

Abstract
Cytochrome P450 3A4 (CYP3A4) is the dominant xenobiotic-metabolizing enzyme in the liver and intestine and is involved in the disposition of more than 50% of drugs. Because of its ability to bind multiple substrates, its reaction kinetics are complex, and its association with the microsomal membrane confounds our understanding of how this enzyme recognizes and recruits diverse substrates. Testosterone (TST) hydroxylation is the prototypical CYP3A4 reaction, displaying positive homotropic cooperativity with three binding sites. Here, exploiting the capability of accelerated molecular dynamics (aMD) to sample events in the millisecond regime, I performed >25-μs aMD simulations in the presence of three TST molecules. These simulations identified high-occupancy surface-binding sites as well as a pathway for TST ingress into the CYP3A4 active site originating in the membrane. Adaptive biasing force analysis of the latter pathway revealed a metastable intermediate that could constitute a third binding site at high TST concentrations. Prompted by the observation that interactions between TST and the G′-helix mobilize the ligand into the active site, a free-energy analysis of TST distribution in the membrane was conducted and revealed that the depth of the G′-helix is optimal for extracting TST. In summary, these simulations confirm separate, but adjacent substrate-binding sites within the enzyme and the existence of an auxiliary TST-binding site. The broader impact of these simulations is that they support a mechanism in which cytochromes P450 directly recruit membrane-solubilized substrates.
Keywords: cytochrome P450, drug metabolism, molecular dynamics, computational biology, substrate specificity, accelerated molecular dynamics, adaptive biasing force, CYP3A4, substrate recognition, testosterone hydroxylation
Introduction
The cytochromes P450 metabolize nearly all drugs and environmental xenobiotics to which humans are exposed. Of the 57 cytochromes P450 encoded by the human genome, cytochrome P450 3A4 (CYP3A4)2 is involved in the clearance of more than half of approved drugs and presents significant challenge to optimizing pharmacokinetics in drug development and avoiding adverse effects in the clinic (1, 2). CYP3A4 enzyme kinetics can demonstrate marked cooperativity (3). Positive cooperativity is characterized by a continuous, positive increase in the rate of catalysis with increasing substrate concentration, whereas positive heterotropic cooperativity occurs when the rate of substrate turnover is enhanced by a second distinct effector molecule. Cooperativity in CYP catalysis was first recognized in microsome preparations (4–6) and later confirmed using purified CYP3A4 with several substrates including testosterone (TST), 17β-estradiol, amitriptyline, and aflatoxin B1 (7). A shift in the regioselectivity of midazolam (MDZ) oxidation in patients by concurrent fluconazole administration supports that heterotropic CYP3A4 interactions are also relevant in vivo (8). Cooperativity in CYP3A4 catalysis has been explained by mechanisms invoking simultaneous occupation of the active site by multiple ligands or binding to nearby allosteric sites (9–15).
The membrane environment can have a profound effect on membrane protein function. With the exception of the N-terminal transmembrane helix, only the F′- and G′-helices of CYP3A4 are purported to interact with the membrane. Notwithstanding an only superficial interaction with the catalytic domain, the membrane imparts dramatic changes in the physical properties of the protein. Incorporation of CYP3A4 into lipid nanodiscs induces a clear increase in its thermal stability. Furthermore, the structural and dynamic changes imparted by the membrane likewise influence ligand binding affinities. Dissociation constants of some ligands decrease by an order of magnitude, and the kinetics of ligand binding are tunable by changing the lipid composition (5–7). In view of the evidence that membrane interactions modulate CYP3A4 function, it is important that they be included in models, experimental or theoretical, aimed at understanding the functional dynamics of this important enzyme.
The amphipathic nature of CYP3A4 substrates has led to speculation that the enzyme harvests them from the membrane; however, neither experimental nor theoretical evidence in direct support of this mechanism are available. Understanding how substrates are recognized by CYP3A4 and the locations of allosteric sites are required to improve predictive tools of CYP3A4 metabolism that would be invaluable to optimizing drug therapy. The ideal computational approach to help fill this knowledge gap would permit simulation of the necessary time scales for a ligand to sufficiently explore the protein surface to identify optimal binding site(s). Accelerated molecular dynamics (aMD) is particularly advantageous for this purpose because, in combination with the recent advances in graphical processing unit–enabled hardware, it can provide access to ligand-binding events that occur on the millisecond time scale without requisite knowledge of the binding pathway (16). Hundreds-of-nanosecond aMD simulations have captured the activation of the M2 and M3 muscarinic G-protein–coupled receptors (17–19) and ligand binding to the M3 receptor (20). In this work, aMD is extended to CYP3A4 to simulate TST binding to the enzyme free of a predefined reaction coordinate. aMD successfully identifies probable locations of auxiliary binding sites that reflect experimental observations and reveals a membrane-embedded pathway for TST entrance into the active site. Free-energy analyses of the ingress pathway provides a structural basis for the kinetic behavior of CYP3A4 mutants and insight into the mechanism for exclusion of progesterone from the active site in CYP3A4 crystal structures.
Results and discussion
Conventional molecular dynamics
The initial configuration for the CYP3A4-membrane system was generated by inserting the N-terminal transmembrane domain into a 75-Å2 POPC bilayer such that the G′-helix was oriented just above the POPC headgroups, or 8 Å above the average position of the phosphate (PO4) units. In the 50-ns simulation that followed, the A-anchor (residues 44–47), F′-helix (residues 217–226), and G′-helix (residues 227–238) associated with the membrane headgroups in 10 ns. A similar orientation of the catalytic domain and time for insertion were observed by Baylon et al. (22) using only the soluble catalytic domain of CYP3A4 and the highly mobile membrane-mimetic (HMMM) representation that replaces native lipids at the aqueous interface with those having truncated tails and the interior with 1,1-dichloroethane (DCE) to mimic the bulk hydrocarbon environment.
Following the initial 50 ns of conventional molecular dynamics (cMD) simulations, three TST molecules were randomly added to the water surrounding the CYP3A4 catalytic domain, and the system was equilibrated for an additional 100 ns in the constant temperature and pressure (NPT) ensemble. Two of the TST molecules were observed to partition into the POPC headgroups. Consistent with the observations of Baylon et al. (22), excluding the N-terminal helix, the A-anchor was observed to insert deeper into the membrane followed by the G′- and F′-helices. The end point of this simulation, the structure of POPC highlighting the relevant fragments, and the positions of the relevant domains are illustrated in Fig. 1. The average positions of the acylglycerol (AG) and PO4 units were 13.8 ± 0.7 and 17.4 ± 0.2 Å above the membrane center, respectively (Fig. 1C). The F′-helix (z = 15.9 Å) sits slightly higher in the membrane than the G′-helix (z = 14.2 Å) and therefore, on average, is bounded below by the AG and above by the PO4 units. The A-anchor (z = 12.9 Å) persists below the AG centers of mass and into the upper boundary of the hydrocarbon tails. Although the relative insertion depths of these domains are consistent with previous simulations, the absolute depths are greater than those reported previously (22). For example, in the simulations reported by Baylon et al. (22), the F′-helix was not observed to penetrate beyond the choline fragment of the headgroups. In the simulation described herein, insertion depths of these domains are converged prior to the addition of TST molecules to the system, so the observed differences are not attributable to the extended simulation time used in the present study. Conversely, the variations in insertions are likely attributable to differences in the properties of the HMMM and a full membrane. First, the DCE used to mimic the hydrophobic tails in the HMMM extends well into the AG layer of the headgroups (21). The result is a membrane-mimetic representation with greater density in this region than in a full membrane representation, thereby posing a density barrier preventing deeper penetration of the protein helices. In addition, the interspersion of DCE also decreases the polarity at this depth relative to the full membrane, imposing an energetic penalty on further penetration of the amphipathic helices. Nevertheless, experience with the HMMM is still limited, and caution should be exerted when interpreting simulations where there is limited experimental data describing the extent of the protein-membrane interaction.
Figure 1.
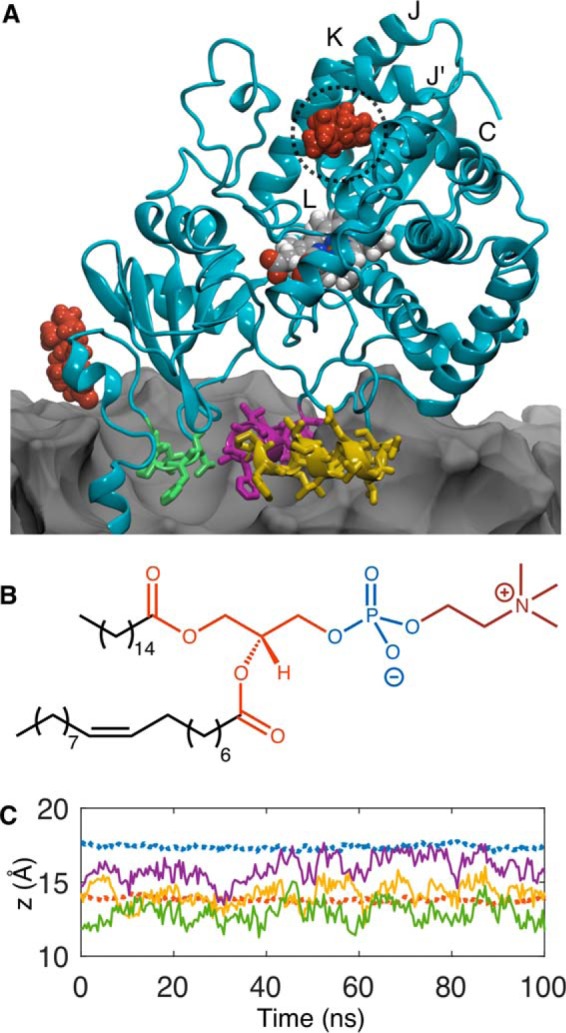
A, end point of the 100-ns cMD simulation following the addition of TST. The A-anchor, F′-helix, and G′-helix are illustrated in green, purple, and yellow, respectively. TST molecules are red, and the POPC membrane, which is partially cut away to reveal the immersed domains, is represented as a gray contour surface. The putative auxiliary TST-binding site is highlighted with a dashed circle. B, structure of POPC illustrating the hydrophobic tails (black), AG (orange), PO4 (blue), and choline (red). C, average distances above the membrane center of lipid components and protein domains in the leaflet bearing the CYP3A4 catalytic domain. The AG and PO4 units are represented by orange and blue dashed lines. Positions of the A-anchor, F′-helix, and, G′-helix are represented with green, purple, and yellow solid lines, respectively.
Accelerated molecular dynamics
Following cMD in the presence of TST, 50 0.5-μs aMD simulations were seeded to extensively sample TST interactions with CYP3A4. Typical of all replicas, the TST distribution from one copy is illustrated in Fig. 2A. It is clear that TST molecules sample the surface of the CYP3A4 catalytic domain as well as partition into and persist in the POPC bilayer. In two replicas, TST entered the active site from the membrane face of the catalytic domain. Snapshots of the TST positions in the window surrounding these binding events are illustrated in Fig. 2B. In both instances, TST initially interacts with the G′-helix and is subsequently captured between the groove formed by the F′-/G′-helices and the B-C loop. To illustrate the timescales of TST binding in each replica, the distances between each TST and the heme iron are plotted over the length of the simulations (Fig. 2, C and D). TST is ensconced in the active site by 150 and 50 ns in the first and second replicas, respectively, with an average iron-TST distance of 5–8 Å (Fig. 2, C and D, blue traces). The length of the simulation in the first replica was extended to 1 μs wherein the TST was observed to remain bound in the active site adjacent to the heme.
Figure 2.

A, distribution of TST molecules (green, blue, and orange) from one 0.5-μs aMD replica. Coordinate positions were recorded every 400 ps. B, snapshots of the two TST molecule positions (pink and orange lines) in the window surrounding their binding events. The POPC membrane is represented as a gray contour surface. C and D, distances between the TST centers of mass and the heme iron versus time for the two replicas where TST was observed to enter the CYP3A4 active site through the membrane. Solid lines represent a 30-ns moving average of the distances. E, volumetric map (red) illustrating the highest TST mass-weighted density over the 25 μs of aMD simulation. Representative TST positions from trajectory snapshots where it was observed to occupy these sites are illustrated in green. Both the TST positions and volumetric map are superimposed onto initial CYP3A4 geometry used in the aMD simulations. F and G, magnified views of the volumetric map highlighting the auxiliary TST-binding sites. H, representative cvSMD pathway for TST egress from CYP3A4.
In the cMD simulations, one TST was found in a crevice bordered by the D-, J′-, K-, and L-helices adjacent to the proximal face of the heme (Fig. 2, C and D). TST persisted in this site for 645 ns in the first replica where concomitant active site binding was observed with the exception that from 225 to 275 ns the TST molecule dissociated from the site, diffused more than 40 Å away, and returned to its original position. Beyond 645 ns, TST moved into a second stable binding site 5 Å from its initial position and remained there until the end of the trajectory. In the second replica, TST remained in the same auxiliary site occupied in the first replica through the entire trajectory. In both, the third TST molecule remained more than 20 Å from the heme iron.
Inspection of the remaining replicas found 28 (56%) where TST was bound in the crevices bordered by the D-, J′-, K-, and L-helices similar to that illustrated in Fig. 1A. TST binding to this site was reversible in many of the aMD replicas, so the frequent occupation of this site was not an artifact of the cMD end point. To obtain a cumulative view of their occupancy over all replicas, the trajectories were superimposed using the Cα atoms, and an atomic weighted density map for TST was generated using the VolMap tool in VMD (23). The density isocontour surface reveals the greatest TST density at two neighboring peripheral sites, bordered by the D-, J′-, K-, and L-helices near the heme but not in the active site (Fig. 2E).
Like the high-occupancy site observed in the simulations, there is experimental evidence for a high-affinity TST-binding site that bears the features of being outside the active site (24). Using the CYP3A4 embedded in POPC nanodiscs, occupation of the high-affinity site induces only a minor shift in the iron spin state (22%), and under catalytic conditions NADPH oxidation is increased severalfold without coupled product formation. Both of these observations are consistent with a TST-binding site that is too far from the heme iron to efficiently displace the iron-coordinated water or undergo hydrogen-atom abstraction by the compound I oxoferryl species.
In both of the auxiliary sites, TST is predominately surrounded by hydrophobic residues (Fig. 2, F and G). Among these, Arg446 lies at the interface of both sites where its aliphatic side chain contacts TST. Using cross-linking mass spectrometry and mutagenesis, Zhao et al. (25) demonstrated that Arg446 contributes to the binding surface and mediates the electrostatic interaction between CYP3A4 and cytochrome b5. Like occupation of the high-affinity site by TST, the superficial interaction with cytochrome b5 can likewise induce a partial spin transition of the heme iron (26). Loss of catalytic activity in the R446A mutant is not surprising because elimination of the cationic binding site would undoubtedly be detrimental to productive interactions with redox partners. However, this mutant is still capable of binding TST albeit with reduced affinity and cooperativity.
Adaptive biasing force simulations of TST binding
Although aMD has pushed the simulation of events in biomolecules into the millisecond regime, reweighting the trajectories to recover free energies from the original ensemble has proven more difficult because the trajectories are dominated by large values of the boost potential, resulting in noisy energy statistics (27). To assign free energies to states along the pathway for TST ingress, the pathway observed in the aMD simulation was reconstructed using constant velocity–steered molecular dynamics (cvSMD) (28, 29). The cvSMD pathway is illustrated in Fig. 2H. cvSMD provides a finely resolved ensemble of structures along the ingress coordinate (ξ) from which a set of equally spaced representatives were selected for an adaptive biasing force (ABF) calculation (30) of the potential of mean force (PMF). Spanning 22 Å, ξ was partitioned into 2-Å bins that extended from TST burial in the CYP3A4 active site to immersion in the POPC headgroups. Each bin was populated with a trajectory snapshot from the cvSMD simulation corresponding to a value of ξ within that bin. Sampling in each bin was initially performed for 100 ns and in 10-ns increments thereafter. The complete PMF (ΔG(ξ)) converged after 150 ns with an overall free-energy change (ΔGU → B) of −12.9 kcal·mol−1. The 100-ns PMF, those from the subsequent 10-ns increments, and the final converged PMF are illustrated in Fig. 3A. Although the PMF is rugged as expected for ligands binding to proteins, it can be divided into three general regions: 1) TST immersed in the POPC headgroups including initial contact with the G′-helix, 2) a metastable intermediate, and 3) TST bound in the active site.
Figure 3.

A, PMF along the reaction coordinate ξ for TST ingress into CYP3A4. The landscape is characterized by three general regions corresponding to TST in the lipid headgroups and making contact with the G′-helix, a binding intermediate, and TST bound in the active site. Dashed lines denote stages of convergence for the PMF, starting from 100 (dotted lines) to 150 ns (solid black line) in 10-ns intervals. B, representative snapshot depicting initial contact of TST with the G′-helix at ξ = 18 Å. C, representative snapshot depicting the binding intermediate at ξ = 10 Å. D, representative snapshot depicting TST bound in the active site at ξ = 0.5 Å.
From total immersion in the POPC headgroups at ξ ≃ 20–22 Å, the initial minimum occurring at ξ ≃ 18 Å corresponds to the first interaction of TST with the G′-helix. In a representative snapshot from ξ ≃ 18 Å (Fig. 3B), the lower face of TST is bounded by the G′-helix, and the upper face is bounded by the AG fragments of POPC. This initial interaction suggests a mechanism in which the G′-helix serves as the initial contact as well as a “ramp” to load TST directly from the membrane.
From the initial interaction at ξ ≃18 Å, TST subsequently encounters small energy barriers (<1 kcal·mol−1) corresponding to movement over the G′-helix before encountering the intermediate configuration at ξ ≃ 10 Å and 4.9 kcal·mol−1 (ΔGU → I) below the unbound state. At this point in the PMF, TST is flanked by the F′- and G′-helices and the B-C loop (Fig. 3C). The origin of the minimum on the PMF is apparently derived from stabilization through close packing of “Phe cluster” residues (Phe108, Phe213, Phe220, Phe241, and Phe304) as well as Ile120, Ile223, and Val240 around TST. The intermediate TST-binding site is reminiscent of that for progesterone (PRG) in crystal structures with CYP3A4; however, there are important differences (31, 32). In Fig. S1, an overlay of the intermediate structure with the CYP3A4-PRG crystal structure shows that PRG sits closer to the F′-helix and is packed against Phe213, Phe219, and Phe220. PRG is precluded from entering the active site by the interdigitation of the Phe108, Phe213, Phe215, Phe241, and Phe304 side chains. By contrast, TST leverages the aperture open in the intermediate, disrupting the Phe cluster with concomitant movements of the B-C, F-F′, and G-G′ loops, thereby resulting in deeper penetration into the entrance channel.
The PRG site has been speculated to be both an allosteric site and/or a result of PRG capture in the entrance channel by crystallography. These and previous MD simulations have established that interactions with the membrane induce remarkable conformational changes in the F′- an G′-helices compared with those observed crystallographically (22, 33, 34). The interaction with the membrane unwinds the N terminus of the G-helix (residues 243–245) and extends the G-G′ loop, causing Phe241 to disengage from the hydrophobic cluster. In light of these conformational changes, it is not surprising that the positions of TST in the intermediate state and PRG in the crystal structure are not superimposable. In the CYP3A4-PRG structures, the G′-helix packs against the same helix of a second CYP3A4 monomer in the unit cell, and the pair are related by an S2 axis perpendicular to the G′-helical axis (Fig. S2). Sevrioukova and Poulos (32) recognized that the interface created by this interaction creates a hydrophobic patch to accommodate PRG. This interaction apparently stabilizes the closed configuration of the Phe cluster, precluding passage of this ligand into the active site. Assuming PRG enters the enzyme using the same mechanism as TST, it is unlikely that the PRG site represents a native transport intermediate but rather a non-native state resulting from distortion of the native potential-energy surface by the crystal lattice.
From the intermediate state, TST surmounts a 2.2 kcal·mol−1 energetic barrier (ΔGI → B) to enter the active site. Conversely, passage of TST back into the intermediate state requires overcoming a 10.6 kcal·mol−1 barrier, and it is thereby kinetically trapped in the active site. The TST α-face is packed against the heme, whereas the side chains of Phe304, Phe213, and Phe215 contact the β-face (Fig. 3D). Orientation of the α-face of TST toward the heme is inconsistent with the dominant 6β-hydroxylated product (35). Nevertheless, this configuration would not afford adequate volume to accommodate O2; hence, TST reorientation would also be required for productive catalysis. Because the TST C6 lies ∼3.7 Å from the heme iron (Fig. 4A), only a minor reorganization of the TST would be required to accommodate O2.
Figure 4.
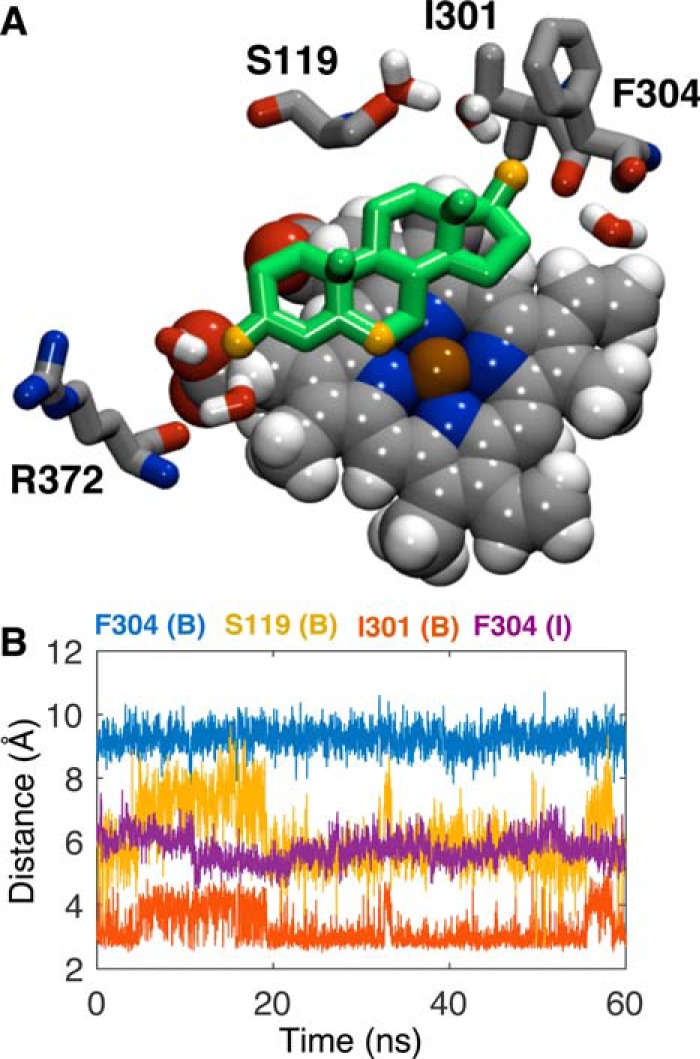
A, representative snapshot depicting TST bound in the active site at ξ = 0. 5 Å illustrating the positions of Ser119, Ile301, Phe304, and Arg372. The 3-keto oxygen atom, 17β-hydroxy oxygen atom, and C6 atom of TST (green) are illustrated in orange. B, distances between the centers of mass of Phe304 phenyl carbon atoms and TST in the bound state (B; blue) and intermediate state (I; purple), Ile301 carbonyl oxygen and 17β-hydroxy oxygen atoms (orange), and Ser119 carbonyl oxygen and 17β-hydroxy oxygen atoms (yellow). Illustrated distances reflect the final 60 ns of simulation in the respective ξ = 0–2 Å and ξ = 8–10 Å bins from the ABF calculations.
The CYP3A4 active site conforms to TST with Phe213 and Phe215 in the F-F′ loop and Phe304 in the I-helix, restraining TST close to the heme. Leu482, Ala305, Arg105, and Ser119 surround the TST periphery, limiting translation (Fig. 3D). In the recent structure obtained with MDZ, the ligand contacted both Leu482 and Ser119 where the latter residue apparently contributes a functionally important hydrogen bond to MDZ (36). The relative positions of TST, Ser119, Ile301, Phe304, and Arg372 are illustrated in Fig. 4A, and the distances between those that directly contact TST and the TST center of mass are plotted in Fig. 4B. In contrast to the CYP3A4 structure with MDZ, hydrogen bonds involving Ser119 were not observed; rather the backbone carbonyl of Ile301 is hydrogen-bonded to the 17β-hydroxyl of TST. At the opposing end, the 3-keto oxygen is hydrogen-bonded to water molecules that bridge the heme propionate to the backbone carbonyl of Arg372.
Numerous residues have been targeted for mutagenesis to identify regions of the protein involved in substrate specificity and kinetic cooperativity (37–40). Mutations have successfully restored hyperbolic reaction kinetics as well as mirrored the stimulation induced by heterotropic effectors. One residue, Phe304, has demonstrated importance in substrate specificity and catalytic cooperativity. The Vmax is increased for PRG hydroxylation, and the Hill coefficient is slightly reduced in the F304A mutant, consistent with a decrease in cooperativity (38). Hyperbolic 6β-hydroxylation kinetics are restored in the F304W mutant, and stimulation by α-naphthoflavone is diminished (41). This evidence supports that Phe304 sits near the interface of the active site as well as an effector-binding site. Indeed, Phe304 does directly interact with TST in both the intermediate and fully bound states (Fig. 3, C and D). In the intermediate state, the mean distance between TST and the Phe304 phenyl is 5.8 ± 0.4 Å and is therefore closely packed against the steroid nucleus. In the bound state, at 9.8 ± 0.4 Å, Phe304 is more distant, although it is positioned to prevent TST from reverting back into the intermediate state (Fig. 4B). To the extent that the active and intermediate sites could be simultaneously occupied at high concentrations, the intermediate site could constitute the third TST-binding site and is consistent with neighboring active and allosteric sites.
ABF simulations of TST in a POPC bilayer
It is clear from examining the distribution in individual aMD trajectories that TST predominately persists among the POPC headgroups (Fig. 2A). Similar results were obtained in umbrella-sampling studies of cortisone in a POPC bilayer where the minimum was also within the AGs (42). Considering that the F′- and G′-helices are likewise inserted at a depth dominated by the AG units and ABF calculations support a mechanism where TST is harvested from this region by the G′-helix, it was hypothesized that the insertion of the F′-G′ region was optimized by evolution to harvest amphipathic substrates from the membrane. This hypothesis was tested using a pair of unrestrained simulations of TST in a POPC bilayer and by calculation of the PMF for TST to traverse the bilayer. First, in separate, symmetrically hydrated (±10 Å) 50-Å2 POPC bilayers, TST was placed at the center of the membrane (among the lipid tails) and at 22 Å above the membrane center (surrounded by water). Following equilibration of the system with TST fixed in these two positions, the TST restraints were released, and the simulations were conducted for 50 ns. The composite-normalized, mass-weighted densities for TST and the membrane headgroup components over the simulations are illustrated in Fig. 5A. Despite migrating to opposing leaflets, from these analyses it is evident that TST almost exclusively persists among the AG units of the POPC bilayer.
Figure 5.
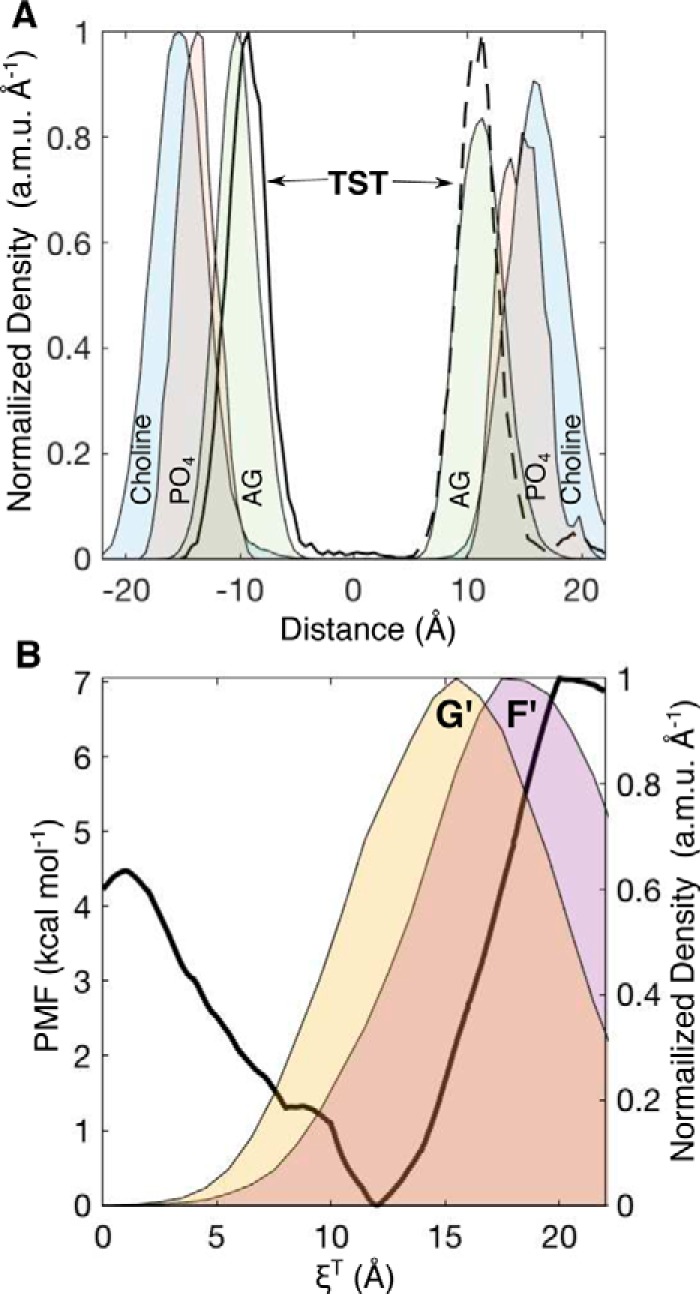
A, normalized, one-dimensional, mass-weighted densities of the choline, PO4, AGs, and TST from two 50-ns simulations where TST was initially positioned at the membrane center (solid line) and in water 22 Å above the membrane center (dashed black line). B, PMF along the reaction coordinate (ξT) for TST entrance into a pristine phospholipid bilayer (left axis). The normalized, one-dimensional, mass-weighted densities (right axis) of the F′- and G′-helices are superimposed as yellow and purple area plots, respectively. a.m.u., atomic mass units.
To determine whether the AG units represented the energy minimum along the one-dimensional reaction coordinate for TST crossing the membrane (ξT), a series of constrained simulations were performed where TST was moved incrementally along the membrane normal between the center and the solvated position to establish initial configurations for an ABF calculation. The resulting PMF for TST crossing the POPC bilayer is illustrated in Fig. 4B. The minimum of the PMF lies at ξT = 12.0 Å above the membrane center, 6.9 kcal·mol−1 below TST in water (ξT = 20–22 Å). The lipid tails present a 4.5 kcal·mol−1 barrier for TST to cross the bilayer. The PMF confirms the result of the unconstrained simulation that dissolution among the AG groups corresponds to the minimum in the PMF. To compare the basin in the PMF with the positions of the F′- and G′-helices, distributions of insertion depths from the 1-μs aMD replica were superimposed in Fig. 5B. The mass density distribution of the G′-helix distance could be fit to a Gaussian centered at 12.8 Å, which nearly matches the position of the PMF basin. Thus, the G′-helix occupies a nearly optimal position for contacting membrane-embedded TST. Because the distributions were derived from long, enhanced-sampling MD simulations, it is not surprising that the distributions of these domains were broad (full width at half-maximum = 11.6 Å), demonstrating that the G′-helix occasionally dips into the upper vicinity of the lipid tails as well as into the choline layer. This behavior would allow CYP3A4 to have great flexibility in the polarity of substrates it could recruit from the membrane. This characteristic would also permit the enzyme to function in membranes with somewhat different and more complex environments akin to those found in vivo compared with the relatively simple model POPC membrane used in these studies. Indeed, the distance separating the preferred dissolution layer for a particular substrate from the optimum position of the G′-helix position in a given membrane environment may influence the kinetics of substrate oxidation by CYP3A4.
Conclusion
The results presented herein represent the first simulation of ligand binding to a cytochrome P450 from solution. The positive homotropic cooperativity in the oxidation of TST by CYP3A4 is well-documented and has been attributed to three TST-binding sites (13, 24, 43). The TST-CYP3A4 pair is an ideal system to capture the structural details of these aspects of cytochrome P450 activity by simulation. aMD frequently and reproducibly identified binding sites in close proximity to the heme that are consistent with the partial heme spin shift and NADPH oxidation without coupled product formation previously observed at low TST concentrations (24). Albeit with lower frequency, aMD also captured the entry of TST into the active site through an entirely membrane-embedded pathway. Free-energy analysis of this path proved it to be a two-step mechanism with an intermediate state where TST rests in the access channel stabilized by residues of the Phe cluster. Taken together with existing site-directed mutagenesis data for this region, it is probable that the active and intermediate sites could be simultaneously occupied at high TST concentrations. Finally, these studies demonstrated that the G′-helix is near optimally positioned within the model membrane to contact TST, although its distribution spans the complete headgroup space as well as into the hydrocarbon tails, affording access to more or less polar substrates. The broader impact of these simulations is that they support a viable mechanism whereby CYP3A4 can recruit substrates directly from the membrane. However, given the vast number of CYP3A4 substrates and their broad range of physicochemical properties, it would be irresponsible to claim that all CYP3A4 substrates use the same mechanism to enter the active site. Nonetheless, it is possible to use the approach described herein to predict how other substrates gain access to the CYP3A4 active site.
Computational methods
System preparation and conventional MD
An N-terminally modified version of the 2.05-Å resolution CYP3A4 structure (Protein Data Bank code 1TQN) was used as the starting point for all simulations (44). Because the extent of the fluctuations observed in the simulations far exceed the structural deviations observed in the available crystal structures, the conclusions are expected to be independent of the crystal structure used to construct the initial model (see Fig. S3). Following removal of crystallographic water, an α-helix with the sequence 1MA-13LLLAVFLVLLYLYGT27 was appended to the N terminus of the protein. This N terminus lacks residues 3–12 of the full-length protein and bears an S18F mutation (underlined). The overwhelming majority of biophysical studies of CYP3A4-ligand interactions in model membranes have been performed with this version of the protein so it was adopted here to avoid confounding comparisons between simulation predictions and past experimental data (24, 45–51). Protonation states of amino acids were assigned using Poisson–Boltzmann-based PROPKA calculations at pH 7.4 (52). Curated CYP3A4 was then inserted into a 75-Å2 POPC membrane constructed using the Membrane Builder plugin of VMD (23). POPC and water molecules overlapping and within 0.8 Å of the protein were removed. The system was solvated with a 10-Å layer of TIP3P water in the ±z directions. Potassium and chloride ions were added to an ionic strength of 0.15 m, thereby ensuring electrical neutrality.
All molecular dynamics procedures were performed with the NAMD 2.12 code (53) using the CHARMM36 (54) force field for POPC and the CHARMM27 (55) force field for the remainder of the system. The complete system was minimized for 10,000 steps followed by equilibration of the lipid tails for 500 ps with the coordinates of the remaining atoms in the system frozen. Melting of the lipid tails around the protein was followed by 2 ns at NPT simulation with a harmonic restraint (5 kcal·mol−1·Å−2) applied to the protein Cα atoms using the Langevin piston method with a target pressure of 1.01325 bars, decay period of 100 fs, and piston temperature of 310 K. After completion of the preparative simulations, the complete CYP3A4 system was equilibrated in the NPT ensemble for an additional 50 ns. This end point served as the initial configuration for the addition of three TST molecules placed randomly in the water on the membrane face bearing the CYP3A4 catalytic domain. Parameters for TST were from the CGenFF force field (56, 57). Following the addition of TST, the system was minimized for 10,000 steps and equilibrated for an additional 100 ns in the NPT ensemble.
Accelerated MD
aMD is a convenient approach to sample rare conformational transitions in proteins by increasing the escape rates from potential energy basins through reduction of energetic barriers (27). In an aMD simulation, when the system potential energy, V(r), is greater than a user-defined value E, the simulation proceeds as in a conventional simulation. Conversely, when V(r) falls below E, a continuous, positive “boost” energy is added such that the simulation is performed on a modified potential energy surface, V*(r).
| (1) |
The value of the boost energy depends on a tuning parameter, α, that determines the extent that energy basins on the modified potential energy surface, V*(r), are elevated, effectively smoothing the potential energy surface.
Fifty replicas based on the end point of the conventional simulation containing TST were created, and their initial velocities were independently randomized at 310 K. aMD simulations were performed in “dual-boost” mode meaning that boost potentials were added to both the dihedral and total system potential energies with Edihed = Vdihed_avg + 0.3Vdihed_avg, αdihed = 0.3Vdihed_avg/5, Etotal = Vtotal_avg + 0.2Natoms, and αtotal = 0.2Natoms. aMD simulations were performed for 0.5 μs in each replica. The simulation was extended to 1 μs in one of the replicas where TST entered the active site.
Steered molecular dynamics
Following identification of the binding pathway in aMD simulations, cvSMD simulations were initiated from the first aMD trajectory snapshot that revealed occupation of the CYP3A4 active site by TST. The binding pathway was approximated by a linear path defined by the TST center of mass and a second point on the cvSMD trajectory where TST was immersed in the lipid headgroups adjacent to the G′-helix. The vector defined by these points served as the pulling vector. A moving harmonic restraint was attached to the TST center of mass and pulled with a velocity of 1.25 × 10−5 Å·fs−1 along the pulling vector. Cα atoms of residues 19, 39, 167, 194, 285, 341, and 417, all distant from the entrance channel, were harmonically restrained (600 kcal·mol−1·Å−2) to avoid translation of the CYP3A4 system. The force on the harmonic constraint was recorded every 400 fs. Steered molecular dynamics simulations were performed for a total of 4 ns and repeated 10 times to generate an ensemble for PMF calculations.
Adaptive biasing force simulations
The constant velocity pulling vector from cvSMD was adopted as the reaction coordinate for the ABF simulations (30). The reaction coordinate was divided into 2-Å bins, and trajectory snapshots with ligand positions as close as possible to the beginning of the bins were selected as initial positions. These frames were minimized for 2000 steps with the TST coordinates frozen. Those Cα atoms restrained in the cvSMD simulations were maintained in the ABF calculations. The ABF data were accrued in bins 0.1 Å wide with upper and lower wall constants of 10 kcal·mol−1·Å−2. Simulations were performed until the overall PMF was observed to converge, defined as a mean PMF difference of less than 0.3 kcal·mol−1. Because the biasing force rapidly varies, no bias was applied during the first 1200 fs steps of ABF simulation to avoid perturbation by non-equilibrium effects.
ABF simulations of TST in a POPC bilayer
A 50-Å2 POPC membrane was constructed using the Membrane Builder plugin of VMD (23) and symmetrically hydrated with an additional 10-Å layer of TIP3P water in the ±z directions. The hydrated membrane was minimized for 5000 steps and equilibrated in the NVT ensemble for 0.5 ns and in the NPT ensemble for 5 ns. In separate copies of the equilibrated membrane, TST was placed at the center (0 Å; in the POPC tails) and in 2-Å increments to 22 Å above the center (in water). With TST's position frozen, the systems were minimized for 10,000 steps and equilibrated for 4 ns in the NVT ensemble. Each constrained simulation served as an initial configuration for an ABF calculation of the PMF for TST entry into the membrane with the bilayer normal as reaction coordinate. The 22-Å reaction coordinate was partitioned into 2-Å bins, and each was populated with the end point geometry from each of the corresponding constrained simulations. Within each of the major bins, ABF data were accrued in bins 0.1 Å wide with upper and lower wall constants of 10 kcal·mol−1·Å−1. The ABF simulations were performed for 50 ns. The equilibrated systems having TST frozen at 0 and 22 Å were used as the initial configurations for two 50-ns NPT simulations where TST was unrestrained. Atomic density profiles were calculated using the Density Profile Tool for VMD (58).
Author contributions
J. C. H. data curation; J. C. H. formal analysis; J. C. H. funding acquisition; J. C. H. investigation; J. C. H. visualization; J. C. H. writing-original draft; J. C. H. writing-review and editing.
Supplementary Material
Acknowledgments
This work was supported by a generous allocation of computational resources from the Ohio Supercomputer Center. I thank William M. Atkins (University of Washington), Kakali Sen (Daresbury Laboratory), and Lisa S. Shock (Virginia Commonwealth University) for insightful discussions and critically reading the manuscript.
This work was supported by National Institutes of Health Grant R01GM114168 (to J. C. H.). The author declares that he has no conflicts of interest with the contents of this article. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- CYP3A4
- cytochrome P450 3A4
- ABF
- adaptive biasing force
- AG
- acylglycerol
- PO4
- phosphate
- aMD
- accelerated molecular dynamics
- cMD
- conventional molecular dynamics
- cvSMD
- constant velocity–steered molecular dynamics
- MDZ
- midazolam
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- TST
- testosterone
- PMF
- potential of mean force
- PRG
- progesterone
- HMMM
- highly mobile membrane-mimetic
- DCE
- 1,1-dichloroethane
- NPT
- constant temperature and pressure
- NVT
- constant temperature and volume.
References
- 1. Rendic S. (2002) Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab. Rev. 34, 83–448 10.1081/DMR-120001392 [DOI] [PubMed] [Google Scholar]
- 2. Rendic S., and Guengerich F. P. (2012) Contributions of human enzymes in carcinogen metabolism. Chem. Res. Toxicol. 25, 1316–1383 10.1021/tx300132k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Conner K. P., Woods C. M., and Atkins W. M. (2011) Interactions of cytochrome P450s with their ligands. Arch. Biochem. Biophys. 507, 56–65 10.1016/j.abb.2010.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lasker J. M., Huang M. T., and Conney A. H. (1982) In vivo activation of zoxazolamine metabolism by flavone. Science 216, 1419–1421 10.1126/science.7089530 [DOI] [PubMed] [Google Scholar]
- 5. Lasker J. M., Huang M. T., and Conney A. H. (1984) In vitro and in vivo activation of oxidative drug metabolism by flavonoids. J. Pharmacol. Exp. Ther. 229, 162–170 [PubMed] [Google Scholar]
- 6. Schwab G. E., Raucy J. L., and Johnson E. F. (1988) Modulation of rabbit and human hepatic cytochrome P-450-catalyzed steroid hydroxylations by α-naphthoflavone. Mol. Pharmacol. 33, 493–499 [PubMed] [Google Scholar]
- 7. Ueng Y. F., Kuwabara T., Chun Y. J., and Guengerich F. P. (1997) Cooperativity in oxidations catalyzed by cytochrome P450 3A4. Biochemistry 36, 370–381 10.1021/bi962359z [DOI] [PubMed] [Google Scholar]
- 8. Yang J., Atkins W. M., Isoherranen N., Paine M. F., and Thummel K. E. (2012) Evidence of CYP3A allosterism in vivo: analysis of interaction between fluconazole and midazolam. Clin. Pharmacol. Ther. 91, 442–449 10.1038/clpt.2011.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shou M., Grogan J., Mancewicz J. A., Krausz K. W., Gonzalez F. J., Gelboin H. V., and Korzekwa K. R. (1994) Activation of CYP3A4: evidence for the simultaneous binding of two substrates in a cytochrome P450 active site. Biochemistry 33, 6450–6455 10.1021/bi00187a009 [DOI] [PubMed] [Google Scholar]
- 10. Hosea N. A., Miller G. P., and Guengerich F. P. (2000) Elucidation of distinct ligand binding sites for cytochrome P450 3A4. Biochemistry 39, 5929–5939 10.1021/bi992765t [DOI] [PubMed] [Google Scholar]
- 11. Oda Y., and Kharasch E. D. (2001) Metabolism of methadone and levo-α-acetylmethadol (LAAM) by human intestinal cytochrome P450 3A4 (CYP3A4): potential contribution of intestinal metabolism to presystemic clearance and bioactivation. J. Pharmacol. Exp. Ther. 298, 1021–1032 [PubMed] [Google Scholar]
- 12. Shou M., Dai R., Cui D., Korzekwa K. R., Baillie T. A., and Rushmore T. H. (2001) A kinetic model for the metabolic interaction of two substrates at the active site of cytochrome P450 3A4. J. Biol. Chem. 276, 2256–2262 10.1074/jbc.M008799200 [DOI] [PubMed] [Google Scholar]
- 13. Roberts A. G., and Atkins W. M. (2007) Energetics of heterotropic cooperativity between α-naphthoflavone and testosterone binding to CYP3A4. Arch. Biochem. Biophys. 463, 89–101 10.1016/j.abb.2007.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kapelyukh Y., Paine M. J., Maréchal J. D., Sutcliffe M. J., Wolf C. R., and Roberts G. C. (2008) Multiple substrate binding by cytochrome P450 3A4: estimation of the number of bound substrate molecules. Drug Metab. Dispos. 36, 2136–2144 10.1124/dmd.108.021733 [DOI] [PubMed] [Google Scholar]
- 15. Woods C. M., Fernandez C., Kunze K. L., and Atkins W. M. (2011) Allosteric activation of cytochrome P450 3A4 by α-naphthoflavone: branch point regulation revealed by isotope dilution analysis. Biochemistry 50, 10041–10051 10.1021/bi2013454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pierce L. C., Salomon-Ferrer R., de Oliveira C. A. F., McCammon J. A., and Walker R. C. (2012) Routine access to millisecond time scale events with accelerated molecular dynamics. J. Chem. Theory Comput. 8, 2997–3002 10.1021/ct300284c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miao Y., Nichols S. E., Gasper P. M., Metzger V. T., and McCammon J. A. (2013) Activation and dynamic network of the M2 muscarinic receptor. Proc. Natl. Acad. Sci. U.S.A. 110, 10982–10987 10.1073/pnas.1309755110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miao Y., Nichols S. E., and McCammon J. A. (2014) Free energy landscape of G-protein coupled receptors, explored by accelerated molecular dynamics. Phys. Chem. Chem. Phys. 16, 6398–6406 10.1039/c3cp53962h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miao Y., and McCammon J. A. (2016) Graded activation and free energy landscapes of a muscarinic G-protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 113, 12162–12167 10.1073/pnas.1614538113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kappel K., Miao Y., and McCammon J. A. (2015) Accelerated molecular dynamics simulations of ligand binding to a muscarinic G-protein-coupled receptor. Q. Rev. Biophys. 48, 479–487 10.1017/S0033583515000153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ohkubo Y. Z., Pogorelov T. V., Arcario M. J., Christensen G. A., and Tajkhorshid E. (2012) Accelerating membrane insertion of peripheral proteins with a novel membrane mimetic model. Biophys. J. 102, 2130–2139 10.1016/j.bpj.2012.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baylon J. L., Lenov I. L., Sligar S. G., and Tajkhorshid E. (2013) Characterizing the membrane-bound state of cytochrome P450 3A4: structure, depth of insertion, and orientation. J. Am. Chem. Soc. 135, 8542–8551 10.1021/ja4003525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 24. Frank D. J., Denisov I. G., and Sligar S. G. (2011) Analysis of heterotropic cooperativity in cytochrome P450 3A4 using α-naphthoflavone and testosterone. J. Biol. Chem. 286, 5540–5545 10.1074/jbc.M110.182055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao C., Gao Q., Roberts A. G., Shaffer S. A., Doneanu C. E., Xue S., Goodlett D. R., Nelson S. D., and Atkins W. M. (2012) Cross-linking mass spectrometry and mutagenesis confirm the functional importance of surface interactions between CYP3A4 and holo/apo cytochrome b5. Biochemistry 51, 9488–9500 10.1021/bi301069r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mak P. J., Im S. C., Zhang H., Waskell L. A., and Kincaid J. R. (2008) Resonance Raman studies of cytochrome P450 2B4 in its interactions with substrates and redox partners. Biochemistry 47, 3950–3963 10.1021/bi800034b [DOI] [PubMed] [Google Scholar]
- 27. Hamelberg D., Mongan J., and McCammon J. A. (2004) Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J. Chem. Phys. 120, 11919–11929 10.1063/1.1755656 [DOI] [PubMed] [Google Scholar]
- 28. Isralewitz B., Gao M., and Schulten K. (2001) Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 11, 224–230 10.1016/S0959-440X(00)00194-9 [DOI] [PubMed] [Google Scholar]
- 29. Ramírez C. L., Martí M. A., and Roitberg A. E. (2016) Steered molecular dynamics methods applied to enzyme cechanism and energetics. Methods Enzymol. 578, 123–143 10.1016/bs.mie.2016.05.029 [DOI] [PubMed] [Google Scholar]
- 30. Comer J., Gumbart J. C., Hénin J., Lelièvre T., Pohorille A., and Chipot C. (2015) The adaptive biasing force method: everything you always wanted to know but were afraid to ask. J. Phys. Chem. B 119, 1129–1151 10.1021/jp506633n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Williams P. A., Cosme J., Vinkovic D. M., Ward A., Angove H. C., Day P. J., Vonrhein C., Tickle I. J., and Jhoti H. (2004) Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 305, 683–686 10.1126/science.1099736 [DOI] [PubMed] [Google Scholar]
- 32. Sevrioukova I. F., and Poulos T. L. (2015) Anion-dependent stimulation of CYP3A4 monooxygenase. Biochemistry 54, 4083–4096 10.1021/acs.biochem.5b00510 [DOI] [PubMed] [Google Scholar]
- 33. Berka K., Paloncýová M., Anzenbacher P., and Otyepka M. (2013) Behavior of human cytochromes P450 on lipid membranes. J. Phys. Chem. B 117, 11556–11564 10.1021/jp4059559 [DOI] [PubMed] [Google Scholar]
- 34. Lonsdale R., Rouse S. L., Sansom M. S., and Mulholland A. J. (2014) A multiscale approach to modelling drug metabolism by membrane-bound cytochrome P450 enzymes. PLoS Comput. Biol. 10, e1003714 10.1371/journal.pcbi.1003714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Waxman D. J., Attisano C., Guengerich F. P., and Lapenson D. P. (1988) Human liver microsomal steroid metabolism: identification of the major microsomal steroid hormone 6 β-hydroxylase cytochrome P-450 enzyme. Arch. Biochem. Biophys. 263, 424–436 10.1016/0003-9861(88)90655-8 [DOI] [PubMed] [Google Scholar]
- 36. Sevrioukova I. F., and Poulos T. L. (2017) Structural basis for regiospecific midazolam oxidation by human cytochrome P450 3A4. Proc. Natl. Acad. Sci. U.S.A. 114, 486–491 10.1073/pnas.1616198114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harlow G. R., and Halpert J. R. (1997) Alanine-scanning mutagenesis of a putative substrate recognition site in human cytochrome P450 3A4. Role of residues 210 and 211 in flavonoid activation and substrate specificity. J. Biol. Chem. 272, 5396–5402 10.1074/jbc.272.9.5396 [DOI] [PubMed] [Google Scholar]
- 38. Domanski T. L., Liu J., Harlow G. R., and Halpert J. R. (1998) Analysis of four residues within substrate recognition site 4 of human cytochrome P450 3A4: role in steroid hydroxylase activity andα-naphthoflavone stimulation. Arch. Biochem. Biophys. 350, 223–232 10.1006/abbi.1997.0525 [DOI] [PubMed] [Google Scholar]
- 39. Harlow G. R., and Halpert J. R. (1998) Analysis of human cytochrome P450 3A4 cooperativity: construction and characterization of a site-directed mutant that displays hyperbolic steroid hydroxylation kinetics. Proc. Natl. Acad. Sci. U.S.A. 95, 6636–6641 10.1073/pnas.95.12.6636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Domanski T. L., He Y. A., Khan K. K., Roussel F., Wang Q., and Halpert J. R. (2001) Phenylalanine and tryptophan scanning mutagenesis of CYP3A4 substrate recognition site residues and effect on substrate oxidation and cooperativity. Biochemistry 40, 10150–10160 10.1021/bi010758a [DOI] [PubMed] [Google Scholar]
- 41. Domanski T. L., He Y. A., Harlow G. R., and Halpert J. R. (2000) Dual role of human cytochrome P450 3A4 residue Phe-304 in substrate specificity and cooperativity. J. Pharmacol. Exp. Ther. 293, 585–591 [PubMed] [Google Scholar]
- 42. Vijayan R., and Biggin P. C. (2008) A steroid in a lipid bilayer: localization, orientation, and energetics. Biophys J. 95, L45–L47 10.1529/biophysj.108.140087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cameron M. D., Wen B., Roberts A. G., Atkins W. M., Campbell A. P., and Nelson S. D. (2007) Cooperative binding of acetaminophen and caffeine within the P450 3A4 active site. Chem. Res. Toxicol. 20, 1434–1441 10.1021/tx7000702 [DOI] [PubMed] [Google Scholar]
- 44. Yano J. K., Wester M. R., Schoch G. A., Griffin K. J., Stout C. D., and Johnson E. F. (2004) The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-Å resolution. J. Biol. Chem. 279, 38091–38094 10.1074/jbc.C400293200 [DOI] [PubMed] [Google Scholar]
- 45. Baas B. J., Denisov I. G., and Sligar S. G. (2004) Homotropic cooperativity of monomeric cytochrome P450 3A4 in a nanoscale native bilayer environment. Arch. Biochem. Biophys. 430, 218–228 10.1016/j.abb.2004.07.003 [DOI] [PubMed] [Google Scholar]
- 46. Nath A., Grinkova Y. V., Sligar S. G., and Atkins W. M. (2007) Ligand binding to cytochrome P450 3A4 in phospholipid bilayer nanodiscs: the effect of model membranes. J. Biol. Chem. 282, 28309–28320 10.1074/jbc.M703568200 [DOI] [PubMed] [Google Scholar]
- 47. Frank D. J., Denisov I. G., and Sligar S. G. (2009) Mixing apples and oranges: analysis of heterotropic cooperativity in cytochrome P450 3A4. Arch. Biochem. Biophys. 488, 146–152 10.1016/j.abb.2009.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mak P. J., Denisov I. G., Grinkova Y. V., Sligar S. G., and Kincaid J. R. (2011) Defining CYP3A4 structural responses to substrate binding. Raman spectroscopic studies of a nanodisc-incorporated mammalian cytochrome P450. J. Am. Chem. Soc. 133, 1357–1366 10.1021/ja105869p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Luthra A., Gregory M., Grinkova Y. V., Denisov I. G., and Sligar S. G. (2013) Nanodiscs in the studies of membrane-bound cytochrome P450 enzymes. Methods Mol. Biol. 987, 115–127 10.1007/978-1-62703-321-3_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McClary W. D., Sumida J. P., Scian M., Paço L., and Atkins W. M. (2016) Membrane fluidity modulates thermal stability and ligand binding of cytochrome P4503A4 in lipid nanodiscs. Biochemistry 55, 6258–6268 10.1021/acs.biochem.6b00715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Treuheit N. A., Redhair M., Kwon H., McClary W. D., Guttman M., Sumida J. P., and Atkins W. M. (2016) Membrane interactions, ligand-dependent dynamics, and stability of cytochrome P4503A4 in lipid nanodiscs. Biochemistry 55, 1058–1069 10.1021/acs.biochem.5b01313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Olsson M. H., Søndergaard C. R., Rostkowski M., and Jensen J. H. (2011) PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 10.1021/ct100578z [DOI] [PubMed] [Google Scholar]
- 53. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D. Jr., and Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 10.1021/jp101759q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mackerell A. D. Jr., Feig M., and Brooks C. L. 3rd (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 10.1002/jcc.20065 [DOI] [PubMed] [Google Scholar]
- 56. Vanommeslaeghe K., and MacKerell A. D. Jr. (2012) Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model 52, 3144–3154 10.1021/ci300363c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vanommeslaeghe K., Raman E. P., and MacKerell A. D. Jr. (2012) Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 52, 3155–3168 10.1021/ci3003649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Giorgino T. (2014) Computing 1-D atomic densities in macromolecular simulations: The density profile tool for VMD. Comput. Phys. Commun. 185, 317–322 10.1016/j.cpc.2013.08.022 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.