

Abstract
Apolipoprotein-E (apoE) is a glycoprotein highly expressed in the brain, where it appears to play a role in lipid transport, β-amyloid clearance, and neuronal signaling. ApoE proteolytic fragments are also present in the brain, but the enzymes responsible for apoE fragmentation are unknown, and the biological activity of specific apoE fragments remains to be determined. Here we utilized SK-N-SH neuroblastoma cells differentiated into neurons with all-trans-retinoic acid (ATRA) to study extracellular apoE proteolysis. ApoE fragments were detectable in culture supernatants after 3 days, and their levels were increased for up to 9 days in the presence of ATRA. The concentration of apoE fragments was positively correlated with levels of the neuronal maturation markers (PSD95 and SMI32). The most abundant apoE fragments were 25- and 28-kDa N-terminal fragments that both contained sialylated glycosylation and bound to heparin. We detected apoE fragments only in the extracellular milieu and not in cell lysates, suggesting that an extracellular protease contributes to apoE fragmentation. Of note, siRNA-mediated knockdown of high-temperature requirement serine peptidase A1 (HtrA1) and a specific HtrA1 inhibitor reduced apoE 25-kDa fragment formation by 41 and 86%, respectively. Recombinant 25-kDa fragment apoE and full-length apoE both stimulated neuritogenesis in vitro, increasing neuroblastoma neurite growth by more than 2-fold over a 6-day period. This study provides a cellular model for assessing apoE proteolysis, indicates that HtrA1 regulates apoE 25-kDa fragment formation under physiological conditions, and reveals a new neurotrophic function for the apoE 25-kDa fragment.
Keywords: apolipoprotein E (ApoE), proteostasis, neurobiology, neurite outgrowth, neurodegeneration, Alzheimer disease, apoE fragmentation, HtrA1, neuritogenesis
Introduction
Apolipoprotein-E (apoE)6 is a ∼35-kDa glycoprotein that is highly expressed in the brain, where it is thought to play a role in lipid transport, β-amyloid (Aβ) clearance, and neuronal signaling (1). ApoE consists of a ∼25-kDa four-helix bundle N-terminal domain and a ∼10-kDa C-terminal domain. A short hinge region containing a single O-linked glycan separates these domains. The APOE genotype is the greatest known genetic risk factor for AD, whereby possession of one or two copies of the APOEϵ4 allele confers a 3- or 10-fold increase, respectively, in AD risk compared with APOEϵ3 homozygotes (2). In contrast, the APOEϵ2 allele is associated with decreased AD risk (2). These APOE genotypes produce the three major apoE isoforms, apoE2, apoE3, and apoE4, which differ in their Cys/Arg composition at positions 112 and 158. ApoE2 contains Cys112 and Cys158; apoE3 contains Cys112 and Arg158; and apoE4 contains Arg112 and Arg158 (3).
ApoE is present in the central nervous system (CNS) as both spherical and discoidal lipoprotein complexes (4) and is strongly expressed in the choroid plexus (5, 6). Astrocytes are thought to be the primary source of apoE in the brain, although microglia and neurons also contribute to the apoE CNS pool under certain circumstances (4, 7–9). Although there are several postulated pathways whereby apoE may affect synaptic plasticity, axon guidance, neuron survival, Aβ homeostasis, and microtubule stability, the exact pathways by which apoE isoforms influence AD remain to be fully resolved (10).
Previous studies indicate that lipidated apoE promotes the extracellular degradation of Aβ by insulin-degrading enzyme, targets Aβ for intracellular degradation in microglia, and facilitates Aβ clearance from the CNS via low-density lipoprotein receptor–related protein-1 (LRP1) (11). It has also been reported that apoE may compete with Aβ for interaction with LRP1 (12) and that apoE directly regulates neuronal Aβ production in an isoform-dependent manner (13). A complete understanding of the mechanisms by which apoE regulates AD risk therefore remains an unresolved question.
We reported previously that apoE is proteolytically cleaved in the human brain (hippocampus, frontal cortex, and cerebellum, both gray and white matter) and that proteolytic processing of apoE3 generates a predominant stable ∼25-kDa fragment (14). The apoE 25-kDa fragment was not recognized by an apoE C-terminal antibody, and, based on the predicted sensitivity of the hinge region to proteolysis, we proposed that the apoE 25-kDa fragment represents the N-terminal four-helix bundle that contains the LDL receptor binding domain (14). Several other groups also detected apoE fragments in the human brain, some of which associate with neurofibrillary tangles and amyloid plaques (15–19). As it is possible that apoE fragments, particularly the stable 25-kDa fragment (14), may possess biological activity, it is important to understand the processes that contribute to the formation of apoE fragments under physiological conditions.
As mentioned above, astrocytes are the main source of apoE in the CNS under normal physiological conditions. However, the cell types responsible for the generation of the apoE 25-kDa fragment are unknown. Previous data suggest that a neuronal chymotrypsin-like protease or aspartic protease may be involved in apoE proteolytic fragmentation (19, 20). Either endogenous neuronal apoE or endocytosed apoE may be processed in the neuron and (re-)secreted as apoE proteolytic fragments. Of potential relevance, apoE re-secretion pathways have been described, at least in cells from peripheral tissues (21). It may also be possible that neurons secrete apoE along with a protease that together result in extracellular apoE fragmentation or that apoE that is normally constitutively secreted by astrocytes (or microglia) is cleaved by a secreted neuronal protease. Consistent with the latter idea, rat hippocampal neurons secrete a serine protease that generates apoE proteolytic fragments with a very similar Mr profile to the fragments seen in human post-mortem brain samples (9). Although the identity of the secreted enzyme responsible for the production of apoE proteolytic fragments by rat hippocampal neurons was not identified (9), a serine protease, high-temperature requirement serine peptidase A1 (HtrA1) has recently been shown to cleave recombinant apoE into fragments, including a predominant N-terminal apoE 25-kDa fragment (22), which again is remarkably similar to the apoE fragmentation pattern seen in human post-mortem brain samples (9, 14).
In the present study, we examined apoE proteolytic fragmentation using the human SK-N-SH neuroblastoma cell line. This study provides a cellular model to assess apoE proteolysis, shows that HtrA1 regulates apoE 25-kDa fragment formation under physiological conditions, and reveals a new neurotrophic function for the apoE 25-kDa fragment.
Results
The SK-N-SH neuroblastoma cell line is an established in vitro model for human neurobiology studies. Chronic treatment of SK-N-SH cells with all-trans-retinoic acid (ATRA) induces a post-mitotic neuronal phenotype with cell monolayers containing a dense neurite network (23, 24). We examined the secretion of apoE from SK-N-SH cells and the possible influence of ATRA-mediated neuronal differentiation. Under standard culture conditions, apoE was clearly detected in cell-conditioned medium (Fig. 1A). Although the majority of apoE (∼80%) was present as full-length apoE, after 3 days in culture, small amounts of apoE fragments were also detected (Fig. 1). Treatment with ATRA time-dependently induced the production of apoE and also strongly increased the production of apoE fragments. Analysis of the apoE size distribution profiles indicated that apoE fragments at 25 and 28 kDa were produced at high levels with ATRA treatment (Fig. 1). After 9 days of ATRA treatment, the levels of the dominant apoE 28- and 25-kDa fragments were increased by ∼30% and 6-fold, respectively (Fig. 1B). There was very little change in the formation of apoE fragments by cells that were cultured for up to 9 days in the absence of ATRA (Fig. S1). In addition, examination of the full-length Western blots indicated that small amounts of apoE fragment at 30 kDa were detectable, and traces of fragments at a mass < 25 kDa (i.e. at ∼21, ∼15, and ∼12 kDa) were also detectable in the cell culture supernatants (Fig. S2).
Figure 1.
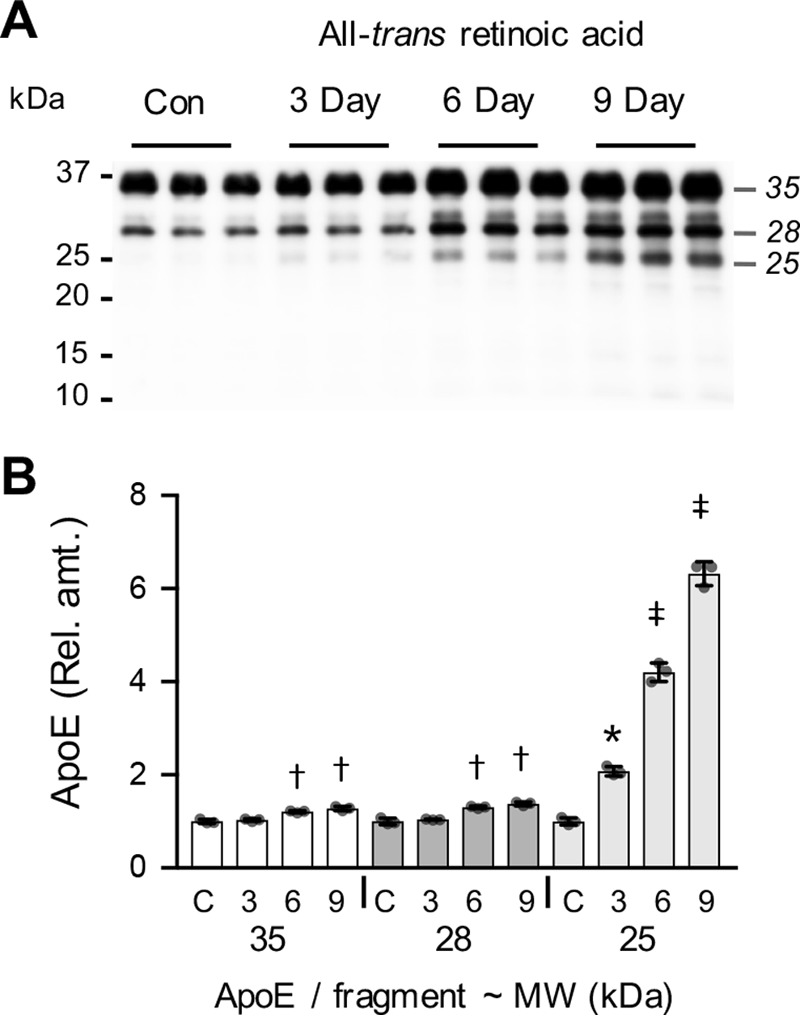
ATRA induces the production of apoE and apoE fragments by SK-N-SH cells. SK-N-SH cells were cultured for 3, 6, or 9 days in the presence of 10 μm ATRA, and the cell culture supernatants were collected and analyzed for apoE by Western blotting. Control (Con) medium from the 3-day cell cultures was analyzed for comparison. Full-length apoE and the major apoE fragments identified by Western blotting are illustrated (A). Quantitative assessment of the full-length apoE (35 kDa) and apoE fragments at 30, 28, and 25 kDa indicate a significant time-dependent increase in all apoE species (B). Data shown in B are derived from one experiment performed in triplicate that is representative of three independently performed experiments. The histogram bars indicate mean values, and error bars indicate S.D. Values in B are relative optical density measurements where the 3-day control sample is defined as 1.0. *, p < 0.05; †, p < 0.01; ‡, p < 0.0001, compared with the control (C) condition; one-way ANOVA with Tukey's post hoc analysis.
To assess the temporal association of apoE production with the development of the SK-N-SH neuronal phenotype, we analyzed cell lysates for apoE and for markers of synapse formation (PSD95) and neurite formation (SMI32). As expected, ATRA strongly increased PSD95 and SMI32 levels over the 9-day experiment (Fig. 2A). Phase-contrast images confirmed that the changes in these differentiation markers were associated with the development of a more neuronal phenotype (Fig. 2D). Consistent with the data in Fig. 1, cellular apoE levels were also time-dependently up-regulated by ATRA treatment (Fig. 2B). Intracellular apoE was detected as a doublet or triplet band that is consistent with previous studies of intracellular apoE maturation and glycosylation (25). Importantly, no apoE fragmentation products were detected in cell lysates at any of the time points examined, in either the presence or absence of ATRA (Fig. S3). These data indicate that apoE is up-regulated in concert with ATRA-induced neuronal differentiation and suggest that apoE proteolytic fragmentation may only occur in the extracellular compartment.
Figure 2.
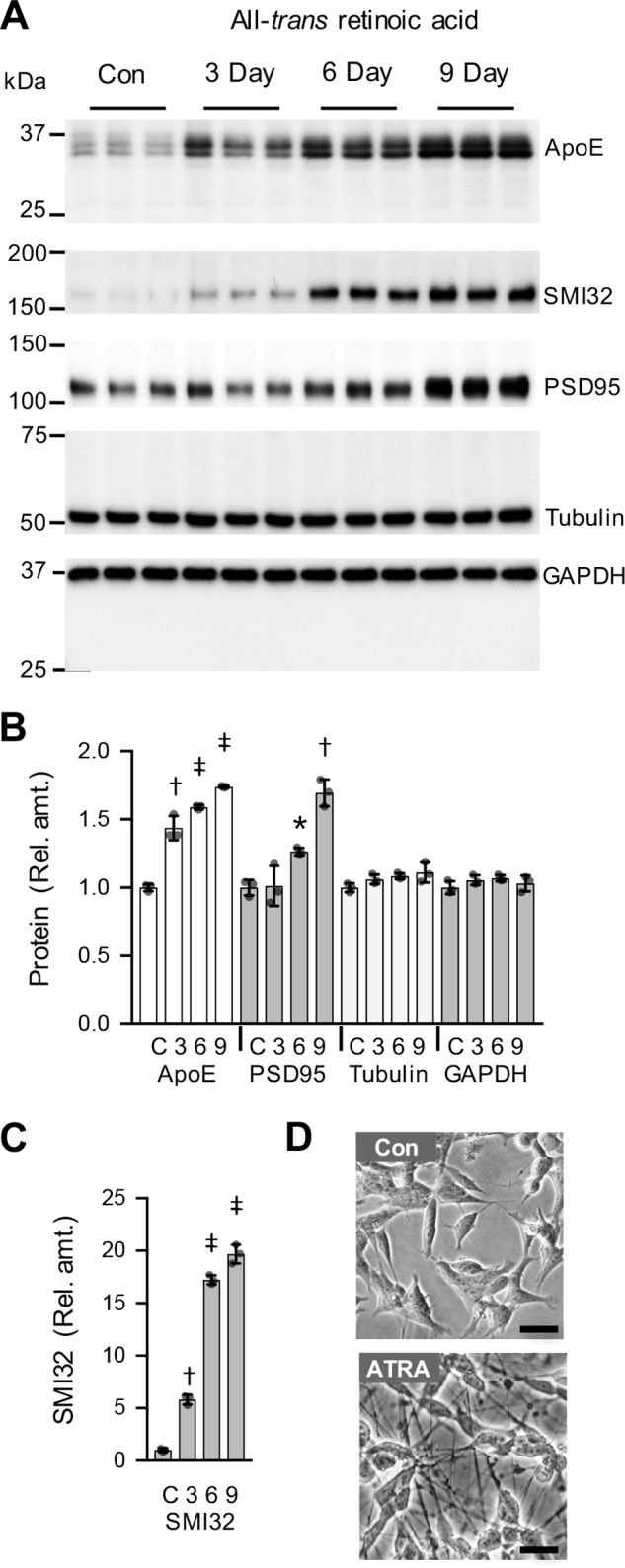
Regulation of SK-N-SH cellular apoE and synaptic marker levels by ATRA. SK-N-SH cells were cultured for 3, 6, or 9 days in the presence of 10 μm ATRA. The cell lysates were analyzed for apoE, non-phosphorylated neurofilament H (SMI32), PSD95, and the housekeeper proteins tubulin and GAPDH by Western blotting (A). Lysates from the 3-day control (Con) cell cultures were analyzed for comparison. Quantitative assessment of the full-length apoE (35 kDa) and the neurofilament and synaptic marker proteins (SMI32 and PSD95) indicate a significant time-dependent increase compared with the housekeeper proteins (B and C). Data in B and C are relative optical density measurements, and the 3-day control sample is defined as 1.0. Phase-contrast images of the 3-day control and 9-day ATRA-treated (ATRA) cells are shown (D). Quantitative data shown are derived from one experiment performed in triplicate representative of three independently performed experiments. The histogram bars represent the mean value, and the error bars represent S.D. values. *, p < 0.05; †, p < 0.01; ‡, p < 0.0001 compared with the control (C) condition; one-way ANOVA with Tukey's post hoc analysis. Scale bars in D, = 25 μm.
Based on our previous results from post-mortem human brain tissue (14), we predicted that the apoE 25–28-kDa fragments generated by ATRA-treated SK-N-SH cells may be N-terminal fragments. A protease-susceptible hinge domain separates the apoE N-terminal and C-terminal domains (Fig. 3A), and this is predicted to contain a number of enzyme cut sites. Using apoE N terminus–specific and C terminus–specific antibodies (recognizing residues 141–160 and 285–299, respectively), we confirmed that the two quantitatively dominant apoE fragments at 28 and 25 kDa were N-terminal fragments (Fig. 3B). Furthermore, the apoE N-terminal fragments both strongly bound to heparin (Fig. 3C), consistent with the known N-terminal heparin-binding domain comprising residues 143–146 (26). These apoE fragments were also reduced slightly in Mr after treatment with neuraminidase, consistent with cleavage of terminal sialic acid residues from the O-linked glycan at Thr194 (Fig. 3D). The calculated molecular mass of the apoE N-terminal peptide, including Thr194, is 22,373 Da. Although the degree of CNS apoE sialylation is variable (25), using the mass of one of the most abundant apoE O-glycan species at Thr194 (i.e. HexNAc-Hex(NeuAc)2) of 947 Da (27), the minimum predicted mass of a glycosylated apoE N-terminal fragment (residues 1–194) would therefore be ∼23.3 kDa (i.e. predicted to migrate to a position just below the 25-kDa PAGE marker protein, which is where the apoE 25-kDa fragment is detected) (Fig. 3B).
Figure 3.
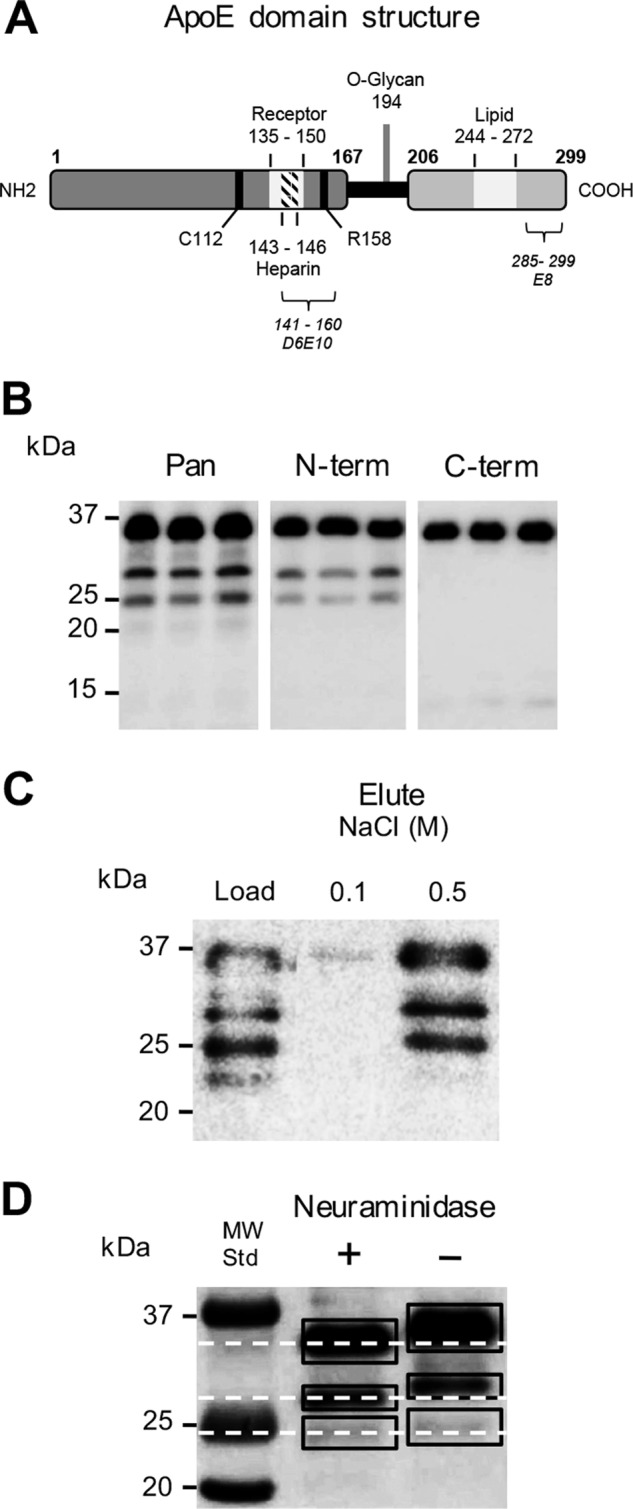
Characterization of apoE fragments. A schematic overview of apoE3 domain structure is shown (A). The LDL receptor-binding region (Receptor), heparin-binding region (Heparin), O-linked glycosylation site (O-Glycan), and lipid-binding domains (Lipid) are depicted. The Cys112 and Arg158 residues of apoE3 indicate the variable positions responsible for the three major apoE isoforms (A). SK-N-SH cells were cultured for 9 days in the presence of 10 μm ATRA, and the cell culture supernatants were collected and analyzed for apoE by Western blotting using a goat polyclonal antibody raised against full-length apoE (Pan), as well as monoclonal antibodies specific for the apoE N terminus (N-term) and C terminus (C-term) (D6E10 and E8, respectively, recognizing epitopes comprising amino acid residues 141–160 and 285–299, respectively, as shown in A). Full-length apoE and the apoE 25- and 28-kDa fragments were detected by the full-length apoE and apoE N terminus antibodies, whereas only full-length apoE was identified by the apoE C terminus antibody (B). The cell culture supernatants were assessed for heparin binding using heparin-affinity chromatography and Western blotting. Full-length apoE and the apoE 25- and 28-kDa fragments were strongly bound to the heparin column and eluted with 0.5 m NaCl (C). The cell culture supernatants were also assessed for the presence of sialylated glycans using neuraminidase treatment and Western blotting. The dashed lines indicate the shift in full-length apoE and apoE fragment size due to desialylation. Full-length apoE and the apoE 25- and 28-kDa fragments all migrated further in the blot after neuraminidase treatment, thereby removal of sialic acid (D). Data are derived from one experiment representative of at least two independently performed experiments.
To gain mechanistic information related to the extracellular formation of apoE N-terminal fragments, a selection of protease inhibitors was added to 9-day ATRA-differentiated SK-N-SH cell cultures for a period of 24 h. It is important to note that the degree of apoE fragmentation detected over the 24-h period used in these experiments is much lower than observed with 3-day culture periods (compare Fig. 4 (A and D) with Fig. 1A). The 24-h period was used to minimize nonspecific effects that the protease inhibitors may have on cellular metabolism. The presence of a broad-spectrum protease inhibitor mixture inhibited the formation of the major apoE N-terminal fragments at 25 kDa by 75% (p = 0.0002) and at 28 kDa by 75% (p = 0.0007) (Fig. 4, A and C, respectively), whereas the levels of full-length secreted or intracellular apoE were unaffected (Fig. 4, A–C). Similarly, the serine protease inhibitor 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF) inhibited the formation of the major apoE N-terminal fragments at 25 kDa by 81% (p = 0.0134) and at 28 kDa by 87% (p = 0.0033) (Fig. 4, D and F), whereas the levels of full-length apoE were unaffected (Fig. 4, D–F). These results suggest that an extracellular serine protease contributes to the production of apoE N-terminal fragments. These results are consistent with a previous study using rat hippocampal neuron-conditioned medium and exogenously added apoE in the presence or absence of a protease inhibitor mixture or the serine protease inhibitor phenylmethylsulfonyl fluoride (9).
Figure 4.

Inhibition of apoE proteolytic fragmentation by protease inhibitors. SK-N-SH cells were cultured for 9 days in the presence of 10 μm ATRA. The culture medium was then replaced with fresh medium alone (Con) or with fresh medium containing either a broad-spectrum PIM or the serine protease inhibitor AEBSF and cultured for a further 24-h period. The addition of PIM inhibited the formation of apoE fragments in the supernatant (S/N) (A). The addition of PIM had no impact on cellular apoE levels (B). A similar result was observed when AEBSF was used to treat the cells (D and E). Quantitative assessment of the full-length apoE (35 kDa) and apoE fragments at 28 and 25 kDa indicates a significant inhibition of apoE fragment formation in the presence of either PIM (C; light gray bars) or AEBSF (F; light gray bars)). Data in C and F are relative optical density measurements, in which the vehicle control (DMSO 1:1000) sample (dark gray bars) is defined as 1.0. Data shown are derived from one experiment performed in triplicate and representative of three independently performed experiments. The histogram bars represent mean values, and the error bars represent S.D. *, p < 0.05; †, p < 0.01 compared with the respective control for each fragment; two-tailed t test.
In a recent study using recombinant proteins, the serine protease HtrA1 was shown to produce apoE N-terminal fragments in an apoE isoform–dependent manner (22). Indeed, the HtrA1-mediated proteolysis of recombinant apoE3 produced a major apoE N-terminal fragment of ∼25 kDa that was shown to encompass amino acid residues 1–195 (22). Intriguingly, in these studies, larger fragments of apoE (e.g. 28 kDa) were not generated (22). Furthermore, the apoE 25-kDa fragment was more rapidly degraded further by HtrA1 to smaller fragments when the apoE4 recombinant protein was assessed, as compared with apoE3 recombinant protein (22). This result is strikingly similar to what has been observed in human post-mortem brain tissues, in which carriers of the APOEϵ3/3 genotype had increased levels of the apoE 25-kDa fragment as compared with APOEϵ4/4 carriers (14).
To test for a possible contribution of HtrA1 in the SK-N-SH–mediated production of the apoE N-terminal 25-kDa fragment, we first assessed the expression of HtrA1 in SK-N-SH cells. Quantitative RT-PCR indicated that HTRA1 mRNA was present in SK-N-SH cells under standard culture conditions and that levels were time-dependently increased by 7.4-fold (p = 0.0003) after 9 days in culture in the presence of ATRA (Fig. S4). This was also confirmed using standard semiquantitative PCR, where a 3.8-fold (p = 0.0003) up-regulation of HTRA1 mRNA level was observed (Fig. S5A). We also detected extracellular HtrA1 protein and found that the levels were significantly increased after 9 days in culture with ATRA treatment (Fig. S5). The HtrA1 Western blots identified two HtrA1 species at ∼50 and 38 kDa (Fig. S5D). This is consistent with previous studies that have shown that the 38 kDa band is an autocatalytic product of the full-length 50-kDa HtrA1 protein (28). Overall, SK-N-SH–secreted HtrA1 protein levels were increased by 2.1-fold (p = 0.0002) after 9 days in culture with ATRA (Fig. S5B).
To assess the role of HtrA1 in the SK-N-SH–mediated production of the apoE N-terminal 25-kDa fragment, we used an siRNA approach. HTRA1 mRNA levels were reduced by >90% relative to both vehicle control and scrambled siRNA (both p < 0.0001) following transfection of the cells with HTRA1 siRNA (Fig. 5, A and B). Similarly, extracellular HtrA1 protein levels were reduced by 91% (p < 0.0001) by HTRA1 siRNA when assessed at day 9 (Fig. 5, C and D). As HtrA1 regulates the growth and differentiation of neurons and other cell types (29–31), we quantified cell confluence using live cell imaging over time and also measured cellular protein levels at day 9. The siRNA-mediated knockdown of HTRA1 reduced cell growth (confluence) and total cell protein by 37 and 39% (p < 0.01), respectively (Fig. S6). We assumed that this decrease in cell growth and consequently cell protein levels would also further decrease apparent HtrA1 levels; therefore, the values for HtrA1 protein level in the HTRA1 siRNA condition illustrated in Fig. 5D have been increased by 39% to compensate for the reduction in cell numbers and total cell protein under this condition.
Figure 5.

Suppression of HtrA1 by siRNA reduces SK-N-SH cellular apoE proteolytic fragmentation. SK-N-SH cells were cultured for up to 9 days in the presence of siRNA targeting HTRA1 (siRNA), vehicle control (Con), or a scrambled siRNA (Scr), and the level of HTRA1 mRNA was determined by quantitative real-time PCR (A). Relative amounts of HTRA1 mRNA and HtrA1 protein were determined by quantitative real-time PCR and Western blotting after 9 days of exposure to siRNA targeting HTRA1 (siRNA), vehicle control (Con), or a scrambled siRNA (Scr) as shown in the histograms (B and D). A representative Western blot of extracellular HtrA1 present in the growth medium collected at day 9 (medium exposed to cells for 72 h, from day 6 to day 9) is shown (C). The same medium was also assessed for apoE fragmentation by Western blotting (E and F). The data in A are derived from one experiment performed in duplicate. Data in B and F are derived from three independently performed experiments performed in either triplicate or quadruplicate. Data in D are derived from one experiment (blot shown in C) representative of three experiments independently performed in either triplicate or quadruplicate. The histogram bars represent mean values, and the error bars represent S.D., except in A, where the error bars represent the range. *, p < 0.05; ‡, p < 0.0001 compared with the control (Con) condition; one-way ANOVA with Tukey's post hoc analysis.
We then measured the amount of apoE 25- and 28-kDa fragments (as a proportion of full-length apoE) under the day 9 siRNA treatment conditions. The apoE Western blots shown in Fig. 5E were loaded with equal amounts of full-length apoE in each lane to compare the fragmentation. The data demonstrate that HTRA1 siRNA selectively reduced the apoE 25-kDa fragment concentration by 41% (p < 0.0001) in the medium, whereas the levels of the apoE 28-kDa fragment were unaffected (Fig. 5, E and F), thus indicating that HtrA1 specifically regulates the production of the apoE 25-kDa fragment under physiological conditions.
Intriguingly, we detected a 25% (p = 0.01) increase in HTRA1 mRNA in the scrambled siRNA condition (Fig. 5B), and this was associated with a similar modest 25% (p = 0.02) increase in apoE 25-kDa fragment concentration (Fig. 5F). Although this was unexpected, as the scrambled siRNA was designed by the manufacturer to be a non-biologically active control, it is nonetheless noteworthy that increased HtrA1 levels were associated with an increased apoE 25-kDa fragment concentration.
As the production of the apoE 25-kDa fragment was only partially, albeit highly significantly, inhibited (∼40%) by the HTRA1 siRNA, and also because this treatment had an impact on cell growth (Fig. S6), we next assessed the role of HtrA1 in apoE 25-kDa fragment formation using HtrA1 boronic acid inhibitor (HBAI; Fig. 6A), as a highly specific HtrA1 inhibitor (22). Based on previous studies (22), we used HBAI at a concentration of 1 μm. When 9-day ATRA-differentiated SK-N-SH cells were treated with HBAI, the formation of the apoE N-terminal 25-kDa fragment was inhibited by 86% (p = 0.0004), whereas the levels of either secreted or cellular full-length apoE were unaffected (Fig. 6, B–D). These data confirm that HtrA1 contributes to the proteolytic formation of apoE 25-kDa fragment under physiological conditions.
Figure 6.
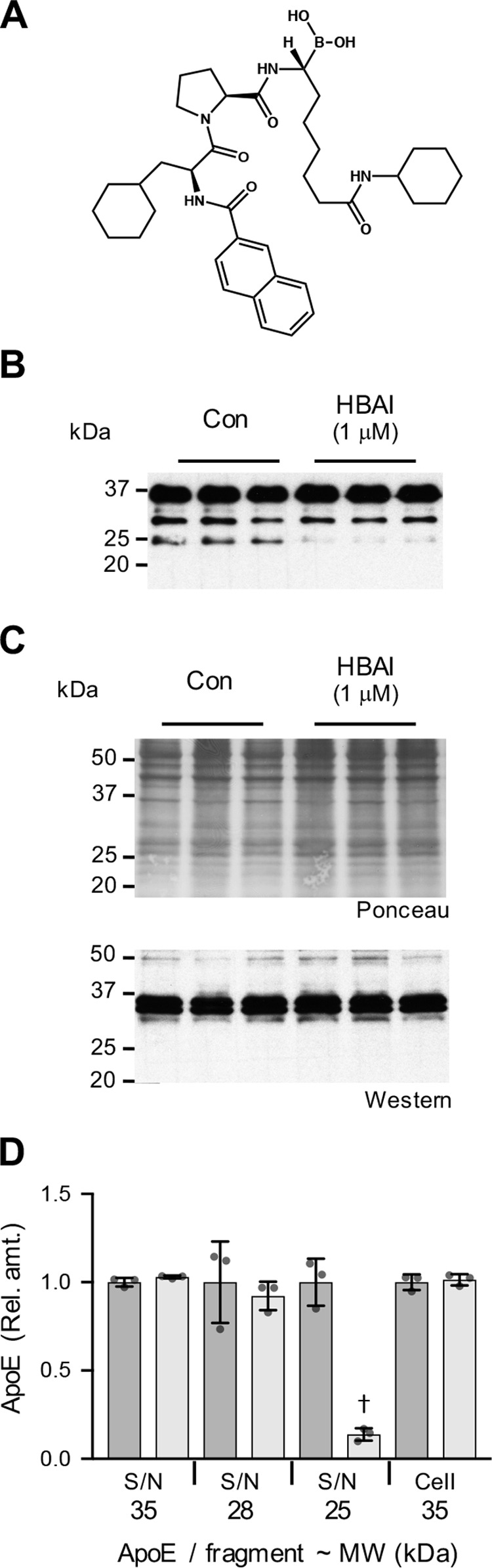
HtrA1 inhibitor reduces SK-N-SH cellular apoE proteolytic fragmentation. The structure of HBAI is shown (A). SK-N-SH cells were cultured for 9 days in the presence of 10 μm ATRA. The culture medium was then replaced with fresh medium alone (Con) or with fresh medium containing either HBAI (1 μm) or vehicle control and cultured for a further 24-h period. The addition of HBAI inhibited the formation of the apoE 25-kDa fragment in the supernatant (S/N) (B). HBAI addition had no impact on cellular apoE levels (C). Quantitative assessments of the full-length apoE (35 kDa) and apoE fragments at 28 and 25 kDa indicate a significant inhibition of apoE 25-kDa fragment formation in the presence of HBAI (light gray bars; D). Data in D are relative optical density measurements in which the vehicle control samples (DMSO (1:1,000), dark gray bars) are defined as 1.0. Data in D are derived from one experiment performed in triplicate (Western blots shown in B and C) and are representative of three independently performed experiments. The histogram bars represent mean values, and the error bars represent S.D. †, p < 0.01 compared with the respective control for each fragment; two-tailed t test.
A previous study demonstrated that recombinant HtrA1 cleaves recombinant apoE to generate a quantitatively major 25-kDa fragment, which was determined by peptide mapping mass spectrometry to encompass apoE amino acid residues 1–195 (22). Based on the fact that this peptide encompasses the apoE N-terminal four-helix bundle and therefore expresses the LDL receptor–binding region (32) and that apoE is well-established to promote neurite outgrowth via interaction with LDL receptor family members (33–37), we next assessed the impact that the recombinant apoE 25-kDa peptide (r-apoE 25; amino acid residues 1–195) has on neuritogenesis in vitro. In these experiments, we used the SH-SY5Y cell line. This cell line was chosen, as it is a subclone of the SK-N-SH line that does not produce detectable apoE and does not produce the high degree of neurite growth that is observed with ATRA-treated SK-N-SH neurons (23). Based on previous studies (35–37) and our own pilot experiments, we used apoE concentrations of 5 and 20 μg/ml.
In the absence of r-apoE and r-apoE 25, SH-SY5Y cells developed a neuronal phenotype after 6 days, with clearly identifiable but relatively short neurites (Fig. 7). Neurite length data were collected in units of mm of total neurites detected/mm2. For the control conditions (Fig. 7B, Con), including 0-, 3-, and 6-day culture, the neurite length value was 1.46 ± 0.09 mm/mm2 (mean ± S.E., n = 30) and remained unchanged over the 6-day period. Under these conditions where ATRA and low serum levels are present (see “Experimental procedures”), the addition of either r-apoE or r-apoE 25 did not have a major impact on cell growth, with confluence levels at a very similar level at day 6 (Fig. 7). However, in agreement with previous studies (35, 37), r-apoE did dose-dependently promote neuritogenesis, with neurite growth increased by 2.4-fold (p < 0.0001) after 6 days of supplementation at 20 μg/ml r-apoE concentration (Fig. 7B). Similarly, the addition of 20 μg/ml r-apoE 25 also strongly promoted neuritogenesis, with neurite growth increased by 2.2-fold (p < 0.0001) after 6 days (Fig. 7B). At the lower dose of 5 μg/ml, r-apoE also significantly promoted neuritogenesis with neurite growth increased by 2.1-fold (p < 0.0001) after 6 days, whereas a non-significant trend for increased neurite growth was observed at the lower dose of r-apoE 25 (Fig. 7B). Overall viability was reduced by 19% (p = 0.0007) after 6 days in the presence of 20 μg/ml r-apoE but was not affected in the other conditions tested (Fig. 7C). The cell culture medium was also collected when the cells were harvested at day 6 (i.e. the medium was present on the cells for 72 h, from day 3 to day 6) and assessed for apoE by Western blotting. As predicted, the medium that was not supplemented with either form of recombinant apoE did not contain detectable apoE (Fig. S7). Interestingly, full-length r-apoE was partly proteolyzed over the 72-h incubation period to generate the apoE 25-kDa fragment (Fig. S7B). In the case of r-apoE 25, no proteolysis was observed (Fig. 7B). A quantitatively minor band (accounting for ∼10% of total r-apoE 25) was present in the r-apoE 25 before addition to the cells, and the level of this minor fragment did not change during the 72-h incubation (Fig. 7B). Overall, these data reveal that the apoE 25-kDa fragment that is derived from HtrA1-mediated proteolysis is a relatively stable fragment that promotes neuritogenesis to a similar degree as reported previously for full-length apoE.
Figure 7.

Recombinant apoE 25-kDa fragment stimulates SH-SY5Y neuritogenesis. Full-length recombinant apoE and apoE 25 (1–195 fragment) were added to SH-SY5Y cells at concentrations of 5 and 20 μg/ml 24 h after plating (day 0), with medium change at day 3 day and final analysis at day 6. Confluence (A) and neurite length (B) were assessed at t = 0 and then every 3 days using an IncuCyte Zoom live cell analysis system. Cell viability was measured at day 6 (C). All data are expressed relative to the respective day 0 control (Con) condition, which is defined as 1.0. Data are derived from two independent experiments performed in quintuplicate. The histogram bars represent mean values, and the error bars represent S.D. †, p < 0.01; ‡, p < 0.0001 compared with the respective day 0 control for each condition (A and B) or †, p < 0.01 compared with the control (Con) condition; one-way ANOVA with Tukey's post hoc analysis. Arrows, neurites (D). Scale bars in D, 100 μm.
Discussion
Herein, we describe a cell culture system that recapitulates the physiologically relevant process of apoE proteolytic fragmentation, a process known to occur in the human brain. Building on recent studies showing that the proteolytic fragmentation of exogenous apoE involves an extracellular neuronal serine protease (9) and that recombinant HtrA1 generates apoE fragments, particularly a major 25-kDa fragment (22) that is very similar to the major apoE fragment detected in the human brain (14), we showed here, using siRNA knockdown of HtrA1 and a highly specific HtrA1 inhibitor, that endogenous apoE is selectively cleaved by endogenous HtrA1 and that the process is linked to neuronal differentiation state. Consistent with our findings, previous studies have also shown that HtrA1 expression is tightly linked with neuronal development both in vitro and in vivo (29, 30) and that HtrA1 expression levels are positively correlated with PSD95 levels during neuronal differentiation (30). In addition, loss of HtrA1-mediated proteolysis of TGF-β was shown to directly prevent neuronal differentiation (30). Intriguingly, similar to our current findings with SK-N-SH neurons, ATRA was reported to induce HtrA1 expression associated with osteogenic differentiation of adipose-derived stromal cells (38). In this case, siRNA-induced knockdown of HtrA1 prevented ATRA-mediated stromal cell osteogenesis (38). These findings raise the possibility that both HtrA1 activity and apoE-mediated signaling may act together to regulate neuronal differentiation.
To explore the relationship between HtrA1 expression and the biological activity of the apoE 25-kDa fragment, we generated a recombinant apoE fragment (spanning residues 1–195) that corresponds to the main apoE fragment generated by HtrA1. Our data indicate potent neuritogenic activity of the apoE 25-kDa fragment that is very similar to that of full-length apoE. There is previous evidence that apoE fragments (and apoE-mimetic peptides) are biologically active, as they regulate neuronal signaling (39, 40) and control either neurodegenerative or neuroprotective pathways and anti-inflammatory pathways, depending on the fragments analyzed (15, 18, 41–54). Some of these previous reports focused on a potentially toxic apoE C-terminal truncated 30-kDa fragment and other shorter thrombolytic and synthetic fragments, which were suggested to be present in the “insoluble fractions” of human AD brain homogenates. In our own studies, we detected a stable apoE 25-kDa fragment that was present in the Tris-soluble fractions of human brain homogenates (14). Because the concentration of this soluble apoE fragment was strongly associated with the apoE3 isoform, we speculated that it may provide a protective function in the brain (14, 55). This is consistent with the findings of our current work, as the stimulation of neuritogenesis by the apoE 25-kDa fragment would also be predicted to represent a protective function in vivo.
It is important to consider the concentrations of apoE 25-kDa fragment and full-length apoE used in our current study (5 and 20 μg/ml) in relation to the levels detected in human tissues. Human CSF and plasma contain apoE at concentrations of ∼10 and 50 μg/ml, respectively (56, 57). Levels of soluble apoE in the human frontal cortex, temporal cortex, and cerebellum are reported to be 95, 122, and 48 μg/g protein, respectively (58). Based on a total protein content of ∼8–10% for human brain tissues (59), it is conceivable that total adult brain apoE would be present at low μg/g wet tissue weight levels (i.e. similar to the concentrations we have used here). It is also noteworthy that as a predominantly secreted protein, extracellular apoE levels in interstitial fluid may be different from total tissue levels derived from the soluble fraction of homogenates. Human brain apoE concentration also varies throughout life; indeed, neonatal brain apoE levels are twice the level found in adults (60), presumably due to the neurotrophic role of apoE during brain development. Soluble brain apoE concentrations are therefore likely to fluctuate, depending on several physiological parameters. It appears that, similar to previous studies focusing on apoE biological functions (35–37), the low μg/ml apoE levels that we have used are likely to be physiologically relevant.
The level of the apoE 25-kDa fragment present in the human brain has not been accurately quantified. However, in a previous Western blot analysis of adult human frontal cortex, temporal cortex, and occipital cortex, we found that the soluble apoE 25-kDa fragment accounted for 20% of total apoE (14). This is very similar to the relative amounts of apoE 25-kDa fragment detected in SK-N-SH neuron culture medium after 9 days of ATRA treatment in the present study. Interestingly, our previous study showed that the concentration of soluble cortical apoE 25-kDa fragment present in APOEϵ3 homozygotes was 3-fold higher than in APOEϵ4 homozygotes (14). It is possible that these differences in neurotrophic apoE 25-kDa fragment concentration over a lifetime may contribute to the increased AD risk associated with the APOEϵ4 genotype. We hasten to add, however, that further work will clearly be required to investigate the potential neurotrophic function of the apoE 25-kDa fragment in vivo.
Based on the knowledge that apoE provides neurotrophic support via its interactions with LDL receptor family members (1, 7, 34, 61, 62), and that the apoE 25-kDa fragment we have identified is stable in vitro and in vivo, it is plausible that this fragment may also promote neuritogenic effects via interaction with LDL receptor family members, as has been demonstrated in previous studies with full-length apoE under numerous experimental settings (35–37, 63). Evidence that the apoE N-terminal fragment interacts with LDL receptor family members is provided by previous studies that have shown the apoE N-terminal domain (the 22-kDa fragment, residues 1–183, associated with dimyristoyl-phosphocholine) is recognized by the LDL receptor (64, 65). Of potential relevance, the injection of apoE N-terminal fragment (residues 1–191) into rabbits indicated a clearance rate from the circulation that is consistent with a receptor-mediated process, for example, via hepatic LRP1 (66), although the fragment was not associated with lipoprotein particles (67). In addition, apoE-derived mimetic peptides that span the LDL receptor–binding region have been used successfully as neuroprotective agents in mice (49, 50, 68, 69).
We also speculate that the apoE 25-kDa fragment may represent a particularly potent neuritogenic agent in vivo, as other functions of full-length apoE that are driven primarily by regions in the C-terminal domain (such as permitting lipoprotein interaction (67, 70), promoting self-aggregation (67, 70), and facilitating Aβ binding and clearance (17)) would be removed or reduced in the apoE 25-kDa fragment, thereby selectively promoting apoE signaling functions via interaction with LDL receptor family members. Previous studies have shown that ApoE knockout mice have cognitive deficits that are more pronounced in females and are exacerbated by age in association with blood-brain barrier dysfunction (71–73). Based on the approach of targeted replacement with either full-length or truncated forms of human apoE in ApoE−/− mice (19, 71, 73), in the future, it would be interesting to use a similar approach to test for the potential protective effect of the apoE 25-kDa fragment on synaptic and cognitive dysfunctions in vivo.
HtrA1 is widely expressed in adult human tissues, including the brain, where it has been localized to neurons, astrocytes, oligodendrocytes, and microglia (74). In addition, HtrA1 expression is regulated spatially and temporally in the developing mouse brain (75) and is known to control neuronal maturation and developmental survival via regulation of the TGF-β signaling pathway by eliciting TGF-β proteolytic cleavage (30). Indeed, HtrA1 is well known to modulate a variety of signaling pathways through proteolytic cleavage (or chaperone-like sequestering) of regulatory proteins (31), which would be in line with a biological signaling function for the apoE 25-kDa fragment. Based on data from our studies and many other laboratories, it is clear that several proteolytic fragments of apoE in addition to the 25-kDa species are generated both in vitro and in vivo, and it is possible that different apoE fragments may have varying biological functions. It is also probable that these other fragments are generated by one or more additional serine proteases, as they have not been detected in studies that focused on proteolytic fragment mapping of digests resulting from incubation of recombinant apoE with recombinant HtrA1 (22).
The role that HtrA1-mediated proteolysis of apoE could play in AD is also not fully established. Previous studies have shown that HtrA1 cleaves Tau and the amyloid precursor protein (76–78), which is predicted to promote protection in the AD context. Furthermore, HtrA1 levels and activity are increased in response to the development of AD pathology (77). It was also recently established that HtrA1 degrades apoE4 more rapidly than apoE3 (22), which may contribute to the overall reduced levels of apoE4 detected in AD brain tissues as compared with apoE3. It is currently unclear how HtrA1 differentially cleaves apoE3 and apoE4 to generate a different fragmentation pattern. The HtrA1 cut site generating the apoE 25-kDa fragment (encompassing amino acids 1–195) was defined using mass spectrometry peptide mapping (22). HtrA1 cuts after apoE Val194, which is typical of an elastase-like serine protease activity. Although HtrA1 activity is thought to be more like a trypsin-like serine protease, regulation via substrate interaction with a PDZ domain may influence the enzyme's protease activity (79, 80). Of potential significance, binding of substrate C-terminal or internal hydrophobic stretches of amino acids (or protein-associated lipid) to the HtrA1 PDZ can modulate protease activity (79, 80), and this is thought to contribute to the processing of unfolded proteins by HtrA1. Intriguingly, the apoE4 N-terminal four-helix bundle is known to be less stable than apoE3 (81). Indeed, previous data indicate that apoE4 may form a molten globule-like state in which the four-helix bundle is partially opened and elongated, thereby exposing the hydrophobic core of the protein (81). This may contribute to the differential processing of apoE3 and apoE4 by HtrA1. Elucidation of the precise structural details of HtrA1-mediated apoE isoform–specific proteolysis clearly requires further study. Nonetheless, there are several plausible routes by which HtrA1 could regulate AD development, and these now appear to include impacts on apoE metabolism.
In conclusion, our study provides a cellular model to assess apoE proteolysis, shows that HtrA1 regulates apoE 25-kDa formation under physiological conditions, and reveals a new neurotrophic function for the apoE 25-kDa fragment.
Experimental procedures
Cell culture and treatments
Cell culture media and additives were from Life Technologies (Melbourne, Australia). Human SK-N-SH and SH-SY5Y neuroblastoma cells (American Type Culture Collection (ATCC (Manassas, VA) catalog nos. HTB-11 and CRL-2266, respectively)) were routinely maintained in Dulbecco's modified Eagle's medium/F-12 (1:1, v/v) growth medium supplemented with 5% (v/v) fetal bovine serum, 2 mm glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Cultures were grown in 75-cm2 flasks at 37 °C in 5% CO2 and plated into multiwell plates for use in experiments. Where indicated, cells were treated with the following compounds (or appropriate vehicle control) for the time intervals indicted in the text and figure legends: ATRA (10 μm; Sigma-Aldrich, Castle Hill, Australia), protease inhibitor mixture (PIM; catalog no. P8340, 1:1000, Sigma-Aldrich), AEBSF (1 mm, catalog no. A8456, Sigma-Aldrich), and HBAI (1 μm) synthesized as described previously (22). SK-N-SH and SH-SY5Y cells were genotyped as described previously (14) and confirmed to be homozygous for the APOEϵ3 genotype, as previously reported (82, 83).
Microscopy
Phase-contrast images of SK-N-SH cells grown for 9 days in 12-well cell culture plates under standard culture conditions or in the presence of 10 μm ATRA were acquired using a Nikon Eclipse 300 inverted cell culture microscope and ×10 objective. Bright-field images of SH-SY5Y cells were taken using an IncuCyte Zoom live cell analysis system with a ×20 objective (Essen BioScience, Ann Arbor, MI). Confluence and neurite length were assessed every 3 days, and images were analyzed using the IncuCyte software.
Western blotting
SK-N-SH cell culture medium and cell lysates were collected at the indicated times. Cell lysates were harvested in lysis buffer (radioimmune precipitation assay lysis buffer (10 mm Tris-Cl (pH 8.0), 1 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mm NaCl, 1 mm phenylmethylsulfonyl fluoirde) with PIM (1:100)) essentially as described previously (8, 84). In brief, bicinchoninic acid (BCA) protein assays were performed on cell lysates, and equal amounts of protein (∼15–50 μg of protein/lane) were separated on reducing SDS-polyacrylamide gels (8, 12, or 15%, depending on the Mr of the protein of interest) and transferred onto 0.45-μm nitrocellulose membranes at 100 V for 30 min. Membranes were Ponceau-stained and scanned before blocking overnight at 22 °C for 1 h in PBS containing 0.1% (w/v) Tween-20 (PBS-T) containing 5% (w/v) nonfat dry milk. The membranes were then probed with the relevant antibodies at 4 °C for 16 h to reveal the bands of interest. Dilutions of antibodies used were as follows: apoE (goat polyclonal Millipore catalog no. 178479, RRID:AB_2057989, 1:10,000), SMI32 (Covance Research Products Inc. catalog no. SMI-312R, RRID:AB_2314906, 1:2,000), PSD95 (Millipore catalog no. 04-1066, RRID:AB_1977415, 1:500), GAPDH (rabbit polyclonal, OSG00032W, Osenses, 1:10,000), β-tubulin isotype III (Sigma-Aldrich catalog no. T8660, RRID:AB_477590, 1:5,000), apoE N-terminal D6E10 (Abcam catalog no. ab1906, RRID:AB_302668, 1:1,000), apoE C-terminal (clone E-8, Santa Cruz Biotechnology, Inc., catalog no. sc-393302, 1:200), and HtrA1 monoclonal antibody (R&D Systems catalog no. MAB2916, RRID:AB_2122710, 1:500, used under non-reducing PAGE conditions). The membranes were washed three times in PBS-T, and signals were detected using species-specific HRP-conjugated secondary antibodies (all from donkey, Santa Cruz Biotechnology, Inc. (1:5,000) or Jackson ImmunoResearch Laboratories (1:5,000)), and the proteins of interest were detected using enhanced chemiluminescence (ECL, Amersham Biosciences or Pierce ECL Plus Western blotting substrate, Thermo Fisher Scientific) and captured by X-ray film or by a gel imager (GE Amersham Biosciences Imager 600). Signal intensity was quantified using ImageJ software (W. S. Rasband, National Institutes of Health, Bethesda, MD).
For quantification of apoE proteolytic fragments, a fixed area was used to separately measure signal intensity from the regions encompassing full-length apoE (∼35 kDa) and apoE fragments at ∼28 and ∼25 kDa. An adjacent blank region was used as a background control. Relative differences between samples were always assessed on the same blots.
Assessment of apoE/apoE fragment desialylation and heparin binding
Where indicated, 9-day ATRA-treated cell culture supernatant samples were treated with neuraminidase (NMDase; catalog no. N3786, Sigma) to probe for sialylation on full-length apoE and apoE fragments. Samples (0.3 ml) were mixed with an equal volume of 1× NMDase reaction buffer, and 6 μl of enzyme solution was added. Sample controls were treated using the same buffers but without NMDase. After incubation at 37 °C for 14 h, all samples were heated at 100 °C for 5 min and then analyzed by 15% SDS-PAGE and Western blotting for apoE detection as described above.
The heparin-binding activity of full-length apoE and apoE fragments was assessed using HiTrap Heparin HP columns (GE Healthcare, Rydalmere, Australia), following the manufacturer's instructions. In brief, the column was equilibrated with 10 ml of 100 mm sodium phosphate buffer (pH 7), and 9 ml of 9-day ATRA-treated cell culture supernatant sample was added after the addition of 1 ml of 100 mm NaCl “load” sample. Heparin-binding proteins were then eluted by the addition of 5 ml of 100 mm NaCl, followed by 5 ml of 500 mm NaCl. The load sample and the eluates were then analyzed by 15% SDS-PAGE and Western blotting for apoE detection as described above.
Quantitative real-time PCR
Analysis of mRNA expression level was carried out using quantitative real-time PCR as previously reported (85). Cells were rinsed twice with ice-cold PBS. Total cellular RNA was extracted with TRIsure reagent (Bioline (Sydney, Australia), catalog no. BIO-38022), according to the manufacturer's instructions. The concentration of the purified RNA was determined by recording the absorbance at 260 nm using a Nanodrop 2000 (Thermo Scientific, Wilmington, DE), and the purity and the integrity of the isolated RNA were checked by determining the A260/A280 ratio (>1.8). Two μg of total RNA was used for reverse transcription with an oligo(dT)18 primer according to the instructions provided with the Tetro cDNA synthesis kit (Bioline, catalog no. BIO-65043).
Conventional PCR amplification for primer pair checking was performed using MyTaq HS reagents (Bioline, catalog no. BIO-25045) and carried out with 30 cycles of denaturation (95 °C, 15 s), annealing (62 °C, 15 s), and extension (72 °C, 20 s), and the PCR products were visualized with a Gel Doc 2000 imager system after electrophoresis in 1.5% agarose gels. Quantitative real-time PCR was carried out in a Roche LightCycler® 480 real-time PCR system or Corbett Rotor Gene 3000 system using the SensiFAST SYBR® No-ROX kit (Bioline, catalog no. BIO-98005), following the manufacturer's protocol. The PCR program was an initial denaturation step at 92 °C for 2 min, followed by 40 cycles of denaturation (92 °C for 10 s), annealing (62 °C for 10 s), and extension (72 °C for 10 s). A final melting step was performed (95 °C for 5 s, 65 °C for 1 min, and acquiring continuously until 97 °C). The primer pairs used were as follows: human HTRA1 (HtrA serine peptidase 1, Gene ID 5654), 5′-GACTACATCCAGACCGACGC-3′ (forward) and 5′-TTTGGCTTTGCTGGACGTGA-3′ (reverse); human GAPDH(glyceraldehyde-3-phosphate dehydrogenase, Gene ID 2597), 5′-GAGCACAAGAGGAAGAGAGAGACCC-3′ (forward) and 5′-GTTGAGCACAGGGTACTTTATTGATGGTACATG-3′ (reverse); and human HPRT1 (hypoxantine phosphoribosyltransferase 1, Gene ID 3251), 5′-ATAAGCCAGACTTTGTTGG-3′ (forward) and 5′-ATAGGACTCCAGATGTTTCC-3′ (reverse). The efficiency of each sample was determined using LinRegPCR (86), and the analysis was performed following the 2−ΔCt method (87).
HtrA1 siRNA experiments
Human HTRA1 siRNA (5′-GAAGUGAUUGGAAUUAACATT-3′; 5′-UGUUAAUUCCAAUCACUUCTT-3′) and scrambled siRNA (5′-GAAAGUUGGAUCAAUAAGUTT-3′; 5′-ACUUAUUGAUCCAACUUUCTT-3′) were designed by and obtained from Sigma-Aldrich and transfected into SK-N-SH cells using Lipofectamine RNAiMAX (Thermo Scientific), following the manufacturer's instructions. SK-N-SH cells were seeded at 100,000 cells/well in a 12-well plate and cultured under the conditions described above for 9 days. HTRA1 siRNA was transfected 24 h after the initial seeding, and media were changed every 3 days until cells were analyzed at day 9 for mRNA and protein expression as described above. Total protein for the cell lysate was quantified using the detergent-compatible Lowry method (DC Protein Assay, Bio-Rad, Gladesville, Australia), following the manufacturer's instructions.
Expression and purification of recombinant human apoE and apoE 25
The APOE3 cDNA clone was kindly provided by Dr. Zhijiang Chen (Salk Institute), and full-length apoE was generated as described previously (22). The apoE 25 (amino acids 1–195) coding sequence was subcloned into the pHisMAL vector with a tobacco etch virus cleavage site between the apoE protein and the His6-MBP for bacterial expression. The apoE 25 protein was then expressed in Escherichia coli strain Rosetta 2 (DE3), and the recombinant protein was isolated from bacterial lysates as described previously (22) and incubated with tobacco etch virus protease at 4 °C for 12 h to cleave off the His6-MBP tag. The cleaved protein mixture was incubated with nickel-nitrilotriacetic acid resin to remove the His6-MBP tag, and the apoE 25 protein was further purified by monoQ and size-exclusion chromatography. Eluted apoE 25–containing fractions were pooled and concentrated using a 10,000 molecular weight cutoff filter (Millipore) and stored at −80 °C.
Assessment of apoE and apoE 25 neurotrophic activities
Full-length recombinant apoE and apoE 25 (1–195 fragment) were added to SH-SY5Y cells at concentrations of 5 and 20 μg/ml 24 h after plating at 10,000 cells/well in a 96-well plate. The concentration of fetal bovine serum in the cell culture media was reduced to 1% (v/v), and ATRA was maintained at 10 μm. Medium changes (including replenishing r-apoE and r-apoE 25) were made at 3 days until the final analysis at day 6. Confluence and neurite length were assessed in five individual wells every 3 days using an IncuCyte Zoom live cell analysis system (as described above). Neurite length was measured as detected by the IncuCyte Zoom software as the total combined length of all neurites detected per mm2. Cell viability was measured at day 6 using the Presto Blue assay (Thermo Scientific), following the manufacturer's instructions.
Statistics
SPSS Statistics software (version 24, SPSS Inc., Chicago, IL) was used for statistical analysis. All data are expressed as -fold increase relative to the control (vehicle-treated) group that was arbitrarily assigned a value of 1.0. Where two groups were compared, a two-tailed t test was used. Where multiple groups were compared, statistical significance was determined using one-way ANOVA for repeated measures. For all ANOVA testing, Tukey's post hoc analysis was used. For both the ANOVA and the t test, p < 0.05 was considered significant.
Author contributions
S.S.M., H.L., K.R., and Q.C. data curation; S.S.M., H.L., K.R., Q.C., A.S., L.O., and B.G. formal analysis; S.S.M., H.L., K.R., Q.C., A.S., and B.G. validation; S.S.M., H.L., K.R., Q.C., A.S., L.O., and B.G. investigation; S.S.M., H.L., K.R., Q.C., A.S., L.O., and B.G. methodology; S.S.M., H.L., K.R., Q.C., A.S., L.O., and B.G. writing-review and editing; H.L., K.R., and Q.C. visualization; Q.C., A.S., L.O., and B.G. resources; A.S., L.O., and B.G. conceptualization; A.S., L.O., and B.G. supervision; A.S., L.O., and B.G. funding acquisition; B.G. writing-original draft; B.G. project administration.
Supplementary Material
This research was supported by National Health and Medical Research Council (NHMRC) of Australia Grant 1079995. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S7.
- apoE
- apolipoprotein E
- Aβ
- amyloid-β peptide
- AD
- Alzheimer's disease
- AEBSF
- 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride
- ATRA
- all-trans-retinoic acid
- BCA
- bicinchoninic acid
- CNS
- central nervous system
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- HBAI
- HtrA1 boronic acid inhibitor
- HtrA1
- high-temperature requirement serine peptidase A1
- LDL
- low-density lipoprotein
- LRP1
- low-density lipoprotein receptor-related protein-1
- NMDase
- neuraminidase
- PSD95
- post-synaptic density protein 95
- SMI32
- non-phosphorylated neurofilament heavy chain marker
- ANOVA
- analysis of variance
- r-apoE
- recombinant apoE
- r-apoE 25
- recombinant apoE 25-kDa peptide
- PIM
- protease inhibitor mixture.
References
- 1. Herz J., and Beffert U. (2000) Apolipoprotein E receptors: linking brain development and Alzheimer's disease. Nat. Rev. Neurosci. 1, 51–58 10.1038/35036221 [DOI] [PubMed] [Google Scholar]
- 2. Corder E. H., Saunders A. M., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Small G. W., Roses A. D., Haines J. L., and Pericak-Vance M. A. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- 3. Rall S. C. Jr., Weisgraber K. H., Innerarity T. L., and Mahley R. W. (1982) Structural basis for receptor binding heterogeneity of apolipoprotein E from type III hyperlipoproteinemic subjects. Proc. Natl. Acad. Sci. U.S.A. 79, 4696–4700 10.1073/pnas.79.15.4696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. LaDu M. J., Gilligan S. M., Lukens J. R., Cabana V. G., Reardon C. A., Van Eldik L. J., and Holtzman D. M. (1998) Nascent astrocyte particles differ from lipoproteins in CSF. J. Neurochem. 70, 2070–2081 [DOI] [PubMed] [Google Scholar]
- 5. Achariyar T. M., Li B., Peng W., Verghese P. B., Shi Y., McConnell E., Benraiss A., Kasper T., Song W., Takano T., Holtzman D. M., Nedergaard M., and Deane R. (2016) Glymphatic distribution of CSF-derived apoE into brain is isoform specific and suppressed during sleep deprivation. Mol. Neurodegener. 11, 74 10.1186/s13024-016-0138-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xu Q., Bernardo A., Walker D., Kanegawa T., Mahley R. W., and Huang Y. (2006) Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 26, 4985–4994 10.1523/JNEUROSCI.5476-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mahley R. W. (1988) Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240, 622–630 10.1126/science.3283935 [DOI] [PubMed] [Google Scholar]
- 8. Elliott D. A., Kim W. S., Jans D. A., and Garner B. (2007) Apoptosis induces neuronal apolipoprotein-E synthesis and localization in apoptotic bodies. Neurosci. Lett. 416, 206–210 10.1016/j.neulet.2007.02.014 [DOI] [PubMed] [Google Scholar]
- 9. Tamboli I. Y., Heo D., and Rebeck G. W. (2014) Extracellular proteolysis of apolipoprotein E (apoE) by secreted serine neuronal protease. PLoS One 9, e93120 10.1371/journal.pone.0093120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao N., Liu C. C., Qiao W., and Bu G. (2018) Apolipoprotein E, receptors, and modulation of Alzheimer's disease. Biol. Psychiatry 83, 347–357 10.1016/j.biopsych.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang Q., Lee C. Y., Mandrekar S., Wilkinson B., Cramer P., Zelcer N., Mann K., Lamb B., Willson T. M., Collins J. L., Richardson J. C., Smith J. D., Comery T. A., Riddell D., Holtzman D. M., et al. (2008) ApoE promotes the proteolytic degradation of Abeta. Neuron 58, 681–693 10.1016/j.neuron.2008.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Verghese P. B., Castellano J. M., Garai K., Wang Y., Jiang H., Shah A., Bu G., Frieden C., and Holtzman D. M. (2013) ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. U.S.A. 110, E1807–E1816 10.1073/pnas.1220484110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang Y. A., Zhou B., Wernig M., and Südhof T. C. (2017) ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell 168, 427–441.e21 10.1016/j.cell.2016.12.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Elliott D. A., Tsoi K., Holinkova S., Chan S. L., Kim W. S., Halliday G. M., Rye K. A., and Garner B. (2011) Isoform-specific proteolysis of apolipoprotein-E in the brain. Neurobiol. Aging 32, 257–271 10.1016/j.neurobiolaging.2009.02.006 [DOI] [PubMed] [Google Scholar]
- 15. Marques M. A., Tolar M., Harmony J. A., and Crutcher K. A. (1996) A thrombin cleavage fragment of apolipoprotein E exhibits isoform-specific neurotoxicity. Neuroreport 7, 2529–2532 10.1097/00001756-199611040-00025 [DOI] [PubMed] [Google Scholar]
- 16. Aizawa Y., Fukatsu R., Takamaru Y., Tsuzuki K., Chiba H., Kobayashi K., Fujii N., and Takahata N. (1997) Amino-terminus truncated apolipoprotein E is the major species in amyloid deposits in Alzheimer's disease-affected brains: a possible role for apolipoprotein E in Alzheimer's disease. Brain Res. 768, 208–214 10.1016/S0006-8993(97)00640-9 [DOI] [PubMed] [Google Scholar]
- 17. Cho H. S., Hyman B. T., Greenberg S. M., and Rebeck G. W. (2001) Quantitation of apoE domains in Alzheimer disease brain suggests a role for apoE in Aβ aggregation. J. Neuropathol. Exp. Neurol. 60, 342–349 10.1093/jnen/60.4.342 [DOI] [PubMed] [Google Scholar]
- 18. Huang Y., Liu X. Q., Wyss-Coray T., Brecht W. J., Sanan D. A., and Mahley R. W. (2001) Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc. Natl. Acad. Sci. U.S.A. 98, 8838–8843 10.1073/pnas.151254698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harris F. M., Brecht W. J., Xu Q., Tesseur I., Kekonius L., Wyss-Coray T., Fish J. D., Masliah E., Hopkins P. C., Scearce-Levie K., Weisgraber K. H., Mucke L., Mahley R. W., and Huang Y. (2003) Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer's disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 100, 10966–10971 10.1073/pnas.1434398100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marques M. A., Owens P. A., and Crutcher K. A. (2004) Progress toward identification of protease activity involved in proteolysis of apolipoprotein e in human brain. J. Mol. Neurosci. 24, 73–80 10.1385/JMN:24:1:073 [DOI] [PubMed] [Google Scholar]
- 21. Heeren J., Grewal T., Jäckle S., and Beisiegel U. (2001) Recycling of apolipoprotein E and lipoprotein lipase through endosomal compartments in vivo. J. Biol. Chem. 276, 42333–42338 10.1074/jbc.M107461200 [DOI] [PubMed] [Google Scholar]
- 22. Chu Q., Diedrich J. K., Vaughan J. M., Donaldson C. J., Nunn M. F., Lee K. F., and Saghatelian A. (2016) HtrA1 proteolysis of ApoE in vitro is allele selective. J. Am. Chem. Soc. 138, 9473–9478 10.1021/jacs.6b03463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Preis P. N., Saya H., Nádasdi L., Hochhaus G., Levin V., and Sadée W. (1988) Neuronal cell differentiation of human neuroblastoma cells by retinoic acid plus herbimycin A. Cancer Res. 48, 6530–6534 [PubMed] [Google Scholar]
- 24. Ciccarone V., Spengler B. A., Meyers M. B., Biedler J. L., and Ross R. A. (1989) Phenotypic diversification in human neuroblastoma cells: expression of distinct neural crest lineages. Cancer Res. 49, 219–225 [PubMed] [Google Scholar]
- 25. Zannis V. I., McPherson J., Goldberger G., Karathanasis S. K., and Breslow J. L. (1984) Synthesis, intracellular processing, and signal peptide of human apolipoprotein E. J. Biol. Chem. 259, 5495–5499 [PubMed] [Google Scholar]
- 26. Saito H., Dhanasekaran P., Nguyen D., Baldwin F., Weisgraber K. H., Wehrli S., Phillips M. C., and Lund-Katz S. (2003) Characterization of the heparin binding sites in human apolipoprotein E. J. Biol. Chem. 278, 14782–14787 10.1074/jbc.M213207200 [DOI] [PubMed] [Google Scholar]
- 27. Nilsson J., Rüetschi U., Halim A., Hesse C., Carlsohn E., Brinkmalm G., and Larson G. (2009) Enrichment of glycopeptides for glycan structure and attachment site identification. Nat. Methods 6, 809–811 10.1038/nmeth.1392 [DOI] [PubMed] [Google Scholar]
- 28. Lorenzi T., Lorenzi M., Altobelli E., Marzioni D., Mensà E., Quaranta A., Paolinelli F., Morroni M., Mazzucchelli R., De Luca A., Procopio A. D., Baldi A., Muzzonigro G., Montironi R., and Castellucci M. (2013) HtrA1 in human urothelial bladder cancer: a secreted protein and a potential novel biomarker. Int. J. Cancer 133, 2650–2661 [DOI] [PubMed] [Google Scholar]
- 29. D'Angelo V., Pecoraro G., Indolfi P., Iannotta A., Donofrio V., Errico M. E., Indolfi C., Ramaglia M., Lombardi A., Di Martino M., Gigantino V., Baldi A., Caraglia M., De Luca A., and Casale F. (2014) Expression and localization of serine protease Htra1 in neuroblastoma: correlation with cellular differentiation grade. J. Neurooncol. 117, 287–294 10.1007/s11060-014-1387-4 [DOI] [PubMed] [Google Scholar]
- 30. Launay S., Maubert E., Lebeurrier N., Tennstaedt A., Campioni M., Docagne F., Gabriel C., Dauphinot L., Potier M. C., Ehrmann M., Baldi A., and Vivien D. (2008) HtrA1-dependent proteolysis of TGF-beta controls both neuronal maturation and developmental survival. Cell Death Differ. 15, 1408–1416 10.1038/cdd.2008.82 [DOI] [PubMed] [Google Scholar]
- 31. Clausen T., Kaiser M., Huber R., and Ehrmann M. (2011) HTRA proteases: regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol. 12, 152–162 10.1038/nrm3065 [DOI] [PubMed] [Google Scholar]
- 32. Wilson C., Wardell M. R., Weisgraber K. H., Mahley R. W., and Agard D. A. (1991) Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science 252, 1817–1822 10.1126/science.2063194 [DOI] [PubMed] [Google Scholar]
- 33. Herz J., and Chen Y. (2006) Reelin, lipoprotein receptors and synaptic plasticity. Nat. Rev. Neurosci. 7, 850–859 10.1038/nrn2009 [DOI] [PubMed] [Google Scholar]
- 34. Holtzman D. M., Herz J., and Bu G. (2012) Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hussain A., Luong M., Pooley A., and Nathan B. P. (2013) Isoform-specific effects of apoE on neurite outgrowth in olfactory epithelium culture. J. Biomed. Sci. 20, 49 10.1186/1423-0127-20-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nathan B. P., Jiang Y., Wong G. K., Shen F., Brewer G. J., and Struble R. G. (2002) Apolipoprotein E4 inhibits, and apolipoprotein E3 promotes neurite outgrowth in cultured adult mouse cortical neurons through the low-density lipoprotein receptor-related protein. Brain Res. 928, 96–105 10.1016/S0006-8993(01)03367-4 [DOI] [PubMed] [Google Scholar]
- 37. Puttfarcken P. S., Manelli A. M., Falduto M. T., Getz G. S., and LaDu M. J. (1997) Effect of apolipoprotein E on neurite outgrowth and β-amyloid-induced toxicity in developing rat primary hippocampal cultures. J. Neurochem. 68, 760–769 [DOI] [PubMed] [Google Scholar]
- 38. Glanz S., Mirsaidi A., López-Fagundo C., Filliat G., Tiaden A. N., and Richards P. J. (2016) Loss-of-function of HtrA1 abrogates all-trans-retinoic acid-induced osteogenic differentiation of mouse adipose-derived stromal cells through deficiencies in p70S6K activation. Stem Cells Dev. 25, 687–698 10.1089/scd.2015.0368 [DOI] [PubMed] [Google Scholar]
- 39. Hoe H. S., Harris D. C., and Rebeck G. W. (2005) Multiple pathways of apolipoprotein E signaling in primary neurons. J. Neurochem. 93, 145–155 10.1111/j.1471-4159.2004.03007.x [DOI] [PubMed] [Google Scholar]
- 40. Gay E. A., Bienstock R. J., Lamb P. W., and Yakel J. L. (2007) Structural determinates for apolipoprotein E-derived peptide interaction with the α7 nicotinic acetylcholine receptor. Mol. Pharmacol. 72, 838–849 10.1124/mol.107.035527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Crutcher K. A., Clay M. A., Scott S. A., Tian X., Tolar M., and Harmony J. A. (1994) Neurite degeneration elicited by apolipoprotein E peptides. Exp. Neurol. 130, 120–126 10.1006/exnr.1994.1191 [DOI] [PubMed] [Google Scholar]
- 42. Clay M. A., Anantharamaiah G. M., Mistry M. J., Balasubramaniam A., and Harmony J. A. (1995) Localization of a domain in apolipoprotein E with both cytostatic and cytotoxic activity. Biochemistry 34, 11142–11151 10.1021/bi00035a020 [DOI] [PubMed] [Google Scholar]
- 43. Tolar M., Keller J. N., Chan S., Mattson M. P., Marques M. A., and Crutcher K. A. (1999) Truncated apolipoprotein E (ApoE) causes increased intracellular calcium and may mediate ApoE neurotoxicity. J. Neurosci. 19, 7100–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tolar M., Marques M. A., Harmony J. A., and Crutcher K. A. (1997) Neurotoxicity of the 22 kDa thrombin-cleavage fragment of apolipoprotein E and related synthetic peptides is receptor-mediated. J. Neurosci. 17, 5678–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chang S., ran Ma T., Miranda R. D., Balestra M. E., Mahley R. W., and Huang Y. (2005) Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 102, 18694–18699 10.1073/pnas.0508254102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wellnitz S., Friedlein A., Bonanni C., Anquez V., Goepfert F., Loetscher H., Adessi C., and Czech C. (2005) A 13 kDa carboxy-terminal fragment of ApoE stabilizes Aβ hexamers. J. Neurochem. 94, 1351–1360 10.1111/j.1471-4159.2005.03295.x [DOI] [PubMed] [Google Scholar]
- 47. Aono M., Bennett E. R., Kim K. S., Lynch J. R., Myers J., Pearlstein R. D., Warner D. S., and Laskowitz D. T. (2003) Protective effect of apolipoprotein E-mimetic peptides on N-methyl-d-aspartate excitotoxicity in primary rat neuronal-glial cell cultures. Neuroscience 116, 437–445 10.1016/S0306-4522(02)00709-1 [DOI] [PubMed] [Google Scholar]
- 48. Laskowitz D. T., Thekdi A. D., Thekdi S. D., Han S. K., Myers J. K., Pizzo S. V., and Bennett E. R. (2001) Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp. Neurol. 167, 74–85 10.1006/exnr.2001.7541 [DOI] [PubMed] [Google Scholar]
- 49. Lynch J. R., Tang W., Wang H., Vitek M. P., Bennett E. R., Sullivan P. M., Warner D. S., and Laskowitz D. T. (2003) APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J. Biol. Chem. 278, 48529–48533 10.1074/jbc.M306923200 [DOI] [PubMed] [Google Scholar]
- 50. Lynch J. R., Wang H., Mace B., Leinenweber S., Warner D. S., Bennett E. R., Vitek M. P., McKenna S., and Laskowitz D. T. (2005) A novel therapeutic derived from apolipoprotein E reduces brain inflammation and improves outcome after closed head injury. Exp. Neurol. 192, 109–116 10.1016/j.expneurol.2004.11.014 [DOI] [PubMed] [Google Scholar]
- 51. Li F. Q., Sempowski G. D., McKenna S. E., Laskowitz D. T., Colton C. A., and Vitek M. P. (2006) Apolipoprotein E-derived peptides ameliorate clinical disability and inflammatory infiltrates into the spinal cord in a murine model of multiple sclerosis. J. Pharmacol. Exp. Ther. 318, 956–965 10.1124/jpet.106.103671 [DOI] [PubMed] [Google Scholar]
- 52. Singh K., Chaturvedi R., Asim M., Barry D. P., Lewis N. D., Vitek M. P., and Wilson K. T. (2008) The apolipoprotein E-mimetic peptide COG112 inhibits the inflammatory response to citrobacter rodentium in colonic epithelial cells by preventing NF-κB activation. J. Biol. Chem. 283, 16752–16761 10.1074/jbc.M710530200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dafnis I., Tzinia A. K., Tsilibary E. C., Zannis V. I., and Chroni A. (2012) An apolipoprotein E4 fragment affects matrix metalloproteinase 9, tissue inhibitor of metalloproteinase 1 and cytokine levels in brain cell lines. Neuroscience 210, 21–32 10.1016/j.neuroscience.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rohn T. T., Catlin L. W., Coonse K. G., and Habig J. W. (2012) Identification of an amino-terminal fragment of apolipoprotein E4 that localizes to neurofibrillary tangles of the Alzheimer's disease brain. Brain Res. 1475, 106–115 10.1016/j.brainres.2012.08.003 [DOI] [PubMed] [Google Scholar]
- 55. Elliott D. A., Weickert C. S., and Garner B. (2010) Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin. Lipidol. 51, 555–573 10.2217/clp.10.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tai L. M., Bilousova T., Jungbauer L., Roeske S. K., Youmans K. L., Yu C., Poon W. W., Cornwell L. B., Miller C. A., Vinters H. V., Van Eldik L. J., Fardo D. W., Estus S., Bu G., Gylys K. H., and Ladu M. J. (2013) Levels of soluble apolipoprotein E/amyloid-β (Aβ) complex are reduced and oligomeric Aβ increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J. Biol. Chem. 288, 5914–5926 10.1074/jbc.M112.442103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wahrle S. E., Shah A. R., Fagan A. M., Smemo S., Kauwe J. S., Grupe A., Hinrichs A., Mayo K., Jiang H., Thal L. J., Goate A. M., and Holtzman D. M. (2007) Apolipoprotein E levels in cerebrospinal fluid and the effects of ABCA1 polymorphisms. Mol. Neurodegener. 2, 7 10.1186/1750-1326-2-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pirttilä T., Soininen H., Heinonen O., Lehtimäki T., Bogdanovic N., Paljärvi L., Kosunen O., Winblad B., Riekkinen P. Sr., Wisniewski H. M., and Mehta P. D. (1996) Apolipoprotein E (apoE) levels in brains from Alzheimer disease patients and controls. Brain Res. 722, 71–77 10.1016/0006-8993(96)00183-7 [DOI] [PubMed] [Google Scholar]
- 59. Banay-Schwartz M., Kenessey A., DeGuzman T., Lajtha A., and Palkovits M. (1992) Protein content of various regions of rat brain and adult and aging human brain. Age 15, 51–54 10.1007/BF02435024 [DOI] [PubMed] [Google Scholar]
- 60. Kim W. S., Wong J., Weickert C. S., Webster M. J., Bahn S., and Garner B. (2009) Apolipoprotein-D expression is increased during development and maturation of the human prefrontal cortex. J. Neurochem. 109, 1053–1066 10.1111/j.1471-4159.2009.06031.x [DOI] [PubMed] [Google Scholar]
- 61. Koch S., Donarski N., Goetze K., Kreckel M., Stuerenburg H. J., Buhmann C., and Beisiegel U. (2001) Characterization of four lipoprotein classes in human cerebrospinal fluid. J. Lipid Res. 42, 1143–1151 [PubMed] [Google Scholar]
- 62. Beffert U., Stolt P. C., and Herz J. (2004) Functions of lipoprotein receptors in neurons. J. Lipid Res. 45, 403–409 10.1194/jlr.R300017-JLR200 [DOI] [PubMed] [Google Scholar]
- 63. Holtzman D. M., Pitas R. E., Kilbridge J., Nathan B., Mahley R. W., Bu G., and Schwartz A. L. (1995) Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc. Natl. Acad. Sci. U.S.A. 92, 9480–9484 10.1073/pnas.92.21.9480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Raussens V., Fisher C. A., Goormaghtigh E., Ryan R. O., and Ruysschaert J. M. (1998) The low density lipoprotein receptor active conformation of apolipoprotein E: helix organization in N-terminal domain-phospholipid disc particles. J. Biol. Chem. 273, 25825–25830 10.1074/jbc.273.40.25825 [DOI] [PubMed] [Google Scholar]
- 65. Fisher C. A., Narayanaswami V., and Ryan R. O. (2000) The lipid-associated conformation of the low density lipoprotein receptor binding domain of human apolipoprotein E. J. Biol. Chem. 275, 33601–33606 10.1074/jbc.M002643200 [DOI] [PubMed] [Google Scholar]
- 66. Beisiegel U., Weber W., Ihrke G., Herz J., and Stanley K. K. (1989) The LDL-receptor-related protein, LRP, is an apolipoprotein E-binding protein. Nature 341, 162–164 10.1038/341162a0 [DOI] [PubMed] [Google Scholar]
- 67. Westerlund J. A., and Weisgraber K. H. (1993) Discrete carboxyl-terminal segments of apolipoprotein E mediate lipoprotein association and protein oligomerization. J. Biol. Chem. 268, 15745–15750 [PubMed] [Google Scholar]
- 68. Wu Y., Pang J., Peng J., Cao F., Vitek M. P., Li F., Jiang Y., and Sun X. (2016) An apoE-derived mimic peptide, COG1410, alleviates early brain injury via reducing apoptosis and neuroinflammation in a mouse model of subarachnoid hemorrhage. Neurosci. Lett. 627, 92–99 10.1016/j.neulet.2016.05.058 [DOI] [PubMed] [Google Scholar]
- 69. Cao F., Jiang Y., Wu Y., Zhong J., Liu J., Qin X., Chen L., Vitek M. P., Li F., Xu L., and Sun X. (2016) Apolipoprotein E-mimetic COG1410 reduces acute vasogenic edema following traumatic brain injury. J. Neurotrauma 33, 175–182 10.1089/neu.2015.3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sakamoto T., Tanaka M., Vedhachalam C., Nickel M., Nguyen D., Dhanasekaran P., Phillips M. C., Lund-Katz S., and Saito H. (2008) Contributions of the carboxyl-terminal helical segment to the self-association and lipoprotein preferences of human apolipoprotein E3 and E4 isoforms. Biochemistry 47, 2968–2977 10.1021/bi701923h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Raber J., Wong D., Buttini M., Orth M., Bellosta S., Pitas R. E., Mahley R. W., and Mucke L. (1998) Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc. Natl. Acad. Sci. U.S.A. 95, 10914–10919 10.1073/pnas.95.18.10914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mulder M., Blokland A., van den Berg D. J., Schulten H., Bakker A. H., Terwel D., Honig W., de Kloet E. R., Havekes L. M., Steinbusch H. W., and de Lange E. C. (2001) Apolipoprotein E protects against neuropathology induced by a high-fat diet and maintains the integrity of the blood-brain barrier during aging. Lab. Invest. 81, 953–960 10.1038/labinvest.3780307 [DOI] [PubMed] [Google Scholar]
- 73. Lane-Donovan C., Wong W. M., Durakoglugil M. S., Wasser C. R., Jiang S., Xian X., and Herz J. (2016) Genetic restoration of plasma apoE improves cognition and partially restores synaptic defects in apoE-deficient mice. J. Neurosci. 36, 10141–10150 10.1523/JNEUROSCI.1054-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. De Luca A., De Falco M., Severino A., Campioni M., Santini D., Baldi F., Paggi M. G., and Baldi A. (2003) Distribution of the serine protease HtrA1 in normal human tissues. J. Histochem. Cytochem. 51, 1279–1284 10.1177/002215540305101004 [DOI] [PubMed] [Google Scholar]
- 75. De Luca A., De Falco M., De Luca L., Penta R., Shridhar V., Baldi F., Campioni M., Paggi M. G., and Baldi A. (2004) Pattern of expression of HtrA1 during mouse development. J. Histochem. Cytochem. 52, 1609–1617 10.1369/jhc.4A6330.2004 [DOI] [PubMed] [Google Scholar]
- 76. Grau S., Baldi A., Bussani R., Tian X., Stefanescu R., Przybylski M., Richards P., Jones S. A., Shridhar V., Clausen T., and Ehrmann M. (2005) Implications of the serine protease HtrA1 in amyloid precursor protein processing. Proc. Natl. Acad. Sci. U.S.A. 102, 6021–6026 10.1073/pnas.0501823102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tennstaedt A., Pöpsel S., Truebestein L., Hauske P., Brockmann A., Schmidt N., Irle I., Sacca B., Niemeyer C. M., Brandt R., Ksiezak-Reding H., Tirniceriu A. L., Egensperger R., Baldi A., Dehmelt L., et al. (2012) Human high temperature requirement serine protease A1 (HTRA1) degrades Tau protein aggregates. J. Biol. Chem. 287, 20931–20941 10.1074/jbc.M111.316232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Poepsel S., Sprengel A., Sacca B., Kaschani F., Kaiser M., Gatsogiannis C., Raunser S., Clausen T., and Ehrmann M. (2015) Determinants of amyloid fibril degradation by the PDZ protease HTRA1. Nat. Chem. Biol. 11, 862–869 10.1038/nchembio.1931 [DOI] [PubMed] [Google Scholar]
- 79. Runyon S. T., Zhang Y., Appleton B. A., Sazinsky S. L., Wu P., Pan B., Wiesmann C., Skelton N. J., and Sidhu S. S. (2007) Structural and functional analysis of the PDZ domains of human HtrA1 and HtrA3. Protein Sci. 16, 2454–2471 10.1110/ps.073049407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Truebestein L., Tennstaedt A., Mönig T., Krojer T., Canellas F., Kaiser M., Clausen T., and Ehrmann M. (2011) Substrate-induced remodeling of the active site regulates human HTRA1 activity. Nat. Struct. Mol. Biol. 18, 386–388 10.1038/nsmb.2013 [DOI] [PubMed] [Google Scholar]
- 81. Morrow J. A., Hatters D. M., Lu B., Hochtl P., Oberg K. A., Rupp B., and Weisgraber K. H. (2002) Apolipoprotein E4 forms a molten globule: a potential basis for its association with disease. J. Biol. Chem. 277, 50380–50385 10.1074/jbc.M204898200 [DOI] [PubMed] [Google Scholar]
- 82. Caillet-Boudin M. L., Dupont-Wallois L., Soulié C., and Delacourte A. (1998) Apolipoprotein E and Tau phosphorylation in human neuroblastoma cells. Neurosci. Lett. 250, 83–86 10.1016/S0304-3940(98)00448-0 [DOI] [PubMed] [Google Scholar]
- 83. Dupont-Wallois L., Soulié C., Sergeant N., Wavrant-de Wrieze N., Chartier-Harlin M. C., Delacourte A., and Caillet-Boudin M. L. (1997) ApoE synthesis in human neuroblastoma cells. Neurobiol. Dis. 4, 356–364 10.1006/nbdi.1997.0155 [DOI] [PubMed] [Google Scholar]
- 84. Li H., Kim W. S., Guillemin G. J., Hill A. F., Evin G., and Garner B. (2010) Modulation of amyloid precursor protein processing by synthetic ceramide analogues. Biochim. Biophys. Acta 1801, 887–895 10.1016/j.bbalip.2010.05.012 [DOI] [PubMed] [Google Scholar]
- 85. Li H., Evin G., Hill A. F., Hung Y. H., Bush A. I., and Garner B. (2012) Dissociation of ERK signalling inhibition from the anti-amyloidogenic action of synthetic ceramide analogues. Clin. Sci. 122, 409–419 10.1042/CS20110257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ruijter J. M., Ramakers C., Hoogaars W. M., Karlen Y., Bakker O., van den Hoff M. J., and Moorman A. F. (2009) Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 37, e45 10.1093/nar/gkp045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Schmittgen T. D., and Livak K. J. (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.