

Abstract
The growing burden of obesity- and aging-related diseases has hastened the search for governing biological processes. Cellular senescence is a stress-induced state of stable growth arrest strongly associated with aging that is aberrantly activated by obesity. The transition of a cell to a senescent state is demarcated by an array of phenotypic markers, and leveraging their context-dependent presentation is essential for determining the influence of senescent cells on tissue pathogenesis. Biomarkers of senescent cells have been identified in tissues that contribute to metabolic disease, including fat, liver, skeletal muscle, pancreata, and cardiovascular tissue, suggesting that pharmacological and behavioral interventions that alter their abundance and/or behavior may be a novel therapeutic strategy. However, contradictory findings with regard to a protective versus deleterious role of senescent cells in certain contexts emphasize the need for additional studies to uncover the complex interplay that defines multi-organ disease processes associated with obesity and aging.
Keywords: Aging, Obesity, Diabetes, Exercise, Senolytics, Inflammation
1. Central challenge: aging- and obesity-related chronic disease
Both the aged and obese populations are steadily growing. By 2030, more than 20% of the population will be age 65 or older (West et al., 2014) and 40% of the population will be obese (Finkelstein et al., 2012) (Fig. 1). Aging and obesity are major risk factors for chronic conditions that make up the leading causes of death, including diabetes, cardiovascular disease, hepatic steatosis, and cancer (Must et al., 1999), and therefore, underlie enormous public health and socioeconomic consequences. Although effective solutions to these challenges have been elusive, the causes are more well-defined. The multitude of complex environmental and behavioral factors driving the obesity epidemic boil down to a fundamental issue: energy imbalance (Swinburn et al., 2011). Regular consumption of high calorie diets and physical inactivity result in excess energy storage and disruption of whole-body metabolism and health. In contrast to obesity, aging is inevitable. However, healthspan extension—delaying the onset of age-related disease, disability, and frailty until late in the lifespan—is heavily influenced by modifiable factors, many of which are common to obesity. Public health campaigns to promote healthy lifestyle choices are critically important, as is the geroscience initiative to define the central biological processes that underlie the multitude of age-related chronic conditions, which may be targeted to improve multiple health parameters. From considerable work, cellular senescence has emerged as a central mechanism of age-related disease that may be prematurely activated by mediators of obesity (Tchkonia et al., 2013a).
Fig. 1.
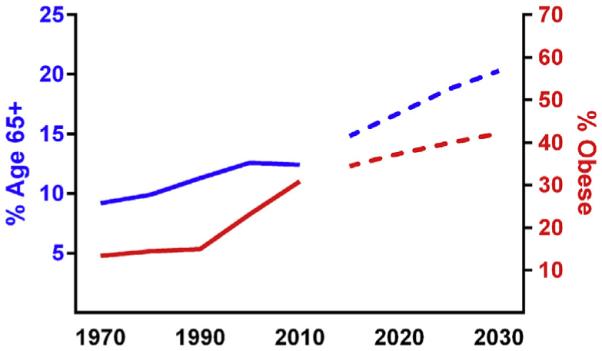
Projected rising trends in obesity and aging. Historical (solid blue line) and forecasted (dotted blue line) percentage of the U.S. population that is 65 years or older (West et al., 2014; United States Census Bureau, 2014), compared to historical (solid red line) and predicted (dotted red line) percentage of the population that is obese (Finkelstein et al., 2012; Ogden et al., 2010).
2. Cellular senescence: aging mechanism activated by nutrient excess
Cellular senescence is an evolutionarily conserved state of stable replicative arrest that occurs in response to diverse forms of cellular stress and damage. Damage accumulation stimulates the activity of cyclin dependent kinase inhibitors p16INK4a and/or p53-p21. These senescence effectors antagonize cyclin dependent kinases to prevent inactivation of retinoblastoma (Rb) protein, ultimately blocking cell cycle progression (Alcorta et al., 1996; Beausejour et al., 2003). Proliferation arrest is accompanied by molecular reprogramming and production of a unique secretome, comprised of cytokines, chemokines, extracellular matrix remodeling factors, and growth factors, termed the senescence-associated secretory phenotype (SASP) (Kuilman and Peeper, 2009). Through secretion of chemokines and cytokines, the SASP is believed to be the means by which senescent cells request clearance by the immune system (Freund et al., 2010). In aging tissues, however, apoptosis-resistant senescent cells persist and accumulate. It is unknown whether impaired immune-mediated removal results from insufficient signals produced by the senescent cells or immunosencence, likely, a combination of the two. Regardless, senescent cells autonomously prevent neoplastic transformation while promoting aging phenotypes, through both SASP signaling and reduced regenerative capacity arising from depletion of the mitotic pool (Campisi, 2005).
Nutrient excess may promote early and aberrant senescent cell accretion by accelerating intrinsic, age-related causes of senescence, including telomere erosion, oxidative and genomic damage, and proteome instability. Somatic cell telomere length is established in early life and declines at relatively equivalent rates across tissues types (Daniali et al., 2013). Tissue expansion in obesity may escalate replicative demands, ultimately leading to telomere attrition (Zannolli et al., 2008; Moreno-Navarrete et al., 2010). Mitochondrial dysfunction, including reduced mitochondrial mass, number (Ritov et al., 2005), and enzymatic oxidative capacity (Mogensen et al., 2007), is a common feature of obesity-related pathology, particularly insulin resistance. Increased metabolic loading is associated with mitochondrial respiration uncoupling, attenuated electron transport chain function, and increased production of reactive oxidative species (ROS). Indeed, increased markers of ROS damage have been identified in adipose tissue (Talior et al., 2003; Furukawa et al., 2004), cardiovascular tissue (Lee et al., 2012; Katunga et al., 2015), and liver (Pessayre, 2007; Matsuzawa-Nagata et al., 2008) of obese humans and mice. In addition, DNA damage robustly activates senescence, and increased frequency of genomic damage makers have been identified in obese individuals with type 2 diabetes (Al-Aubaidy and Jelinek, 2011). Similarly, compromised proteasome and endoplasmic reticulum function contributes to obesity-related metabolic deficiency (Otoda et al., 2013; Ozcan et al., 2004). While exacerbating inherent governors of cellular aging, energy excess in obesity may also drive and maintain senescence by disrupting other metabolic processes, including glucose utilization (Dorr et al., 2013) and nucleotide production (Aird et al., 2013).
2.1. Biomarkers of cellular senescence
Biological features of senescent cells are readily manipulated and leveraged as detection biomarkers in preclinical and human studies (Fig. 2). Protein and transcript assessment of senescence effectors, including p16INK4a, p53, and p21, is a first-line profiling strategy. Evidence suggests that p53-21 signaling initiates senescence, while p16INK4a signaling is required for maintenance of senescence (Alcorta et al., 1996; Beausejour et al., 2003). The notion that p16INK4a is a primary gatekeeper of a permanent senescent state that increases throughout aging led to the generation of several transgenic reporter and cell-deletion mouse models (p16INK-ATTAC (Baker et al., 2011), p16-LUC (Burd et al., 2013), p16-3MR (Demaria et al., 2014)). Results derived from these mice have supported the conclusion that p16INK4a-positive cells markedly accumulate in numerous tissues with chronological age (Krishnamurthy et al., 2004; Liu et al., 2009a) and in accelerated aging models (Gregg et al., 2012; Baker et al., 2004). Suicide transgene mediated deletion of p16INK4a-expressing cells improves multiple aging phenotypes in progeroid mice (Baker et al., 2011) and extends life-span in wild-type mice (Baker et al., 2016), further implicating the central role of this senescence factor in tissue aging.
Fig. 2.

Biomarkers of senescence. Senescent cells display features unique to non-senescent counterparts, and identification of multiple markers is important for confirming a senescent identity. Cell cycle is arrested (identifiable by a loss of proliferative markers) through the activity of cyclin dependent kinase inhibitors, including p16INK4a and/or p53/p21. This may be induced by several forms of cellular dysfunction, such as mitochondria-generated ROS, telomere attrition, and DNA damage, which may be unresolvable within telomeres (TAF). Transition to a senescent state may be accompanied by changes in morphology and nuclear dynamics, such as the formation of senescence-associated hetero-chromatin foci (SAHF) and reductions in the nuclear lamina marker lamin B1. Senescent cells develop a unique secretome, the senescence-associated secretory phenotype (SASP), which is compositionally context dependent and imparts potent paracrine effects.
In addition to cyclin dependent kinase inhibitor activation and abundance, senescent cells are characterized by various phenotypic markers, which differentially present based on induction mechanism (e.g. DNA damage, oncogene activation, chemotherapeutic agent) and state (e.g. senescence initiation versus maintenance). Expression and secretion of the SASP (Sharpless and Sherr, 2015; Acosta et al., 2008; Kuilman et al., 2008) and mitochondrial dysfunction (Passos et al., 2010; Henson et al., 2014; Correia-Melo et al., 2016) are consequences of senescence, plausible causes of paracrine senescence, and deterministic of senescent state stabilization. Monitoring of temporal changes in the secretome, mitochondrial dynamics, and ROS, therefore, divulge sequential mechanistic events. Senescence-associated β-galactosidase (SA-β-gal) positivity reflects lysosomal βwhich potently induces senescence-galactosidase activity at pH 6 (Dimri et al., 1995), likely indicative of altered proteolytic function, and is widely used to detect senescent cells. Replicative limits are reached by telomere erosion in the absence of compensatory telomerase activity, which potently induces senescence (Allsopp and Harley, 1995; Bodnar et al., 1998). Accrual of γH2AX and 53BP1 foci demarcates DNA damage response activation (Zou et al., 2004; d'Adda di Fagagna et al., 2003). Telomeric DNA appears to preferentially harbor damage, likely due to shelterin complex-mediated inhibition of repair. Thus, colocalization of γH2AX or 53BP1 with telomeric repeats, also known as telomere associated foci (TAF), is an increasingly utilized senescence indicator (Hewitt et al., 2012). Reduced Ki67 staining or bromodeoxyuridine (BrdU) incorporation can be leveraged to confirm a lack of proliferation in response to stimulation (Berkenkamp et al., 2014). Accretion of senescence associated heterochromatin foci (SAHF), detectable by 4′-6-Diamidino-2-phenylindole (DAPI) staining and enriched in lysine 9 trimethylated histone H3 (H3K9Me3), among other histone modifications, reflects chromatin remodeling that represses proliferative transcriptional events (Adams, 2007). Senescent cells often undergo morphological changes, including flattening, enlarging, or elongating (Bayreuther et al., 1988). Nuclear morphology changes are associated with a loss of lamin B1 signal, indicative of nuclear lamina alterations in senescent cells (Freund et al., 2012).
Differential combinations of these markers have been useful for identifying senescent cells in discrete tissues and in differing disease and aging contexts. However, the relative impact of senescent cell abundance on a given tissue and whole-body homeostasis is perceived to be highly variable and remains to be elucidated. For example, the impact of 10% versus 20% senescent β-cells on pancreatic function or 10% senescent β-cells versus 10% senescent adipocytes on whole-body homeostasis is not yet known. The secretory phenotype also varies in distinct tissue and disease contexts. Nevertheless, senescent cells and the SASP have been implicated in aging- and obesity-related health deterioration in multiple metabolic tissues, including adipose tissue, liver, skeletal muscle, pancreata, the microbiome, and the cardiovascular system (Fig. 3).
Fig. 3.

Senescent tissues in obesity-related disease. Senescent phenotypes have been identified in metabolically active tissues, including visceral adipose, pancreas, liver, cardiovascular tissue, and skeletal muscle. Obesity-related changes in the microbiota also induce senesecence in the liver.
2.2. Adipose tissue
Adipose tissue is the largest energy storage depot and a powerful endocrine organ. Coaction of these roles influences whole-body homeostasis through regulation of energy balance, satiety, insulin sensitivity, angiogenesis, blood pressure, and immune function (Galic et al., 2010). In obesity, adipocyte hypertrophy leading to tissue expansion is accompanied by a proinflammatory profile mediated by production of cytokines and chemokines and infiltration of macrophages. This phenomenon is particularly apparent in visceral, relative to subcutaneous, adipose tissue (Tchkonia et al., 2013b). Visceral adipose tissue accumulation is strongly linked to chronic disease risk (Fox et al., 2007), with waisthip ratio (a surrogate measure of visceral adiposity) proposed as a better predictor of obesity-related mortality than other common clinical measures, including blood pressure, cholesterol levels, or body mass index (Welborn et al., 2003). The potent inflammatory nature of visceral adipose tissue may be governed, in part, by cellular senescence (Tchkonia et al., 2010).
A seminal study by Minamino et al. demonstrated a mechanistic relationship between diet-induced metabolic disease and adipose senescence. Using mice that overexpress agouti peptide to drive excessive calorie intake and glucose and insulin intolerance, they showed that diet-induced obesity resulted in increased SA-β-gal and expression of p53 and p21 in adipose tissue, concordant with increased inflammatory and oxidative stress markers. p53 upregulation and deficiency exacerbated and diminished these outcomes, respectively (Minamino et al., 2009). Similarly, we recently identified significant upregulation of p16, p53, p21, and p16-GFP reporter transcript in visceral adipose tissue of middle-aged mice fed a high calorie diet for 4 months. Senescence effector activation was associated with increased SA-β-gal activity, with 12% of adipose cells staining positively, presentation of the SASP, and systemic metabolic disease. Exercise initiated at the same time as the high calorie diet or after accumulation of senescent burden ameliorated these effects (Schafer et al., 2016). In the absence of obesity, age-related adipose tissue atrophy is prevented by transgenic clearance of senescent cells (Xu et al., 2015a). Correspondingly, pharmacological inhibition of JAK signaling, an established mediator of the SASP, reduces adipose expression of proinflammatory SASP components (e.g. IL6, CXCL1, MIP-1βConsistent with mouse studies), which are believed to be important contributors to aging-related chronic sterile inflammation. The reduced pro-inflammatory molecular phenotype of adipose tissue is associated with improvements in physical function in 24 month old wild-type mice (Xu et al., 2015b). The culmination of these findings suggest that senescence and the SASP play a mechanistic role in age- and obesity-related adipose deterioration, including inflammatory and metabolic deficiencies, which may be prevented or rescued by exercise.
Consistent with mouse studies, p53, SA-β-gal, γH2AX, and inflammatory SASP factors are significantly increased in adipose extracts of individuals with type 2 diabetes (Minamino et al., 2009). Visceral adipose from obese individuals also contains higher levels of SASP components, including IGFBP3, IL6, and MCP-1 (Villaret et al., 2010; Pou et al., 2007). Preadipocytes, progenitors that comprise 15–50% of adipose tissue, have been implicated as a primary cell type prone to senescence (Xu et al., 2015b; Stout et al., 2014); however, adipocytes may also comprise a significant portion of the senescent-positive cellular pool in adipose (Minamino et al., 2009). Through quantification of γH2AX-positive and Ki67-negative cells with validation by SA-β-gal staining, a recent study identified more than 10% of preadipocytes isolated from older adults as senescent, while less than 4% of preadipocytes isolated from young adults were senescent (Xu et al., 2015b). Emerging evidence, therefore, suggests that visceral adipose may be uniquely susceptible to acquiring a senescent phenotype, which is mechanistically linked to systemic metabolic and functional decline in response to nutrient excess and chronological aging.
2.3. Liver
Ectopic accumulation of fat in peripheral organs is a key feature of metabolic disease. Hepatic steatosis, also known as nonalcoholic fatty liver disease, is highly prevalent in overweight older adults (Bertolotti et al., 2014), and several lines of evidence suggests that cellular senescence may exert causal influence. Hepatocytes in nonalcoholic fatty livers display elevated levels of γH2AX and p21, as well as shorter telomeres, and p21 levels correlate with type 2 diabetes and fibrosis stage (Aravinthan et al., 2013). Similarly, in cirrhotic livers, telomere length and senescent burden, determined by SA-β-gal staining, correlate with fibrosis severity (Wiemann et al., 2002). Cholangiocytes within liver specimens of individuals with primary sclerosing cholangitis, an idiopathic fibro-inflammatory disease, express p16INK4a, γH2AX, and SASP components (IL-6, IL-8, CCL2, and PAI-1) (Tabibian et al., 2014).
Results from biomarker-based human studies are supported by findings derived from animal models. High-fat feeding in obesity-prone rats increases hepatic expression of p16INK4a and p21 and decreases retinoblastoma protein phosphorylation, concomitant with elevation of triacylglycerol levels (Zhang et al., 2012). p53 expression increases in the liver of obese mice (Minamino et al., 2009). Likewise, high calorie consumption in mice increases hepatic p21 expression, liver weight, triglycerides, ceramides, and ectopic fat accumulation, and exercise ameliorates these outcomes (Schafer et al., 2016). Hepatic senescence may also drive malignancy through a high fat diet and microbiota-driven mechanism (Yoshimoto et al., 2013), a phenomena described in a subsequent section. The damaging effects of high-fat diet on liver health appear to be transmissible to offspring and involve senescence. The livers of 12-week-old pups of dams fed a high-fat diet show increased p16INK4a and inflammatory marker expression, reduced Rb protein phosphorylation, and attenuated activation of antioxidant defense genes (Zhang et al., 2011). Moreover, longer-lived, diet restricted mice exhibit fewer senescent hepatocytes than ad libitum-fed counterparts (Jurk et al., 2014).
Hepatic studies have also supported the hypothesis that senescence may be paracrinely propagated (Tabibian et al., 2014; Acosta et al., 2013; Nelson et al., 2012). Although results point to a negative role for senescence in liver disease, senescence may also restrict liver fibrosis, thereby providing essential benefit. In a toxin-induced liver damage model, mice deficient in p53 or p16INK4a display larger fibrotic areas than wild-type counterparts (Krizhanovsky et al., 2008). Similarly, IL22, which is produced by hepatic stellate cells following liver damage to resolve fibrosis, induces hepatic stellate cell senescence (Kong et al., 2012). Cumulatively, results in humans and rodents suggest the effects of senescence on liver health are context specific, and additional studies are required to determine the conditions in which hepatic senescent cells may serve as viable therapeutic targets.
2.4. Skeletal muscle
Skeletal muscle is a key metabolic organ. It is the primary site of insulin-mediated glucose disposal, the largest glycogen reservoir, and a major determinant of energy expenditure (Tzankoff and Norris, 1978). A decrease in skeletal muscle quantity is an early hallmark of aging, and alterations in skeletal muscle quality are consequences of both aging and obesity. Indeed, skeletal muscle mass progressively declines from the fourth decade of life and, concurrently, accumulates lipids within, and adipocytes between, skeletal muscle cells (Delmonico et al., 2009). Similar to liver, skeletal muscle is a prominent site of ectopic lipid accumulation in obese individuals (Goodpaster et al., 2000). Aging- and obesity-associated changes in skeletal muscle mass and composition contribute to systemic metabolic decline (Goodpaster and Wolf, 2004; LeBrasseur et al., 2011; White and Lebrasseur, 2014).
The biological mechanisms that underlie aging-associated muscle loss are not well defined, but are undoubtedly multifactorial. Similarly, the contribution of senescent skeletal muscle cells to sarcopenia is not yet clear. In part, this may reflect the predominantly post-mitotic state of skeletal muscle relative to other metabolic organs. Jeyapalan and colleagues failed to observe differences in 53BP1 foci in skeletal muscle nuclei between young and very old baboons (Jeyapalan et al., 2007). In contrast, the prevalence of this senescence biomarker in mitotic dermal fibroblasts increased from 3% in young animals to over 30% in old animals. An analysis of satellite cells, the mitotically competent adult muscle stem cells that reside beneath the basal lamina of mature muscle fibers, revealed that proliferative capacity and telomere length decreases during the first two decades of human life, but are then extremely stable throughout adulthood and into very late life (Decary et al., 1997). Even so, recent work has demonstrated that, at least in very old mice (28- to 32-months), satellite cells are prone to senesce through autophagy failure, which compromises skeletal muscle regeneration (Sousa-Victor et al., 2014; Garcia-Prat et al., 2016). Specifically, the investigators observed that the transition from quiescence to pre-senescence in satellite cells results from derepression of p16INK4a. In response to injury, aged satellite cells failed to activate, enter the cell cycle, and proliferate, and instead became senescent. They further showed that genetic silencing of p16INK4a by shRNA prevented senescence-associated gene expression and growth arrest, and restored regenerative capacity. The anti-rejuvenative effects of senescence have been further supported by Cosgrove et al., who demonstrated that p38 inhibition, a damage-activated MAPK signaling factor upstream of p53, in aged, hydrogel-cultured muscle satellite cells improves transplant engraftment efficiency and muscle function in aged mice (Cosgrove et al., 2014). Furthermore, clearance of senescent muscle cells in progeroid mice increases muscle fiber diameter and improves exercise capacity on a treadmill test (Baker et al., 2011). Thus, senescence of skeletal muscle cells per se does not appear to significantly contribute to the age-related loss of skeletal muscle mass, but may be a determinant of compromised late-life regenerative capacity.
While the accumulation of adipocytes in the skeletal muscle of older adults and obese individuals is widely recognized, until recently, the source of these cells was not understood. Muscle-derived satellite cells (Asakura et al., 2001), side population cells (rare, heterogeneous multipotent stem cells) (Uezumi et al., 2006), and mesenchymal stem cells (MSCs) (da Silva Meirelles et al., 2006) have all been shown to have adipogenic differentiation potential in vitro. However, Uezumi and colleagues nicely demonstrated that a specific subset of MSCs (CD31−CD45−PDGFRα+) isolated from skeletal muscle uniquely express master regulators of adipogenesis (C/EBPα and PPARγ), but not myogenic markers, and can robustly differentiate into lipid-containing adipocytes in vitro and in vivo (Uezumi et al., 2010). The propensity for these cells to become senescent in response to aging- and obesity-associated stresses is unknown but is an intriguing area for future investigation. As discussed above, preadipocytes, which share a similar cell-surface molecular phenotype to MSCs found in muscle (CD31−CD45−CD29+CD34+Sca-1+) (Uezumi et al., 2010), are highly prone to senescence and have a robust SASP that contributes to metabolic dysregulation and potentially sarcopenia. Skeletal muscle cells exhibit impaired insulin signaling and GLUT4 translocation when co-cultured with adipocytes (Dietze et al., 2002) and exhibit insulin resistance when exposed to the conditioned medium of adipocytes (Sell et al., 2008). MCP-1 has been identified as a potential causal factor (Sell et al., 2006). Whether targeted removal of senescence-prone fat cells from skeletal muscle or suppression of their SASP provides a means to improve the metabolic state of skeletal muscle in the context of aging or obesity remains to be determined.
2.5. Pancreas
Diabetes is characterized by insufficient production and secretion of insulin. Insulin-producing β-cell replication in pancreatic islets declines throughout normal aging in both mice (Teta et al., 2005) and humans (Meier et al., 2008). In obesity, β-cell populations initially expand to cope with increased systemic glucose and peripheral insulin resistance. However, islet mass ultimately decreases, due to β-cell apoptosis, leading to insufficient insulin production (Butler et al., 2003; Gepts, 1965; Gepts and De Mey, 1978). Insulitis, infiltration of immune cells within islets, is another common feature in type 2 diabetes, and is associated with increased levels of cytokines and chemokines (Boni-Schnetzler et al., 2008), an effect that may be exacerbated by free fatty acid exposure (Boni-Schnetzler et al., 2009). The origin of the proinflammatory islet milieu is unknown; SASP signaling is a potential source.
Considerable evidence suggests that p16INK4a-induced senescence limits compensatory β-cell proliferation in response to metabolic loading, directly contributing to type 2 diabetes pathogenesis. In islets, p16INK4a expression, which is regulated by a Bmi-1 polycomb complex, increases with age. High-fat diet consumption in young mice with low p16INK4a and high Bmi-1 expression drives β-cell proliferation, and contrastingly, islet expansion is limited in older mice with higher pancreatic p16INK4a expression (Tschen et al., 2009). Ablation of Bmi-1 increases p16INK4a levels, prevents β-cell proliferation and islet expansion, and drives hypoinsulinemia and glucose intolerance with preserved insulin sensitivity (Dhawan et al., 2009). Overexpression of p16INK4a decreases islet proliferation, and p16INK4a deficiency enhances islet proliferation following toxin-induced β-cell death (Krishnamurthy et al., 2006). Similarly, cyclin-dependent kinase 4 (Cdk4) activity, which is requisite for β-cell proliferation, is inhibited by p16INK4a (Krishnamurthy et al., 2006; Tsutsui et al., 1999). Mice lacking Cdk4 activity develop insulin-deficient diabetes, and contrastingly, mice expressing a modified Cdk4 that cannot be inhibited by p16INK4a display pancreatic hyperplasia (Rane et al., 1999). The islets of 12-month-old mice with high-fat diet-induced glucose intolerance display SA-β-gal positivity and significantly lower Ki67 staining than younger or control-fed counterparts (Sone and Kagawa, 2005). In contrast, a novel role for p16INK4a in age-related β-cell maturation was recently reported by Helman and colleagues, who demonstrated that p16INK4a induction improves glucose stimulated insulin secretion (Helman et al., 2016).
Several genome-wide association studies have linked the CDKN2A locus, which encodes p16INK4a, to type 2 diabetes (Wei et al., 2015; van Hoek et al., 2008; Zeggini et al., 2007; Scott et al., 2007). However, a study examining associations between diabetes-associated polymorphisms and β-cell function and insulin sensitivity in healthy adults failed to identify a relationship between CDKN2A and metabolic impairments (Pascoe et al., 2007). Therefore, it is currently unclear whether CDKN2A mutations confer type 2 diabetes susceptibility through diminished peripheral glucose handling and insulin sensitivity or islet endocrine dysfunction that manifests after pathology initiation, potentially in response to sustained nutrient excess. The culmination of previous explorations highlight a critical role for aging- and obesity-regulated senescence in pancreatic health and type 2 diabetes risk.
2.6. Cardiovascular
Cardiovascular disease is strongly linked to obesity and aging and remains the leading cause of death in developed countries. Many risk factors for development of cardiovascular disease are well defined and have robust pharmacotherapeutic strategies for mitigation (e.g., hypercholesterolemia, hypertension, chronic inflammation), yet morbidity and mortality due to cardiovascular diseases continues to climb. While the local and systemic pro-inflammatory milieu promoted by cellular senescence represents an intuitive contributor to accelerated development of cardiovascular disease, conflicting clinical and experimental findings have left the field in a highly contentious state.
Early studies in humans reported polymorphisms in senescence-associated CDKN2A-encoding locus (Chr9p21) to be strongly associated with development of atherosclerotic disease (Helgadottir et al., 2007; McPherson et al., 2007). Specifically, one of the most common SNP variants was associated with reduced expression of INK4/ARF variants such as p15INK4b, p16INK4a, ARF, and ANRIL in peripheral blood T-cells—a finding, at least at first glance, is inconsistent with a causal role for senescence in cardiovascular disease (Liu et al., 2009b). In aged or diseased cardiovascular tissues, however, numerous studies have reported increases in markers of senescent cells, including p16INK4a (Krishnamurthy et al., 2004; Roos et al., 2013), SA-βgal (Vasile et al., 2001; Minamino et al., 2003), and TAF (Roos et al., 2016).
Genetic inactivation of senescence effectors accelerates the development and complexity of atherosclerotic plaques (Merched and Chan, 2004; Guevara et al., 1999) and increases fibrosis following transverse aortic constriction (Meyer et al., 2016). These findings mirror conclusions derived from studies of senescence effector inactivation in liver, which support restrictive function in fibrotic pathogenesis (Krizhanovsky et al., 2008). While at face value these results argue against reducing or preventing formation of senescent cells as a strategy to treat cardiovascular disease, interpretation is confounded by the fact that genetic inactivation approaches also affect regulation of cell cycle checkpoints and, in general, result in dramatically augmented cellular proliferation in affected tissues (as most of these strains are known to have far more aggressive cancer phenotypes).
An alternate and more clinically relevant strategy for understanding the role of senescent cells in cardiovascular pathogenesis is to use genetic or pharmacological strategies to kill senescent cells. We recently reported that suicide-gene mediated clearance of p16INK4a-positive cells in chronologically aged p16INK-ATTAC mice improves endothelial function, attenuates age-associated cardiomyocyte hypertrophy, and prevents cardiac dysfunction induced by chronic β-agonist administration. We also demonstrated that a single dose of a “senolytic” drug cocktail (described further in a subsequent section) improves vascular endothelial and cardiac function in chronologically aged mice (Zhu et al., 2015). Chronic reductions in TAF+ senescent cells are associated with attenuated endothelial dysfunction in aged mice, improved vascular smooth muscle sensitivity to nitric oxide donors in hypercholesterolemic mice, and reduced vascular calcification in hypercholesterolemic mice (Roos et al., 2016). Critically, our group also found that the beneficial cardiovascular effects of chronic exercise training are closely associated with reductions in visceral adipose tissue senescence, suggesting that reducing senescent cell burden at non-cardiovascular sites may have significant benefits through systemic or paracrine factors (Schafer et al., 2016). Collectively, conflicting reports highlight the need for additional studies to understand the mechanistic contribution of senescent cells in individual pathogenic contexts; however, our recent findings demonstrate that targeting senescent cells is a viable strategy to improve age- and obesity-related cardiovascular phenotypes.
2.7. Microbiota
Scientific understanding of dynamic host-microbiota interplay is still in its infancy. Recent studies, however, have highlighted the powerful impact of diet on shaping the microbiota, with early results suggesting that dietary choices may induce senescence through microbiota-driven mechanisms. A compelling report by Yoshimoto et al. showed that obesity induced by diet or genetic manipulation perturbed the murine gut microbiome, effectually increasing production of deoxycholic acid (DCA). Hepatic stellate cell senescence was induced by elevated DCA exposure and promoted liver tumorigenesis through the SASP (Yoshimoto et al., 2013). A Drosophila study demonstrated that inhibition of the JAK/STAT SASP-regulation pathway in midgut cells, which mediate microbiota composition, extended lifespan and improved parameters of functional decline, including metaplasia and commensal dysbiosis (Li et al., 2016); whether a comparable host-commensal relationship is conserved in other model organisms or humans remains to be seen. Indeed, future work is expected to provide mechanistic insights on the interplay between host and microbiota aging and the influence of metabolic excess.
2.8. Therapeutic opportunities
The involvement of senescence in a multitude of chronic conditions has prompted major drug development efforts. In an initial study on senolytics, drugs that selectively kill senescent cells, Zhu et al. interrogated gene expression profiles to identify pro-survival and anti-apoptotic pathways specifically activated in senescent cells that may be targeted for their removal. Components of ephrin survival-regulating dependence receptor pathway, phosphatidylinositol-4,5-bisphosphate 3-kinase delta catalytic subunit (PI3KCD), p21, plasminogen-activated inhibitor-2 (PAI-2), and BCL-xL were validated as viable targets through RNA interference. Based on reported utility in targeting these factors, candidate senolytics were selected and tested for their ability to kill senescent cells in vitro. Dasatinib (D) and quercetin (Q), a tyrosine kinase inhibitor and flavonol, respectively, delivered in combination effectively killed senescent preadipocytes and HUVEC cells. Administration of DQ to aged, radiation-exposed, or Ercc1−/Δ progeroid mice reduced senescent cell burden and improved healthspan parameters (Zhu et al., 2015), and as previously described, DQ-mediated senescent cell clearance improved vascular phenotypes in aged and hyper-cholesterolemic mice (Roos et al., 2016). Subsequent studies have identified navitoclax, a Bcl-2 family inhibitor, as a senolytic in vitro (Leverson et al., 2015; Zhu et al., 2016).
SASP blockade is another strategy for attenuating the negative effects of senescent cells. mTOR inhibition extends lifespan in model organisms through regulation of several metabolic processes (Johnson et al., 2013), but SASP suppression appears be one of the means by which mTOR inhibition exerts anti-aging benefits. Specifically, rapamycin attenuates SASP transcription, transcript stability, and translation, ultimately leading to secretome depletion (Laberge et al., 2015; Herranz et al., 2015). NF-κB is a master regulator of SASP expression (Chien et al., 2011), and rapamycin-mediated suppression of IL1A translation diminishes NF-κB transcriptional activity (Laberge et al., 2015). Moreover, in vitro rapamycin treatment allows cells to maintain a degree of proliferative potential following senescence induction (Demidenko et al., 2009). Given the established secretome role in senescence maintenance, SASP suppression is a likely mechanism underlying rapamycin-induced escape from irreversible arrest. As previously discussed, pharmacological inhibition of the SASP-regulatory JAK pathway also confers healthspan benefits, which appear to be mediated by attenuation of adipose senescence and inflammation (Xu et al., 2015b).
Behavior modification is also expected to have potent effects on senescent burden, particularly in the context of nutrient excess. Voluntary running wheel exercise prevents and mitigates visceral adipose senescence and SASP resulting from high calorie feeding (Schafer et al., 2016) and, similarly, reduces aortic senescence markers (Werner et al., 2009). Calorie restriction (CR), which extends health- and life-span, decreases p16-and SA-β-gal-positive senescent cell burden in the kidney of aged rats, coincident with altered glomerular volume and fibrosis (Ning et al., 2013). Reduced senescence factors have also been demonstrated in the intestine and liver of mice maintained on a CR diet (Wang et al., 2010). Future studies aimed at understanding the timing, dosing, cell-type specificity, and systemic effects (beneficial and deleterious) of senolytic, SASP-inhibition, and behavioral interventions will be instrumental for moving targeted approached towards therapeutic viability. However, intriguing early reports demonstrate amelioration of senescence and the SASP as a promising strategy for aging- and obesity-associated conditions.
3. Conclusions and future studies
An increasing body of evidence supports the premise that cellular senescence is induced in multiple tissues by nutrient excess. This transition is important for preventing malignant events in response to oxidative and genomic stress, telomere erosion, and proteome instability brought on by metabolic loading. However, the consequence is premature accumulation of senescent cells. Senescent cells limit regeneration of metabolic tissues and through the SASP, modify tissue architecture and cause sterile inflammation. Paracrine SASP signaling causes senescent transmission to neighboring cells, and influences metabolic homeostasis in distal tissues.
Currently, the essential stimuli and kinetics defining the genesis of senescent cells and their contribution to tissue- and disease-specific changes are unclear and warrant further investigation. In obesity and aging studies, it is common to focus on one organ or pathology type. However, this approach limits information that may be gleaned about the temporal order of senescence induction. We recently profiled senescence markers in multiple tissues known to be metabolically sensitive, including adipose, liver, heart, skeletal muscle, and pancreas, in middle-aged mice maintained on a high fat and sugar diet. Visceral adipose displayed the strongest senescent signature, and to a lesser degree subcutaneous adipose and liver also exhibited increased senescence factors (Schafer et al., 2016). Thus, it appears that visceral adipose may be an early tissue driven to senesce by nutrient excess. Similarly, ectopic fat deposition in skeletal muscle and liver is a feature of obesity-related metabolic disease, and may reflect reprograming of resident cells to adipogenic differentiation states (Schafer et al., 2016; Goodpaster et al., 2000; Uezumi et al., 2010). It is currently unknown whether these cells, like preadipocytes, possess increased senescence propensity. Given the mechanistic role of adipose tissue inflammation in obesity-related metabolic disease, we speculate that targeting senescent adipose cells is a viable option for attenuating the whole-organism inflammatory milieu in obesity.
While investigations in adipose tissue point to a clear negative role for cellular senescence in obesity-related metabolic disease, the picture is more complex for liver, skeletal muscle, pancreas, and cardiovascular tissue. Cellular senescence biomarkers are readily identifiable in the livers of individuals with nonalcoholic fatty liver disease and cirrhosis (Aravinthan et al., 2013; Wiemann et al., 2002), and in the livers of high-fat fed mice, a senescent phenotype is accompanied by ectopic accumulation of fat, triglycerides, and ceramides (Schafer et al., 2016). Similarly, obesity-mediated microbiota perturbation leads to production of metabolites that promote liver cancer through a SASP-dependent mechanism (Yoshimoto et al., 2013). In contrast, genetic ablation of p53 or p16INK4a function exacerbates liver fibrosis (Krizhanovsky et al., 2008). Comparable phenomena have been observed in cardiac tissue, wherein, accumulation of senescence markers is associated with vascular endothelial and cardiac impairments, and clearance of senescent cells improves cardiovascular dysfunction (Roos et al., 2016). Antithetically, defective senescence effector function intensifies cardiovascular disease (Merched and Chan, 2004; Guevara et al., 1999; Meyer et al., 2016). Since p16INK4a, p53, and p21 exert well-known pleiotropic functions, which convey both anti-proliferative and pro-aging effects, context is essential for interpreting results. Indeed, considerable results suggest that targeting senescent cells through suicide-gene or pharmacological approaches in adulthood or following disease initiation may attenuate pathology, while systemic or life-long inactivation of senescence effectors is expected to bestow negative outcomes. A newer area of interest is senescence of post-mitotic cells; whether this is a critical feature of obesity- or aging-associated changes in cardiac or skeletal muscle remains to be examined. In the pancreas, p16INK4a-mediated senescence limits β-cell expansion and is believed to underlie insufficient insulin production in obesity-related type 2 diabetes (Tschen et al., 2009; Dhawan et al., 2009). New results, however, suggest that increases in pancreatic p16INK4a expression are associated with enhanced glucose stimulated insulin secretion and improved glucose tolerance in mice with genetically-induced diabetes (Helman et al., 2016). Thus, contradictory findings in metabolically active tissues emphasize the importance of additional mechanistic studies to understand the genesis of senescent phenotypes and their effects on organ heath and function. This work will be essential for determining contexts appropriate for targeted therapies, including senolytics, SASP-inhibitors, and behavioral strategies.
Acknowledgements
The authors acknowledge the support of the Glenn Foundation for Medical Research and NIH/NIA grants AG041122, AG053832, HL111121, TR000954, AG49182, and AG13925. We thank Matthew Moore, Mayo Clinic Center for Innovation, for generating figures.
References
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d'Adda di Fagagna F, Bernard D, Hernando E, Gil J. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L, Gil J. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. cell Biol. 2013;15:978–990. doi: 10.1038/ncb2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD. Remodeling of chromatin structure in senescent cells and its potential impact on tumor suppression and aging. Gene. 2007;397:84–93. doi: 10.1016/j.gene.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird KM, Zhang G, Li H, Tu Z, Bitler BG, Garipov A, Wu H, Wei Z, Wagner SN, Herlyn M, Zhang R. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 2013;3:1252–1265. doi: 10.1016/j.celrep.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Aubaidy HA, Jelinek HF. Oxidative DNA damage and obesity in type 2 diabetes mellitus. Eur. J. Endocrinol./Eur. Fed. Endocr. Soc. 2011;164:899–904. doi: 10.1530/EJE-11-0053. [DOI] [PubMed] [Google Scholar]
- Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 1996;93:13742–13747. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allsopp RC, Harley CB. Evidence for a critical telomere length in senescent human fibroblasts. Exp. Cell Res. 1995;219:130–136. doi: 10.1006/excr.1995.1213. [DOI] [PubMed] [Google Scholar]
- Aravinthan A, Scarpini C, Tachtatzis P, Verma S, Penrhyn-Lowe S, Harvey R, Davies SE, Allison M, Coleman N, Alexander G. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J. Hepatology. 2013;58:549–556. doi: 10.1016/j.jhep.2012.10.031. [DOI] [PubMed] [Google Scholar]
- Asakura A, Komaki M, Rudnicki M. Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation. 2001;68:245–253. doi: 10.1046/j.1432-0436.2001.680412.x. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, van Deursen JM. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayreuther K, Rodemann HP, Hommel R, Dittmann K, Albiez M, Francz PI. Human skin fibroblasts in vitro differentiate along a terminal cell lineage. Proc. Natl. Acad. Sci. U. S. A. 1988;85:5112–5116. doi: 10.1073/pnas.85.14.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkenkamp B, Susnik N, Baisantry A, Kuznetsova I, Jacobi C, Sorensen-Zender I, Broecker V, Haller H, Melk A, Schmitt R. In vivo and in vitro analysis of age-associated changes and somatic cellular senescence in renal epithelial cells. PloS One. 2014;9:e88071. doi: 10.1371/journal.pone.0088071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti M, Lonardo A, Mussi C, Baldelli E, Pellegrini E, Ballestri S, Romagnoli D, Loria P. Nonalcoholic fatty liver disease and aging: epidemiology to management. World J. Gastroenterology. 2014;20:14185–14204. doi: 10.3748/wjg.v20.i39.14185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Boni-Schnetzler M, Thorne J, Parnaud G, Marselli L, Ehses JA, Kerr-Conte J, Pattou F, Halban PA, Weir GC, Donath MY. Increased interleukin (IL)- 1beta messenger ribonucleic acid expression in beta -cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J. Clin. Endocrinol. Metabolism. 2008;93:4065–4074. doi: 10.1210/jc.2008-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni-Schnetzler M, Boller S, Debray S, Bouzakri K, Meier DT, Prazak R, Kerr-Conte J, Pattou F, Ehses JA, Schuit FC, Donath MY. Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology. 2009;150:5218–5229. doi: 10.1210/en.2009-0543. [DOI] [PubMed] [Google Scholar]
- Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, Bardeesy N, Castrillon DH, Beach DH, Sharpless NE. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell. 2013;152:340–351. doi: 10.1016/j.cell.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, Lowe SW. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes & Dev. 2011;25:2125–2136. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, Rushton MD, Charles M, Jurk D, Tait SW, Czapiewski R, Greaves L, Nelson G, Bohlooly YM, Rodriguez-Cuenca S, Vidal-Puig A, Mann D, Saretzki G, Quarato G, Green DR, Adams PD, von Zglinicki T, Korolchuk VI, Passos JF. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016;35:724–742. doi: 10.15252/embj.201592862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, Blau HM. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014;20:255–264. doi: 10.1038/nm.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- da Silva Meirelles L, Chagastelles PC, Nardi NB. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci. 2006;119:2204–2213. doi: 10.1242/jcs.02932. [DOI] [PubMed] [Google Scholar]
- Daniali L, Benetos A, Susser E, Kark JD, Labat C, Kimura M, Desai K, Granick M, Aviv A. Telomeres shorten at equivalent rates in somatic tissues of adults. Nat. Commun. 2013;4:1597. doi: 10.1038/ncomms2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decary S, Mouly V, Hamida CB, Sautet A, Barbet JP, Butler-Browne GS. Replicative potential and telomere length in human skeletal muscle: implications for satellite cell-mediated gene therapy. Hum. Gene Ther. 1997;8:1429–1438. doi: 10.1089/hum.1997.8.12-1429. [DOI] [PubMed] [Google Scholar]
- Delmonico MJ, Harris TB, Visser M, Park SW, Conroy MB, Velasquez-Mieyer P, Boudreau R, Manini TM, Nevitt M, Newman AB, Goodpaster BH. Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am. J. Clin. Nutr. 2009;90:1579–1585. doi: 10.3945/ajcn.2009.28047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dolle ME, Hoeijmakers JH, de Bruin A, Hara E, Campisi J. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell. 2014;31:722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell cycle. 2009;8:1888–1895. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- Dhawan S, Tschen SI, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes & Dev. 2009;23:906–911. doi: 10.1101/gad.1742609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietze D, Koenen M, Rohrig K, Horikoshi H, Hauner H, Eckel J. Impairment of insulin signaling in human skeletal muscle cells by co-culture with human adipocytes. Diabetes. 2002;51:2369–2376. doi: 10.2337/diabetes.51.8.2369. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U. S. A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Dabritz JH, Lisec J, Lenze D, Gerhardt A, Schleicher K, Kratzat S, Purfurst B, Walenta S, Mueller-Klieser W, Graler M, Hummel M, Keller U, Buck AK, Dorken B, Willmitzer L, Reimann M, Kempa S, Lee S, Schmitt CA. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature. 2013;501:421–425. doi: 10.1038/nature12437. [DOI] [PubMed] [Google Scholar]
- Finkelstein EA, Khavjou OA, Thompson H, Trogdon JG, Pan L, Sherry B, Dietz W. Obesity and severe obesity forecasts through 2030. Am. J. Prev. Med. 2012;42:563–570. doi: 10.1016/j.amepre.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu CY, Vasan RS, Murabito JM, Meigs JB, Cupples LA, D'Agostino RB, Sr., O'Donnell CJ. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation. 2007;116:39–48. doi: 10.1161/CIRCULATIONAHA.106.675355. [DOI] [PubMed] [Google Scholar]
- Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 2010;16:238–246. doi: 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. cell. 2012;23:2066–2075. doi: 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investigation. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galic S, Oakhill JS, Steinberg GR. Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010;316:129–139. doi: 10.1016/j.mce.2009.08.018. [DOI] [PubMed] [Google Scholar]
- Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, Sandri M, Munoz-Canoves P. Autophagy maintains stemness by preventing senescence. Nature. 2016;529:37–42. doi: 10.1038/nature16187. [DOI] [PubMed] [Google Scholar]
- Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14:619–633. doi: 10.2337/diab.14.10.619. [DOI] [PubMed] [Google Scholar]
- Gepts W, De Mey J. Islet cell survival determined by morphology. An immunocytochemical study of the islets of Langerhans in juvenile diabetes mellitus. Diabetes. 1978;27(Suppl. 1):251–261. doi: 10.2337/diab.27.1.s251. [DOI] [PubMed] [Google Scholar]
- Goodpaster BH, Wolf D. Skeletal muscle lipid accumulation in obesity, insulin resistance, and type 2 diabetes. Pediatr. Diabetes. 2004;5:219–226. doi: 10.1111/j.1399-543X.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- Goodpaster BH, Theriault R, Watkins SC, Kelley DE. Intramuscular lipid content is increased in obesity and decreased by weight loss. Metabolism. 2000;49:467–472. doi: 10.1016/s0026-0495(00)80010-4. [DOI] [PubMed] [Google Scholar]
- Gregg SQ, Gutierrez V, Robinson AR, Woodell T, Nakao A, Ross MA, Michalopoulos GK, Rigatti L, Rothermel CE, Kamileri I, Garinis GA, Stolz DB, Niedernhofer LJ. A mouse model of accelerated liver aging caused by a defect in DNA repair. Hepatology. 2012;55:609–621. doi: 10.1002/hep.24713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guevara NV, Kim HS, Antonova EI, Chan L. The absence of p53 accelerates atherosclerosis by increasing cell proliferation in vivo. Nat. Med. 1999;5:335–339. doi: 10.1038/6585. [DOI] [PubMed] [Google Scholar]
- Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, Masson G, Gudbjartsson DF, Magnusson KP, Andersen K, Levey AI, Backman VM, Matthiasdottir S, Jonsdottir T, Palsson S, Einarsdottir H, Gunnarsdottir S, Gylfason A, Vaccarino V, Hooper WC, Reilly MP, Granger CB, Austin H, Rader DJ, Shah SH, Quyyumi AA, Gulcher JR, Thorgeirsson G, Thorsteinsdottir U, Kong A, Stefansson K. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, Swisa A, Condiotti R, Granit RZ, Nevo Y, Fixler Y, Shreibman D, Zamir A, Tornovsky-Babeay S, Dai C, Glaser B, Powers AC, Shapiro AM, Magnuson MA, Dor Y, Ben-Porath I. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 2016;22:412–420. doi: 10.1038/nm.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson SM, Lanna A, Riddell NE, Franzese O, Macaulay R, Griffiths SJ, Puleston DJ, Watson AS, Simon AK, Tooze SA, Akbar AN. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8(+) T cells. J. Clin. investigation. 2014;124:4004–4016. doi: 10.1172/JCI75051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, Georgilis A, Montoya A, Wolter K, Dharmalingam G, Faull P, Carroll T, Martinez-Barbera JP, Cutillas P, Reisinger F, Heikenwalder M, Miller RA, Withers D, Zender L, Thomas GJ, Gil J. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015;17:1205–1217. doi: 10.1038/ncb3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012;3:708. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 2007;128:36–44. doi: 10.1016/j.mad.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–345. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, Saretzki G, Fox C, Lawless C, Anderson R, Hewitt G, Pender SL, Fullard N, Nelson G, Mann J, van de Sluis B, Mann DA, von Zglinicki T. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014;2:4172. doi: 10.1038/ncomms5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katunga LA, Gudimella P, Efird JT, Abernathy S, Mattox TA, Beatty C, Darden TM, Thayne KA, Alwair H, Kypson AP, Virag JA, Anderson EJ. Obesity in a model of gpx4 haploinsufficiency uncovers a causal role for lipid-derived aldehydes in human metabolic disease and cardiomyopathy. Mol. Metab. 2015;4:493–506. doi: 10.1016/j.molmet.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Feng D, Wang H, Hong F, Bertola A, Wang FS, Gao B. Inter-leukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. 2012;56:1150–1159. doi: 10.1002/hep.25744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investigation. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nature reviews. Cancer. 2009;9:81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, Limbad C, Demaria M, Li P, Hubbard GB, Ikeno Y, Javors M, Desprez PY, Benz CC, Kapahi P, Nelson PS, Campisi J. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015;17:1049–1061. doi: 10.1038/ncb3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBrasseur NK, Walsh K, Arany Z. Metabolic benefits of resistance training and fast glycolytic skeletal muscle. American journal of physiology. Endocrinol. metabolism. 2011;300:E3–E10. doi: 10.1152/ajpendo.00512.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R, Margaritis M, Channon KM, Antoniades C. Evaluating oxidative stress in human cardiovascular disease: methodological aspects and considerations. Curr. Med. Chem. 2012;19:2504–2520. doi: 10.2174/092986712800493057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, Kovar P, Tanaka A, Bruncko M, Sheppard GS, Wang L, Gierke S, Kategaya L, Anderson DJ, Wong C, Eastham-Anderson J, Ludlam MJ, Sampath D, Fairbrother WJ, Wertz I, Rosenberg SH, Tse C, Elmore SW, Souers AJ. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax) Cell Death Dis. 2015;6:e1590. doi: 10.1038/cddis.2014.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Qi Y, Jasper H. Preventing age-related decline of gut compartmentalization limits microbiota dysbiosis and extends lifespan. Cell Host Microbe. 2016;19:240–253. doi: 10.1016/j.chom.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Ibrahim JG, Thomas NE, Sharpless NE. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging cell. 2009;8:439–448. doi: 10.1111/j.1474-9726.2009.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Mohlke KL, Ibrahim JG, Thomas NE, Sharpless NE. INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PloS One. 2009;4:e5027. doi: 10.1371/journal.pone.0005027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa-Nagata N, Takamura T, Ando H, Nakamura S, Kurita S, Misu H, Ota T, Yokoyama M, Honda M, Miyamoto K, Kaneko S. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism Clin. Exp. 2008;57:1071–1077. doi: 10.1016/j.metabol.2008.03.010. [DOI] [PubMed] [Google Scholar]
- McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, Boerwinkle E, Hobbs HH, Cohen JC. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57:1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merched AJ, Chan L. Absence of p21Waf1/Cip1/Sdi1 modulates macrophage differentiation and inflammatory response and protects against atherosclerosis. Circulation. 2004;110:3830–3841. doi: 10.1161/01.CIR.0000148681.01282.89. [DOI] [PubMed] [Google Scholar]
- Meyer K, Hodwin B, Ramanujam D, Engelhardt S, Sarikas A. Essential role for premature senescence of myofibroblasts in myocardial fibrosis. J. Am. Coll. Cardiol. 2016;67:2018–2028. doi: 10.1016/j.jacc.2016.02.047. [DOI] [PubMed] [Google Scholar]
- Minamino T, Yoshida T, Tateno K, Miyauchi H, Zou Y, Toko H, Komuro I. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation. 2003;108:2264–2269. doi: 10.1161/01.CIR.0000093274.82929.22. [DOI] [PubMed] [Google Scholar]
- Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, Nojima A, Nabetani A, Oike Y, Matsubara H, Ishikawa F, Komuro I. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009;15:1082–1087. doi: 10.1038/nm.2014. [DOI] [PubMed] [Google Scholar]
- Mogensen M, Sahlin K, Fernstrom M, Glintborg D, Vind BF, Beck-Nielsen H, Hojlund K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2007;56:1592–1599. doi: 10.2337/db06-0981. [DOI] [PubMed] [Google Scholar]
- Moreno-Navarrete JM, Ortega F, Sabater M, Ricart W, Fernandez-Real JM. Telomere length of subcutaneous adipose tissue cells is shorter in obese and formerly obese subjects. Int. J. Obes. 2010;34:1345–1348. doi: 10.1038/ijo.2010.49. [DOI] [PubMed] [Google Scholar]
- Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. JAMA. 1999;282:1523–1529. doi: 10.1001/jama.282.16.1523. [DOI] [PubMed] [Google Scholar]
- Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11:345–349. doi: 10.1111/j.1474-9726.2012.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning YC, Cai GY, Zhuo L, Gao JJ, Dong D, Cui S, Feng Z, Shi SZ, Bai XY, Sun XF, Chen XM. Short-term calorie restriction protects against renal senescence of aged rats by increasing autophagic activity and reducing oxidative damage. Mech. ageing Dev. 2013;134:570–579. doi: 10.1016/j.mad.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Carroll MD. Prevalence of Overweight, Obesity, and Extreme Obesity Among Adults: United States, Trends 1960–1962 through 2007–2008. National Center for Health Statistics. 2010 [Google Scholar]
- Otoda T, Takamura T, Misu H, Ota T, Murata S, Hayashi H, Takayama H, Kikuchi A, Kanamori T, Shima KR, Lan F, Takeda T, Kurita S, Ishikura K, Kita Y, Iwayama K, Kato K, Uno M, Takeshita Y, Yamamoto M, Tokuyama K, Iseki S, Tanaka K, Kaneko S. Proteasome dysfunction mediates obesity-induced endoplasmic reticulum stress and insulin resistance in the liver. Diabetes. 2013;62:811–824. doi: 10.2337/db11-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Pascoe L, Tura A, Patel SK, Ibrahim IM, Ferrannini E, Zeggini E, Weedon MN, Mari A, Hattersley AT, McCarthy MI, Frayling TM, Walker M. Common variants of the novel type 2 diabetes genes CDKAL1 and HHEX/IDE are associated with decreased pancreatic beta-cell function. Diabetes. 2007;56:3101–3104. doi: 10.2337/db07-0634. [DOI] [PubMed] [Google Scholar]
- Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, Saretzki G, Rudolph KL, Kirkwood TB, von Zglinicki T. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010;6:347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessayre D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterology Hepatology. 2007;22(Suppl. 1):S20–S27. doi: 10.1111/j.1440-1746.2006.04640.x. [DOI] [PubMed] [Google Scholar]
- Pou KM, Massaro JM, Hoffmann U, Vasan RS, Maurovich-Horvat P, Larson MG, Keaney JF, Jr., Meigs JB, Lipinska I, Kathiresan S, Murabito JM, O'Donnell CJ, Benjamin EJ, Fox CS. Visceral and subcutaneous adipose tissue volumes are cross-sectionally related to markers of inflammation and oxidative stress: the Framingham Heart Study. Circulation. 2007;116:1234–1241. doi: 10.1161/CIRCULATIONAHA.107.710509. [DOI] [PubMed] [Google Scholar]
- Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat. Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- Roos CM, Hagler M, Zhang B, Oehler EA, Arghami A, Miller JD. Transcriptional and phenotypic changes in aorta and aortic valve with aging and MnSOD deficiency in mice. American journal of physiology. Heart Circulatory Physiology. 2013;305:H1428–H1439. doi: 10.1152/ajpheart.00735.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, Kirkland JL, Miller JD. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016:1–5. doi: 10.1111/acel.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer MJ, White TA, Evans G, Tonne JM, Verzosa GC, Stout MB, Mazula DL, Palmer AK, Baker DJ, Jensen MD, Torbenson MS, Miller JD, Ikeda Y, Tchkonia T, van Deursen JM, Kirkland JL, LeBrasseur NK. Exercise prevents diet-induced cellular senescence in adipose tissue. Diabetes. 2016;65(6):1606–1615. doi: 10.2337/db15-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J, Buchanan TA, Watanabe RM, Valle TT, Kinnunen L, Abecasis GR, Pugh EW, Doheny KF, Bergman RN, Tuomilehto J, Collins FS, Boehnke M. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sell H, Dietze-Schroeder D, Kaiser U, Eckel J. Monocyte chemotactic protein-1 is a potential player in the negative cross-talk between adipose tissue and skeletal muscle. Endocrinology. 2006;147:2458–2467. doi: 10.1210/en.2005-0969. [DOI] [PubMed] [Google Scholar]
- Sell H, Eckardt K, Taube A, Tews D, Gurgui M, Van Echten-Deckert G, Eckel J. Skeletal muscle insulin resistance induced by adipocyte-conditioned medium: underlying mechanisms and reversibility. Am. J. Physiol. Endocrinol. Metab. 2008;294:E1070–E1077. doi: 10.1152/ajpendo.00529.2007. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nature reviews. Cancer. 2015;15:397–408. doi: 10.1038/nrc3960. [DOI] [PubMed] [Google Scholar]
- Sone H, Kagawa Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia. 2005;48:58–67. doi: 10.1007/s00125-004-1605-2. [DOI] [PubMed] [Google Scholar]
- Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, Perdiguero E, Munoz-Canoves P. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506:316–321. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- Stout MB, Tchkonia T, Pirtskhalava T, Palmer AK, List EO, Berryman DE, Lubbers ER, Escande C, Spong A, Masternak MM, Oberg AL, LeBrasseur NK, Miller RA, Kopchick JJ, Bartke A, Kirkland JL. Growth hormone action predicts age-related white adipose tissue dysfunction and senescent cell burden in mice. Aging. 2014;6:575–586. doi: 10.18632/aging.100681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinburn BA, Sacks G, Hall KD, McPherson K, Finegood DT, Moodie ML, Gortmaker SL. The global obesity pandemic: shaped by global drivers and local environments. Lancet. 2011;378:804–814. doi: 10.1016/S0140-6736(11)60813-1. [DOI] [PubMed] [Google Scholar]
- Tabibian JH, O'Hara SP, Splinter PL, Trussoni CE, LaRusso NF. Cholangiocyte senescence by way of N-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology. 2014;59:2263–2275. doi: 10.1002/hep.26993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talior I, Yarkoni M, Bashan N, Eldar-Finkelman H. Increased glucose uptake promotes oxidative stress and PKC-delta activation in adipocytes of obese, insulin-resistant mice. American journal of physiology. Endocrinol. Metabolism. 2003;285:E295–E302. doi: 10.1152/ajpendo.00044.2003. [DOI] [PubMed] [Google Scholar]
- Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, Khosla S, Jensen MD, Kirkland JL. Fat tissue, aging, and cellular senescence. Aging Cell. 2010;9:667–684. doi: 10.1111/j.1474-9726.2010.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 2013;123:966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchkonia T, Thomou T, Zhu Y, Karagiannides I, Pothoulakis C, Jensen MD, Kirkland JL. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab. 2013;17:644–656. doi: 10.1016/j.cmet.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes. 2009;58:1312–1320. doi: 10.2337/db08-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui T, Hesabi B, Moons DS, Pandolfi PP, Hansel KS, Koff A, Kiyokawa H. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol. Cell. Biol. 1999;19:7011–7019. doi: 10.1128/mcb.19.10.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzankoff S, Norris AH. Longitudinal changes in basal metabolic rate in man. J. Appl. Physiol. 1978;33:536–539. doi: 10.1152/jappl.1978.45.4.536. [DOI] [PubMed] [Google Scholar]
- Uezumi A, Ojima K, Fukada S, Ikemoto M, Masuda S, Miyagoe-Suzuki Y, Takeda S. Functional heterogeneity of side population cells in skeletal muscle. Biochem. Biophys. Res. Commun. 2006;341:864–873. doi: 10.1016/j.bbrc.2006.01.037. [DOI] [PubMed] [Google Scholar]
- Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010;12:143–152. doi: 10.1038/ncb2014. [DOI] [PubMed] [Google Scholar]
- United States Census Bureau 2014. (Projections)
- van Hoek M, Dehghan A, Witteman JC, van Duijn CM, Uitterlinden AG, Oostra BA, Hofman A, Sijbrands EJ, Janssens AC. Predicting type 2 diabetes based on polymorphisms from genome-wide association studies: a population-based study. Diabetes. 2008;57:3122–3128. doi: 10.2337/db08-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasile E, Tomita Y, Brown LF, Kocher O, Dvorak HF. Differential expression of thymosin beta-10 by early passage and senescent vascular endothelium is modulated by VPF/VEGF: evidence for senescent endothelial cells in vivo at sites of atherosclerosis. FASEB J. official Publ. Fed. Am. Soc. Exp. Biol. 2001;15:458–466. doi: 10.1096/fj.00-0051com. [DOI] [PubMed] [Google Scholar]
- Villaret A, Galitzky J, Decaunes P, Esteve D, Marques MA, Sengenes C, Chiotasso P, Tchkonia T, Lafontan M, Kirkland JL, Bouloumie A. Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes. 2010;59:2755–2763. doi: 10.2337/db10-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Maddick M, Miwa S, Jurk D, Czapiewski R, Saretzki G, Langie SA, Godschalk RW, Cameron K, von Zglinicki T. Adult-onset, short-term dietary restriction reduces cell senescence in mice. Aging. 2010;2:555–566. doi: 10.18632/aging.100196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Cai C, Feng S, Lv J, Li S, Chang B, Zhang H, Shi W, Han H, Ling C, Yu P, Chen Y, Sun N, Tian J, Jiao H, Yang F, Li M, Wang Y, Zou L, Su L, Li J, Li R, Qiu H, Shi J, Liu S, Chang M, Lin J, Chen L, Li WD. Tox and CDKN2A/B gene polymorphisms are associated with type 2 diabetes in han chinese. Sci. Rep. 2015;5:11900. doi: 10.1038/srep11900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welborn TA, Dhaliwal SS, Bennett SA. Waist-hip ratio is the dominant risk factor predicting cardiovascular death in Australia. Med. J. Aust. 2003;179:580–585. doi: 10.5694/j.1326-5377.2003.tb05704.x. [DOI] [PubMed] [Google Scholar]
- Werner C, Furster T, Widmann T, Poss J, Roggia C, Hanhoun M, Scharhag J, Buchner N, Meyer T, Kindermann W, Haendeler J, Bohm M, Laufs U. Physical exercise prevents cellular senescence in circulating leukocytes and in the vessel wall. Circulation. 2009;120:2438–2447. doi: 10.1161/CIRCULATIONAHA.109.861005. [DOI] [PubMed] [Google Scholar]
- West LA, Cole S, Goodkind D, He W. 65+ in the United States: 2010. United States Census Bureau; 2014. [Google Scholar]
- White TA, Lebrasseur NK. Myostatin and sarcopenia: opportunities and challenges - a mini-review. Gerontology. 2014;60:289–293. doi: 10.1159/000356740. [DOI] [PubMed] [Google Scholar]
- Wiemann SU, Satyanarayana A, Tsahuridu M, Tillmann HL, Zender L, Klempnauer J, Flemming P, Franco S, Blasco MA, Manns MP, Rudolph KL. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J. official Publ. Fed. Am. Soc. Exp. Biol. 2002;16:935–942. doi: 10.1096/fj.01-0977com. [DOI] [PubMed] [Google Scholar]
- Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, Sepe A, Johnson KO, Stout MB, Giorgadze N, Jensen MD, LeBrasseur NK, Tchkonia T, Kirkland JL. Targeting senescent cells enhances adipogenesis and metabolic function in old age. eLife. 2015:4. doi: 10.7554/eLife.12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, White TA, Johnson KO, Stout MB, Mezera V, Giorgadze N, Jensen MD, LeBrasseur NK, Kirkland JL. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. U. S. A. 2015;112:E6301–E6310. doi: 10.1073/pnas.1515386112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Honda K, Ishikawa Y, Hara E, Ohtani N. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- Zannolli R, Mohn A, Buoni S, Pietrobelli A, Messina M, Chiarelli F, Miracco C. Telomere length and obesity. Acta Paediatr. 2008;97:952–954. doi: 10.1111/j.1651-2227.2008.00783.x. [DOI] [PubMed] [Google Scholar]
- Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney AS, McCarthy MI, Hattersley AT. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Strakovsky R, Zhou D, Zhang Y, Pan YX. A maternal high-fat diet represses the expression of antioxidant defense genes and induces the cellular senescence pathway in the liver of male offspring rats. J. Nutr. 2011;141:1254–1259. doi: 10.3945/jn.111.139576. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhou D, Strakovsky R, Zhang Y, Pan YX. Hepatic cellular senescence pathway genes are induced through histone modifications in a diet-induced obese rat model. American journal of physiology. Gastrointest. Liver Physiology. 2012;302:G558–G564. doi: 10.1152/ajpgi.00032.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, O'Hara SP, LaRusso NF, Miller JD, Roos CM, Verzosa GC, LeBrasseur NK, Wren JD, Farr JN, Khosla S, Stout MB, McGowan SJ, Fuhrmann-Stroissnigg H, Gurkar AU, Zhao J, Colangelo D, Dorronsoro A, Ling YY, Barghouthy AS, Navarro DC, Sano T, Robbins PD, Niedernhofer LJ, Kirkland JL. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14(4):644–658. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, Pirtskhalava T, Giorgadze N, Johnson KO, Giles CB, Wren JD, Niedernhofer LJ, Robbins PD, Kirkland JL. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016;15(3):428–435. doi: 10.1111/acel.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Sfeir A, Gryaznov SM, Shay JW, Wright WE. Does a sentinel or a subset of short telomeres determine replicative senescence? Mol. Biol. Cell. 2004;15:3709–3718. doi: 10.1091/mbc.E04-03-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]