kati W. M., Kohlbrenner W. E., Liu Y., Masse S. V., Middleton T., Molla A., Montgomery D. A., Beno D. W. A., Stewart K. D., Stoll V. S., Kempf D. J. Bioorg. Med. Chem. Lett. 2008;18:3887. doi: 10.1016/j.bmcl.2008.06.043. [DOI] [PubMed] [Google Scholar]
- Voets M., Antes I., Scherer C., Müller-Vieira U., Biemel K., Marchais-Oberwinkler S., Hartmann R. W. J. Med. Chem. 2006;49:2222. doi: 10.1021/jm060055x. [DOI] [PubMed] [Google Scholar]
- Silva Jr L. F., Siqueira F. A., Pedrozo E. C., Vieira F. Y. M., Doriguetto A. C. Org. Lett. 2007;9:1433. doi: 10.1021/ol070027o. [DOI] [PubMed] [Google Scholar]
- , and references therein. ; (a) For reviews, see: Pape A. R., Kaliappan K. P., Kündig E. P., Chem. Rev., 2000, 100 , 2917 . [DOI] [PubMed] [Google Scholar]; (b) Rawson D. J., Meyers A. I. J. Org. Chem. 1991;56:2292. [Google Scholar]; (c) Meyers A. I., Brown J. D., Laucher D. Tetrahedron Lett. 1987;28:5279. [Google Scholar]; (d) Meyers A. I., Lutomski K. A., Laucher D. Tetrahedron. 1988;44:3107. [Google Scholar]; (e) Amurrio D., Khan K., Kündig E. P. J. Org. Chem. 1996;61:2258. [Google Scholar]; (f) Ahmed A., Clayden J., Rowley M. Chem. Commun. 1998:297. [Google Scholar]; (g) Tomioka K., Shindo M., Koga K. Tetrahedron Lett. 1990;31:1739. [Google Scholar]; (h) Tomioka K., Inoue I., Shindo M., Koga K. Tetrahedron Lett. 1990;31:6681. [Google Scholar]; (i) Shindo M., Koga K., Asano Y., Tomioka K. Tetrahedron. 1999;55:4955. [Google Scholar]; (j) Tomioka K., Shindo M., Koga K. J. Am. Chem. Soc. 1989;111:8266. [Google Scholar]
- Reviews: ; (a) Segura J. L., Martin N. Chem. Rev. 1999;99:3199. doi: 10.1021/cr990011e. [DOI] [PubMed] [Google Scholar]; (b) Quinkert G., Stark H. Angew. Chem., Int. Ed. 1983;22:637. [Google Scholar]; (c) Yang N. C., Rivas C. J. J. Am. Chem. Soc. 1961;83:2213. [Google Scholar]; (d) Arnold B. J., Mellows S. M., Sammes P. G. J. Chem. Soc., Perkin Trans. 1. 1973:1266. [Google Scholar]; (e) Macdonald D. I., Durst T. J. Org. Chem. 1986;51:4749. [Google Scholar]; (f) Charlton J. L., Koh K. J. Org. Chem. 1992;57:1514. [Google Scholar]; (g) Sano H., Asanuma D., Kosugi M. Chem. Commun. 1999:1559. [Google Scholar]; (h) Nicolaou K. C., Gray D. L. F. Angew. Chem., Int. Ed. 2001;40:761. [PubMed] [Google Scholar]; (i) Nicolaou K. C., Gray D. L. F., Tae J. Angew. Chem., Int. Ed. 2001;40:3675. doi: 10.1002/1521-3773(20011001)40:19<3675::aid-anie3675>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]; (j) Nicolaou K. C., Gray D. L. F., Tae J. Angew. Chem., Int. Ed. 2001;40:3679. doi: 10.1002/1521-3773(20011001)40:19<3679::aid-anie3679>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]; (k) Nicolaou K. C., Gray D. L. F., Tae J. J. Am. Chem. Soc. 2004;126:613. doi: 10.1021/ja030498f. [DOI] [PubMed] [Google Scholar]
- Kanai G., Miyaura N., Suzuki A. Chem. Lett. 1993;22:845. [Google Scholar]
- Asao N., Kasahara T., Yamamoto Y. Angew. Chem., Int. Ed. 2003;42:3504. doi: 10.1002/anie.200351390. [DOI] [PubMed] [Google Scholar]
- (a) Dzik W. I., Xu X., Zhang X. P., Reek J. N. H., de Bruin B. J. Am. Chem. Soc. 2010;132:10891. doi: 10.1021/ja103768r. [DOI] [PubMed] [Google Scholar]; (b) Lu H., Dzik W. I., Xu X., Wojtas L., de Bruin B., Zhang X. P. J. Am. Chem. Soc. 2011;133:8518. doi: 10.1021/ja203434c. [DOI] [PubMed] [Google Scholar]; (c) Dzik W. I., Zhang X. P., de Bruin B. Inorg. Chem. 2011;50:9896. doi: 10.1021/ic200043a. [DOI] [PubMed] [Google Scholar]; (d) Dzik W. I., Reek J. N. H., de Bruin B. Chem.–Eur. J. 2008;14:7594. doi: 10.1002/chem.200800262. [DOI] [PubMed] [Google Scholar]; (e) Lyaskovskyy V., de Bruin B. ACS Catal. 2012;2:270. [Google Scholar]
- (a) Doyle M. P. Angew. Chem., Int. Ed. 2009;48:850. doi: 10.1002/anie.200804940. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhu S., Ruppel J. V., Lu H., Wojtas L., Zhang X. P. J. Am. Chem. Soc. 2008;130:5042. doi: 10.1021/ja7106838. [DOI] [PubMed] [Google Scholar]; (c) Zhu S., Perman J. A., Zhang X. P. Angew. Chem., Int. Ed. 2008;47:8460. doi: 10.1002/anie.200803857. [DOI] [PubMed] [Google Scholar]; (d) Fantauzzi S., Gallo E., Rose E., Raoul N., Caselli A., Issa S., Ragaini F., Cenini S. Organometallics. 2008;27:6143. [Google Scholar]; (e) Xu X., Zhu S., Cui X., Wojtas L., Zhang X. P. Angew. Chem., Int. Ed. 2013;52:11857. doi: 10.1002/anie.201305883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Ruppel J. V., Kamble R. M., Zhang X. P. Org. Lett. 2007;9:4889. doi: 10.1021/ol702265h. [DOI] [PubMed] [Google Scholar]; (b) Lu H., Li C., Jiang H., Lizardi C. L., Zhang X. P. Angew. Chem., Int. Ed. 2014;53:7028. doi: 10.1002/anie.201400557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Jiang J., Xu D., Luo Q., Wang H., Chen J., Li H., Wang Y., Wan X. Angew. Chem., Int. Ed. 2015;54:1231. doi: 10.1002/anie.201408874. [DOI] [PubMed] [Google Scholar]
- Paul N. D., Chirila A., Lu H., Zhang X. P., de Bruin B. Chem.–Eur. J. 2013;19:12953. doi: 10.1002/chem.201301731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul N. D., Mandal S., Otte M., Cui X., Zhang X. P., de Bruin B. J. Am. Chem. Soc. 2014;136:1090. doi: 10.1021/ja4111336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B. G., Chirila A., Tromp M., Reek J. N. H., de Bruin B. J. Am. Chem. Soc. 2016;138:8968. doi: 10.1021/jacs.6b05434. [DOI] [PubMed] [Google Scholar]
- For selected reviews about C-H functionalisation by carbene insertion: ; (a) Caballero A., Pérez P. J. Chem. Soc. Rev. 2013;42:8809. doi: 10.1039/c3cs60120j. [DOI] [PubMed] [Google Scholar]; (b) Davies H. M. L., Morton D. Chem. Soc. Rev. 2011;40:1857. doi: 10.1039/c0cs00217h. [DOI] [PubMed] [Google Scholar]; (c) Lu H.-J., Zhang X. P. Chem. Soc. Rev. 2011;40:1899. doi: 10.1039/c0cs00070a. [DOI] [PubMed] [Google Scholar]
- Uncatalysed thermal decomposition of substrate 1a is unselective, producing many different products (among which ∼20% of the carbene dimerization product). Product 2a was not detected in these mixtures
- Chirila A., Das B. G., Paul N. D., de Bruin B. ChemCatChem. 2017;9:1413. doi: 10.1002/cctc.201601568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reviews: ; (a) Boutagy J., Thomas R. Chem. Rev. 1974;74:87. [Google Scholar]; (b) Stec W. J. Acc. Chem. Res. 1983;16:411. [Google Scholar]
- Bryan C. S., Lautens M. Org. Lett. 2010;12:2754. doi: 10.1021/ol100844v. [DOI] [PubMed] [Google Scholar]
- Considering the results obtained in Table 4, diene formation for substrate 1l could perhaps be expected, but was not observed. This is because the C-H bond of the alkyl substituent cannot rotate towards the methide for the required [1,7]-hydride shift, since it is constrained in the cyclohexyl ring (Scheme 5)
- While the major E-isomer of 1o transforms selectively to the E-aryl diene product, the non-productive Z-isomer of 1o participates in a Buchner-type ring expansion of a benzene solvent molecule to produce side product 4 (see ESI for structure and characterization). As a result, product 3o was obtained as a mixture containing ∼10% of compound 4. It proved difficult to isolate pure 3o, clean from 4
- (a) Bäckvall J.-E., Chinchilla R., Nájera C., Yus M. Chem. Rev. 1998;98:2291. doi: 10.1021/cr970326z. [DOI] [PubMed] [Google Scholar]; (b) Mundal D. A., Lutz K. E., Thomson R. J. Org. Lett. 2009;11:465. doi: 10.1021/ol802585r. [DOI] [PubMed] [Google Scholar]; (c) Baldry P. J. J. Chem. Soc., Perkin Trans. 1. 1975:1913. [Google Scholar]; (d) Baldry P. J. J. Chem. Soc., Perkin Trans. 2. 1980:805. [Google Scholar]; (e) Um J. M., Xu H., Houk K. N., Tang W. J. Am. Chem. Soc. 2009;131:6664. doi: 10.1021/ja9016446. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yayli N., Sivrikaya S. Ö., Yaşar A., Üçüncü O., Güleç C., Kolayli S., Küçük M., Çelik E. J. Photochem. Photobiol., A. 2005;175:22. [Google Scholar]
- Xia Y., Wang J. Chem. Soc. Rev. 2017;46:2306. doi: 10.1039/c6cs00737f. [DOI] [PubMed] [Google Scholar]
- Transition metal diazo adducts like B are sometimes named 'carbenoids'. For a discussion on the naming of carbenoids and (transition)metal carbenes (or metallocarbenes), see: ; (a) Caballero A. and Pérez P. J., Chem.–Eur. J ., 10.1002/chem.201702392. [DOI]; (b) Maxwell J. L., Brown K. C., Bartley D. W., Kodadek T. Science. 1992;256:1544. doi: 10.1126/science.256.5063.1544. [DOI] [PubMed] [Google Scholar]
- Intramolecular HAT (viaTS2) first requires rotation around the Caromatic–Cvinylic single bond of C to produce Crotated, with its allylic hydrogen atoms closer to the carbene radical moiety. Crotated is computed to be only +1 kcal mol–1 less stable than C. See ESI.
- Unlike the copper, silver and gold systems recently reported by Pérez and coworkers, HAT viaTS2 does not involve active participation of the gem-ester moiety: Fructos M. R., Besora M., Braga A. A. C., Díaz-Requejo M. M., Maseras F., Pérez P. J., Organometallics, 2017, 36 , 172 . [Google Scholar]
- (a) Heimgartner H., Ulrich L., Hansen H.-J., Schmid H. Helv. Chim. Acta. 1971;54:2313. [Google Scholar]; (b) Heimgartner H., Zsindely J., Hansen H.-J., Schmid H. Helv. Chim. Acta. 1973;56:2924. [Google Scholar]
- Heating the kinetic product 3m for a prolonged period does not lead to spontaneous ring-closure to produce the corresponding thermodynamic 1,2-dihydronaphthalene product (2). Heating 3m in the presence of NaOMe only led to trans-esterification
- Trapping of the corresponding o-QDM intermediate by TEMPO to produce compound 6 cannot be fully excluded
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.