

Abstract
Supramolecular materials composed of proteins and peptides have been receiving considerable attention toward a range of diseases and conditions from vaccines to drug delivery. Owing to the relative newness of this class of materials, the bulk of work to date has been preclinical. However, examples of approved treatments particularly in vaccines, dentistry, and hemostasis demonstrate the translational potential of supramolecular polypeptides. Critical milestones in the clinical development of this class of materials and currently approved supramolecular polypeptide therapies are described in this study. Additional examples of not‐yet‐approved materials that are steadily advancing toward clinical use are also featured. Spherical assemblies such as virus‐like particles, designed protein nanoparticles, and spherical peptide amphiphiles are highlighted, followed by fiber‐forming systems such as fibrillizing peptides, fiber‐forming peptide‐amphiphiles, and filamentous bacteriophages.
Keywords: commercialization, peptides, self‐assembly, translation, vaccines
Supramolecular peptide biomaterials are being advanced toward clinical applications. This progress report describes self‐assembled polypeptide biomaterials that have been translated into clinical applications, along with those making steady progress toward patients. Although the majority of new supramolecular polypeptide materials have yet to be translated, the regulatory approval and commercialization of several examples is encouraging for the field.
1. Introduction
Over the past few decades, researchers have built an expansive toolbox of self‐assembling peptides and proteins that offer unique advantages over traditional small molecules and polymers for a range of biomedical applications. The first examples of synthetic self‐assemblies were bioinspired derivatives of native proteins, but an increasing familiarity with the design rules governing supramolecular assembly has more recently facilitated the de novo design of this class of materials. The ability to create predictable and engineerable supramolecular structures has led to the implementation of several of these materials within biomedical applications. In this review, we highlight self‐assembling polypeptide materials that have been clinically translated. We will also discuss the advantageous properties and features that have enabled their clinical implementation. For recent reviews on preclinical work relating to this class of materials including self‐assembling immunomodulating materials,1, 2 self‐assembled materials for cell delivery,3, 4 and self‐assembled biomaterials as a whole,5, 6 the reader is referred to the review articles indicated.
Properly designed polypeptides can self‐assemble into a range of predictable structures including nanofibers, micelles, nanoparticles, and extended networks. These architectures are finding utilization in biomedical applications ranging between immunomodulation, drug delivery, tissue regeneration, defect repair, cell delivery, and combinations thereof. Many self‐assembling systems have been investigated in preclinical research, but among these, relatively few have been successfully translated into approved devices and therapeutics. Those that are in current clinical use tend to possess the following features that have enabled their success:
-
‐
Chemical definition—Highly specified control over material composition, assembly properties, and bioactivity, in contrast with biologically sourced materials
-
‐
Manufacturability—The extent to which the platform can be manufactured with relative ease at minimum cost and with maximum reproducibility
-
‐
Tunability—The ability to adjust the amount of multiple selected active components in a material with precision and reproducibility. Synthetic supramolecular systems tend to feature this property well beyond naturally derived materials.
Selected platforms are discussed based on these features, because they have had a meaningful translation into clinical practice, or because they have had steady progress toward clinical translation. These include spherical assemblies such as virus‐like particles, designed protein nanoparticles, and peptide amphiphiles (Figure 1 ); and elongated structures such as β‐sheet nanofibers, fiber‐forming peptide amphiphiles, and filamentous phage (Figure 2 ). A timeline of their development is shown in Figure 3 .
Figure 1.
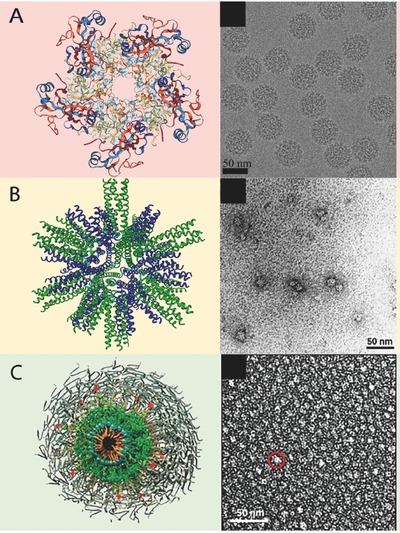
Compiled computer generated models and transmission electron microscopy images of spherical supramolecular assemblies. A) Virus‐like particle. B) Designed protein nanoparticle. C) Peptide amphiphile micelle. (A, left) Adapted with permission.7 Copyright 2007, ASBMB. (A, right) Adapted with permission.8 Copyright 2017, Elsevier. (B) Adapted with permission.9 Copyright 2006, Elsevier. (C) Adapted with permission.10
Figure 2.
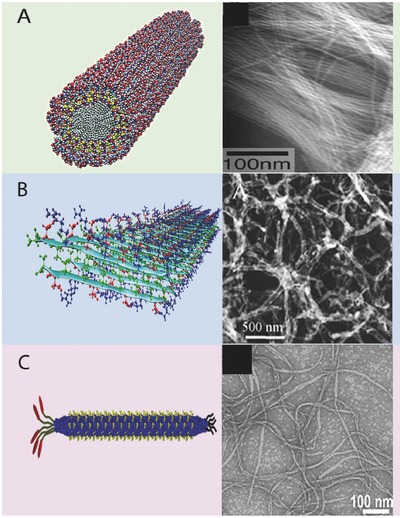
Compiled computer generated models and transmission electron microscopy images of nanofibrillar supramolecular assemblies. A) Peptide amphiphile nanofiber. B) Beta sheet nanofiber. C) Filamentous phage. (A, left) Adapted with permission.11 Copyright 2001, AAAS. (A, right) Adapted with permission.12 Copyright 2002, National Academy of Science. (B, left) Adapted with permission.13 Copyright 2013, American Chemical Society. (B, right) Adapted with permission.14 (C, left) Adapted with permission.15 Copyright 2014, Elsevier. (C, right) Adapted with permission.16
Figure 3.
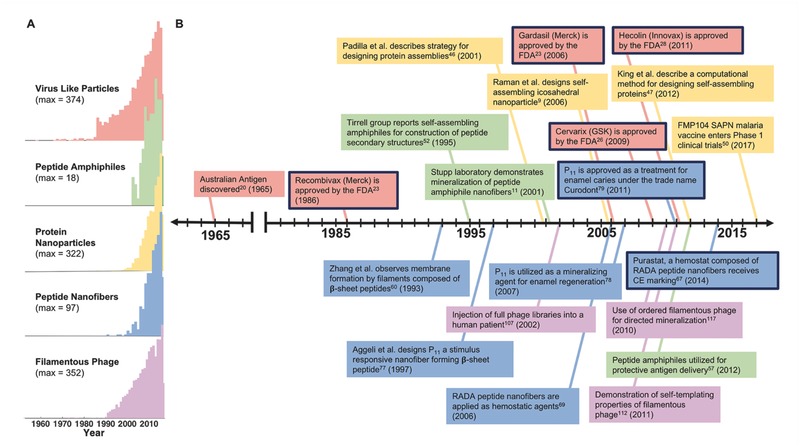
Timeline of clinical development of supramolecular polypeptide materials. A) Citations per year of therapeutic materials in each of the listed classes of polypeptide‐based supramolecular materials. B) A timeline representing major events in the progression of self‐assembling polypeptide therapeutics. Each family of materials is color‐coded to match the citation data. Events in which a technology was approved for clinical use are outlined in black. Citation reports were generated using Web of Science and search terms indicating the therapeutic or clinical use of the relevant platforms.
2. Virus‐Like Particles
Some of the first engineered supramolecular structures to be translated effectively have been virus‐like particles (VLPs), multiprotein constructs that self‐assemble to mimic the organization, structure, and immunogenicity of native viruses but that lack infectious genetic materials (Figure 1A).7, 8 Their development has preceded other supramolecular materials discussed here (see timeline in Figure 3). VLPs are potent immunogens, able to stimulate B cell/antibody responses, CD4+ T‐cell responses, and cytotoxic T‐cell responses.17, 18 Owing to their lack of a viral genome, VLP‐based vaccines circumvent some risks associated attenuated or inactivated live viruses, highlighting an advantage of supramolecular systems: they are more compositionally defined than the analogous biological structures, viruses. They also have advantages over subunit vaccines based on viral proteins or peptides conjugated to carrier proteins, which commonly require higher and more frequent dosing and adjuvants to be as effective as inactivated or attenuated viruses.19 As alternatives to these previous platforms, VLPs were developed to display an array of epitopes that mimic the surface of native viruses more effectively than subunit or peptide vaccines, thus improving their immunogenic properties.20 VLPs, like viruses, come in a range structures including those with a single capsid protein, multiple capsid proteins, or those without lipid envelopes.21 VLPs consisting of multiple capsid proteins are expressed and assembled via subsequent processing or by co‐expression of polycistronic genes within a cell.
Several additional aspects of VLPs have contributed to their clinical translation. Whereas the first translated VLP‐based vaccines raised antibodies against the naturally occurring virus capsid proteins of the assembled structure, VLPs can also be used as vehicles to display heterologous antigens associated with other infectious diseases. This modularity of the platform is described below, in the discussion of chimeric and decorated VLPs. The manufacturability of VLPs depends upon the complexity of the platform. For example, GlaxoSmithKline's (GSK's) Cervarix (human papillomavirus, HPV), Engerix (hepatitis B virus), Merck & Co.'s Recombivax HB (hepatitis B virus), and Gardasil (human papillomavirus) are generated based on a single capsid protein—a feature that accelerated expression optimization and subsequent approval and commercialization. However, as platforms become increasingly complex, they become increasingly more challenging to manufacture at a large scale. Multi‐subunit, chimeric, and other types of VLPs must overcome these challenges. Fortunately, a large assortment of expression systems (bacterial, yeast, insect, plant, mammalian, and cell‐free) have been developed for the production of new VLP platform candidates.
The idea of using noninfectious virus particles to develop prophylactic human vaccines first became attractive when noninfectious VLPs composed of the surface antigen from the hepatitis B virus (HBsAg, also known as the Australia antigen), were discovered in human sera in 1965.20 The first VLP‐based human vaccine consisted of HBsAg VLPs derived from human plasma and was licensed in the United States in 1981 (Figure 3). Subsequently, after the advent of HIV/AIDS rendered plasma‐derived products more challenging, the HBsAg particles were produced recombinantly in yeast, and the first recombinant human vaccine, Recombivax HB, was licensed by Merck in 1986.22 Three years later in 1989, Engerix‐B, a similar HBV vaccine, was licensed by GlaxoSmithKline. It took another 17 years before the next recombinant VLP‐based vaccine was licensed for human use.13
Gardasil, licensed by Merck in 2006, is a vaccine against HPV that protects against HPV‐related diseases such as cervical cancer. In 1991, several laboratories concurrently observed that recombinantly expressed HPV L1 and L2 virus capsid proteins self‐assembled into VLPs resembling the native virion structure.23 In early 1992, based on this observation, Merck Research Laboratories (MRL) initiated an HPV vaccine program. At the time, a growing number of HPV genotypes were identified and associated with benign genital warts and or precursor lesions to cervical cancer. MRL devised a strategy to produce a quadrivalent HPV L1 VLP that would protect against cervical cancers (caused by HPV16 and 18) as well as genital warts (caused by HPV 6 and 11).
Several concurrent studies in the 1990s were additionally critical to the success of HPV vaccines. One important advancement was the development of an in vitro method of producing HPV virions. The HPV lifecycle is linked to human epithelial tissue differentiation which is difficult to achieve in vitro. Kreider et al. overcame this challenge was overcome by developing a complex xenograft model in which human foreskin epithelial tissue was infected with HPV 11 and grown under the renal capsule of athymic mice. This method allowed for the production of infectious HPV 11.23 In 1995, animal challenge studies were published that continued to advance HPV vaccines. Two studies demonstrated measurable serum antibody responses to VLP immunization and subsequent protection in later challenges.23 The third demonstrated the importance of maintaining the native VLP structure for optimal vaccine efficacy.24 Another key contribution to the success of MRL's HPV vaccine came from work developing a manufacturing process for HBsAg production in Saccharomyces cerevisiae. It provided a foundation for the development of methods to design, express, and purify large amounts of HPV VLPs.23
In 1997, HPV 11 L1 VLPs were evaluated for the first time in humans as a monovalent vaccine during a Phase I clinical trial to demonstrate safety and immunogenicity. The monovalent vaccine was chosen owing to the availability of robust models for the evaluation of immunogenicity, and because models for the other three HPV types had not yet been developed. The Phase 1 study outcomes were promising, with no reported significant adverse effects. A second study tested the ability of monovalent HPV 16 L1 VLP efficacy in preventing HPV infection, providing the first demonstration that vaccination with HPV VLPs could prevent disease. These favorable results supported the development of the multivalent vaccine and spurred the advancement of the program. In 2006, after impressive efficacy data and an acceptable safety profile, the first HPV VLP vaccine, Gardasil, was licensed (see timeline in Figure 3).23, 25
Another HPV VLP human vaccine for the prevention of cervical cancer, Cervarix, was developed by GlaxoSmithKline and licensed in the United States in 2009. This vaccine includes HPV types 16 and 18 and has been produced using insect cells infected with recombinant baculovirus.26 Cervarix demonstrated a 90.4% efficacy against cervical intraepithelial neoplasia lesions containing HPV 16 and 18 and indicated cross‐protection with the HPV types 31 and 45, leading to the protection against 80% of cervical cancers.27 Hecolin (Xiamen Innovax Biotech), a recombinant VLP‐based vaccine for prophylactic use against hepatitis E virus infection, was licensed in China in 2011.28 In the past decade, VLP vaccines have played a critical role in the improvement of human health and continue to be applied to new diseases. VLP‐based vaccines are currently in clinical trials for the treatment of many infectious diseases: HBV, HIV, HPV, human parvovirus, influenza virus A, norwalk virus, ebola virus, and severe acute respiratory syndromerelated coronavirus. The inclusion of preclinical testing to this list broadens it even further, to diseases including chikungunya virus, west nile virus, and mumps virus.29
As mentioned in the introduction, one of the key strategic strengths of supramolecular systems is the modularity that they tend to possess. In VLPs, this modularity is exemplified by chimeric VLPs, in which epitopes of choice from various diseases are inserted into the particle‐forming proteins. Provided that the new epitope can be inserted in a way that does not disrupt self‐assembly, and that presents the epitope on the surface of the particle so that it is both antigenic and immunogenic, the modularity of the system is maintained.20 By being able to insert epitopes of choice, the range of therapeutic applications becomes quite broad. For example, several clinical studies are currently evaluating chimeric VLP vaccines for the treatment of noninfectious diseases such as cancer (melanoma),30 neurodegenerative diseases (Alzheimer's disease),31 autoimmune diseases (allergic rhinoconjunctivitis and asthmas),32 and other disorders. Another approach for including chosen epitopes/antigens is separately expressing the VLP and target protein and conjugating them together. This approach is often preferable and necessary due to differences in optimal expression conditions for each component. Thus, postproduction methods have been developed to link the VLP and target antigen. This is achieved through genetic manipulation, coupling via supramolecular or covalent bonds, or by encapsulation of cargo by disassembling and reassembling purified VLPs in the presence of the desired molecule.33 Coupling chemistries range from the use of bifunctional crosslinkers, click chemistry, sortase‐mediated attachment, polyhistidine/NTA‐Ni2+, or affinity‐tag interactions to conjugate the VLP and target antigen34 Notably, these postproduction‐modified VLPs have led to several platforms currently being tested in clinical trials.35, 36, 37
Although a range of chemical cross‐linking strategies have been developed for VLPs,38, 39 conjugation of target antigens with multiple reactive sites can lead to heterogeneous coupling or unfavorable epitope display.38 The Bachmann group addressed this challenge using the SpyTag‐SpyCatcher system.40 SpyTag is a peptide that forms a spontaneous and irreversible isopeptide bond with SpyCatcher, its protein partner.40 The SpyCatcher‐VLP platform is expressed in Escherichia coli and mixed with SpyTag‐Antigen to form “Plug‐and‐Display” decorated VLPs, another highly modular, tunable approach that allows for incorporation of a wide variety of antigens to the VLP surface. The group reported that SpyCatcher‐VLPs decorated with the CIDR or Pfs25 antigens generated a robust antibody response are only a single immunization without requiring adjuvant. This platform shows promise in application such as drug delivery, enzyme scaffolds, biosensors, and cancer immunotherapy,41 and it mitigates the need for complex chimeric protein expression or the incorporation of unnatural amino acids, as would be necessary in copper‐catalyzed azide‐alkyne cycloaddition (click chemistry) or other chemoselective bioconjugation reactions.
VLPs, owing to their structural definition and flexibility in formulation, also make useful experimental tools for studying basic aspects of immunity. They usually range in diameter from 20–200 nm, an optimal size for drainage to lymph nodes and subsequent interactions with B cells. Whereas the differentiation of naïve B cells into memory B cells has been extensively studied at the cellular and molecular level, the fate of memory B cells upon antigen re‐encounter has been comparatively less well‐studied. The Bachmann group used a Qβ VLP as a model system and were able to track VLP‐specific B cells via flow cytometry and histology to follow naïve and memory B cell responses.42 They unexpectedly found that, during secondary B cell responses, secondary plasma cells are generated, whereas naïve B cells are recruited into a parallel primary B cell response. This phenomenon allows a plasticity of the memory B cell repertoire upon multiple antigenic exposures.
Shortcomings of VLPs include their requirement for a continuous and well‐regulated cold chain, which negatively impacts their distribution to the developing world. To address this challenge, the Chackerian group has developed a VLP‐based vaccine candidate that is compatible with spray drying, thus enhancing its stability over a broad range of temperatures.43 This platform targets a highly conserved, broadly neutralizing epitope from the HPV minor capsid protein, L2. Not only does this vaccine elicit high‐titer and long‐lasting antibody immune responses,44 but the spray dried VLPs were highly immunogenic in a mouse model after being stored for 14 months at either room temperature or 37 °C. Other constraints that VLPs are subject to include the relatively small size of epitopes that can be accommodated within the particles. For example, large antigens such as HIV envelope and influenza hemagglutinin proteins are too large to be packaged within native VLPs. The practicality of VLP platforms is also limited by manufacturing considerations.21 For example, VLPs derived from E. coli, the most widely used and most efficient expression system in the biotechnology industry, have a high degree of heterogeneity in their physical properties, including the shape and diameter of the particles. Such particles require post‐purification disassembly and reassembly in optimized conditions. In addition, it may be possible to produce highly immunogenic VLP preparations, but the antigen might not be viable in the VLP context until stable formulations can be developed. Formulations must be resistant to aggregation upon exposure to low salt and protein concentration, as well as protection against surface adsorption and aggregation as a result of heat stress and physical agitation in order to achieve the multiple‐year stability required for a marketed vaccine.20 Newer expression systems in conjunction with innovative purification strategies could determine the pace for the next generation of VLP‐based vaccine candidates. For example, yeast, insect, and mammalian expression systems have been used to circumvent the limitations of bacterial expression such as sub‐optimal pH and lipid compositions within the bacteria. Additionally, the advancement of high‐throughput biophysical and structural analyses of recombinant VLPs may play a key role in the assessment of VLP candidates. Electron microscopy, electrospray differential mobility analysis, atomic force microscopy, X‐ray crystallography, and dynamic light scattering provide quantitative structural data for each vaccine candidate and play a valuable role in ensuring product robustness from the early clinical development stage and beyond.20
3. Designed Protein Nanoparticles
As an alternative to particles inspired by natural virus capsid proteins, fully designed protein nanoparticles have received considerable interest. Although “bottom‐up” approaches for designing nanomaterials were popularized over 30 years ago,45 the development of supramolecular polypeptide materials into successful medical technologies was initially slow, hindered by the sheer diversity of possible self‐assembled structures and the lack of design rules. In 2001, the Yeates group developed an approach based on molecular symmetry to fabricate protein assemblies having a range of predictable architectures.46 Their strategy was a breakthrough in rational self‐assembling protein design.
In the past few years, the capacity to model and predict protein structures and energetics has increased along with computing power, leading to the computational design of de novo self‐assembling protein nanoparticles.47 The Baker group used naturally occurring oligomeric proteins as building blocks to design cage‐like assemblies with accuracy. Recently, they generated a hyperstable 60‐subunit protein icosahedron via symmetric modelling coupled with computational protein–protein interface design.48 This structure is robust to genetic fusions, making it a notably modular platform that could be used in multivalent epitope display as well as drug delivery.
Taking inspiration from nature has also proven invaluable in the design of mechanically and chemically stable nanoparticles. In 2006, Raman et al. designed a self‐assembling nanoparticle based on virus capsids via superposition of different protein oligomerization domains onto the symmetry axes of an icosahedron shown in Figure 1B. The monomer building block consisted of a protein chain made of two coiled coils connected by a short linker region. The association between the coiled coils caused the assembly of monomers into a roughly spherical nanoparticle.9
The self‐assembling protein nanoparticle's versatile and flexible design allow for optimization of biophysical and immunological properties making them a desirable vaccine platform. Whereas synthetic peptides are usually not sufficiently immunogenic and require adjuvants, the Burkhard group has developed a self‐assembling protein nanoparticle platform that displays both B and T cell epitopes to produce a vaccine with self‐adjuvanting qualities.49 This platform is currently under development to improve a malaria vaccine, “RTS's” that is based on the circumsporozoite protein of Plasmodium falciparum.50 The self‐assembled protein nanoparticles are able to stimulate high titer, high avidity antibodies and present CD8+ T‐cell eptiopes to stimulate IL‐2 and IFN‐γ producing long‐term memory T‐cells in mouse models. The vaccine candidate FMP014, based on this platform, is currently undergoing phase 1 clinical trials.
A principal advantage of designed protein nanoparticles is their chemical definition. The ability to use computational methods to design nanoparticles of different geometries and sizes opens the door to applications such as drug delivery, in which the shape and size of the delivery vehicle are crucial. However, the manufacturability of these platforms, similar to VLPs, is a challenge due to their multi‐subunit nature and the need to be recombinantly expressed.
4. Spherically Assembled Peptide Amphiphiles
Micellar nanocarriers composed of amphiphilic molecules have had particular success in the pharmaceutical realm as a tool to increase bioavailability, retention, and solubility of various drugs.51 However, in this review, we shall be focusing on peptide amphiphiles (PAs) as they relate to our theme of the clinical translation of peptide‐based materials. Such structures self‐assemble from molecules composed of a hydrophobic domain, usually an alkyl chain, and a hydrophilic peptide domain.4, 52 PA assembly is driven by hydrophobic–hydrophobic interactions in water, and bioactivity is programmed into the hydrophilic peptide head groups (see Figure 1C). For a comprehensive review on PAs used in biomedical applications, the reader is directed to Acar et al.6
Targeting, diagnostic, and theranostic platforms have been derived from peptide amphiphile micelles (PAMs). To increase the size of hydrophobic head groups and push systems toward a micellar packing morphology, the micelles consist of a biologically active peptide attached to a hydrophobic alkyl tail via a bulky polyethylene glycol (PEG) spacer. The PEG spacer allows for enhanced blood circulation times while retaining the packing parameters necessary for micelle formation.6 These molecules are shown to circulate through the blood stream without causing blockage, and are cleared via the reticulo‐endothelial system and renal system with 90% clearance and no toxicity after 7 d.53
PAMs are currently in preclinical development for both cancer and atherosclerosis diagnostic applications. For cancer applications, fluorescently‐labeled PAs with the fibrin‐binding peptide CREKA were used to target glioblastoma cells. Upon intravenous administration to GL261 glioma‐bearing mice, nontargeting micelles passively accumulated at the fibrin deposits characteristic of tumor vasculature. These micelles displayed enhanced tumor homing as early as 1 h after administration without inducing cytotoxicity or tissue damage.54 Toward a therapy for atherosclerosis, the CREKA targeting peptide, an antithrombin peptide called hirulog, and fluorescence molecules were assembled into theranostic micelles.55 This fibrin‐targeting micelle could also be functionalized by the addition of the gadolinium chelator diethylenetriaminepentaacetic acid, allowing for plaque localization and visualization using T1‐weighted magnetic resonance imaging (MRI) imaging.56 Similar PAMs have been designed for the targeting of monocytes as a strategy to diagnose atherosclerosis via the areas of heightened immunological activity characterized by the disease.10
Similarly to previously described vaccine nanoparticles, PA micelles are able to elicit either humoral or cell‐mediated immunity without additional adjuvant. An antigen is simply conjugated to the tail domain of the molecule prior to self‐assembly. Black et al. was able to assemble cylindrical micelles from monomers consisting of a dialkyl tail conjugated to a peptide containing the known cytotoxic T‐cell epitope from the model tumor antigen ovalbumin.57 These diC16‐OVA micelles were able to stimulate OVA‐specific T‐cells, offering in vivo protection from tumors without any additional adjuvant. This observation spurred additional immunological studies to expand upon the potential of peptide amphiphile micelle vaccine platforms. In 2016, Barrett et al. used a Group A Streptococcus (GAS) B‐cell antigen coupled to a diC16 tail which drove self‐assembly of cylindrical micelles, to induce a micelle‐mediated immune response (without adjuvant) that was stronger than seen with a conventional gold‐standard vaccine formulation.58 Although spherically assembled peptide amphiphiles have not yet reached regulatory approval for clinical applications, their versatility, modularity, and demonstrated success in preclinical work is encouraging.
5. Fiber‐Forming Platforms
Clinically translatable supramolecular materials are not limited to spherical morphologies. Extended structures with high aspect ratios have also seen considerable development toward a variety of medical technologies (Figure 2A–C). Prominent among these have been fibrillar assemblies of peptides and their derivatives. These structures have been investigated over the past 20 years and have made progress toward therapies in hemostasis, dentistry, wound healing, and immunology.
While studying structural biology in Alexander Rich's research group, S. Zhang discovered a self‐assembling β‐sheet peptide based on the DNA‐binding protein, zuotin.59 The peptide formed amphiphilic tapes with two distinct surfaces, one hydrophobic and the other hydrophilic, and it also contained complementary charged residues that additionally favored this β‐sheet folding (see Figure 2B). Following this discovery, a mimic of the native peptide was designed by mutating the charged and hydrophobic residues, but leaving the pattern intact.60 This mimic demonstrated that self‐assembly of peptides with these motifs was not a sequence‐specific anomaly, but could be recreated in similar systems, forming the first steps toward design rules for fibrillizing peptides. The designer peptide was shown to also form macroscopic membranes and support the attachment of mammalian cells, demonstrating its utility as a biomaterial.61 Subsequently, it was found that the original synthetic sequence RADA16‐II (RARADADA)2 and its modified form RADA16‐I (RADA)4 spontaneously formed hydrogels of entangled nanofibers in salt‐containing solutions and cell culture media.62 Mixtures of peptides bearing multiple bioactive groups could be incorporated into a single macroscopic gel by dosing in various amounts of monomeric peptides to the gel mixture. Additionally, the incorporation of ligands or epitopes within the materials simply required extension of the peptide at either terminus with the desired sequence, thus lowering the barrier of synthetic difficulty for biological researchers. These gels have been subsequently developed toward neuronal regeneration, cytokine delivery, as biotinylated scaffolds for versatile protein delivery, and other applications.62, 63, 64
The release of proteins and small molecules from RADA hydrogels largely depends on the size of the protein cargo regardless of charge and hydrophilic character, allowing a wide variety of biomolecules to be delivered in these gels.65 Besides physical entrapment, a biotin‐sandwich approach has been used to tether insulin‐like growth factor 1 (IGF‐1) to gels for long term localization of bioactive IGF‐1 in the scaffold.63 Biotinylation provides the advantage of modularity within the hydrogel delivery system, as any biotinylated protein can be immobilized to biotinylated fibers via a streptavidin linkage. Biotinylated fibers have been investigated for myocardial regeneration following infarction and showed success in rats when injected into infarcted hearts along with cardiac progenitor cells.66
Although RADA nanofibers have been investigated preclinically for a wide range of medical applications, they have had the most clinical success as a hemostatic agent. Marketed under the trade name Purastat by 3‐D Matrix Ltd.,67 the product is composed solely of peptide dissolved in sterile water, which forms fibers when in contact with biological fluids. This self‐assembly provides a physical blockage in order to limit bleeding at the site of application. Although this peptide's use as a hemostat is still being actively developed as a component of layer‐by‐layer wound dressings,68 the original RADA16‐I peptide has been useful as a surgical hemostat for multiple surgeries because of the dense mesh it creates at neutral pH. The RADA16‐I peptide which became Purastat was first tested as a hemostatic agent in 2006 and demonstrated hemostasis in rats by stopping bleeding in under 15 s when applied as a 3%–4% aqueous solution to wounds in skin, brain, spinal cord, femoral artery, and liver.69 This rapid induction of hemostasis circumvented the need for components of the clotting cascade to activate the hemostat, it avoided the use of heat or pyrogenic substances, and it was effective in the presence of anti‐coagulant therapies70 without causing pronounced tissue responses.71 Although the peptide is slightly acidic, it did not cause significant inflammation in any animal or clinical studies, even when applied to the brain.72 Also named TDM‐621, the RADA16‐I peptide was first tested as a hemostat in human surgeries in Japan during cardiovascular procedures,73 and later during endoscopic mucosal resection,74 with no treatment‐related adverse events in either trial. Purastat is limited to relatively low pressure hemorrhages in comparison to major arterial injuries,75 but has general utility as a versatile, biodegradable hemostat. Clinical trials to monitor the post‐market performance of Purastat for vascular surgery (NCT03103282), and to test the use of Purastat during endoscopic submucosal dissection are scheduled to begin soon (NCT02833558). Purastat has already achieved CE marking in Europe and has currently received medical device product registration approval in a number of countries including Thailand, Mexico, and Indonesia, and Australia.
Initial studies of the RADA family of peptides inspired the development of general design principles for forming nanofibers, during which other peptides were shown to have similar self‐assembling properties. Notably, peptide P11, developed by Aggeli et al., would eventually lead to the development of the enamel regeneration product Curodont. The first sequence designed by Aggeli et al. in 1997 was a mimic of a β‐sheet transmembrane protein.76, 77 This peptide was designed de novo based on patterns found in the transmembrane protein and was shown to form high aspect‐ratio fibrils that tangled to form hydrogels independently of pH in a manner similar to the protein‐inspired peptide on which it was based.77 Currently, this peptide is on the market in the European Union (EU) and Switzerland in the form of a treatment for dental enamel caries, and it functions by creating a local environment that enhances enamel mineralization.77 Upon injection, monomeric peptides assemble into nanotapes to create a 3D matrix that in some respects resembles the matrix environment necessary for enamel deposition.78
A major design improvement preceding the final Curodont peptide sequence was the introduction of pH‐sensitivity. In 2003, Aggeli et al. rationally modified the original peptide sequence so that it was negatively charged at pH > 8, thus creating electrostatic repulsion and allowing the peptides to remain in a monomeric, unassembled state.78 Upon switching to more acidic pH, however, the acidic residues became protonated and monomers assembled in to β‐sheet tapes.79 This pH‐sensitivity allows for in situ assembly of peptides into nanofibers, which is hypothesized to improve the efficacy of Curodont as the material is able to completely fill demineralized defects of varying shapes and sizes. This pH‐sensitive assembly was specifically explored in the context of enamel remineralization in 2007, where simulated intraoral conditions were employed to assess the performance of self‐assembled scaffolds which were administered in a basic solution, allowed to fibriliize, and then incubated under cyclic pH.78 These studies were completed on enamel lesions formed on extracted human teeth and indicated that the P11 scaffold caused hydroxyapatite mineralization where the peptide solution was applied.78 Thus, positive and significant results were obtained without the need for specialized application methods or a poorly translatable animal model, a significant advantage for the development of polypeptide biomaterials toward dental applications. After the enamel restorative properties of P11 were developed in preclinical models, Credentis was founded in 2010, Curodont was launched as the company's first product, and its safety and efficacy were verified in a clinical trial to treat dental caries.78 In 2012 it received market approval (CE‐label) for medical devices in the EU. In keeping with the original peptide design, the final formulation of Curodont is composed of only a monomeric 11‐amino acid peptide (P11) dissolved in water, with no additional bioactive agents.80 Following a single application, the size and color of lesions are significantly improved, as observed one month after treatment.80, 81
Peptide nanofibers have also increasingly received attention in immunological contexts, an area in which our group is active. However, because the focus here is on clinical development and these materials have not yet reached clinical trials, these applications will be only briefly described, and the reader is referred to other recent reviews for more expansive descriptions of this burgeoning field.1, 2, 82, 83 After the discovery that β‐sheet fibrillized peptides can raise strong antibody responses without the requirement of supplemental adjuvant,84 these materials have been investigated preclinically toward a range of diseases and conditions including malaria,85 Staphylococus aureus infections,86 influenza,87 West Nile virus, cancer,88 and cocaine abuse.89 Immunogenic peptide nanofibers are produced by co‐assembling fibrillizing peptides extended with specific epitopes or haptens along with other fibrillizing peptides bearing T‐cell epitopes. They stimulate antibody/B‐cell responses, CD4 T‐cell responses, and CD8 T‐cell responses90 which can be raised in tunable magnitudes without causing inflammation.85, 86, 89
Other recent extensions of fibrillizing peptide technologies have included assemblies containing both peptides and other therapeutically active compounds. In these drug–peptide formulations, hydrophobic, π–π, and electrostatic interactions induce the assembly of the molecules into fibrillar networks much in the same way as pure peptide systems.91, 92 A recently described example is a strategy in which the US food and drug administration (FDA)‐approved anticancer drug Pemetrexed was conjugated to a four‐amino acid peptide and used both as an MRI contrast agent and to form drug depots near tumor sites.93 The dual function of the material (contrast and depot formation) was possible because the peptide‐drug conjugate formed fibrillar hydrogels at high concentrations to form the drug depot, while lower concentrations in the circulation acted as the MRI contrast agent. A range of other strategies have likewise employed fibrillizing peptides to alter the delivery or pharmacokinetics of various drugs. For example, Hartgerink and co‐workers showed that several different multivalent drug molecules such as clodronate, heparin, and suramin could be used to stabilize β‐sheet fibrillar hydrogels by shielding the surface charges on the peptides that would otherwise inhibit gelation, thus forming drug depots.94 Naphthalene‐modified peptides have been explored to carry hydrophobic drugs such as Curcumin, which require carrier transport due to their low solubility.95 These examples highlight considerable future potential for peptide nanofibers toward clinical applications.
6. Peptide Amphiphile Nanofibers
Alongside the use of β‐sheet peptides, peptide amphiphiles composed of a peptide head group and an alkyl tail are a related class of materials that have seen considerable interest for creating supramolecular nanofibers (see Figure 2A). Although the self‐assembly of peptide amphiphiles into spherical structures had been known for some time,96 the landmark 2001 paper by Hartgerink, Stupp, and colleagues catalyzed much interest in the ability of this class of molecule to form nanofibrous materials for specific biomedical applications.11 In this report, nanofibers spontaneously assembled from peptide amphiphiles into parallel bundles and promoted mineralization of hydroxyapatite.11 Since then, fibrillar peptide amphiphile materials have been explored in a broad range of medical applications ranging between wound healing,97 bone healing,98 the delivery of proteins,99 nervous tissue repair,100 and others. Additional chemistries have been developed to stabilize the materials. For example, toward applications where mechanical integrity is necessary, adhesive groups have been incorporated into the hydrophilic heads to render the fibrous gels self‐healing after they are strained mechanically.101 A recent example of clinically directed peptide amphiphile nanofibers used them to deliver bioactive proteins such as bone morphogenic protein (BMP) to induce osteogenesis in an environment mimicking native bone growth.98 Other examples have included nerve repair, which benefitted from the bioactivity and parallel alignment of fibrous scaffolds,102 and burn injuries, where heparin‐mimetic gels induced the formation of vascularized, collagen‐rich tissues.97 Peptide amphiphiles have also been used to deliver bioactive cargo outside of the regenerative context, including the electrostatic complexation of antisense oligonucleotides to a cationic peptide head to form a depot for sustained release of oligonucleotide.103 Because networks of peptide‐based fibers are morphologically similar to the extracellular matrix, they have also been highly useful as in vitro culture materials104 and can be utilized as cell delivery vehicles, as demonstrated in the transplantation of islet cells105 and bone marrow‐derived pro‐angiogenic cells cultured prior to transplantation.106 Although work with fibrous peptide amphiphiles has remained largely preclinical to date, is anticipated that the coming years will see many of these applications brought forward into approved therapeutics.
7. Nanofibers Formed from Filamentous Phage
Filamentous bacteriophages (see Figure 2C) represent an additional step in the progression of nanofiber‐forming biomaterials. Although their production is considerably different than the chemical synthesis of PAs and short peptides, their elongated morphology and polyamino acid composition are similar in some respects, and can be used to endow phage‐based materials with similar properties to the other fiber‐forming platforms discussed above. Filamentous phages have highly engineerable coat proteins which allow the high density surface display of selected proteins. M13 phage are naturally filamentous, and they resemble peptide and peptide‐amphiphile nanofibers in morphology as they are less than 10 nm in diameter yet almost 1 µm in length. Because M13 bacteriophages are unable to infect mammalian cells, there is negligible risk of virulent infection when using these viruses in medical applications. For these reasons, they have been historically used as antimicrobial agents and are currently approved for use in food products, but have had seen limited use in clinical trials as therapeutics. A few examples exist where the tissue‐targeting and tumor‐homing abilities of full phage libraries were tested in humans.107, 108
Bacteriophages were initially investigated as nanomaterials when they were observed to form liquid crystals, and they proved useful for the templating of inorganic structures by incorporating metal binding peptides on the viral coat proteins.109, 110 Following the discovery of this functionality, their liquid crystalline behavior was utilized to form aligned matrices for neural cell culture by displaying Arg‐Gly‐Asp (RGD) and Ile‐Lys‐Val‐Ala‐Val (IKVAV) peptides on the phage surface.111 This work included the use of standard phage display to select optimal 8‐amino acid sequences for receptor binding, and the resultant filamentous phages were produced in E. coli, a relatively simple manufacturing process that is likely to be scalable.
In another recent example of preclinical materials development using filamentous phages, their self‐templating properties proved useful for controlling the direction of osteoblast growth using the orientation of the phage‐based substrate.112 The use of both self‐templated and fabricated directionality in phage substrates has allowed for the directional growth of human fibroblasts,113 proliferation and elongation of neural progenitor cells,114 and stimulated the differentiation of mesenchymal stem cells into osteoblasts when the phages displayed an osteogenic peptide on their surface.115 As in vivo injectable materials, phages have been used preclinically as carriers for magnetic nanoparticles for targeted imaging of cancerous tumors by displaying a high density of targeting ligands and metal binding peptides on the phage surface.116 A related study used a modified approach by conjugating a streptavidin‐linked fluorophore to phages displaying tumor‐targeting and streptavidin‐binding peptides on their surface for targeted cancer imaging without the use of metal particles.15 These studies demonstrated the importance of directional patterning in combination with the display of specific peptides on the phages. Moreover, the incorporation of multiple bioactive phage populations into a single material only requires adjustment of the mixture of various deposited phages, so in this way these materials feature the modularity characteristic of supramolecular systems.
Similar to other nanofibrous materials, filamentous phage materials have received significant attention for inducing osteogenesis because they can form collagen‐mimetic bundles for the mineralization of hydroxyapatite similarly to PA and β‐sheet peptide nanofibers. Display of anioinic peptides caused parallel assembly of individual phage in the presence of cations, followed by formation of oriented crystals when counterions were introduced owing to the local supersaturation of inorganic ions.117 This approach closely resembled the demonstration of PA mineralization presented by Stupp and coworkers and illustrates conservation of biological processes across materials platforms.11 Since the demonstration of phage assembly mineralization, osteogenesis studies have expanded to include presentation of a hydroxyapatite nucleating protein, Dentin Matrix Protein‐1, as an alternative moiety for inducing crystal formation.118 Tobacco mosiac virus (TMV), another rod‐like virus was shown to cause differentiation of mesenchymal stem cells into bone cells as culture on the TMV coated substrate caused an upregulation of BMP‐2, osteocalcin, and calcium sequestration, which are all markers of bone development.119 Recently, RGD‐bearing phages were 3D‐printed into a ceramic scaffold to induce osteogenesis and angiogenesis concurrently without the addition of exogenous vascular endothelial growth factor.16 Because of the scale at which phage can be produced, they may have manufacturing advantages over other designer materials.
8. Conclusions and Future Directions
Over the past 50 years, supramolecular assemblies of peptides and proteins have developed from single vaccines to a broad range of technologies that spans the breadth of biomaterials applications as drug delivery vehicles, scaffolds for tissue regeneration, and other therapeutics.1, 2, 5, 6 Currently, several have achieved regulatory approval for clinical use (Table 1 ), primarily in the vaccine space. The advancement of these platforms can be attributed to many factors. Peptides and proteins are more economical than ever to produce, and manufacturing efficiencies continue to be developed. The design rules for each subclass of materials has been significantly mapped in recent years. And strategies have been optimized for incorporating disease‐specific ligands, epitopes, or other moieties within each platform. We expect the coming decades to witness the implementation of many new examples of supramolecular polypeptide therapies.
Table 1.
A collection of engineered recombinant and synthetic self‐assembling protein and peptide biomaterials that have been clinically translated in the United States and Europe
| Technology | Type | Disease target | Manufacturer | Ref. |
|---|---|---|---|---|
| Recombivax‐HB | VLP | Hepatitis B Virus | Merck | 120 |
| Engerix‐B | VLP | Hepatitis B Virus | GlaxoSmithKline | 120 |
| GenHevac B | VLP | Hepatitis B Virus | Pasteur‐Merieux Aventis | 120 |
| Hepavax‐Gene | VLP | Hepatitus B Virus | Crucell | 120 |
| Gardasil | VLP | Human Papilloma Virus | Merck | 25 |
| Cervarix | VLP | Human Papilloma Virus | GlaxoSmithKline | 25 |
| Curodont | Betasheet Fiber | Enamel Regeneration | Credentis | 78 |
| Purastat | Betasheet Fiber | Hemostasis | 3D Matrix Medical Technology | 67 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The immunogenic features of peptide assemblies are advantageous for the development of synthetic vaccines and other engineered immunotherapies, a topic which has been recently reviewed by our group and others,1, 2, 5, 82, 83 but it is also important to note for other assemblies containing high densities of protein or peptide ligands/epitopes that such multivalent displays may induce unwanted immune responses. It remains to be seen whether such responses can be tolerated in specific applications, or if they could even be turned in the favor of the material's clinical performance. Interestingly, neither Curodont78 nor Purastat67 contain specific ligands/epitopes, possibly avoiding immunogenicity that may be observed in trials of other nanofibrous materials containing additional protein or peptide functional components.
Additional future work may focus on investigating the biodistribution and pharmacokinetics of preclinical self‐assembling materials. Due to the dynamic nature of these materials, it is important to understand how the in vivo environment affects their long‐term structural organization, retention, and clearance. In order to be clinically translated, these platforms must also be capable of large‐scale production and stable storage. Unlike traditional small molecules, these materials must generally be kept in monomeric or otherwise stable states of assembly prior to administration. Each of these issues represents important considerations that have not yet been fully worked out for supramolecular materials.
With several self‐assembling platforms being discovered and developed over the past 50 years, it is worth considering the cross‐roads that lay ahead: should we focus on the development of the promising platforms discussed here, or should we continue searching for new, novel platforms? On the one hand, several of the aforementioned platforms have shown encouraging preclinical data and are being investigated in new disease models. On the other, the discovery of a new, more efficacious and versatile platforms could spur unforeseen therapies based on the self‐assembling concept. Either way, we predict that self‐assembling peptide and protein technologies will continue making strides in the preclinical and clinical realms.
Conflict of Interest
J.H.C. is an inventor on 4 submitted or awarded patents describing supramolecular polypeptide biomaterials for healthcare uses.
Acknowledgements
K.M.H. and C.N.F. contributed equally to this work. Research on supramolecular peptide biomaterials in our group has been supported by the National Institutes of Health under grant numbers 5R01EB009701; 5R01AI118182; 5R21CA196434; and 7R21AR066244). This Progress Report's contents are solely the responsibility of the authors and do not necessarily represent the official views of these agencies.
Biographies
Kelly Hainline is a doctoral student in Joel Collier's lab at Duke University. She received her B.E. in Biomedical Engineering with a minor in Materials Science and Engineering from Vanderbilt University in 2016. Her research focuses on the integration of functional proteins into supramolecular assemblies for 3D cell culture and immunotherapy applications.

Chelsea N. Fries is a doctoral student pursuing her Ph.D. in Joel Collier's group at Duke University in the Biomedical Engineering Department. Her research focuses on the molecular design of peptide‐based materials for immunological applications. She received her B.S. in Biomedical Engineering from Northwestern University with a minor in Chemistry.

Joel H. Collier is an Associate Professor at Duke University in the Biomedical Engineering Department. His research focuses on designing novel biomolecular materials for applications within immunotherapies, 3D cell culture, and tissue repair. He received his B.S. in Materials Science from Rice University and his Ph.D. in Biomedical Engineering from Northwestern University. In 2016 he moved from the Surgery Department at the University of Chicago to Duke Biomedical Engineering.

Hainline K. M., Fries C. N., Collier J. H., Adv. Healthcare Mater. 2018, 7, 1700930 10.1002/adhm.201700930
References
- 1. Kelly S. H., Shores L. S., Votaw N. L., Collier J. H., Adv. Drug Delivery Rev. 2017, 114, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wen Y., Collier J. H., Curr. Opin. Immunol. 2015, 35, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Branco M. C., Schneider J. P., Acta Biomater. 2009, 5, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cui H., Webber M. J., Stupp S. I., Biopolymers 2010, 94, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Webber M. J., Appel E. A., Meijer E. W., Langer R., Nat. Mater. 2016, 15, 13. [DOI] [PubMed] [Google Scholar]
- 6. Acar H., Srivastava S., Chung E. J., Schnorenberg M. R., Barrett J. C., LaBelle J. L., Tirrell M., Adv. Drug Delivery Rev. 2016, 110, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bishop B., Dasgupta J., Klein M., Garcea R. L., Christensen N. D., Zhao R., Chen X. S., J. Biol. Chem. 2007, 282, 31803. [DOI] [PubMed] [Google Scholar]
- 8. Guan J., Bywaters S. M., Brendle S. A., Ashley R. E., Makhov A. M., Conway J. F., Christensen N. D., Hafenstein S., Structure 2017, 25, 253. [DOI] [PubMed] [Google Scholar]
- 9. Raman S., Machaidze G., Lustig A., Aebi U., Burkhard P., Nanomed.: Nanotechnol., Biol. Med. 2006, 2, 95. [DOI] [PubMed] [Google Scholar]
- 10. Chung E. J., Mlinar L. B., Nord K., Sugimoto M. J., Wonder E., Alenghat F. J., Fang Y, Tirrell M., Adv. Healthcare Mater. 2015, 4, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hartgerink J. D., Benaish E., Stupp S. I., Science 2001, 294, 1684. [DOI] [PubMed] [Google Scholar]
- 12. Hartgerink J. D., Beniash E., Stupp S. I., Proc. Natl. Acad. Sci. USA 2002, 99, 5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cormier A. R., Pang X., Zimmerman M. I., Zhou H.‐X., Paravastu A. K., ACS Nano 2013, 7, 7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sieminski A. L., Semino C. E., Gong H., Kamm R. D., J. Biomed. Mater. Res. 2008, 87A, 494. [DOI] [PubMed] [Google Scholar]
- 15. Jin H.‐E., Farr R., Lee S.‐W., Biomaterials 2014, 35, 9236. [DOI] [PubMed] [Google Scholar]
- 16. Wang J., Yang M., Zhu Y., Wang L., Tomsia A. P., Mao C., Adv. Mater. 2014, 26, 4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schirmbeck R., Böhm W., Reimann J., Intervirology 1996, 39, 111. [DOI] [PubMed] [Google Scholar]
- 18. Murata K., Lechmann M., Qiao M., Gunji T., Alter H. J., Liang T. J., Proc. Natl. Acad. Sci. USA 2003, 100, 6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Noad R., Roy P., Trends Microbiol. 2003, 11, 438. [DOI] [PubMed] [Google Scholar]
- 20. Zhao Q., Li S., Yu H., Xia N., Modis Y., Trends Biotechnol. 2013, 31, 654. [DOI] [PubMed] [Google Scholar]
- 21. Grgacic E. V. L., Anderson D. A., Methods 2006, 40, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McAleer W. J., Buynak E. B., Maigetter R. Z., Wampler D. E., Miller W. J., Hilleman M. R., Nature 1984, 307, 178. [DOI] [PubMed] [Google Scholar]
- 23. Bryan J. T., Buckland B., Hammond J., Jansen K. U., Curr. Opin. Chem. Biol. 2016, 32, 34. [DOI] [PubMed] [Google Scholar]
- 24. Jansen K. U., Rosolowsky M., Schultz L. D., Markus H. Z., Cook J. C., Donnelly J. J., Martinez D., Ellis R. W., Shaw A. R., Vaccine 1995, 13, 1509. [DOI] [PubMed] [Google Scholar]
- 25. Shi L., Sings H. L., Bryan J. T., Wang B., Wang Y., Mach H., Kosinski M., Washabaugh M. W., Sitrin R., Barr E., Clin. Pharmacol. Ther. 2007, 81, 259. [DOI] [PubMed] [Google Scholar]
- 26. Monie A., Hung C.‐F., Roden R., Wu T.‐C., Biol.: Targets Ther. 2008, 2, 107. [PMC free article] [PubMed] [Google Scholar]
- 27. Dubin G., Harper D. M., Franco E. L., Wheeler C. M., Moscicki A.‐B., Romanowski B., Roteli‐Martins C. M., Jenkins D., Schuind A., Costa Clemens S. A., Dubin G., Lancet 2006, 367, 1247. [DOI] [PubMed] [Google Scholar]
- 28. Wu T., Li S.‐W., Zhang J., Ng M.‐H., Xia N.‐S., Zhao Q., Hum. Vaccines Immunother. 2014, 8, 823. [DOI] [PubMed] [Google Scholar]
- 29. Shirbaghaee Z., Bolhassani A., Biopolymers 2015, 105, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Braun M., Jandus C., Maurer P., Hammann‐Haenni A., Schwarz K., Bachmann M. F., Speiser D. E., Romero P., Eur. J. Immunol. 2011, 42, 330. [DOI] [PubMed] [Google Scholar]
- 31. Chackerian B., Rangel M., Hunter Z., Peabody D. S., Vaccine 2006, 24, 6321. [DOI] [PubMed] [Google Scholar]
- 32. Senti G., Johansen P., Haug S., Bull C., Gottschaller C., Müller P., Pfister T., Maurer P., Bachmann M. F., Graf N., Kündig T. M., Clin. Exp. Allergy 2009, 39, 562. [DOI] [PubMed] [Google Scholar]
- 33. Naskalska A., Pyrc K., Pol. J. Microbiol. 2015, 64, 3. [PubMed] [Google Scholar]
- 34. Frietze K. M., Peabody D. S., Chackerian B., Curr. Opin. Virol. 2016, 18, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klimek L., Willers J., Hammann‐Haenni A., Pfaar O., Stocker H., Mueller P., Renner W. A., Bachmann M. F., Clin. Exp. Allergy 2011, 41, 1305. [DOI] [PubMed] [Google Scholar]
- 36. Ambühl P. M., Tissot A. C., Fulurija A., Maurer P., Nussberger J., Sabat R., Nief V., Schellekens C., Sladko K., Roubicek K., Pfister T., Rettenbacher M., Volk H.‐D., Wagner F., Müller P., Jennings G. T., Bachmann M. F., J. Hypertens. 2007, 25, 63. [DOI] [PubMed] [Google Scholar]
- 37. Tissot A. C., Maurer P., Nussberger J., Sabat R., Pfister T., Ignatenko S., Volk H.‐D., Stocker H., Müller P., Jennings G. T., Wagner F., Bachmann M. F., Lancet 2008, 371, 821. [DOI] [PubMed] [Google Scholar]
- 38. Smith M. T., Hawes A. K., Bundy B. C., Curr. Opin. Biotechnol. 2013, 24, 620. [DOI] [PubMed] [Google Scholar]
- 39. Sapsford K. E., Algar W. R., Berti L., Gemmill K. B., Casey B. J., Oh E., Stewart M. H., Medintz I. L., Chem. Rev. 2013, 113, 1904. [DOI] [PubMed] [Google Scholar]
- 40. Zakeri B., Fierer J. O., Celik E., Chittock E. C., Schwarz‐Linek U., Moy V. T., Howarth M., Proc. Natl. Acad. Sci. USA 2012, 109, E690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brune K. D., Leneghan D. B., Brian I. J., Ishizuka A. S., Bachmann M. F., Draper S. J., Biswas S., Howarth M., Sci. Rep. 2016, 6, 1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zabel F., Kündig T. M., Bachmann M. F., Curr. Opin. Virol. 2013, 3, 357. [DOI] [PubMed] [Google Scholar]
- 43. Saboo S., Tumban E., Peabody J., Wafula D., Peabody D. S., Chackerian B., Muttil P., Mol. Pharmaceutics 2016, 13, 1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tumban E., Muttil P., Escobar C. A. A., Peabody J., Wafula D., Peabody D. S., Chackerian B., Vaccine 2015, 33, 3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Drexler K. E., Proc. Natl. Acad. Sci. USA 1981, 78, 5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Padilla J. E., Colovos C., Yeates T. O., Proc. Natl. Acad. Sci. USA 2001, 98, 2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. King N. P., Sheffler W., Sawaya M. R., Vollmar B. S., Sumida J. P., Andre I., Gonen T., Yeates T. O., Baker D., Science 2012, 336, 1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hsia Y., Bale J. B., Gonen S., Shi D., Sheffler W., Fong K. K., Nattermann U., Xu C., Huang P.‐S., Ravichandran R., Yi S., Davis T. N., Gonen T., King N. P., Baker D., Nature 2016, 535, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kaba S. A., Brando C., Guo Q., Mittelholzer C., Raman S., Tropel D., Aebi U., Burkhard P., Lanar D. E., J. Immunol. 2009, 183, 7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Heppner D. G., Kester K. E., Ockenhouse C. F., Tornieporth N., Ofori O., Lyon J. A., Stewart V. A., Duboise P., Lanar D. E., Krzych U., Moris P., Angov E., Cummings J. F., Leach A., Hall B. T., Dutta S., Schwenk R., Hillier C., Ogutu B, Ware L. A., Nair L., Darko C. A., Withers M. R., Ogutu B., Polhemus M. E., Fukuda M., Pichyangkul S., Gettyacamin M., Diggs C., Soisson L., Milman J., Dubois M.‐C., Garcon N., Tucker K., Wittes J., Plowe C. V., Thera M. A., Duombo O. K., Pau M. G., Goudsmit J., Ballou W. R., Cohen J., Vaccine 2005, 23, 2243. [DOI] [PubMed] [Google Scholar]
- 51. Torchilin V. P., Pharm. Res. 2006, 24, 1. [DOI] [PubMed] [Google Scholar]
- 52. Berndt P., Fields G. B., Tirrell M., J. Am. Chem. Soc. 1995, 117, 9515. [Google Scholar]
- 53. Chung E. J., Mlinar L. B., Sugimoto M. J., Nord K., Roman B. B., Tirrell M., Nanomed.: Nanotechnol., Biol. Med. 2015, 11, 479. [DOI] [PubMed] [Google Scholar]
- 54. Chung E. J., Cheng Y., Morshed R., Nord K., Han Y., Wegscheid M. L., Auffinger B., Wainwright D. A., Lesniak M. S., Tirrell M. V., Biomaterials 2014, 35, 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peters D., Kastantin M., Kotamraju V. R., Karmali P. P., Gujraty K., Tirrell M., Ruoslahti E., Proc. Natl. Acad. Sci. USA 2009, 106, 9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yoo S. P., Pineda F., Barrett J. C., Poon C., Tirrell M., Chung E. J., ACS Omega 2016, 1, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Black M., Trent A., Kostenko Y., Lee J. S., Olive C., Tirrell M., Adv. Mater. 2012, 24, 3845. [DOI] [PubMed] [Google Scholar]
- 58. Barrett J. C., Ulery B. D., Trent A., Liang S., David N. A., Tirrell M. V., ACS Biomater. Sci. Eng. 2016, 3, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang S., Lockshin C., Herbert A., Winter E., Rich A., EMBO J. 1992, 11, 3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang S., Holmes T., Lockshin C., Rich A., Proc. Natl. Acad. Sci. 1993, 90, 3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang S., Holmes T., DiPersio M., Hynes R., Su X., Rich A., Biomaterials 1995, 16, 1385. [DOI] [PubMed] [Google Scholar]
- 62. Holmes T., de Lacalle S., Su X., Liu G., Rich A., Zhang S., Proc. Natl. Acad. Sci. USA 2000, 97, 6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Davis M. E., Hsieh P. C. H., Takahashi T., Song Q., Zhang S., Kamm R. D., Grodzinsky A., Anversa P., Lee R., Proc. Natl. Acad. Sci. USA 2006, 103, 8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gelain F., Unsworth L. D., Zhang S., J. Controlled Release 2010, 145, 231. [DOI] [PubMed] [Google Scholar]
- 65. Koutsopoulos S., Unsworth L. D., Nagai Y., Zhang S., Proc. Natl. Acad. Sci. USA 2009, 106, 4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Padin‐Iruegas M. E., Misao Y., Davis M. E., Segers V. F. M., Esposito G., Tokunou T., Urbanek K., Hosoda T., Rota M., Anversa P., Leri A., Lee R. T., Kajstura J., Circulation 2009, 120, 876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pioche M., Camus M., Rivory J. R. M., Leblanc S., Lienhart I., Barret M., Chaussade S., Saurin J.‐C., Prat F., Ponchon T., Endosc. Int. Open 2016, 04, E415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hsu B. B., Conway W., Tschabrunn C. M., Mehta M., Perez‐Cuevas M. B., Zhang S., Hammond P. T., ACS Nano 2015, 9, 9394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. G Ellis‐Behnke R., Liang Y.‐X., Tay D. K. C., Kau P. W. F., Schneider G. E., Zhang S., Wu W., So K.‐F., Nanomed.: Nanotechnol., Biol., Med. 2006, 2, 207. [Google Scholar]
- 70. Csukas D., Urbanics R., Moritz A., Ellis‐Behnke R., Nanomed.: Nanotechnol., Biol., Med. 2015, 11, 2025. [DOI] [PubMed] [Google Scholar]
- 71. Song H., Zhang L., Zhao X., Macromol. Biosci. 2010, 10, 33. [DOI] [PubMed] [Google Scholar]
- 72. Xu F.‐F., Wang Y.‐C., Sun S., Ho A. S. W., Lee D., Kiang K. M. Y., Zhang X.‐Q., Lui W.‐M., Liu B.‐Y., Wu W.‐T., Leung G. K. K., Clin. Transl. Sci. 2015, 8, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Masuhara H., Fujii T., Watanabe Y., Koyama N., Tokuhiro K., Ann. Thorac. Cardiovasc. Surg. 2012, 18, 444. [DOI] [PubMed] [Google Scholar]
- 74. Yoshida M., Goto N., Kawaguchi M., Koyama H., Kuroda J., Kitahora T., Iwasaki H., Suzuki S., Kataoka M., Takashi F., Kitajima M., J. Gastroenterol. Hepatol. 2014, 29, 77. [DOI] [PubMed] [Google Scholar]
- 75. Jukes A., Murphy J., Vreugde S., Psaltis A., Wormald P., J. Neurol. Surg. B 2016, 38, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Aggeli A., Boden N., Cheng Y., Findlay J., Knowles P. F., Kovatchev P., Turnbull P., Biochemistry 1996, 35, 16213. [DOI] [PubMed] [Google Scholar]
- 77. Aggeli A., Bell M., Boden N., Keen J. N., Knowles P. F., McLeish T., Pitkeathly M., Radford S. E., Nature 1997, 386, 259. [DOI] [PubMed] [Google Scholar]
- 78. Kirkham J., Firth A., Vernals D., Boden N., Robinson C., Shore R. C., Brookes S. J., Aggeli A., J. Dent. Res. 2007, 86, 426. [DOI] [PubMed] [Google Scholar]
- 79. Aggeli A., Bell M., Carrick L. M., Fishwick C. W. G., Harding R., Mawer P. J., Radford S. E., Strong A. E., Boden N., J. Am. Chem. Soc. 2003, 125, 9619. [DOI] [PubMed] [Google Scholar]
- 80. Brunton P. A., Davies R. P. W., Burke J. L., Smith A., Aggeli A., Brookes S. J., Kirkham J., Br. Dent. J. 2013, 215, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jablonski‐Momeni A., Heinzel‐Gutenbrunner M., J. Orofacial Orthop. 2014, 75, 175. [DOI] [PubMed] [Google Scholar]
- 82. Tostanoski L. H., Jewell C. M., Adv. Drug Delivery Rev. 2017, 114, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mora‐Solano C., Collier J. H., J. Mater. Chem. B 2014, 2, 2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rudra J. S., Tian Y. F., Jung J. P., Collier J. H., Proc. Natl. Acad. Sci. USA 2010, 107, 622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rudra J. S., Mishra S., Chong A. S., Mitchell R. A., Nardin E. H., Nussenzweig V., Collier J. H., Biomaterials 2012, 33, 6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pompano R. R., Chen J., Verbus E. A., Han H., Fridman A., McNeely T., Collier J. H., Chong A. S., Adv Healthc Mater 2014, 3, 1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chen J., Pompano R. R., Santiago F. W., Maillat L., Sciammas R., Sun T., Han H., Topham D. J., Chong A. S., Collier J. H., Biomaterials 2013, 34, 8776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Huang Z.‐H., Shi L., Ma J.‐W., Sun Z.‐Y., Cai H., Chen Y.‐X., Zhao Y.‐F., Li Y.‐M. a., J. Am. Chem. Soc. 2012, 134, 8730. [DOI] [PubMed] [Google Scholar]
- 89. Rudra J. S., Ding Y., Neelakantan H., Ding C., Appavu R., Stutz S., Snook J. D., Chen H., Cunningham K. A., Zhou J., ACS Chem. Neurosci. 2016, 7, 546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chesson C. B., Huelsmann E. J., Lacek A. T., Kohlhapp F. J., Webb M. F., Nabatiyan A., Zloza A., Rudra J. S., Vaccine 2014, 32, 1174. [DOI] [PubMed] [Google Scholar]
- 91. Caplan M. R., Moore P. N., Zhang S., Kamm R. D., Lauffenburger D. A., Biomacromolecules 2000, 1, 627. [DOI] [PubMed] [Google Scholar]
- 92. Caplan M. R., Schwartzfarb E. M., Zhang S., Kamm R. D., Lauffenburger D. A., Biomaterials 2002, 23, 219. [DOI] [PubMed] [Google Scholar]
- 93. Lock L. L., Li Y., Mao X., Chen H., Staedtke V., Bai R., Ma W., Lin R., Li Y., Liu G., Cui H., ACS Nano 2017, 11, 797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kumar V. A., Shi S., Wang B. K., Li I.‐C., Jalan A. A., Sarkar B., Wickremasinghe N. C., Hartgerink J. D., J. Am. Chem. Soc. 2015, 137, 4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Liu J., Liu J., Xu H., Zhang Y., Chu L., Liu Q., Song N., Yang C., Int. J. Nanomedicine. 2014, 9, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yu Y.‐C., Berndt P., Tirrell M., Fields G. B., J. Am. Chem. Soc. 1996, 118, 12515. [Google Scholar]
- 97. Yergoz F., Hastar N., Cimenci C. E., Ozkan A. D., Tekinay T., Guler M. O., Tekinay A. B., Biomaterials 2017, 134, 117. [DOI] [PubMed] [Google Scholar]
- 98. Lee S. S., Hsu E. L., Mendoza M., Ghodasra J., Nickoli M. S., Ashtekar A., Polavarapu M., Babu J., Riaz R. M., Nicolas J. D., Nelson D., Hashmi S. Z., Kaltz S. R., Earhart J. S., Merk B. R., McKee J. S., Bairstow S. F., Shah R. N., Hsu W. K., Stupp S. I., Adv. Healthcare Mater. 2014, 4, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Choe S., Veliceasa D., Bond C. W., Harrington D. A., Stupp S. I., McVary K. T., Podlasek C. A., Acta Biomater. 2016, 32, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Li A., Hokugo A., Yalom A., Berns E. J., Stephanopoulos N., McClendon M. T., Segovia L. A., Spigelman I., Stupp S. I., Jarrahy R., Biomaterials 2014, 35, 8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ceylan H., Urel M., Erkal T. S., Tekinay A. B., Dana A., Guler M. O., Adv. Funct. Mater. 2012, 23, 2081. [Google Scholar]
- 102. Zhan X., Gao M., Jiang Y., Zhang W., Wong W. M., Yuan Q., Su H., Kan X., Dai X., Zhang W., Guo J., Wu W., Nanomed.: Nanotechnol., Biol., Med. 2013, 9, 305. [Google Scholar]
- 103. Bulut S., Erkal T. S., Toksoz S., Tekinay A. B., Tekinay T., Guler M. O., Biomacromolecules 2011, 12, 3007. [DOI] [PubMed] [Google Scholar]
- 104. Koutsopoulos S., Zhang S., Acta Biomater. 2013, 9, 5162. [DOI] [PubMed] [Google Scholar]
- 105. Uzunalli G., Tumtas Y., Delibasi T., Yasa O., Mercan S., Guler M. O., Tekinay A. B., Acta Biomater. 2015, 22, 8. [DOI] [PubMed] [Google Scholar]
- 106. Tongers J., Webber M. J., Vaughan E. E., Sleep E., Renault M.‐A., Roncalli J. G., Klyachko E., Thorne T., Yu Y., Marquardt K.‐T., Kamide C. E., Ito A., Misener S., Millay M., Liu T., Jujo K., Qin G., Losordo D. W., Stupp S. I., Kishore R., J. Mol. Cell. Cardiol. 2014, 74, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Arap W., Kolonin M. G., Trepel M., Lahdenranta J., Cardo‐Vila M., Giordano R. J., Mintz P. J., Ardelt P. U., Yao V. J., Vidal C. I., Chen L., Flamm A., Valtanen H., Weavind L. M., Hicks M. E., Pollock R. E., Botz G. H., Bucana C. D., Koivunen E., Cahill D., Troncoso P., Baggerley K. A., Pentz R. D., Do K.‐A., Logothetis C. J., Pasqualini R., Nat. Med. 2002, 8, 121.11821895 [Google Scholar]
- 108. Krag D. N., Shukla G., Shen G.‐P., Pero S., Ashikaga T., Fuller S., Weaver D. L., Burdette‐Radoux S., Thomas C., Cancer Res. 2006, 66, 7724. [DOI] [PubMed] [Google Scholar]
- 109. Lee S.‐W., Mao C., Flynn C. E., Belcher A. M., Science 2002, 296, 892. [DOI] [PubMed] [Google Scholar]
- 110. Huang Y., Chiang C.‐Y., Lee S. K., Gao Y., Hu E. L., Yoreo J. D., Belcher A. M., Nano Lett. 2005, 5, 1429. [DOI] [PubMed] [Google Scholar]
- 111. Merzlyak A., Indrakanti S., Lee S.‐W., Nano Lett. 2009, 9, 846. [DOI] [PubMed] [Google Scholar]
- 112. Chung W.‐J., Oh J.‐W., Kwak K., Lee B. Y., Meyer J., Wang E., Hexemer A., Lee S.‐W., Nature 2011, 478, 364. [DOI] [PubMed] [Google Scholar]
- 113. Yoo S. Y., Chung W.‐J., Kim T. H., Le M., Lee S.‐W., Soft Matter 2011, 7, 363. [Google Scholar]
- 114. Chung W.‐J., Merzlyak A., Yoo S. Y., Lee S.‐W., Langmuir 2010, 26, 9885. [DOI] [PubMed] [Google Scholar]
- 115. Zhu H., Cao B., Zhen Z., Laxmi A. A., Li D., Liu S., Mao C., Biomaterials 2011, 32, 4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ghosh D., Lee Y., Thomas S., Kohli A. G., Yun D. S., Belcher A. M., Kelly K. A., Nat. Nanotechnol. 2012, 7, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. He T., Abbineni G., Cao B., Mao C., Small 2010, 6, 2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Xu H., Cao B., George A., Mao C., Biomacromolecules 2011, 12, 2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lee L. A., Muhammad S. M., Nguyen Q. L., Sitasuwan P., Horvath G., Wang Q., Mol. Pharmaceutics 2012, 9, 2121. [DOI] [PubMed] [Google Scholar]
- 120. Roldão A., Mellado M. C. M., Castiljo L. R., Carrondo M. J. T., Alves P. M., Expert Rev. Vaccines 2010, 9, 1149. [DOI] [PubMed] [Google Scholar]