

Abstract
Expression of the cell-surface glycoprotein MHC class I polypeptide-related sequence A (MICA) is induced in dangerous, abnormal, or “stressed” cells, including cancer cells, virus-infected cells, and rapidly proliferating cells. MICA is recognized by the activating immune cell receptor natural killer group 2D (NKG2D), providing a mechanism by which immune cells can identify and potentially eliminate pathological cells. Immune recognition through NKG2D is implicated in cancer, atherosclerosis, transplant rejection, and inflammatory diseases, such as rheumatoid arthritis. Despite the wide range of potential therapeutic applications of MICA manipulation, the factors that control MICA expression are unclear. Here we use metabolic interventions and metabolomic analyses to show that the transition from quiescent cellular metabolism to a “Warburg” or biosynthetic metabolic state induces MICA expression. Specifically, we show that glucose transport into the cell and active glycolytic metabolism are necessary to up-regulate MICA expression. Active purine synthesis is necessary to support this effect of glucose, and increases in purine nucleotide levels are sufficient to induce MICA expression. Metabolic induction of MICA expression directly influences NKG2D-dependent cytotoxicity by immune cells. These findings support a model of MICA regulation whereby the purine metabolic activity of individual cells is reflected by cell-surface MICA expression and is the subject of surveillance by NKG2D receptor-expressing immune cells.
Keywords: immunology; innate immunity; metabolism; nucleoside/nucleotide metabolism; purine; Warburg effect; natural killer cells (NK cells); purine metabolism, glucose metabolism; immunometabolism; immunosurveillance; NKG2D ligands
Introduction
The capacity to recognize and eliminate harmful elements while safeguarding healthy “self” cells is a key characteristic of the mature human immune system. This capacity arises, in large part, through the successful passage of lymphocytes through a series of immune self-tolerance mechanisms. Immune self-tolerance poses a challenge to the immune recognition of damaged or stressed self cells. The circumvention of such tolerance mechanisms using PD1-PDL1 inhibition has led to major recent clinical success with cancer immune therapies (1). The natural killer group 2D (NKG2D)2 receptor-ligand system functions to overcome tolerance and eliminate stressed self cells even in the presence of functioning self-tolerance (2).
In humans, the NKG2D ligands are a group of eight cell-surface proteins, of which the highly polymorphic major histocompatibility class I chain-related protein A (MICA) is the most extensively studied. Generally, healthy quiescent cells do not express ligands for NKG2D (3, 4), but cell-surface expression of MICA is switched on by events such as malignant transformation (5), viral infection (6, 7), proliferative cell activation (8, 9), and some pro-inflammatory stimuli (10, 11). MICA-expressing cells are then targeted by immune cells (including natural killer (NK) cells, NKT cells, and cytotoxic CD8+ T cells) that express the activating NKG2D receptor; this interaction can result in direct cytotoxicity (12), co-stimulation (6, 13, 14), or cytokine secretion (15), depending on the context.
Evidence from observational human studies and mouse models implicates immune recognition through NKG2D in a range of autoimmune and inflammatory conditions, including rheumatoid arthritis (16), inflammatory bowel disease (17), and atherosclerosis (11). The expression of MICA on human allogeneic organ transplants has been linked to transplant rejection (18), and advanced-stage cancers are associated with loss of MICA expression (5). Hence, the ability to manipulate MICA expression in vivo has many potential clinical applications: up-regulation of MICA could promote cancer immunity, and down-regulation could be beneficial in autoimmune disease or transplantation.
Multiple factors have been associated with changes in MICA expression, including activation of the DNA damage response pathway (19), Toll-like receptor (TLR) stimulation (10), histone deacetylation (20), heat shock transformation (21), ionizing radiation (22), growth factor pathway activation (23), cell-surface shedding (24), and microRNA expression (25). In addition, a number of gene-regulatory elements and transcription factors are known to play a role in MICA induction (11, 26). However, an integrated understanding of the mechanisms determining MICA expression remains elusive.
MICA expression in human primary cells or tissue samples is found in settings independently associated with high metabolic activity (increased glucose uptake, glycolysis, high lactate output, and proportionate reduction in TCA cycle metabolism, or “Warburg metabolism” (27–31)). This state of “activated metabolism” can be considered as a biosynthetic state, where enhanced glycolytic flux generates intermediate substrates for biomolecule synthesis (32). High-energy purine nucleotides, such as ATP, are among the downstream products.
Here, we show that glucose metabolism leading to the generation of high-energy purine nucleotides, a process at the core of the Warburg effect, induces cell-surface expression of MICA. We demonstrate that MICA induction by high-energy purine nucleotides is associated with increased NKG2D-dependent cellular immunogenicity and susceptibility to NK cell cytotoxicity, supporting our hypothesis that NKG2D provides a mechanism for immune oversight of metabolically activated cells.
Results
Glucose induces MICA expression
We hypothesized that the transition from quiescent to “activated” or Warburg metabolism plays an important role in NKG2D ligand induction. To test this hypothesis, we used glucose restriction to model quiescent versus activated metabolism and observed a direct correlation between the glucose concentration of culture medium and cell-surface expression of MICA in human embryonic kidney (HEK)-293T cells, cervical cancer cells (HeLa), fibrosarcoma cells (HT1080), and breast cancer cells (MCF7) (Fig. 1).
Figure 1.

Glucose induces MICA expression. 293T (human embryonic kidney), HeLa (cervical cancer), HT1080 (fibrosarcoma), and MCF7 (breast cancer) cells were cultured for 48 h in medium containing 5 mm glucose that was then replaced with fresh medium containing either 0, 2.5, 5, 12.5, or 25 mm glucose. The cells were cultured for a further 48 h in these conditions before cell-surface MICA expression was measured by flow cytometry. MICA expression rose with the glucose concentration. The dotted histogram represents the isotype control sample, and the glucose concentration is indicated by the gray scale.
We undertook further characterization in HEK-293T cells (Fig. 2, A–M). In keeping with a Warburg phenotype, cells cultured in high (25 mm) glucose concentrations consumed more glucose and produced more lactate than cells cultured in low (5 mm) glucose (Fig. 2, A and B). Compared with cells cultured in 5 mm glucose, intracellular glucose concentrations increased 2.13-fold (p < 0.05) (Fig. 2C), and total RNA concentrations were higher (p < 0.005) in cells cultured in 25 mm glucose (Fig. 2D). The difference in cellular proliferation rate was ∼2.2-fold across a range of glucose concentrations, but unlike MICA expression, the relationship was not directly proportional with maximum proliferation rates observed at intermediate glucose concentrations (Fig. 2E). The distribution of cell-cycle phases was not significantly altered by glucose concentration (Fig. 2, F and G). We used an inducible lentiviral eGFP construct to confirm that the capacity for gene expression remained intact in low glucose conditions (Fig. 2, H and I). Detected endogenous MICA levels varied directly with glucose concentration in permeabilized cells, suggesting that the observed effect of glucose is not due to a change in the distribution of MICA between different cellular compartments (Fig. 2, J and K). Changes in osmolality did not influence MICA cell-surface levels (Fig. 2, L and M). Glucose-induced MICA expression was observed at the cell surface in the presence of the matrix metalloprotease inhibitor MMPI III, demonstrating that the effects we observe are not due to changes in MICA shedding (Fig. S1, A and B). In addition, MICA mRNA transcript levels were increased over 4-fold in cells cultured in 25 mm glucose (Fig. S1C).
Figure 2.

Glucose restriction replicates quiescent to activated metabolism transition. A, 293T cells cultured in low (5 mm) glucose consumed less glucose than cells cultured in high (25 mm) glucose (p < 0.0001). B, lactate production was ∼4 times higher in cell cultured in high glucose compared with low glucose (p < 0.0001). C, intracellular glucose concentrations measured by nanoflow LC-MS were 2.13-fold higher in cells cultured in high glucose (p < 0.05). D, the RNA concentration per cell, reduced by culture with the RNA synthesis inhibitor actinomycin D (ActD), is 13% higher in cells cultured in 25 mm glucose compared with cells cultured in 5 mm glucose (p < 0.005). E, cellular proliferation measured by cell counting or CFSE dilution was maximal at intermediate glucose concentrations, unlike MICA expression. F, cell-cycle profiles were similar for cells cultured in 5 or 25 mm glucose. G, no significant difference was observed between cell-cycle phases in these conditions. H, MICA expression is induced by high (25 mm) glucose in cells transfected with inducible eGFP (p < 0.0001), but eGFP itself is not induced by high glucose. I, the induction of eGFP expression by doxycycline is equally efficient at low (5 mm) and high (25 mm) glucose. J, total cell MICA levels measured by flow cytometry of permeabilized cells is proportional to glucose concentration and cell-surface MICA. K, proliferating cell nuclear antigen (PCNA), used as a control for adequate permeabilization, is expressed independently of glucose concentration. L, glucose, mannitol, and sodium chloride each raise culture serum osmolality. M, change in culture medium osmolality does not affect MICA expression. Error bars, 95% confidence interval.
Glucose transport and metabolism are necessary for MICA induction
Next we investigated how glucose might alter MICA expression. Glucose is primarily transported into cells through the GLUT family of transporters (33). Two inhibitors of these transporters, cytochalasin B and phloretin, prevented glucose-induced MICA expression (GIME) in a dose-dependent manner (Fig. 3, A and B). Mannose, an alternative substrate for glycolysis transported via the same GLUT transporters, also caused MICA expression (Fig. 3C). A third glycolytic substrate, fructose, had a limited effect on MICA expression in untransfected HEK-293T cells (Fig. 3C), but fructose-induced MICA expression was observed in HEK-293T cells transfected with GLUT5, the fructose transporter (Fig. 3, D and E). 2-Deoxyglucose is a glucose analogue readily transported into the cell and phosphorylated by hexokinase to generate 2-deoxyglucose 6-phosphate. Whereas 2-deoxyglucose 6-phosphate inhibits downstream enzymes in proximal glycolysis, it can induce the expression of genes directly regulated by glucose 6-phosphate (34). However, we found that unlike glucose, 2-deoxyglucose did not induce the expression of MICA (Fig. 3F) and inhibited its expression in high-glucose conditions (Fig. 3G), an effect that was not overcome by the addition of mannose (Fig. 3H). Together, these experiments suggest that the effect of glucose on MICA expression is dependent on the transport of glucose into the cell and its metabolism through glycolysis.
Figure 3.

The transport of glycolytic substrates into the cell is necessary for their effect on MICA expression. A, cytochalasin B, an inhibitor of GLUT-mediated glucose transport, inhibits cell-surface MICA expression in HEK-293T cells cultured in 25 mm glucose. B, phloretin, a second GLUT transporter inhibitor, also reduces MICA expression in 25 mm glucose. C, mannose, an alternative substrate for glycolysis, induces MICA expression. The ability of fructose to induce MICA expression is limited in normal cells. D, mRNA for the fructose transporter GLUT5 is not detected in HEK-293T cells but is present in GLUT5-transfected cells. E, fructose-induced MICA expression is observed in GLUT5-transfected cells. F, the glucose analogue 2-deoxyglucose (2DG), which is not a substrate for glycolysis, does not induce MICA expression. G, 2DG inhibits MICA expression in 25 mm glucose. H, mannose does not overcome the inhibition of glucose-induced MICA expression by 2DG. RT+, with reverse transcriptase; RT−, without reverse transcriptase; dH2O, deionized H2O. Error bars, 95% confidence interval.
Purine nucleotides are necessary for glucose-induced MICA expression and sufficient to induce MICA expression
Glycolysis produces many intracellular metabolites. The proximal metabolites of glycolysis are essential for de novo nucleotide synthesis (Fig. 4). We hypothesized that nucleotide synthesis might mediate GIME. Because the de novo synthesis of the purine nucleobase is directly dependent on the supply of proximal glycolytic metabolites, we first tested this hypothesis by treating cells cultured in high glucose (25 mm) with two inhibitors of de novo purine synthesis, 6-diazo-oxo-norleucine (DON) and azaserine. Both compounds prevented GIME (Fig. 5, A and B). The specificity of this effect on de novo purine synthesis was tested using hypoxanthine, aminopterin, and thymidine (HAT)-selected cells. Whereas cells grown in standard culture medium depend on de novo purine synthesis, HAT-selected cells use the salvage pathway exclusively for new purine nucleotide synthesis. Azaserine inhibited GIME only in cells grown in standard culture medium and had no effect on HAT-selected cells (Fig. 5, C and D), consistent with a specific action on de novo purine synthesis. DON probably has additional off-target inhibitory effects. The addition of a purine salvage pathway substrate to azaserine-treated cells in high glucose caused dose-dependent MICA expression (Fig. 5E), confirming that purine synthesis is necessary for and can control GIME. This finding was reproducible with a range of salvage pathway substrates, including the purine nucleobases adenine and guanine, purine nucleosides including inosine, and deoxynucleosides (Fig. S2, A–C).
Figure 4.

Nucleotide synthesis, structure, and nomenclature. A, ribose 5-phosphate (R5P) is produced by the metabolism of glucose 6-phosphate (G6P) through the pentose phosphate pathway (PPP). Phosphoribosyl pyrophosphate (PRPP) is essential for both salvage purine synthesis and de novo purine synthesis. The inhibitors DON and azaserine inhibit enzymes in the de novo purine synthesis pathway proximal to the intermediate AICA-Rt. The nucleoside AICA-Rs is readily transported across the cell membrane and is phosphorylated by adenosine kinase to the de novo synthesis pathway intermediate AICA-Rt. B, nucleobases and nucleosides are readily transportable across the cell membrane. Nucleotides (phosphonucleosides) have low cell membrane permeability. C, de novo and salvage purine synthesis both depend on PRPP. The carbon 5′-phosphate moiety of PRPP, originally added to glucose by hexokinase, defines purine synthesis pathway intermediates as nucleotides. In de novo synthesis, the purine ring is built directly onto PRPP. In contrast, the pyrimidine nucleobase is synthesized independently of PRPP, which is added to the complete nucleobase to form a pyrimidine nucleotide. GAR, glycinamide ribonucleotide; NTP, nucleotide triphosphate; AK, adenosine kinase; arrows represent multistep metabolic pathways.
Figure 5.

An intact purine synthesis pathway is necessary for MICA induction. A, DON, an inhibitor of proximal de novo purine synthesis, prevents GIME. B, azaserine, a second de novo pathway inhibitor, also blocks GIME. C, azaserine, DON, and the GLUT transporter inhibitor phloretin each prevent GIME in HEK-293T cells. D, HAT-selected HEK-293T cells produce purine nucleotides exclusively through the salvage pathway. Azaserine is unable to prevent GIME in HAT-selected cells, showing that inhibition of GIME by azaserine depends on its action in inhibiting de novo purine synthesis. DON, like phloretin, appears to have additional off-target actions. E, the salvage pathway substrate hypoxanthine rescues MICA expression in azaserine-treated cells in 25 mm glucose. F, similarly, AICA-Rs also rescues MICA expression in azaserine-treated cells. G, AICA-Rs, which can be converted to AICA-Rt by adenosine kinase, is sufficient to induce MICA expression even in conditions of low glucose. Error bars, 95% confidence interval.
5-Aminoimidazole-4-carboxamide ribonucleotide (AICA-Rt) is a de novo pathway purine intermediate nucleotide composed of a phosphorylated ribose sugar and an incomplete purine nucleobase. The inhibitor azaserine blocks de novo purine synthesis before the point of AICA-Rt generation. AICA ribonucleoside (AICA-Rs) is the equivalent intermediate nucleoside, composed of an unphosphorylated ribose moiety and the same incomplete purine nucleobase. AICA-Rs can be transported into the cell and phosphorylated by adenosine kinase to generate AICA-Rt. We tested the ability of AICA-Rs to act as a salvage pathway substrate in vitro by culturing cells in the presence of high glucose, azaserine, and increasing amounts of AICA-Rs and observed AICA-Rs-dependent induction of MICA expression (Fig. 5F). To determine whether AICA-Rs was sufficient to induce MICA expression, cells were cultured in 5 mm glucose with increasing doses of AICA-Rs, and AICA-Rs–dependent MICA induction was observed (Fig. 5G). Similarly, the purine nucleosides adenosine and guanosine, but not pyrimidine nucleosides cytidine and thymidine, were sufficient to induce MICA expression in 5 mm glucose (Fig. S2D). The purine nucleosides adenosine and inosine, but not purine nucleobases hypoxanthine and adenine, were sufficient to induce MICA expression in low-glucose conditions (Fig. S2E). Whereas azaserine prevented GIME, it had no effect on AICA-Rs, adenosine, or inosine-induced MICA expression, consistent with the hypothesis that purine nucleosides induce MICA expression downstream and independently of de novo purine synthesis (Fig. S2F). To generate new purine nucleotides, AICA-Rs must be phosphorylated to the de novo pathway intermediate nucleotide AICA-Rt by adenosine kinase inside the cell. Inhibition of adenosine kinase prevented AICA-Rs–induced MICA expression, demonstrating that AICA-Rt, but not AICA-Rs, causes MICA induction (Fig. S2, G–I). There was no correlation between cell proliferation and MICA expression under these different metabolic conditions (Fig. S3).
Intracellular purine nucleotide and tricarboxylic acid cycle intermediates are associated with increased MICA expression
These observations suggested that cellular conditions associated with the production of energy-rich phosphorylated purine nucleotides were pivotal in MICA expression at the cell surface. We tested this association by measuring intracellular metabolites and cell-surface MICA expression in a range of conditions, using capillary electrophoresis time-of-flight mass spectrometry (CE-TOFMS), liquid chromatography time-of-flight mass spectrometry (LC-TOFMS), and flow cytometry (Fig. 6A). Increased intracellular concentrations of purine nucleotides were observed when the cell-surface MICA expression increased. Furthermore, concentrations of TCA intermediates, necessary for maintaining purine nucleotides in their phosphorylated state, were also associated with high MICA expression (Fig. 6, B and C). ATP demonstrated the strongest linear correlation with MICA expression (Table 1). Interrogation of the complete data sets also indicated a metabolic signature that is associated with MICA expression. Principal component analysis clearly separated a low-MICA expression cluster, and two high-MICA expression clusters: an active de novo or salvage purine synthesis cluster and a purine nucleoside cluster (Fig. S4A). Metabolite enrichment analysis for predicted metabolite sets showed significant enrichment in pathways supporting nucleotide and TCA cycle product synthesis, including citrate synthase, ATP synthase, nucleoside-diphosphatase, and glutamate transport (Fig. S4B). A heat map of the full metabolome data set demonstrated Ward clustering according to MICA expression across a diverse set of metabolic conditions (Fig. S5). MICA expression was associated with increased high-energy purine compounds (ATP, GTP) and TCA cycle metabolites.
Figure 6.

High-energy purine nucleotides and TCA cycle intermediates correlate strongly with MICA expression. A, intracellular metabolite concentrations, measured by CE-TOFMS and LC-TOFMS, in a range of metabolic conditions, are shown in this heat map. Signal strength is depicted as a percentage of the maximum for each compound. B, the strongest linear correlations between intracellular metabolite concentrations and cell-surface MICA expression were noted for purine nucleotides, including ATP and TCA cycle intermediates. C, cell-surface levels of MICA measured by flow cytometry. Error bars, 95% confidence interval.
Table 1.
Linear correlation between intracellular metabolite concentrations and cell surface MICA expression
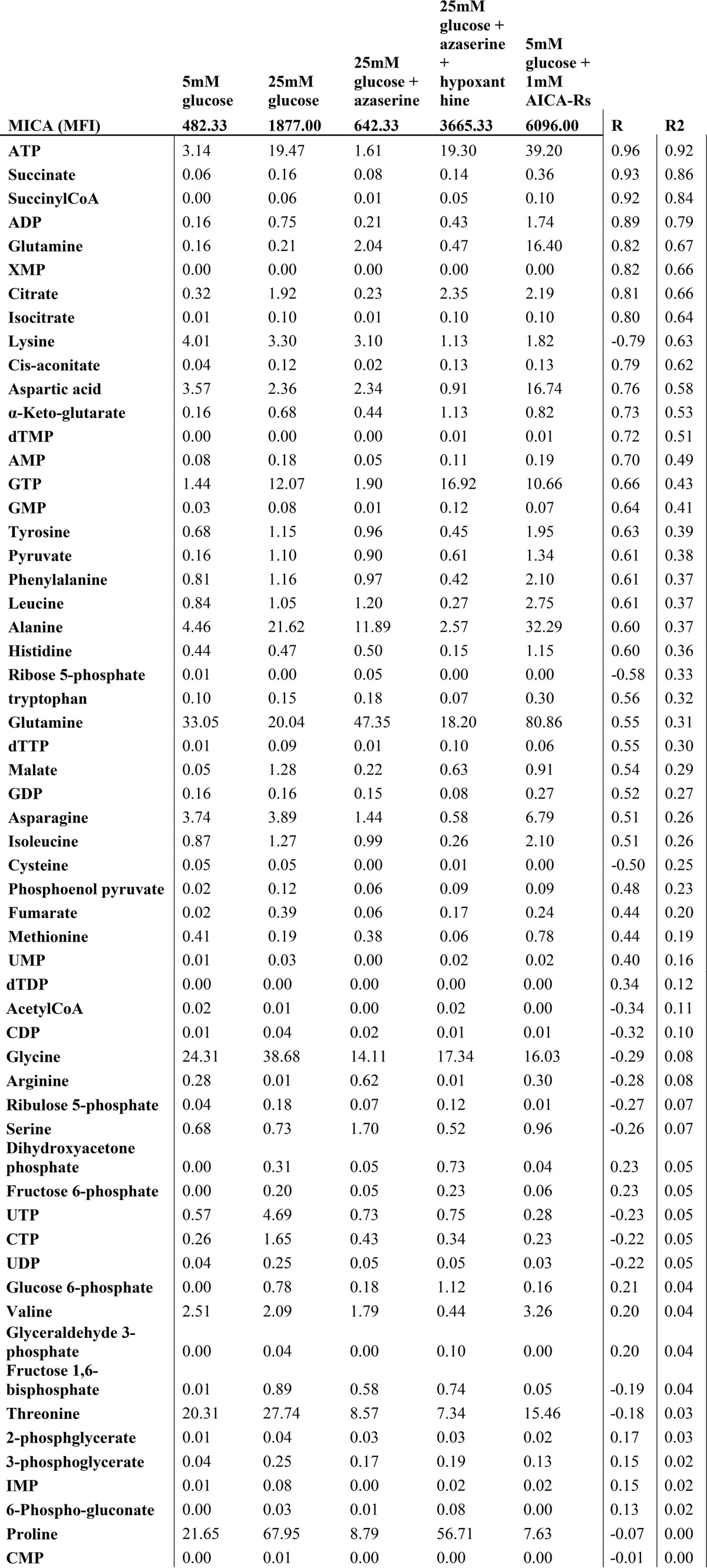
Glucose- and purine-induced MICA expression increase NKG2D-dependent cellular immunogenicity
We performed chromium-release cytotoxicity assays to test the functional significance of the altered MICA expression seen with metabolic changes. However, first we measured the effect of glucose and purine nucleotides on the other six NKG2D ligands and confirmed that expression of MICB, ULBP1, ULBP2, and ULBP3 were similarly responsive to metabolic changes (Fig. 7, A and B). Chromium-release assays demonstrated a significant increase in NK cell cytotoxicity toward cells cultured in glucose, AICA-Rs, adenosine, or inosine (Fig. 7, C–E). This increased killing was significantly reduced by preincubation with an NKG2D receptor–blocking antibody (Fig. 7, F–H).
Figure 7.

Cellular metabolism affects cellular immunogenicity. A, in addition to affecting MICA expression, glucose, azaserine, and azaserine/hypoxanthine have a similar effect on MICB, ULBP1, ULBP2, and ULBP3. B, AICA-Rs, adenosine, and inosine each induce the expression of ULBP2 and ULBP3 even in 5 mm glucose. As multiple NKG2D ligands are similarly affected, we used anti-NKG2D receptor for blocking assays. C, cells cultured in 25 mm glucose or in 25 mm glucose with azaserine and hypoxanthine were killed more effectively than cells cultured in 5 mm glucose or in 25 mm glucose with azaserine. D, cells cultured in 5 mm glucose with AICA-Rs or adenosine were more susceptible to killing than cells cultured in 5 mm glucose alone. E, similarly, cells cultured in 5 mm glucose with inosine were more susceptible to NK cell cytotoxicity than cells cultured in 5 mm glucose only. F–H, in conditions described in C–E, the addition of a blocking NKG2D antibody prevented a significant amount of induced killing, consistent with an NKG2D-dependent killing mechanism. Error bars, 95% confidence interval.
Glucose-induced MICA expression is observed in primary cells
The impact of changes in metabolism on NKG2D ligand expression was evaluated in primary human fibroblast cells during infection with human cytomegalovirus (CMV). CMV infection elicits increased glucose consumption and lactate production; these effects are prevented by azaserine and restored by the salvage pathway substrate hypoxanthine (Fig. S6, A and B). A raised glucose concentration was associated with an increase in the CMV-induced cell-surface expression of MICA, MICB, ULBP1, and ULBP2 (Fig. S6C). The induction of these NKG2D ligands was significantly higher in 25 mm glucose, was limited by azaserine, and was rescued in the presence of azaserine by hypoxanthine (p < 0.001) (Fig. S6D).
Discussion
The focus of immunometabolism studies has been largely on the considerable influence of metabolism on the function of immune cells (35). Here we show that metabolism also affects cells that are potential targets for immune cells; glucose uptake and metabolism to purine nucleotides drives cell-surface MICA expression and NKG2D-dependent cytotoxic killing of these cells by immune cells.
These results are consistent with observational studies of NKG2D ligand expression in disparate settings from malignancy (5) to viral infection (6, 7) and physiological cell proliferation (8, 9). Each of these cellular processes is supported by enhanced biosynthesis, whether dysregulated, pathological, or physiological. Enhanced biosynthesis, the primary physiological outcome of “activated” or Warburg metabolism, can confer a survival advantage to individual cells by producing biological substrates (lipids, nucleotides, and amino acids) to generate the complex molecules (membranes, nucleic acids, and proteins) that support cell maintenance, function, and division. Dysregulation of biosynthesis is a challenge to the integrity and immunity of multicellular organisms, as it may support inappropriate cell survival or pathophysiological cellular processes. Thus, dysregulated enhanced biosynthesis may advantage a cancer cell, so detection and destruction of such cells by the immune system would be desirable.
Warburg metabolism describes a metabolic phenotype of increased glucose consumption, lactate production, biosynthesis, and an increased ratio of glycolysis to TCA cycle metabolism. This metabolic state supports viral replication (31) and proliferation of both healthy (36) and malignant (37) cells. The molecular events that control transition between quiescent and activated metabolic phenotypes remain unclear (38, 39). Our data demonstrate that glucose restriction replicates the biomolecular and metabolomic characteristics of quiescent and activated metabolic states, with increased glucose consumption, lactate production, intracellular ribonucleic acid levels, and TCA cycle intermediate and high-energy purine nucleotide concentrations. The inhibition of de novo purine synthesis replicates the glucose-restricted condition in both metabolic profile and MICA expression, whereas rescue of the purine salvage pathway restores both metabolic profile and MICA expression to the levels seen with high glucose. Supplying cells directly with the purine nucleotide substrate AICA-Rs induces MICA expression and increases cellular purine nucleotide concentrations without an increase in early glycolytic pathway intermediates, suggesting that the purine output from glycolysis is key to MICA induction. Whereas cell proliferation is observed in several settings in which MICA is expressed (malignant transformation, lymphocyte activation), our data suggest that MICA expression in response to metabolic change can occur in the absence of cellular proliferation.
In healthy individuals, interstitial glucose concentrations typically range from 4.2 to 9.3 mm over a 24-h period (40). In diabetic patients, plasma glucose concentrations can be higher, but glucose levels are generally controlled by diet, insulin, or other drugs because persistent changes substantially beyond the normal range can lead to diabetic ketoacidosis, which carries a high mortality. Raised glucose levels in diabetes have a limited effect on intracellular glucose concentration because of the lack of insulin, which is required to promote glucose uptake into cells. In contrast, glucose concentrations in the solid tumor microenvironment may be 10-fold lower than in healthy tissues (41). Overall, our findings indicate that extracellular glucose will only influence MICA levels in cells that can take up and metabolize the glucose through the purine synthesis pathway. Notably, CMV infection, which is associated with NKG2D ligand induction, is known to both up-regulate the GLUT4 glucose transporter (42) and induce Warburg metabolism (27).
The complexity of gene-regulatory networks means that a broad array of cellular interventions may alter the expression of a given gene. Previous studies of MICA regulation show that the DNA damage response pathway (19), TLR stimulation (10), histone deacetylation (20), heat shock transformation (21), ionizing radiation (22), growth factor pathway activation (23), and microRNA expression (25) can each induce MICA expression. Whereas TLR stimulation has been shown to induce a Warburg phenotype (28), the relationship between cellular metabolism and these other stimuli is not well-defined. Further characterization of the cellular metabolic state in these settings is necessary to assess the role of cellular nucleotide concentrations in NKG2D ligand induction under these conditions.
Patterns of NKG2D ligand expression differ between cell types. Whether individual NKG2D ligands are independently regulated for distinct immunological functions or there is redundancy in the ligand repertoire remains unclear. We show that where we observe MICA expression, it is dependent on intracellular purine nucleotide concentrations. Our data also show that the effect of cellular metabolism on MICA expression applies to other NKG2D ligands that are expressed in this cellular context.
T cell immunity, a relatively recent evolutionary development, provides adaptive immune oversight of the cellular proteome, marking somatic cells producing viral or abnormal self proteins as T cell targets (Fig. S7). In the cytosol, continuously sampled cellular proteins are degraded to peptides and displayed at the cell surface in the peptide-binding cleft of the major histocompatibility complex (MHC) class I molecule. Cells displaying abnormal peptide-MHC class I complexes, recognized by interaction with the T cell receptor (TCR), are susceptible to elimination by T cells through direct cytotoxicity or cytokine production. This potent adaptive T cell response is checked by an elaborate set of mechanisms collectively referred to as immune self-tolerance, preventing the activation of T cells by self protein-MHC class I complexes. In contrast to TCR surveillance of peptides, our data demonstrate that NKG2D-mediated immune recognition of MICA at the cell surface allows innate immune oversight of nucleotide biosynthesis. Thus, both TCR- and NKG2D- mediated immunity are triggered principally by distinct biosynthetic consequences of viral infection and malignant transformation. This functional homology is reflected in the structural homology of MHC class I molecules and MICA; MHC class I proteins bind peptides in a peptide-binding groove, but MICA does not, and structural studies have shown that the groove is narrowed and closed (43, 44). Immune oversight of nucleotide synthesis permits the early detection and elimination of transformed or excessively proliferating cells, which may demonstrate a “normal” or self proteome, toward which adaptive immunity is self-tolerant.
This insight into the molecular control of MICA underlines the importance of metabolic surveillance in cellular immunity and outlines a potential route to therapeutic advancement in cancer, autoimmune disease, and transplantation.
Experimental procedures
Cell lines and standard culture medium
293T, HeLa, MCF7, and HT1080 cell lines were cultured in glucose-free Dulbecco's modified Eagle's medium, supplemented with 10% fetal calf serum, 2 mm pyruvate, penicillin/streptomycin solution, and glucose at the concentration indicated. NK92 cells were cultured in Roswell Park Memorial Institute 1640 culture medium (RPMI) supplemented with 10% fetal calf serum, 2 mm pyruvate, penicillin/streptomycin solution, and interleukin-2 (200 units/ml). Primary human adult dermal fibroblasts were purchased from Life Technologies, Inc. and cultured in RPMI with 10% fetal calf serum, 2 mm glutamine, and penicillin/streptomycin solution.
Chemicals and reagents
Glucose (catalog no. 49152), fructose (catalog no. F3510), mannose (catalog no. M6020), mannitol (catalog no. 17311), azaserine (catalog no. 11430), 6 diazo-oxo-norleucine (catalog no. D2141), 2-deoxyglucose (catalog no. D8375), phloretin (catalog no. P7912), cytochalasin B (catalog no. C6762-1mg), carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) (catalog no. 21888), all nucleosides and nucleobases, and 50× hypoxanthine-thymidine solution (catalog no. A9666) were purchased from Sigma-Aldrich. AICA-Rs (catalog no. 9944S) was purchased from New England Biolabs (Ipswich, MA). Adenosine kinase inhibitor (catalog no. 116890, CAS 214697-26-4) was purchased from Merck-Millipore (Darmstadt, Germany), and iodotubercidin was from Abcam (Cambridge, UK).
Glucose and lactate measurement
Measurements of glucose, lactate, and osmolality in cell culture media were performed by the Department of Clinical Biochemistry, John Radcliffe Hospital, Oxford. Glucose and lactate concentrations were determined using the glucose hexokinase II reagent kit (catalog no. 04903429, Bayer Healthcare, Leverkusen, Germany) and the lactate reagent kit (catalog no. 07109944, Bayer Healthcare), respectively. Osmolality was calculated on a VITECH 3320 micro-osmometer. Intracellular glucose and lactate concentrations were determined by a liquid chromatography/mass spectrometry quadrupole time-of-flight nanoflow method, as described previously (45).
Antibodies
Antibodies against MICA (2C10, IgG1), ULBP4 (6E6, IgG2B), and ULBP5 (6D10, IgM) were purchased from Santa Cruz Biotechnology (Heidelberg, Germany); MICB (MAB1599, IgG2b), ULBP1 (MAB1380, IgG2a), ULBP2 (MAB1298, IgG2a), and ULBP3 (MAB1517, IgG2a) were purchased from R&D Systems (Minneapolis, MN); podoplanin (NZ-1, IgG1) was purchased from AngioBio. HLA-ABC (W6/32, IgG2a), anti-PCNA (14-9910-80, IgG2a), and all isotype control antibodies were purchased from eBioscience (Hatfield, UK). The Alexa Fluor secondary antibodies 647-conjugated goat anti-mouse IgM (A-21238) and 647-conjugated goat anti-mouse IgM (A-21236) were purchased from Invitrogen.
Flow cytometry
Flow cytometry analysis was carried out on a BD Biosciences FacsCanto flow cytometer, using BD FacsDiva acquisition software, and results were analyzed using FlowJo (Ashland, OR).
For cell-surface staining, cells were washed with phosphate-buffered saline with 0.03% azide (PBSA). Primary and secondary antibody staining were carried out by standard protocols. Propidium iodide (PI; 4 μg/ml) was added to the final resuspension buffer to determine cell viability.
Total cell MICA was measured by flow cytometry of permeabilized cells. Cells were fixed by resuspension in 2% paraformaldehyde for 30 min and permeabilized with 0.05% saponin in PBSA for 30 min. The standard staining protocol was then followed, but with 0.05% saponin added to each of the buffers. Unless otherwise stated, experiments were conducted at least in triplicate. The histograms show average mean fluorescence intensity of three biological replicate samples, and error bars represent the 95% confidence interval.
Cell-cycle analysis
Cells were fixed in 1% paraformaldehyde for 60 min on ice, permeabilized in 70% ethanol, and resuspended in staining solution (40 μg/ml PI, 100 μg/ml RNase A in PBS) for 30 min at 37 °C. The cell-cycle analysis was conducted by flow cytometry with PI fluorescence measured in linear mode. Doublets were excluded by initial PI width-area gating. PI voltage was adjusted to center the G1 population on the 50,000-V area level, and cell-cycle parameter modeling was performed using FlowJo software.
Carboxyfluorescein diacetate succinimidyl ester (CFSE) proliferation assay
For adherent cells, the culture medium was replaced with CFDA-SE (5 μm). Cells were incubated at 37 °C for 10 min, and the CFDA-SE solution was replaced with fresh culture medium, as indicated. Cells were cultured for a further 48 h, before measuring CFSE fluorescence by flow cytometry through a FITC filter.
Cloning and transfection
The Consensus Coding Sequence Database (CCDS) was used to identify the reference sequence for GLUT5 expression cloning. Primers were designed to include a Kozak sequence (forward, TTAATAAGCTTGCCATGGAGCAACAGGATCAGAG; reverse, TTAATCTCGAGTCACTGTTCCGAAGTGACAGGTG). The gene was cloned from MCF7 cell cDNA and ligated into the pCDNA3.1 expression vector. Cells were transfected using a standard polyethyleneimine protocol. The doxycycline-inducible eGFP lentiviral expression vector was generated by cloning eGFP from the expression vector pEGFP-N1 between the XhoI and MluI sites of a modified pTripZ vector. The modified pTripZ vector was made by excising Turboprop using the AgeI and MluI restriction sites and inserting the following modified multiple cloning site: forward primer, CCGGTATCGATGAATTCTTCGAACTCGAGATTAATAAGCTTA; reverse primer, CGCGTAAGCTTATTAATCTCGAGTTCGAAGAATTCATCGATA. This plasmid was used to generate a lentiviral vector using standard protocols. Infected cells were selected with puromycin and induced with doxycycline at a concentration of 2 μg/ml.
Reverse transcription-PCR
RNA extraction was carried out using TRIzol and the Purelink RNA minikit, including DNase I digestion using the Purelink DNase I kit (Life Technologies), according to the manufacturer's protocol. RNA concentration was measured by Nanodrop (Wilmington, DE). The following PCR primers were used: GLUT1, GTGCAGCAGCCTGTGTATGC and GGCCACGATGCTCAGATAGG; GLUT2, CTGTGCTGGGTTCCTTCCAG and AAGGGGTTGGTTTTGGGTTC; GLUT3, TACAGCGATGGGGACACAGAAG and CCAGAGAGACGTGAGCAGCAC; GLUT4, CCCTGGTCCTTGCTGTGTTC and AAAAGATGGCCACGGAGAGG; GLUT5, GAATTCATGGAAGACTT and GCCATCTACGTTTGCAA (46).
Chromium release cytotoxicity assay
1 × 106 target cells for each test condition were washed in fresh culture medium and pelleted. These target cell pellets were resuspended in 50 μl of chromium-51 (0.05 mCi)/sample and incubated for 1 h at 37 °C. Labeled cells were washed twice with fresh RPMI and transferred to a 96-well plate, with 5000 cells in 50 μl of fresh culture medium per well. NK92 cells were resuspended in fresh NK cell medium and serially diluted to generate the effector/target ratios described, at a volume of 100 μl/well. Background lysis was measured by adding 100 μl of NK92 medium only, and maximal lysis was measured by adding 100 μl of 5% Triton-X. Effector and target cells were mixed by gentle pipetting and co-incubated at 37 °C for 1 h. The reaction plate was then centrifuged, and 25 μl of supernatant from each well was added to 150 μl of scintillation fluid in a fresh 96-well plate. Scintillation counts were measured on a Microbeta TriLux liquid scintillation counter. To measure NKG2D-specific killing, effector NK92 cells were initially resuspended in 2 ml of NK92 culture medium, and an anti-NKG2D antibody (BD Biosciences, catalog no. 552866) was added at a concentration of 5 μg/ml. The NK92 cells were incubated at 37 °C for 30 min before being resuspended in RPMI and added to target cells as described previously.
Metabolite analysis
The concentrations of intracellular anionic and cationic metabolites were measured by CE-TOFMS and LC-TOFMS (47, 48). Samples were prepared as described previously (49).
Statistical analysis
Histogram bars represent mean values, and error bars represent the 95% confidence interval of the mean. Statistical significance and p values were calculated using t tests unless otherwise specified. Correlation between cell-surface MICA expression and intracellular metabolite concentration was calculated using the correlation coefficient and coefficient of determination between mean values measured in parallel. *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001, unless otherwise specified. Metabolomic analysis was undertaken using R and algorithms implemented in Metaboanalyst version 3.0 (50).
Author contributions
M. T. M. and C. A. O. conceived the and wrote the paper. M. T. M., C. A. O., A. V., G. M., T. K. H., J. A., and T. S. designed the experiments. M. T. M., A. V., G. M., T. H., N. A. J., and T. S. performed the experiments. M. T. M. and C. A. O. wrote the original manuscript draft. M. T. M., C. A. O., A. V., G. M., J. A., and N. A. J. reviewed and edited the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Thue Schwartz and fellow members of the Novo Nordisk Foundation Immunometabolism consortium for advice and helpful discussions.
This work was supported by the Medical Research Council Grants G116/165 and G0901998, the National Institute for Health Research Oxford Comprehensive Biomedical Research Centre Program, and Novo Nordisk Foundation Grant NNF15CC0018346. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6.
- NKG2D
- natural killer group 2D
- NK
- natural killer
- DON
- 6-diazo-oxonorleucine
- MHC
- major histocompatibility complex
- MICA
- MHC class I polypeptide-related sequence A
- TLR
- T cell receptor
- TCA
- trichloroacetic acid
- HEK
- human embryonic kidney
- eGFP
- enhanced green fluorescent protein
- GIME
- glucose-induced MICA expression
- HAT
- hypoxanthine, aminopterin, and thymidine
- AICA-Rt
- 5-aminoimidazole-4-carboxamide ribonucleotide
- AICA-Rs
- AICA ribonucleoside
- CE-TOFMS
- capillary electrophoresis time-of-flight mass spectrometry
- LC-TOFMS
- liquid chromatography time-of-flight mass spectrometry
- CMV
- cytomegalovirus
- TCR
- T cell receptor
- RPMI
- Roswell Park Memorial Institute 1640 culture medium
- PI
- propidium iodide
- CFDA-SE
- carboxyfluorescein diacetate succinimidyl ester
- 2DG
- 2-deoxyglucose
- CFSE
- carboxyfluorescein diacetate succinimidyl ester.
References
- 1. Topalian S. L., Drake C. G., and Pardoll D. M. (2015) Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461 10.1016/j.ccell.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Raulet D. H., Gasser S., Gowen B. G., Deng W., and Jung H. (2013) Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 31, 413–441 10.1146/annurev-immunol-032712-095951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bauer S., Groh V., Wu J., Steinle A., Phillips J. H., Lanier L. L., and Spies T. (1999) Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285, 727–729 10.1126/science.285.5428.727 [DOI] [PubMed] [Google Scholar]
- 4. Wu J., Song Y., Bakker A. B. H., Bauer S., Spies T., Lanier L. L., and Phillips J. H. (1999) An activating immunoreceptor complex formed by NKG2D and DAP10. Science 285, 730–732 10.1126/science.285.5428.730 [DOI] [PubMed] [Google Scholar]
- 5. McGilvray R. W., Eagle R. A., Watson N. F. S., Al-Attar A., Ball G., Jafferji I., Trowsdale J., and Durrant L. G. (2009) NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin. Cancer Res. 15, 6993–7002 10.1158/1078-0432.CCR-09-0991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Groh V., Rhinehart R., Randolph-Habecker J., Topp M. S., Riddell S. R., and Spies T. (2001) Costimulation of CD8αβ T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat. Immunol. 2, 255–260 10.1038/85321 [DOI] [PubMed] [Google Scholar]
- 7. Eagle R. A., Traherne J. A., Ashiru O., Wills M. R., and Trowsdale J. (2006) Regulation of NKG2D ligand gene expression. Hum. Immunol. 67, 159–169 10.1016/j.humimm.2006.02.015 [DOI] [PubMed] [Google Scholar]
- 8. Cerboni C., Zingoni A., Cippitelli M., Piccoli M., Frati L., and Santoni A. (2007) Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK-cell lysis. Blood 110, 606–615 10.1182/blood-2006-10-052720 [DOI] [PubMed] [Google Scholar]
- 9. Cerboni C., Ardolino M., Santoni A., and Zingoni A. (2009) Detuning CD8+ T lymphocytes by down-regulation of the activating receptor NKG2D: role of NKG2D ligands released by activated T cells. Blood 113, 2955–2964 10.1182/blood-2008-06-165944 [DOI] [PubMed] [Google Scholar]
- 10. Kloss M., Decker P., Baltz K. M., Baessler T., Jung G., Rammensee H.-G., Steinle A., Krusch M., and Salih H. R. (2008) Interaction of monocytes with NK cells upon Toll-like receptor-induced expression of the NKG2D ligand MICA. J. Immunol. 181, 6711–6719 10.4049/jimmunol.181.10.6711 [DOI] [PubMed] [Google Scholar]
- 11. Lin D., Lavender H., Soilleux E. J., and O'Callaghan C. A. (2012) NF-κB regulates MICA gene transcription in endothelial cell through a genetically inhibitable control site. J. Biol. Chem. 287, 4299–4310 10.1074/jbc.M111.282152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cosman D., Müllberg J., Sutherland C. L., Chin W., Armitage R., Fanslow W., Kubin M., and Chalupny N. J. (2001) ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 14, 123–133 10.1016/S1074-7613(01)00095-4 [DOI] [PubMed] [Google Scholar]
- 13. Ehrlich L. I. R., Ogasawara K., Hamerman J. A., Takaki R., Zingoni A., Allison J. P., and Lanier L. L. (2005) Engagement of NKG2D by cognate ligand or antibody alone is insufficient to mediate costimulation of human and mouse CD8+ T cells. J. Immunol. 174, 1922–1931 10.4049/jimmunol.174.4.1922 [DOI] [PubMed] [Google Scholar]
- 14. Rajasekaran K., Xiong V., Fong L., Gorski J., and Malarkannan S. (2010) Functional dichotomy between NKG2D and CD28-mediated co-stimulation in human CD8+ T cells. PLoS One 5, e12635 10.1371/journal.pone.0012635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. André P., Castriconi R., Espéli M., Anfossi N., Juarez T., Hue S., Conway H., Romagné F., Dondero A., Nanni M., Caillat-Zucman S., Raulet D. H., Bottino C., Vivier E., Moretta A., and Paul P. (2004) Comparative analysis of human NK cell activation induced by NKG2D and natural cytotoxicity receptors. Eur. J. Immunol. 34, 961–971 10.1002/eji.200324705 [DOI] [PubMed] [Google Scholar]
- 16. Andersson A. K., Sumariwalla P. F., McCann F. E., Amjadi P., Chang C., McNamee K., Tornehave D., Haase C., Agersø H., Stennicke V. W., Ahern D., Ursø B., Trowsdale J., Feldmann M., and Brennan F. M. (2011) Blockade of NKG2D ameliorates disease in mice with collagen-induced arthritis: a potential pathogenic role in chronic inflammatory arthritis. Arthritis Rheum. 63, 2617–2629 10.1002/art.30460 [DOI] [PubMed] [Google Scholar]
- 17. Ito Y., Kanai T., Totsuka T., Okamoto R., Tsuchiya K., Nemoto Y., Yoshioka A., Tomita T., Nagaishi T., Sakamoto N., Sakanishi T., Okumura K., Yagita H., and Watanabe M. (2008) Blockade of NKG2D signaling prevents the development of murine CD4 + T cell-mediated colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G199–G207 [DOI] [PubMed] [Google Scholar]
- 18. Suárez-Alvarez B., López-Vázquez A., Baltar J. M., Ortega F., and López-Larrea C. (2009) Potential role of NKG2D and its ligands in organ transplantation: new target for immunointervention. Am. J. Transplant. 9, 251–257 10.1111/j.1600-6143.2008.02526.x [DOI] [PubMed] [Google Scholar]
- 19. Gasser S., Orsulic S., Brown E. J., and Raulet D. H. (2005) The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436, 1186–1190 10.1038/nature03884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andresen L., Jensen H., Pedersen M. T., Hansen K. A., and Skov S. (2007) Molecular regulation of MHC class I chain-related protein A expression after HDAC-inhibitor treatment of Jurkat T cells. J. Immunol. 179, 8235–8242 10.4049/jimmunol.179.12.8235 [DOI] [PubMed] [Google Scholar]
- 21. Groh V., Bahram S., Bauer S., Herman A., Beauchamp M., and Spies T. (1996) Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc. Natl. Acad. Sci. U.S.A. 93, 12445–12450 10.1073/pnas.93.22.12445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim J.-Y., Son Y.-O., Park S.-W., Bae J.-H., Chung J. S., Kim H. H., Chung B.-S., Kim S.-H., and Kang C.-D. (2006) Increase of NKG2D ligands and sensitivity to NK cell-mediated cytotoxicity of tumor cells by heat shock and ionizing radiation. Exp. Mol. Med. 38, 474–484 10.1038/emm.2006.56 [DOI] [PubMed] [Google Scholar]
- 23. Vantourout P., Willcox C., Turner A., Swanson C. M., Haque Y., Sobolev O., Grigoriadis A., Tutt A., and Hayday A. (2014) Immunological visibility: posttranscriptional regulation of human NKG2D ligands by the EGF receptor pathway. Sci. Transl. Med. 6, 231ra49–231ra49 10.1126/scitranslmed.3007579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Waldhauer I., Goehlsdorf D., Gieseke F., Weinschenk T., Wittenbrink M., Ludwig A., Stevanovic S., Rammensee H.-G., and Steinle A. (2008) Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 68, 6368–6376 10.1158/0008-5472.CAN-07-6768 [DOI] [PubMed] [Google Scholar]
- 25. Stern-Ginossar N., Gur C., Biton M., Horwitz E., Elboim M., Stanietsky N., Mandelboim M., and Mandelboim O. (2008) Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat. Immunol. 9, 1065–1073 10.1038/ni.1642 [DOI] [PubMed] [Google Scholar]
- 26. Venkataraman G. M., Suciu D., Groh V., Boss J. M., and Spies T. (2007) Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J. Immunol. 178, 961–969 10.4049/jimmunol.178.2.961 [DOI] [PubMed] [Google Scholar]
- 27. Landini M. P. (1984) Early enhanced glucose uptake in human cytomegalovirus-infected cells. J. Gen. Virol. 65, 1229–1232 10.1099/0022-1317-65-7-1229 [DOI] [PubMed] [Google Scholar]
- 28. Krawczyk C. M., Holowka T., Sun J., Blagih J., Amiel E., DeBerardinis R. J., Cross J. R., Jung E., Thompson C. B., Jones R. G., and Pearce E. J. (2010) Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749 10.1182/blood-2009-10-249540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang T., Marquardt C., and Foker J. (1976) Aerobic glycolysis during lymphocyte proliferation. Nature 261, 702–705 10.1038/261702a0 [DOI] [PubMed] [Google Scholar]
- 30. Warburg O., Wind F., and Negelein E. (1927) The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 10.1085/jgp.8.6.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu Y., Clippinger A. J., and Alwine J. C. (2011) Viral effects on metabolism: changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol. 19, 360–367 10.1016/j.tim.2011.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cairns R. A., Harris I. S., and Mak T. W. (2011) Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95 10.1038/nrc2981 [DOI] [PubMed] [Google Scholar]
- 33. Thorens B., and Mueckler M. (2010) Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 298, E141–E145 10.1152/ajpendo.00712.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li M. V., Chen W., Harmancey R. N., Nuotio-Antar A. M., Imamura M., Saha P., Taegtmeyer H., and Chan L. (2010) Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem. Biophys. Res. Commun. 395, 395–400 10.1016/j.bbrc.2010.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Geiger R., Rieckmann J. C., Wolf T., Basso C., Feng Y., Fuhrer T., Kogadeeva M., Picotti P., Meissner F., Mann M., Zamboni N., Sallusto F., and Lanzavecchia A. (2016) l-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 167, 829–842.e13 10.1016/j.cell.2016.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Frauwirth K. A., Riley J. L., Harris M. H., Parry R. V., Rathmell J. C., Plas D. R., Elstrom R. L., June C. H., and Thompson C. B. (2002) The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777 10.1016/S1074-7613(02)00323-0 [DOI] [PubMed] [Google Scholar]
- 37. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- 38. Hirschey M. D., DeBerardinis R. J., Diehl A. M. E., Drew J. E., Frezza C., Green M. F., Jones L. W., Ko Y. H., Le A., Lea M. A., Locasale J. W., Longo V. D., Lyssiotis C. A., McDonnell E., Mehrmohamadi M., et al. (2015) Dysregulated metabolism contributes to oncogenesis. Semin. Cancer Biol. 35, S129–S150 10.1016/j.semcancer.2015.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vander Heiden M. G., Cantley L. C., and Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Freckmann G., Hagenlocher S., Baumstark A., Jendrike N., Gillen R. C., Rössner K., and Haug C. (2007) Continuous glucose profiles in healthy subjects under everyday life conditions and after different meals. J. Diabetes Sci. Technol. 1, 695–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hirayama A., Kami K., Sugimoto M., Sugawara M., Toki N., Onozuka H., Kinoshita T., Saito N., Ochiai A., Tomita M., Esumi H., and Soga T. (2009) Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 69, 4918–4925 10.1158/0008-5472.CAN-08-4806 [DOI] [PubMed] [Google Scholar]
- 42. Yu Y., Maguire T. G., and Alwine J. C. (2011) Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J. Virol. 85, 1573–1580 10.1128/JVI.01967-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bjorkman P. J., Saper M. A., Samraoui B., Bennett W. S., Strominger J. L., and Wiley D. C. (1987) Structure of the human class I histocompatibility antigen, HLA-A2. Nature 329, 506–512 10.1038/329506a0 [DOI] [PubMed] [Google Scholar]
- 44. Li P., Willie S. T., Bauer S., Morris D. L., Spies T., and Strong R. K. (1999) Crystal structure of the MHC class I homolog MIC-A, a γδ T cell ligand. Immunity 10, 577–584 10.1016/S1074-7613(00)80057-6 [DOI] [PubMed] [Google Scholar]
- 45. Valli A., Rodriguez M., Moutsianas L., Fischer R., Fedele V., Huang H.-L., Van Stiphout R., Jones D., McCarthy M., Vinaxia M., Igarashi K., Sato M., Soga T., Buffa F., Mccullagh J., et al. (2015) Hypoxia induces a lipogenic cancer cell phenotype via HIF1α-dependent and -independent pathways. Oncotarget 6, 1920–1941 10.18632/oncotarget.3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reinicke K., Sotomayor P., Cisterna P., Delgado C., Nualart F., and Godoy A. (2012) Cellular distribution of glut-1 and glut-5 in benign and malignant human prostate tissue. J. Cell. Biochem. 113, 553–562 10.1002/jcb.23379 [DOI] [PubMed] [Google Scholar]
- 47. Soga T., Ohashi Y., Ueno Y., Naraoka H., Tomita M., and Nishioka T. (2003) Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J. Proteome Res. 2, 488–494 10.1021/pr034020m [DOI] [PubMed] [Google Scholar]
- 48. Soga T., Igarashi K., Ito C., Mizobuchi K., Zimmermann H.-P., and Tomita M. (2009) Metabolomic profiling of anionic metabolites by capillary electrophoresis mass spectrometry. Anal. Chem. 81, 6165–6174 10.1021/ac900675k [DOI] [PubMed] [Google Scholar]
- 49. Adam J., Hatipoglu E., O'Flaherty L., Ternette N., Sahgal N., Lockstone H., Baban D., Nye E., Stamp G. W., Wolhuter K., Stevens M., Fischer R., Carmeliet P., Maxwell P. H., Pugh C. W., et al. (2011) Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524–537 10.1016/j.ccr.2011.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xia J., Sinelnikov I. V., Han B., and Wishart D. S. (2015) MetaboAnalyst 3.0–making metabolomics more meaningful. Nucleic Acids Res. 43, W251–W257 10.1093/nar/gkv380 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.