

Abstract
PRKR-like endoplasmic reticulum kinase (PERK) is one of the major sensor proteins that detect protein folding imbalances during endoplasmic reticulum (ER) stress. However, it remains unclear how ER stress activates PERK to initiate a downstream unfolded protein response (UPR). Here, we found that PERK's luminal domain can recognize and selectively interact with misfolded proteins but not with native proteins. Screening a phage-display library, we identified a peptide substrate, P16, of the PERK luminal domain and confirmed that P16 efficiently competes with misfolded proteins for binding this domain. To unravel the mechanism by which the PERK luminal domain interacts with misfolded proteins, we determined the crystal structure of the bovine PERK luminal domain complexed with P16 to 2.8-Å resolution. The structure revealed that PERK's luminal domain binds the peptide through a conserved hydrophobic groove. Substitutions within hydrophobic regions of the PERK luminal domain abolished the binding between PERK and misfolded proteins. We also noted that peptide binding results in major conformational changes in the PERK luminal domain that may favor PERK oligomerization. The structure of the PERK luminal domain–P16 complex suggested stacking of the luminal domain that leads to PERK oligomerization and activation via autophosphorylation after ligand binding. Collectively, our structural and biochemical results strongly support a ligand-driven model in which the PERK luminal domain interacts directly with misfolded proteins to induce PERK oligomerization and activation, resulting in ER stress signaling and the UPR.
Keywords: unfolded protein response (UPR), structural biology, crystallography, stress response, signaling, crystal structure, ER stress activation, eukaryotic translation initiation factor 2α kinase 3, PERK, stress response
Introduction
A number of exogenous and endogenous factors such as UV radiation, reactive oxygen species, hypoxia, protein mutations, and nutrient starvation may disturb the protein maturation in endoplasmic reticulum (ER)3 and lead to ER stress. To resolve the imbalance in protein folding homeostasis, eukaryotic cells develop the evolutionarily conserved, ER-specific unfolded protein response (UPR) (1–5). This delicate UPR signaling cascade relies on three independent ER-resident stress sensors: double-stranded RNA-dependent protein kinase (PRK)-like ER kinase (PERK), inositol requiring 1 (IRE1), and activating transcription factor 6 (ATF6). The mechanism remains unclear how the sensor proteins are activated during ER stress, particularly for PERK. It was proposed that under non-stressed conditions, these ER sensor proteins are inactivated by binding to immunoglobulin-binding protein (BiP). During ER stress, the increasing amounts of misfolded proteins cause the dissociation of BiP from the sensor proteins (6–8). Other data suggested the ligand-driven model that the misfolded protein may interact directly with the sensor proteins PERK and IRE1 to induce the protein oligomerizations, which can lead to the activations of PERK and IRE1 (1, 9–12).
The UPR may alleviate ER stress by regulating a number of transcription pathways. The activation of PERK can shut down the global protein translations to reduce the ER protein input (1, 13). The activations of IRE1 may promote protein folding by over-expressing ER molecular chaperones (14, 15). ATF6, when activated, will undergo the proteolytic cleavage in Golgi apparatus (3). Collectively, activation of UPR may restore homeostasis of the ER. If UPR fails to rescue the ER stress, the cell may go through apoptosis (16).
Both PERK and IRE1 are type I ER transmembrane proteins. During ER stress, the misfolded protein may induce the oligomerizations of the N-terminal ER luminal domains of PERK and IRE1. The cytosolic kinase domain of PERK, upon oligomerization, can be activated by autophosphorylation. The PERK cytosolic kinase domain will then recruit and phosphorylate the substrate protein eIF2α. The phosphorylation of eukaryotic initiation factor (eIF) 2α by PERK will shut off the overall protein translation (17).
It remains unclear how ER stress induces the oligomerizations of PERK luminal domains. Our data have indicated that the PERK luminal domain can selectively interact with misfolded proteins but not native proteins, which support the ligand-driven hypothesis that the binding of the PERK to the misfolded proteins will induce the oligomerizations of these ER stress sensor proteins and initiate the UPR signaling (18, 19). The crystal structure of the PERK luminal domains is available (19, 20). However, the structure of PERK luminal domains cannot provide a clear picture to explain how PERK interacts with misfolded proteins. In this study, we have identified the peptide substrate P16 for PERK luminal domain. The peptide substrate P16 can compete with the denatured model proteins to bind with the PERK luminal domain, which indicates this peptide may represent a genuine peptide substrate for the PERK luminal domain. We determined the crystal structure for the PERK luminal domain complexed with its peptide substrate P16 to 2.8-Å resolution. The structure showed that the PERK luminal domain utilized a conserved hydrophobic groove located at the C-terminal domain to recognize and interact with a broad range of misfolded proteins. Structure-based mutagenesis studies demonstrated that mutations of the conserved hydrophobic residues within the groove significantly reduced the interactions between PERK and the misfolded proteins. The complex structure of the PERK luminal domain and the peptide substrate further demonstrates that peptide binding to the PERK luminal domain may prime the PERK luminal domain into the conformation, which is favorable for oligomerization. The complex structure also suggests a stacking model for PERK luminal domains to oligomerize. Taken together, the data provided solid evidence for the ligand-driven hypothesis that the misfolded protein can directly bind the PERK luminal domain to activate ER stress signaling.
Results and discussion
PERK luminal domain can selectively bind the denatured proteins and suppress protein aggregations
As an ER stress sensor protein, PERK has been proposed to be directly activated by ER misfolded proteins (9, 18, 19). To examine PERK's ability to interact with the misfolded proteins, we have established an ELISA using the model protein rhodanese. To detect the bound PERK luminal domain in ELISA, we produced an N-terminal His-tagged recombinant bovine PERK luminal domain. In ELISA, the chemically denatured model proteins were coated on the plate and the bound PERK luminal domain can be detected by anti-His tag antibody. We have also generated a pulldown assay to confirm direct binding between the PERK luminal domain and the denatured rhodanese. In the assay, the GST–PERK luminal domain fusion protein was immobilized on the beads and the bound denatured rhodanese was detected by Western blotting. The data from both assays clearly showed that the PERK luminal domain selectively recognize and bind the misfolded proteins but not native proteins (Fig. 1, a and b).
Figure 1.

bPERK-LD can directly interact with the denatured model proteins and suppress the heat- or chemical-induced protein aggregations. a, direct binding between bovine PERK luminal domain and the denatured rhodanese shown by ELISA. The chemical denatured rhodanese (labeled as dRho) at various concentrations (0 in background, 3 and 10 μg/ml) were coated on the plate. Native rhodanese (labeled as nRho) was coated on the plate as well. The blank well was utilized as control (labeled as background). After blocking the plate by BSA, bovine PERK luminal domain at 35 μg/ml was added into the wells. After washing, the bound PERK luminal domain was detected by using anti-His tag antibody (see “Experimental procedures” for details). The OD450 readings of the individual experiments are shown in scattered dots. b, the direct binding between bovine PERK luminal domain and the denatured rhodanese shown by a pulldown assay. 100 μg of GST–bPERK-LD was mixed with 20 μl of glutathione-Sepharose 4B beads. By washing the beads with PBS buffer, different amounts of the native rhodanese (nRho) or denatured rhodanese (dRho) were applied. The beads were thoroughly washed with PBST, and then treated with the elution buffer. The eluted samples were resolved by SDS-PAGE and Western immunoblotted with antibodies against rhodanese. Naked beads with denatured rhodanese (lane 1) and purified GST (100 μg total) with denatured rhodanase (lane 2) were utilized as controls. The bands were quantified using the very right one as a reference of 100%. c, the protein aggregation suppression assay for bovine PERK luminal domain by using ADH as the model protein. 5 μm PERK LD and 10 μm ADH were utilized in this reaction. Heat-induced ADH aggregation was monitored by light scattering at OD320 at 5-min intervals. The OD320 readings are shown in vertical axis (as the percentage of the maximum value of buffer controls) and time in minutes is indicated in horizontal axis. ADH only and bPERK-LD only in PBS were used as the negative controls. 5 μm BSA mixed with 10 μm ADH was used as another control. d, protein aggregation suppression assay for the PERK luminal domain using insulin (Ins) as the model protein. Insulin (100 μm) was mixed at 25 °C with bPERK-LD (10 μm). The aggregation was induced by addition of 20 mm DTT and turbidity was monitored at 320 nm. Insulin only and bPERK-LD only were used as the negative controls. The standard derivations of three independent experiments are indicated in the bars.
A common feature for molecular chaperones is the ability to suppress the misfolded protein aggregations induced by heat or chemical denaturing (21, 22). Because the PERK luminal domain can selectively bind misfolded proteins but not native proteins, it would be interesting to examine whether the PERK luminal domain can function to suppress protein aggregations. Here the data from the protein aggregation suppression assay demonstrate that the PERK luminal domain can efficiently protect denatured model proteins alcohol dehydrogenase (ADH) and insulin from aggregating (Fig. 1, c and d). In the assay, the aggregation of ADH was heat-induced and the aggregation of insulin was caused by addition of DTT. The PERK luminal domain suppresses the protein aggregations, whereas the control protein BSA has little effect (Fig. 1c). The data indicated that the PERK luminal domain acts as a molecular chaperone to suppress the model protein aggregations independently of BiP. It is highly likely that the PERK luminal domain functions to suppress protein aggregations by direct binding to the exposed hydrophobic stretches in the denatured proteins. Our data suggest the ligand-driven activation model for PERK, in which the direct binding of the misfolded proteins with PERK luminal domain may induce the oligomerization and activation of the PERK signaling pathway.
Identification of the peptide substrate for PERK luminal domain
To identify a peptide substrate for the PERK luminal domain, we screened the 12-mer phage display library (PhD-12 phage display library, New England Biolabs). After three cycles of bio-panning, 20 colonies of the bound phages by PERK luminal domain were randomly chosen for subsequent DNA extraction and sequencing. Table 1 lists the sequences of the peptide substrate candidates of PERK luminal domains revealed by peptide library screenings. The affinities between these peptide substrate candidates and PERK luminal domains were then measured by utilizing the isothermal titration calorimetry (ITC) technique. These peptide substrate candidates for the PERK luminal domain were synthesized and purified to more than 95% homogeneity (GenScript). The ITC experiments were carried out by injecting the peptide solutions into buffers containing purified bovine PERK luminal domains. Of the six peptide substrate candidates obtained from screening the peptide display library, one peptide with the sequence of ADPQPWRFYAPR showed significant binding affinity to the PERK luminal domain with the dissociation constant Kd of ∼0.2 μm (Fig. 2a). The ITC studies also indicated that one PERK luminal domain monomer can bind one peptide substrate molecule. The ITC studies did not show that other peptides had measurable affinities to the PERK luminal domain. ITC data also showed that purified recombinant human and mouse PERK luminal domain can interact with the identified peptide substrate P16 with similar affinities (Fig. S1, a and b).
Table 1.
Peptide substrate candidate sequences for bovine PERK luminal domain identified by 12-mer phage peptide library screening
The numbers following the sequences indicate the redundancy of the peptide sequence from the screening. The underlined sequence P16 represents a genuine peptide substrate for PERK luminal domain.
| Sequence | ||
|---|---|---|
| P16 | ADPQPWRFYAPR | X9 |
| P22 | NLGYDTASRYKN | X3 |
| P19 | QPYNIAKFASWS | X2 |
| P11 | MGGVSAAVAWST | X2 |
| P3 | YNKIKTLIQDYT | X2 |
| P25 | SWSTSGQQPSPP | X2 |
Figure 2.

Identification of peptide substrate P16 for bovine PERK luminal domain. a, ITC data of bovine PERK luminal domain with peptide P16. The top panel shows the heat release data for injecting the buffer containing peptide P16 in the buffer containing PERK luminal domain. Twenty injections were performed. The lower panel shows the data fitting for the released heat from the reactions with the standard model curve. b, ELISA competition assays showing the identified peptide substrate P16 can inhibit the binding capability of PERK luminal domain to the denatured model proteins luciferase (top panel) and rhodanese (lower panel) in a dose-dependent manner. 100 μl of denatured model proteins at 20 μg/ml was used to coat the plate. After washing, 100 μl of PERK luminal domain (labeled as PERK in the figure) at 1 μm was mixed with the peptide substrate P16 at 0, 10, and 100 μm, and added into the wells. The bound PERK luminal domain can be detected using anti-His tag antibody. The control 12-mer peptide GGGSGGGSGGGS (labeled as ctl peptide) showed no apparent inhibition for the binding. c, the fluorescence polarization assay to measure direct binding between PERK luminal domain and the peptide substrate P16. 10 nm 5-FAM-labeled P16 (final concentration) was added into the PERK luminal domains in 20 mm Hepes, pH 7.2, 150 mm NaCl. The concentrations of PERK luminal domain ranged from 1 to 1000 nm. After a 15-min incubation, the fluorescence polarization signals were measured. To test whether the denatured model protein rhodanese can compete with the peptide substrate P16 to bind with bovine PERK luminal domain, 10 and 100 nm denatured rhodanese were added into the reaction, respectively, and the fluorescence polarization signals were measured.
To confirm that the identified peptide substrate P16 of the PERK luminal domain interacts with PERK luminal domains through the same binding sites as the misfolded polypeptides, we tested whether the peptide P16 could compete with denatured rhodanese to bind the PERK luminal domain by using the ELISA. The chemically denatured rhodanese was coated on the plate, purified recombinant bovine PERK luminal domain and the peptide substrate P16 were mixed at various molar ratios and incubated in the wells. The bound PERK luminal domain was detected by antibody. The data from this ELISA competition assay revealed that the identified peptide substrate P16 can inhibit the binding capability of PERK luminal domains to the denatured model proteins luciferase and rhodanese in a dose-dependent manner (Fig. 2b). The control 12-mer peptide GGGSGGGSGGGS with undetectable binding affinity to the PERK luminal domain by ITC studies showed no apparent competition.
To further examine whether the identified peptide P16 functions as the peptide substrate for PERK luminal domains, we labeled the synthetic peptide P16 at the N terminus by FITC (GenScript) and carried out a competition experiment by use of the fluorescence polarization assay. The data clearly indicated that the fluorescently labeled peptide P16 can readily interact with the PERK luminal domain. Addition of the denatured model protein rhodanese into the reaction can effectively reduce the binding between the PERK luminal domain and fluorescently labeled peptide P16, presumably the denatured rhodanese can compete with P16 to interact with the PERK luminal domain (Fig. 2c).
The data from two competition assays strongly suggest that the identified peptide P16 may bind the PERK luminal domain in a similar fashion as the denatured proteins. Therefore, by utilizing the combination of peptide display library screening, ITC studies, and the competition assays, we have successfully identified a peptide substrate P16 for PERK luminal domain.
The crystal structure of PERK luminal domain complexed with its peptide substrate
We have determined the crystal structure of the bovine PERK luminal domain complexed with the peptide substrate P16 to 2.8-Å resolution. The complex structure was determined by use of a molecular replacement method using the human PERK luminal domain as the search model (Table 2) (19). The bovine PERK luminal domain forms a homodimer in the crystal structure and two PERK dimers are present in each asymmetric unit. A total of 24 β-strands (named as B1–B24 from N-ter to C-ter) and one short α-helix can be found in one complex monomer. The bovine PERK luminal domain monomer contains primarily two domains, a dimerization domain and a peptide-binding domain (Fig. 3a). The peptide-binding domain in the complex molecule is further composed of a β-sandwich subdomain and β-hairpin subdomain. Three layers of β-sheets are present in the β-sandwich subdomain. B1, B2, B3, B18, and B19 form the first layer, B14, B15, B16, and B17 constitute the second layer, and B20, B21, and B24 construct the third layer.
Table 2.
Data collection and structure determination of bovine PERK luminal domain complexed with the peptide substrate P16
| Native | |
|---|---|
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 60.75 161.55 104.78 |
| α, β, γ (°) | 90.00 106.84 90.00 |
| Wavelength(Å) | 1.000 |
| Resolution (Å) | 43.31–2.80 (2.85 – 2.80)a |
| Rsym or Rmerge | 0.081 (0.437) |
| I/σI | 20.8 (2.1) |
| Completeness (%) | 91.5 (94.5) |
| Redundancy | 3.0 (2.9) |
| Refinement | |
| Resolution (Å) | 2.80 |
| No. reflections | 43,538 (3090) |
| Rwork/Rfree | 0.242 (0.325)/0.293 (0.313) |
| No. atoms | |
| Protein | 7268 |
| Ligand/ion | |
| Water | 173 |
| B-factors | |
| Protein | 55.05 |
| Ligand/ion | |
| Water | 58.35 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.710 |
a Highest-resolution shell is shown in parentheses.
Figure 3.
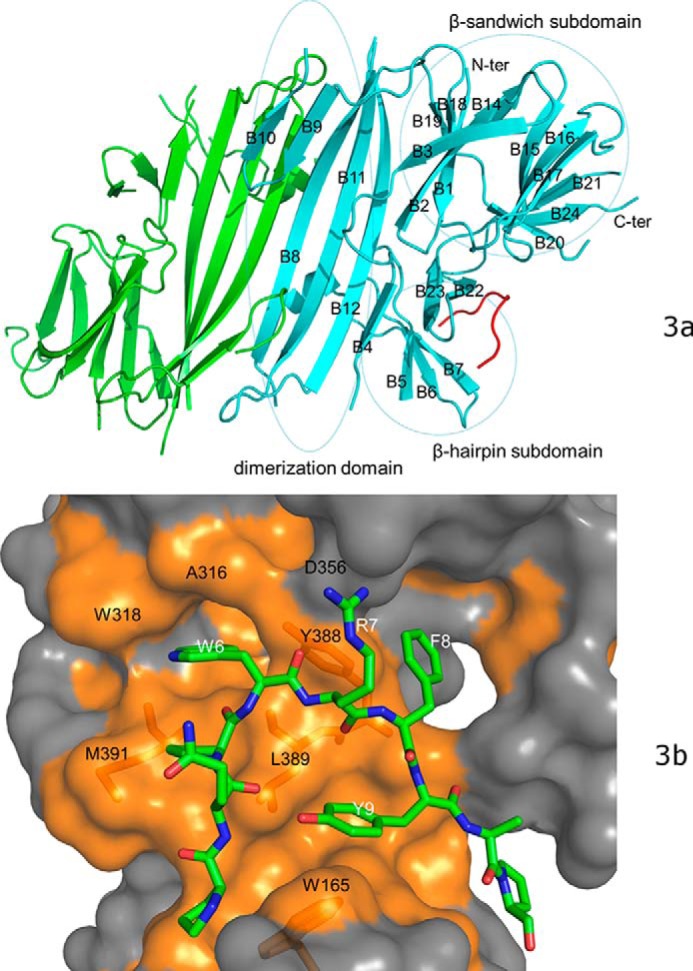
The crystal structure of bovine PERK luminal domain complexed with the peptide substrate P16 determined to 2.8-Å resolution. a, the PERK luminal domain homodimer structure. One of the monomers is ligand-free (in green) and the other is complexed with the peptide substrate P16 (in cyan). The peptide substrate P16 is shown in red. The N terminus and C terminus of the protein are labeled. The 24 β-strands in the complex monomer are labeled as B1–B24. The dimerization domain, β-sandwich subdomain, and β-hairpin subdomain are circled and labeled. b, the surface drawing for the PERK luminal domain complexed with the peptide substrate P16. The PERK luminal domain is shown in surface drawing and the peptide substrate P16 is shown in stick mode. The hydrophobic patches within the peptide-binding groove of the PERK luminal domain is shown in gold. The conserved residues from the peptide-binding grooves that are involved in binding the peptide substrate are labeled in black. Residues Trp-165, Tyr-388, Leu-389, and Met-391 of the PERK luminal domain are shown under the semi-transparent surface. The residues from the peptide substrate P16 that make major contacts with the PERK luminal domain are labeled in white.
In the electron density map, 11 residues of the bound 12-mer peptide substrate P16 can be identified in a large groove located in the peptide-binding domain of PERK. The peptide substrate forms a U-shaped loop in the peptide-binding groove that is constituted by the β-hairpins formed by B6 and B7, B22 and B23 from the β-hairpin subdomain (Fig. 3a). B20 and the loop between B15 and B16 from the β-sandwich subdomain are also involved in constituting the peptide-binding groove. The peptide substrate P16 binds the PERK luminal domain primarily through hydrophobic interactions. The bulky side chains of residues Trp-6, Phe-8, and Tyr-9 from the peptide P16 are positioned toward the bottom of the peptide-binding groove and mediate the hydrophobic interactions (Fig. 3b, supplemental Fig. S2). Trp-6 of peptide substrate P16 is inserted into a hydrophobic pocket formed by residues Ala-316, Trp-318, Tyr-388, Leu-389, and Met-391. Phe-8 from P16 interacts with the side chain of Tyr-388 of the PERK luminal domain. The side chain of Tyr-9 is folded in the center of the U-shaped loop of P16 and makes close contact with Leu-389 and Trp-165 of PERK luminal domain. The polar residues Gln-4 and Arg-7 from P16 face the opening of the peptide-binding groove. The electron density map for the side chains of the solvent-exposed residues Gln-4 and Arg-7 of P16 are not as well defined in the refined structure, indicating that these residues may exhibit certain flexibility in the complex structure (Fig. S2).
It is well known that molecular chaperones bind with misfolded proteins through hydrophobic interactions (23–26). Our complex crystal structure showed that the PERK luminal domain contained a large hydrophobic peptide-binding groove that can accommodate a stretch of peptide of about 10 residues. The peptide-binding groove of the PERK luminal domain is formed by residues Trp-165, Ala-316, Trp-318, Tyr-388, Leu-389, and Met-391. These residues are very well conserved in sequence alignment among multiple species (Fig. S3). It is highly likely that the PERK luminal domain could utilize this peptide-binding groove to interact with the misfolded proteins to suppress protein aggregations. The direct binding of misfolded proteins to the PERK luminal domain may induce the oligomerization of PERK and activate the ER stress signaling.
Conformational changes of PERK luminal domain upon ligand binding
The complex structure of the PERK luminal domain and the peptide substrate P16 indicated that the PERK luminal domain undergoes major conformational changes upon peptide binding. Two PERK luminal domain homodimers are present in one asymmetric unit. Interestingly, three PERK monomers are complexed with P16, whereas the other monomer is not. This provides us an excellent opportunity to compare the ligand-free and ligand-bound structures of the PERK luminal domain (Fig. 4a). In the ligand-free PERK luminal domain monomer, a large portion of the peptide-binding groove is not visible in the electron density map due to high flexibility. The peptide substrate binding apparently stabilizes the PERK peptide-binding domain and the complete peptide-binding groove can be identified in the structure. In the β-sandwich subdomain, the third layer of β-sheet formed by B20, B21, and B24 is completely missing in the ligand-free monomer, whereas it is clearly present in the substrate-bound monomer. The β-strands B14 and B17 in the second layer of the β-sandwich subdomain are missing in the ligand-free monomer and are visible in the ligand-bound monomer. In the β-hairpin subdomain, the loop between B6 and B7 is flexible in the ligand-free monomer and is stabilized and visible in the ligand-bound monomer. Of the six conserved hydrophobic residues located at the bottom of the peptide-binding groove that are important for peptide substrate binding, four (Trp-165, Ala-316, Trp-318, and Tyr-388) are missing in the ligand-free conformation.
Figure 4.

The conformational changes of PERK luminal domain after peptide substrate binding that mediate the PERK oligomerization. a, the superimposition of the peptide-binding domains for the ligand-free monomer (in green) and the peptide-binding monomer (in cyan). The peptide substrate is in red. The β-strands where major conformational changes occur after the peptide binding are labeled. The orientation of this panel is similar to that in Fig. 3b. b, the stacking format revealed by the crystal packing for the PERK luminal domains complexed with peptide substrates. The middle PERK luminal domain complex monomer (cyan) is stacked by two neighboring complex monomers that are translated by 1 unit cell along the a axis. The stacking interactions involve two β-strand formations on either side of the middle monomer complex. On the left side, B3 and B17 (underlined) from the middle monomer form β-strands with B21 and B13 from the left monomer complex (in gold). On the right side, B13 and B21 (underlined) from the middle monomer complex form β-strands with B17 and B3 from the right monomer complex (in silver). This stacking format will allow PERK molecules to oligomerize along the stacking direction (shown in double-ended line) to both ends by any number. The orientation for the middle monomer in this panel is similar to that in panel a and Fig. 3b. c, this panel is generate by rotating b by ∼90° along the horizontal axis.
The crystal structure of the PERK luminal domain complexed with the peptide substrate suggests an induced fit model for PERK to interact with the misfolded proteins. The peptide-binding groove exhibits high flexibility as the peptide substrate-free conformation, whereas peptide substrate binding can stabilize the peptide-binding groove to accommodate the peptide ligand. The complex structure reveals that the PERK luminal domain directly interacts with misfolded proteins via hydrophobic interactions. As an ER stress sensor protein, it is reasonable that PERK can recognize and interact with a wide range of misfolded proteins. The flexible nature of the peptide-binding groove for the PERK luminal domain may be important for PERK to interact with various misfolded proteins. A number of molecular chaperones including trigger factor, Hsp90, GroEL, Hsp40, and sHsp have been reported to contain flexible regions to interact with the client proteins (23, 26–32). It is likely that the PERK peptide-binding groove may adopt significant plasticity depending on the size of its peptide ligand. This cleaver mechanism may enable the PERK luminal domain to sense, recognize, and interact with a broad range of misfolded protein.
Structural implications for ligand-driven PERK oligomerization
Biochemical and structural data have clearly shown that the PERK luminal domain has the capability to interact with the misfolded proteins. The direct binding between PERK and the misfolded protein may tether the PERK homodimers to oligomerize and initiate the ER stress signaling (18). The complex structure of the PERK luminal domain and the peptide substrate P16 further demonstrates that peptide binding to the PERK luminal domain may prime the PERK luminal domain in the conformation, which is favorable for oligomerization. The peptide substrate binding to the PERK luminal domain stabilize the third layer of β-sheet formed by B20, B21, and B24 in the β-sandwich subdomain, which is otherwise completely flexible. In the crystal packing, the stabilized B21 establishes β-strand formation with B3 from the neighbor PERK luminal homodimer (Fig. 4, b and c). In addition, residues 297–301 from the complex monomer extend away and form β-strand B13 with the B17 from the other PERK luminal homodimer. B13 is also completely missing in the ligand-free PERK monomer. Very interestingly, in the crystal structure, one complexed PERK luminal domain is sandwiched by two PERK complexes, which are translated by 1 unit cell along the a axis (Fig. 4, b and c). This leads to stacking of the PERK luminal domains in the crystal. All the molecules that are involved in the stacking are complexed with the peptide substrates. In contrast, the ligand-free monomer in the complex structure is not involved in any major molecular contact in the crystal packing. It is reasonable to postulate that the stacking format of the PERK luminal domain complexes revealed in the crystal structure may mimic the oligomerization of the activated PERK molecules in ER stress signaling.
Therefore, peptide binding to the PERK luminal domain may facilitate the oligomerization of PERK through two mechanisms. 1) The exposed hydrophobic stretches of the misfolded proteins may provide multiple binding sites for the PERK luminal domain and the direct binding of PERK to the misfolded protein can cause PERK to oligomerize. 2) The peptide substrate binding to the PERK luminal domain results in a stabilized complex structure conformation, which is more likely to form oligomers via the stacking format shown in the crystal structure. The combined mechanisms may allow PERK to stack on each other along the misfolded proteins to initiate ER stress signaling.
Structure-based mutagenesis
The complex structure of the PERK luminal domain and peptide substrate P16 indicates that the conserved hydrophobic peptide-binding groove may play central roles in mediating the interactions between PERK and misfolded proteins. To further confirm this observation, we have generated missense mutations W165S, Y388S/L389S/M391S, and W165S/Y388S/L389S/M391S to alter the hydrophobic surface at the peptide-binding groove. The recombinant wildtype and mutant bovine PERK luminal domain protein were produced and purified. All of the proteins existed as homodimers in solution as shown by gel filtration profiles, and the CD spectrums of these proteins were very similar (Fig. S4), indicating that the proteins were probably correctly folded.
First, we measured the binding affinities between PERK luminal domain mutants and peptide substrate P16 by use of an ITC technique. All three mutants exhibited much reduced abilities to interact with P16 (Fig. S5). Due to low heat release signals, none of the data that fits can generate reliable dissociation constants for these mutants. Second, the abilities of these proteins to interact with the denatured model protein rhodanese were examined by use of ELISA. The abilities of PERK luminal domain mutants W165S, Y388S/L389S/M391S, and W165S/Y388S/L389S/M391S to bind the denatured proteins were reduced by ∼70–80% (Fig. 5a). The data clearly showed that the mutations at the conserved hydrophobic residues located at the bottom of the peptide-binding groove of the PERK luminal domain compromised the ability of PERK to interact with the denatured model protein. We have also constructed mutations V310A, L345A, and F357S to change the hydrophobic residues located on other parts of the surface of the β-sandwich domain. None of these mutants showed a reduced binding ability to the peptide substrate P16 and the denatured rhodanese compared with the wildtype PERK luminal domain (data not shown). Next, we examined the in vivo activity of the PERK mutant to sense the ER stress by use of a PERK knockout cell line. We have constructed the mouse PERK mutation W165S/Y383S/L384S/M386S, which corresponds to the bovine PERK mutation W165S/Y388S/L389S/M391S based on sequence alignment. The wildtype full-length mouse PERK and PERK mutant W165S/Y383S/L384S/M386S were transfected into the PERK knockout cell line and ER stress was induced by addition of 5 μg/ml of tunicamycin. The data showed that the phosphorylation levels of PERK and eIF2α were reduced by ∼50% for the PERK mutant W165S/Y383S/L384S/M386S during ER stress (Fig. 5b). The mutagenesis data from biochemical and cell line studies strongly suggest that the identified peptide-binding groove in the complex structure of the PERK luminal domain and the peptide substrate P16 represents the binding site for PERK to interact with misfolded proteins. Changes at the critical sites within the peptide-binding groove of PERK may result in loss of function of PERK to interact with misfolded proteins and to initiate the ER stress signaling.
Figure 5.

The structure-based mutagenesis studies. a, the mutations within the conserved hydrophobic peptide-binding groove of the PERK luminal domain comprise the abilities for the PERK luminal domain to interact with the denatured rhodanese as shown by the ELISA. The chemical denatured rhodanese at 2 μg/ml was coated on the plate. The blank well was utilized as control (labeled as background). After blocking the plate by BSA, bovine PERK luminal domain and its mutants at 1 μm (35 μg/ml) were added into the wells. After washing, the bound PERK luminal domain was detected by using anti-His tag antibody (see “Experimental procedures” for details). The OD450 readings are shown in bars. The S.D. of three independent experiments are indicated in the bars. b, the mouse PERK mutation W165S/Y383S/L384S/M386S exhibited reduced capability to sense the ER stress by use of the PERK knock-out cell line. The mouse wtPERK and PERK W165S/Y383S/L384S/M386S were transiently transfected into the PERK knock-out cell line and ER stress was induced by addition of 5 μg/ml of tunicamycin. The phosphorylated PERK (p-PERK), total PERK, phosphorylated eIF2α (p-eIF2α), and total eIF2α after ER stress were detected by Western blot. The bands were quantified using the wildtype data as a reference of 100%. The data showed that the phosphorylation levels of PERK and eIF2α were reduced by ∼50% for the PERK mutant W165S/Y383S/L384S/M386S during ER stress.
Interestingly, in the human PERK luminal domain structure, the peptide-binding groove is occupied by a short C-terminal helix from another PERK homodimer, which mediates the tetramerization of the PERK luminal domain in the crystal structure (20). Our complex structure and mutagenesis data argue that the physiological role of the peptide-binding groove of the PERK luminal domain is to interact with misfolded proteins to induce oligomerization, but not to directly mediate tetramerization. Several lines of evidence support our hypothesis. 1) The mutagenesis data strongly support that the conserved hydrophobic residues in the peptide-binding groove of PERK are responsible for interacting with denatured proteins. 2) In the ligand-free mouse and bovine PERK luminal domain structures, the peptide-binding groove is empty and the tetramer formation is not observed (Fig. 3a).4 (20). If the peptide-binding groove serves to mediate tetramerization of the PERK luminal domain, one would imagine the mechanism should be conserved among species. 3) In the ligand-free human PERK luminal domain structure, the tetramer is formed by domain swapping between two homodimers. This format excludes the possibility for other types of oligomerization except tetramers. Our bovine complex structure of the PERK luminal domain and peptide substrate suggests a stacking format for PERK oligomerization along the misfolded proteins, which allows any type of oligomer formation.
Experimental procedures
Protein expression and purification
Bovine PERK luminal domain (bPERK-LD) (residues 105–421) was cloned into pET28b and pET41b, generating pET28b-bPERK-LD (His-tagged bPERK-LD) and pET41b-bPERK-LD (GST-bPERK-LD fusion protein). Protein expressions were induced by 0.5 mm isopropyl 1-thio-β-d-galactopyranoside when the Escherichia coli Rosetta(DE3) cells reached A600 of 0.6. Cells were harvested, washed in 100 ml of binding buffer (10 mm Tris-HCl, pH 7.5, 150 mm NaCl), and then lysed by sonication on ice. The lysates were clarified by centrifugation and then the supernatant was loaded onto the nickel-nitrilotriacetic acid column pre-equilibrated with binding buffer. The resin was washed with ice-cold washing buffer (20 mm Tris-HCl, pH 7.9, 0.5 m NaCl, 60 mm imidazole) and the His-tagged bPERK-LD was eluted by elution buffer (20 mm Tris-HCl buffer, pH 7.9, 0.5 m NaCl, and 400 mm EDTA). The final eluant was subsequently loaded onto a Superdex 200 column (Amersham Biosciences) pre-equilibrated with 10 mm Tris-HCl, pH 7.5, and 150 mm NaCl for further purification. The GST-bPERK-LD was purified using the glutathione-Sepharose 4B beads following the manufacturers protocol (GE Healthcare).
Mutations were introduced into bPERK-LD using the NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs, Beverly, MA). All mutant proteins were expressed and purified as the wildtype bPERK-LD.
ELISA
The abilities of the bovine PERK luminal domain and its mutations to interact with denatured model proteins were tested by the ELISA. The model protein, such as rhodanese, was denatured at 1 mg/ml by use of the denaturing buffer (50 mm Tris-HCl, pH 8.0, 6 m guanidine, 0.1% (v/v) 2-mercaptoethanol). The denatured rhodanase was then diluted to 1–20 μg/ml and immobilized on the EIA/RIA plate (Corning Inc., Corning, NY) for 1 h at room temperature. The coated plate was washed three times by using 200 μl of PBST (PBS with 0.2% Tween 20). To block the wells, 200 μl of 1% BSA in PBS was incubated in wells for 1 h at room temperature. The His-tagged bPERK-LD (or the mutants) (35 μg/ml) was then applied onto the wells for 1 h. After extensive washing by PBST, the bound bPERK-LD (or the mutant) can be detected by HRP-conjugated His6 epitope tag antibody (Thermal Fisher Scientific, Madison, WI). OD450 readings were measured after TMP was added into the wells as HRP substrates.
ELISA was also utilized to examine whether peptide substrate P16 can compete with the denatured model protein to bind the bPERK-LD. The denatured rhodanase at 10 μg/ml was coated on the plate. The wells were then washed three times by 200 μl of PBST. 200 μl of 1% BSA in PBS was used to block the uncoated surface. The bPERK-LD at 35 μg/ml (1 μm) was first mixed with various amounts of the peptide substrate P16, and then applied onto the wells. After extensive washing by PBST, the bound bPERK-LD was detected by HRP-conjugated His6 epitope tag antibody. OD450 readings were measured after TMP was added into the wells as HRP substrates.
Pulldown assay
The direct binding between PERK luminal domain and denatured protein was determined by the pulldown assay. Purified GST-bPERK-LD (100 μg total) was mixed with 20 μl of glutathione-Sepharose beads in PBS buffer and incubated for 10 min. The mixture was centrifuged at 1000 × g for 1 min. After washing with 500 μl of PBS buffer, the beads were resuspended with 1 ml of PBS buffer. The denatured or native rhodanese at different amounts were mixed with the beads. After incubation for 10 min at room temperature, the mixture was centrifuged at 1000 × g for 1 min and then the supernatant was removed. The beads were washed three times with PBST and proteins were then eluted by 50 μl of elution buffer containing 50 mm Tris-HCl, pH 8.0, and 10 mm reduced glutathione. To determine whether the denatured rhodanese can bind to the GST or the beads, two control experiments were performed: 1) GST (100 μg) was used to saturate the beads, then the denatured rhodanese was applied accordingly; and 2) the denatured rhodanese was mixed with the naked beads directly.
Eluted protein samples were resolved by SDS-PAGE and then electrophoretically transferred to polyvinylidene difluoride membrane (Millipore). The membrane was blocked in 10 ml of Odyssey blocking buffer (LI-COR Biosciences) for 1 h at room temperature. The anti-rhodanese goat polyclonal IgG was diluted 1:2,000 in 10 ml of Odyssey blocking buffer containing 0.2% Tween 20 (v/v) and incubated for 1 h at room temperature. Blots were then incubated with a 1:10,000 dilution of donkey anti-goat IRDye 800CW (LI-COR Biosciences) in 10 ml of Odyssey blocking buffer for 1 h at room temperature. The membranes were imaged with an Odyssey infrared imaging system (LI-COR Biosciences).
Aggregation suppression assay
The aggregation suppression activity of the PERK luminal domain was determined by monitoring the heat-induced aggregation of ADH and insulin in the presence or absence of bPERK-LD. ADH (10 μm) was pre-mixed with bPERK-LD (5 μm). The mixtures were incubated at 55 °C and the heat-induced protein aggregation was monitored by light scattering at OD320. ADH in PBS was used as the negative control. BSA 5 μm mixed with ADH 10 μm was used as another control.
The insulin aggregation assay was carried out in PBS buffer. Insulin (100 μm) was preincubated at 25 °C with bPERK-LD (10 μm). The aggregation reaction was started by the addition of dithiothreitol (DTT) to a final concentration of 20 mm and the turbidity was monitored at 320 nm. Insulin in PBS was used as the control.
Phage peptide display library screening
The peptide substrates of bPERK-LD were screened by using the Ph.D.TM-12 phage display peptide library (New England Biolabs, Beverly, MA). The bPERK-LD was first coated on a sterile polystyrene Petri dish. Then 10 μl of the original phage library in 1 ml of TBS buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl) with 0.1% Tween 20 was added to the dish and incubated for 1 h at room temperature. The dish was then extensively washed 10 times with TBS buffer and 0.1% Tween 20 to minimize the nonspecific interactions between bPERK-LD and the phage library. The bound phages were then eluted by 1 ml of TBS buffer with 0.25 mg/ml of bPERK-LD protein. The eluted phages were amplified by infecting the E. coli host strain ER2738. In the next three rounds of panning, ∼2 × 1011 pfu was put in the incubation with bPERK-LD. The Tween 20 concentration in the washing buffer was raised to 0.5% in the fourth panning. After the fourth panning, 20 phages were randomly selected and their DNA encoding the peptide substrate were sequenced.
ITC
Measurement of direct binding between bPERK-LD (or the mutants) and the peptide substrate was performed by use of a MicroCal ITC colorimeter (MicroCal, Northampton, MA) at 25 °C. bPERK-LD (or the mutants) and the peptide were dialyzed against the same buffer (10 mm Tris-HCl, pH 7.5, 100 mm NaCl). The reaction cell contained 400 μl of 0.1 mm bPERK-LD (or the mutants), and the injection syringe was filled with 140 μl of 1 mm peptide substrate. Each titration experiment was performed by using 20 injections of 2 μl with 4-s durations and a 150-s interval between injections. All data were analyzed by using the MicroCal ITC analysis software. Peptide substrate was injected into the buffer as control experiments. The heat releases from the control experiments were subtracted from the experimental data before the data were utilized for Kd fitting.
Fluorescence polarization assay
The fluorescence polarization assay was established to measure the direct binding between PERK luminal domain and the 5-FAM-labeled peptide substrate P16. The N terminally 5-FAM-labeled peptide P16 was synthesized (GeneScript). 10 nm labeled P16 (final concentration) was added into PERK luminal domains in 20 mm Hepes, pH 7.2, 150 mm NaCl. The concentrations of PERK luminal domain ranged from 1 to 1000 nm. After a 15-min incubation, the fluorescence polarization signals were recorded by use of the Envision plate reader (PerkinElmer Life Sciences). To test whether the denatured model protein rhodanese can compete with the peptide substrate P16 to bind with bovine PERK luminal domain, 10 and 100 nm denatured rhodanese were added into the reaction, respectively, and the fluorescence polarization signals were measured.
Cell culture treatments and ER stress detections
The mouse PERK knock-out cell line PERK-KO-DR was purchased from ATCC and cultured at 37 °C with 5% CO2 in Dulbecco's modified Eagle's medium, 0.1 mm non-essential amino acids, and 10% fetal calf serum. The full-length mouse PERK and mouse PERK mutation W165S/Y383S/L384S/M386S were cloned into vector CD510B (System Biosciences) by using the Gibson assembly kit (New England Biolabs). The PERK protein expressions were under the control of cytomegalovirus promoters in CD510B. The cells were grown to 80% confluence by use of the 6-well plate. The transient expressions of PERK proteins in PERK-KO-DR cells were performed by use of the FuGENE6 transfection reagents (Promega). Two days after transfections, the ER stress was induced by addition of 5 μg/ml of tunicamycin. Six hours later, the cells were lysed using lysis buffer containing 20 mm Tris-HCl, pH 7.6, 80 mm KCl, 5 mm EDTA, 1 mm EGTA, 0.5% Nonidet P-40, and protease inhibitors. Protein lysates were centrifuged at 17,000 rpm for 10 min at 4 °C and supernatants were taken for protein quantification by use of BCA assay. Equal amounts of proteins (10 μg) were analyzed in SDS-PAGE and Western blotting. The phosphorylated PERK (p-PERK), total PERK, phosphorylated eIF2α (p-eIF2α), and total eIF2α after ER stress were detected by Western blot analysis using rabbit mAb from Cell Signaling Technologies. The IRDye 680RD goat anti-rabbit secondary antibody (Li-Cor) was utilized to develop the polyvinylidene difluoride membrane after blotting. The experiments were carried out twice and representative data were shown in Fig. 5b.
Crystallization and structure determination
The bPERK-LD and its peptide substrate P16 were mixed together in an ∼1:6.5 molar ratio in Tris buffer (10 mm Tris-HCl, pH 7.5, 100 mm NaCl) to produce the bPERK-LD–peptide substrate complex. The complex was concentrated to 10 mg/ml. Crystals were successfully obtained by vapor diffusion using the mother liquid of 0.1 m MES, pH 6.0, and 1% PEG4000, and then were flash frozen at 100 K in a nitrogen gas stream in the cryoprotectant consisting of mother solution supplemented with 30% ethylene glycol. The crystals were diffracted X-ray to 2.8 Å using synchrotron beamline SER-CAT at APS. The atomic coordinates of the human PERK luminal domain structure (PDB code 5SV7) were used as the search model for the molecular replacement method by program PHASER (33). The model was manually built by use of COOT (34). The structure refinement was carried out by use of Phenix (35). The structure was refined by the simulated annealing protocol starting from an annealing temperature of 3000 K. The model was manually examined and adjusted after each cycle of refinement. In the final cycles of refinement, we tightened up the geometry restrains and ADP restrains by reducing the scale factors of X-ray/stereochemistry weight and X-ray/ADP weight. The water molecules were manually examined individually. The crystal is heavily twinned as indicated by Xtriage of Phenix program. The twin fraction is 0.46 with the twin operator of h,-k,-h-l. The final coordinates and structure factors of bPERK-LD complexed with the peptide substrate P16 have been deposited to Protein Data Bank with an accession number of 5V1D.
Author contributions
P. W. data curation; P. W., J. L., and J. T. investigation; B. S. conceptualization; B. S. supervision; B. S. funding acquisition; B. S. validation; B. S. methodology; B. S. writing-original draft; B. S. project administration; B. S. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Dr. David Ron for very helpful discussions and suggestions. We are grateful to the staff scientists in APS beamline SER-CAT for help in data collection.
This work was supported by National Institutes of Health NIGMS Grant R01 GM080261. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5.
The atomic coordinates and structure factors (code 5V1D) have been deposited in the Protein Data Bank (http://wwpdb.org/).
P. Wang, J. Li, J. Tao, and B. Sha, unpublished data.
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- PERK
- double-stranded RNA-dependent protein kinase (PRK)-like ER kinase
- IRE1
- inositol requiring 1
- ATF6
- activating transcription factor 6
- BiP
- immunoglobulin-binding protein
- eIF
- eukaryotic initiation factor
- ADH
- alcohol dehydrogenase
- ITC
- isothermal titration calorimetry
- bPERK-LD
- bovine PERK luminal domain
- HRP
- horseradish peroxidase
- TBS
- Tris-buffered saline.
References
- 1. Ron D., and Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- 2. Schroder M., and Kaufman R. J. (2005) The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789 10.1146/annurev.biochem.73.011303.074134 [DOI] [PubMed] [Google Scholar]
- 3. Rutkowski D. T., and Kaufman R. J. (2007) That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem. Sci. 32, 469–476 10.1016/j.tibs.2007.09.003 [DOI] [PubMed] [Google Scholar]
- 4. Wek R. C., Jiang H. Y., and Anthony T. G. (2006) Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34, 7–11 10.1042/BST0340007 [DOI] [PubMed] [Google Scholar]
- 5. Walter P., and Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 6. Bertolotti A., Zhang Y., Hendershot L. M., Harding H. P., and Ron D. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332 10.1038/35014014 [DOI] [PubMed] [Google Scholar]
- 7. Ma K., Vattem K. M., and Wek R. C. (2002) Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic reticulum stress. J. Biol. Chem. 277, 18728–18735 10.1074/jbc.M200903200 [DOI] [PubMed] [Google Scholar]
- 8. Amin-Wetzel N., Saunders R. A., Kamphuis M. J., Rato C., Preissler S., Harding H. P., and Ron D. (2017) A J-protein co-chaperone recruits BiP to monomerize IRE1 and repress the unfolded protein response. Cell 171, 1625–1637.e1613 10.1016/j.cell.2017.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Credle J. J., Finer-Moore J. S., Papa F. R., Stroud R. M., and Walter P. (2005) On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 102, 18773–18784 10.1073/pnas.0509487102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou J., Liu C. Y., Back S. H., Clark R. L., Peisach D., Xu Z., and Kaufman R. J. (2006) The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. U.S.A. 103, 14343–14348 10.1073/pnas.0606480103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gardner B. M., and Walter P. (2011) Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 333, 1891–1894 10.1126/science.1209126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karagöz G. E., Acosta-Alvear D., Nguyen H. T., Lee C. P., Chu F., and Walter P. (2017) An unfolded protein-induced conformational switch activates mammalian IRE1. Elife 6, e30700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang M., and Kaufman R. J. (2014) The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 14, 581–597 10.1038/nrc3800 [DOI] [PubMed] [Google Scholar]
- 14. Kang S. W., Rane N. S., Kim S. J., Garrison J. L., Taunton J., and Hegde R. S. (2006) Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell 127, 999–1013 10.1016/j.cell.2006.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hollien J., and Weissman J. S. (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107 10.1126/science.1129631 [DOI] [PubMed] [Google Scholar]
- 16. Lin J. H., Li H., Yasumura D., Cohen H. R., Zhang C., Panning B., Shokat K. M., Lavail M. M., and Walter P. (2007) IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949 10.1126/science.1146361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harding H. P., Zhang Y., and Ron D. (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 10.1038/16729 [DOI] [PubMed] [Google Scholar]
- 18. Cui W., Li J., Ron D., and Sha B. (2011) The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr. D Biol. Crystallogr. 67, 423–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang P., Li J., and Sha B. (2016) The ER stress sensor PERK luminal domain functions as a molecular chaperone to interact with misfolded proteins. Acta Crystallogr. D Struct. Biol. 72, 1290–1297 10.1107/S2059798316018064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carrara M., Prischi F., Nowak P. R., and Ali M. M. (2015) Crystal structures reveal transient PERK luminal domain tetramerization in endoplasmic reticulum stress signaling. EMBO J. 34, 1589–1600 10.15252/embj.201489183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tao J., Petrova K., Ron D., and Sha B. (2010) Crystal structure of P58(IPK) TPR fragment reveals the mechanism for its molecular chaperone activity in UPR. J. Mol. Biol. 397, 1307–1315 10.1016/j.jmb.2010.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li J., and Sha B. (2005) Structure-based mutagenesis studies of the peptide substrate binding fragment of type I heat-shock protein 40. Biochem. J. 386, 453–460 10.1042/BJ20041050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li J., Qian X., and Sha B. (2003) The crystal structure of the yeast Hsp40 Ydj1 complexed with its peptide substrate. Structure 11, 1475–1483 10.1016/j.str.2003.10.012 [DOI] [PubMed] [Google Scholar]
- 24. Chen L., and Sigler P. B. (1999) The crystal structure of a GroEL/peptide complex: plasticity as a basis for substrate diversity. Cell 99, 757–768 10.1016/S0092-8674(00)81673-6 [DOI] [PubMed] [Google Scholar]
- 25. Zhu X., Zhao X., Burkholder W. F., Gragerov A., Ogata C. M., Gottesman M. E., and Hendrickson W. A. (1996) Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272, 1606–1614 10.1126/science.272.5268.1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sha B., Lee S., and Cyr D. M. (2000) The crystal structure of the peptide-binding fragment from the yeast Hsp40 protein Sis1. Structure Fold Des. 8, 799–807 10.1016/S0969-2126(00)00170-2 [DOI] [PubMed] [Google Scholar]
- 27. Kaiser C. M., Chang H. C., Agashe V. R., Lakshmipathy S. K., Etchells S. A., Hayer-Hartl M., Hartl F. U., and Barral J. M. (2006) Real-time observation of trigger factor function on translating ribosomes. Nature 444, 455–460 10.1038/nature05225 [DOI] [PubMed] [Google Scholar]
- 28. Lakshmipathy S. K., Gupta R., Pinkert S., Etchells S. A., and Hartl F. U. (2010) Versatility of trigger factor interactions with ribosome-nascent chain complexes. J. Biol. Chem. 285, 27911–27923 10.1074/jbc.M110.134163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Horwich A. L., Fenton W. A., Chapman E., and Farr G. W. (2007) Two families of chaperonin: physiology and mechanism. Annu. Rev. Cell Dev. Biol. 23, 115–145 10.1146/annurev.cellbio.23.090506.123555 [DOI] [PubMed] [Google Scholar]
- 30. Southworth D. R., and Agard D. A. (2011) Client-loading conformation of the Hsp90 molecular chaperone revealed in the cryo-EM structure of the human Hsp90:Hop complex. Mol. Cell 42, 771–781 10.1016/j.molcel.2011.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Taipale M., Jarosz D. F., and Lindquist S. (2010) HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 11, 515–528 10.1038/nrm2918 [DOI] [PubMed] [Google Scholar]
- 32. Jaya N., Garcia V., and Vierling E. (2009) Substrate binding site flexibility of the small heat shock protein molecular chaperones. Proc. Natl. Acad. Sci. U.S.A. 106, 15604–15609 10.1073/pnas.0902177106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 35. Adams P. D., Afonine P. V., Bunkczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.