

Summary
Dietary supplements such as vitamins and minerals are widely used in the hope of improving health, but may have unidentified risks and side effects. In particular, a pathogenic link between dietary supplements and specific oncogenes remains unknown. Here we report that chondroitin-4-sulfate (CHSA), a natural glycosaminoglycan approved as a dietary supplement used for osteoarthritis, selectively promotes tumor growth potential of BRAF V600E-expressing human melanoma cells in patient- and cell line-derived xenograft mice, and confers resistance to BRAF inhibitors. Mechanistically, chondroitin sulfate glucuronyltransferase (CSGlcA-T) signals through its product CHSA to enhance casein kinase 2 (CK2)-PTEN binding and consequent phosphorylation and inhibition of PTEN, which requires CHSA chains and is essential to sustain AKT activation in BRAF V600E-expressing melanoma cells. However, this CHSA-dependent PTEN inhibition is dispensable in cancer cells expressing mutant NRAS or PI3KCA, which directly activate the PI3K-AKT pathway. These results suggest that dietary supplements may exhibit oncogene-dependent pro-tumor effects.
Graphical abstract
The pathogenic links between dietary supplements and oncogenic mutations remain unknown. In this article, Lin et al. demonstrate that chondroitin-4-sulfate, a dietary supplement widely used for osteoarthritis, selectively promotes BRAF-V600E melanoma growth and confers resistance to BRAF inhibitors, suggesting that the generally “safe” dietary supplements may exhibit oncogene-specific pro-tumor effects.
Introduction
The majority of American adults use dietary supplements, which generally include vitamins, minerals, amino acids, fatty acids, fiber, herbs, or other dietary components (Kantor et al., 2016; Knapik et al., 2016). Dietary supplements are intended for ingestion to meet essential nutritional requirements that are not fulfilled through diet alone. For example, essential vitamins and minerals may be taken as dietary supplements to improve the absorption and consumption of these nutrients (Rautiainen et al., 2016; Trivedi and Salvo, 2016). However, dietary supplements are not drugs and thus do not require approval from the US Food and Drug Administration (FDA), which only monitors their safety. Although dietary supplements are generally considered safe, like drugs, these agents may have risks and side effects (Brown, 2017). For example, concerns include high risk of drug interaction problems for cancer patients to take dietary supplements during chemotherapy treatment; an increased cancer risk associated with vitamin A. In addition, antioxidants are widely used in diets and food supplements, which are believed to lower cancer risk by battling radical oxidative species (ROS). However, antioxidants may interfere with certain cancer treatments such as radiation therapy that induce cancer cell death by producing ROS (Schwingshackl et al., 2017; Sprouse and van Breemen, 2016). Recent studies have shown that dietary supplementation with the antioxidants N-acetylcysteine and vitamin E accelerated tumor progression with reduced survival in mouse models of lung cancer, likely by reducing p53 expression (Sayin et al., 2014), and that N-acetylcysteine promoted the metastatic potential but not cell proliferation potential, of melanoma cells in vivo (Le Gal et al., 2015). Moreover, the potential chronic effects of dietary supplements on the pathogenesis and development of human diseases remain unclear. Most importantly, the biological consequences of taking dietary supplements have not been studied in populations with consideration of their specific genetic backgrounds. In particular, the pathogenic links between dietary supplements and specific oncogenic mutations remain unknown. Therefore, studies to determine whether and how dietary supplements promote oncogenesis induced by specific oncogenic mutations will be informative to provide guidance for individuals to select dietary supplements with low cancer risk based on their particular genetic backgrounds.
Chondroitin sulfate is a dietary supplement commonly used for the treatment of osteoarthritis, usually in combination with other ingredients such as glucosamine (Clegg et al., 2006). Chondroitin sulfate is an important structural component of cartilage, which is a sulfated glycosaminoglycan (GAG) composed of a chain of alternating sugars including N-acetylgalactosamine (GalNAc) and glucuronic acid (GlcA) with over 100 disaccharide repeating units (Henrotin et al., 2010). There are four types of chondroitin sulfates including A, C, D and E based on sites of sulfated carbon in sugars. For example, chondroitin-4-sulfate (CHSA) and chondroitin-6-sulfate (CHSC) contain GalNAc with sulfated carbon 4 and 6, respectively, and represent the two major isoforms of chondroitin sulfate in human (Lamari and Karamanos, 2006). Chondroitin sulfate chains usually form part of a proteoglycan by attaching to serine residues of core proteins through a tetrasaccharide bridge in a fixed pattern: chondroitin sulfate-GlcA-Gal-Gal-Xyl-protein (Carney and Muir, 1988; Silbert and Sugumaran, 2002). Commercial dietary chondroitin sulfate supplements are usually manufactured from animal sources including shark and cow cartilage. Orally administered chondroitin sulfate is well absorbed, leading to a significant increase in plasma concentration of chondroitin sulfate with a half-life of 12-24 hours (Conte et al., 1995). In addition to osteoarthritis, which may have a local deficiency or degradation of chondroitin sulfate (Henrotin et al., 2010; Monfort et al., 2008), chondroitin sulfate is also taken orally for other health problems such as joint pain caused by breast cancer treatment, and Kashin-Beck disease that is a chronic type of osteochondropathy (Greenlee et al., 2013; Henrotin et al., 2010).
Chondroitin sulfate has been shown to have a favorable long-term safety profile when taken orally for up to 6 years (Hathcock and Shao, 2007), and is approved in Europe as a slow-acting drug for the treatment of osteoarthritis with only minor side effects such as nausea, mild stomach pain and diarrhea (Clegg et al., 2006; Jordan et al., 2003). Although increased chondroitin sulfate levels in the periacinar and peritumoral fibromuscular stromal tissue were suggested to correlate with progression or recurrence of prostate cancer (Ricciardelli et al., 1997), clinical studies have shown that there was no association between chondroitin sulfate use and the risk of prostate cancer (Brasky et al., 2011). However, these studies were not controlled for populations with specific genetic backgrounds. Thus, the potential pathogenic effects of chronic treatment with chondroitin sulfate on specific human cancer types with particular oncogenic mutational state remain unknown.
Melanoma is one of the most common human cancers, which accounts for approximately 87,110 newly diagnosed patients in 2017 with ∼9,000 death each year (https://www.cancer.org/cancer/melanoma-skin-cancer/about/key-statistics.html). More than 50% of melanomas express oncogenic BRAF V600E mutant, which represents a therapeutic target due to its pathogenic role (Bollag et al., 2012; Gibney et al., 2013; Johnson and Sosman, 2013). We recently performed a systematic RNAi-based screen to identify metabolic vulnerabilities specifically required by oncogenic BRAF V600E mutant, but not other oncogenes such as NRAS Q61R/K, in human melanoma (Kang et al., 2015). The top candidates include a chondroitin N-acetylgalactosaminyltransferase CSGlcA-T, which transfers glucuronic acid (GlcUA) from UDP-GlcUA to N-acetylgalactosamine residues on the non-reducing end of the elongating chondroitin polymer (Rohrmann et al., 1985). Since CSGlcA-T is important for chondroitin polymerization (Izumikawa et al., 2008) and chondroitin sulfate was reported to enter cells through endocytosis (Smedsrod et al., 1985), herein we tested a hypothesis that the widely used dietary supplement chondroitin-4-sulfate may selectively promote BRAF V600E melanoma growth.
Results
Dietary supplement chondroitin-4-sulfate promotes BRAF V600E melanoma growth, which requires the presence of PTEN
We found that oral gavage of chondroitin-4-sulfate (CHSA) resulted in increased growth rates, masses and sizes of tumors in two patient-derived xenograft (PDX) mouse models of melanoma including PDX_TM01612 and PDX_TM01386, where NSG mice were inoculated with human primary melanoma tissues expressing BRAF V600E and wild type PTEN (Figure 1A, left and middle panels; Figure S1A; left panel). Treatment with CHSA resulted in elevated circulating CHSA levels in serum as well as increased intracellular CHSA levels in tumors in these PDX mice (Figure 1A, right panels). In contrast, CHSA treatment did not affect tumor growth rates, masses or sizes in PDX_TM01149 mice inoculated with control primary melanoma tissues expressing wild type BRAF and PTEN (Figure 1B, left and middle panels; Figure S1B, left panel), or in PDX_TM00943 mice inoculated with primary melanoma tissues expressing BRAF V600E but with PTEN deletion (Figure 1C, left and middle panels; Figure S1C, left panel), despite increased serum and intracellular CHSA levels (Figure 1B and 1C, respectively, right panels). Consistent with these findings, CHSA treatment resulted in elevated phosphorylation levels of AKT and its downstream factors BAD and GSK3β, but not MEK1 or ERK1/2 (Figure 1D; Figure S1D, right panel), as well as increased tumor cell proliferation rates assessed by increased immunohistochemistry (IHC) staining of Ki-67 (Figure S1A, right 2 panels) in PDX_TM01612 and PDX_TM01386 tumors derived from BRAF V600E/PTEN-expressing primary melanoma tissues, but not control PDX_TM01149 (BRAF/PTEN; Figures 1D; S1B, right 2 panels), or PDX_TM00943 (BRAF V600E/PTEN del; Figures 1E; S1C, right 2 panels) tumors. These data suggest that the dietary supplement CHSA promotes BRAF V600E melanoma growth, which is likely mediated through enhanced AKT activation by attenuating PTEN.
Figure 1. Dietary supplement chondroitin-4-sulfate (CHSA) selectively promotes BRAF V600E-positive melanoma PDX tumor growth in the presence of PTEN.

(A-C) Xenograft tumor growth (left), tumor weight (middle), mice serum and xenograft tumor CHSA level (right) in NSG mice inoculated with human melanoma BRAF V600E-positive and PTEN-expressing PDX model TM01612 (A, upper panels) and TM01386 (A, lower panels), BRAF wild type and PTEN expressing PDX model TM01149 (B), and BRAF V600E-positive PDX model TM00943 with PTEN deletion (C) and fed with 100mg/kg/day CHSA, or control PBS by daily oral gavage.
(D) Western blot showing AKT, MEK1, and ERK1/2 phosphorylation in tumor lysates obtained from melanoma PTEN-expressing BRAF V600E-positive PDX model TM01612 (left), TM01386 (middle), and PTEN expressing BRAF wild type PDX model TM01149 (right).
(E) Western blot showing AKT, MEK1, and ERK1/2 phosphorylation in tumor lysates obtained from melanoma BRAF V600E-positive PDX model TM00943 with PTEN deletion (left). Right panel shows the PTEN deletion status in TM00943 tumor lysates compared with PTEN wild type expressing melanoma A375 cells as positive control.
Data are representative of two experimental replications. For (A-C), data reflect a single cohort experiment (A, upper: n >=9 tumors from 8 mice per group, lower: n=5 tumors from 8 mice per group; B, n>=14 tumors from 8 mice per group; n=8 tumors from 8 mice per group; C, n=8 tumors from 8 mice per group.) Data are mean ± SEM for tumor growth and mean ± s.d. for tumor weight; p values were obtained by a two-way ANOVA test for tumor growth rates and a two-tailed Student's test for tumor masses (*0.01 < p < 0.05; ***p<0.001; ns, not significant).
See also Figure S1.
CHSA potentiates tumor growth potential and confers drug-resistance to BRAF V600E/PTEN melanoma cells
We next performed additional animal experiments using xenograft mice injected with human melanoma cells. In particular, since activation of the AKT pathway (Delmas et al., 2015; Hu et al., 2013; Perna et al., 2015) and loss of PTEN (Paraiso et al., 2011) have been reported to confer BRAF inhibitor resistance to melanoma cells and mouse models, we hypothesized that CHSA treatment might also mediate BRAF inhibitor resistance in xenograft mice injected with BRAF V600E-expressing melanoma cells. Consistently, we found that oral gavage of CHSA not only increased tumor growth rates, masses and sizes in mice with A375 (Figure 2A, upper left two panels; Figure S2A, upper left panels) or SK-MEL-5 (Figure 2A, lower left two panels; Figure S2A, lower left panels) tumor xenografts expressing BRAF V600E and wild type PTEN, but also reversed the inhibitory effects of BRAF mutant inhibitor PLX4032 on tumor growth and size in these mice (Figure 2A, left two panels; Figure S2A, left panels). Consistent with these results, CHSA treatment resulted in increased serum and intracellular CHSA levels (Figure 2A, middle panels) that correlate with elevated and rescued AKT phosphorylation levels (Figure 2A, right panels) and cell proliferation rates assessed by IHC staining of Ki-67 (Figure S2A, left panels), compared to control mice treated with vehicle or PLX4032, respectively. In contrast, CHSA treatment did not affect growth rates, masses, or sizes of tumors in xenograft mice injected with control HMCB human melanoma cells harboring NRAS Q61K mutant (Figure 2B, left; Figure S2B, left), where increased serum and intracellular CHSA levels (Figure 2B, middle) did not alter phosphorylation levels of AKT, MEK1 or ERK1/2 (Figure 2B, right) or cell proliferation rates assessed by Ki-67 IHC staining (Figure S2B, right).
Figure 2. CHSA specifically promotes tumor growth and confers drug resistance to BRAF V600E/PTEN expressing melanoma cells.

(A-B) Xenograft tumor growth and tumor weight (left 2 panels), CHSA level in mice serum and xenograft tumor (middle upper and lower panels, respectively), and Western blot of tumor lysates showing AKT, MEK1, and EKR1/2 phosphorylation (right) in nude mice inoculated with human melanoma BRAF V600E-positive A375 (A, upper panels) and SK-MEL-5 (A, lower panels) cells, or NRAS-Q61K HMCB cells (B) and fed with CHSA with or without BRAF-V600E inhibitor as indicated.
(C) Xenograft tumor growth and tumor weight (left 2 panels), CHSA level in mice serum and xenograft tumor (middle panels), and Western blot of tumor lysates showing PTEN knock out status, AKT, MEK1, and EKR1/2 phosphorylation in nude mice inoculated with BRAF V600E-positive A375 cell line with PTEN CRISPR/Cas9 knockout.
Data are representative of two experimental replications. For (A-D), data reflect a single cohort experiment (A, upper: n >=9 tumors from 5 mice per group, lower: n=8 tumors from 5 mice per group; B, n=10 tumors from 5 mice per group; C, n=10 tumors from 5 mice per group.) Data are mean ± SEM for tumor growth and mean ± s.d. for tumor weight; p values were obtained by a two-way ANOVA test for tumor growth rates and a two-tailed Student's test for tumor masses (*0.01 < p < 0.05; **0.001 < p < 0.01; ***p<0.001; ns, not significant).
See also Figure S2.
In order to determine whether PTEN presence is required, we generated isogenic A375 cells with stable knockout (KO) of PTEN by CRISPR/Cas9 (Figure S2C). We found that PTEN deletion eliminated CHSA-dependent enhancement of tumor growth rates, masses and sizes, AKT phosphorylation, and cell proliferation rates in mice with A375/PTEN-KO tumor xenografts (Figures 2C and S2D).
Knockdown of CSGlcA-T selectively inhibits BRAF V600E-dependent transformation in melanoma cells expressing PTEN
We next examined whether the chondroitin N-acetylgalactosaminyltransferase CSGlcA-T contributes to BRAF V600E-dependent transformation in melanoma cells, which is important for chondroitin polymerization (Izumikawa et al., 2008) and was identified as a “synthetic lethal” partner of BRAF V600E in human melanoma (Kang et al., 2015). We found that both mRNA and protein expression levels of CSGlcA-T were upregulated (Figure 3A, left and middle panels, respectively), which correlate with increased intracellular CHSA levels (Figure 3A, right), in diverse human melanoma cells, compared to control immortalized human melanocyte Mel-ST cells. In contrast, CSGlcA-T protein levels were not significantly altered among diverse leukemia, lung cancer, and head and neck cancer cells compared to corresponding normal control cells (Figure S3A, upper). Consistent with these findings, data analysis results using Oncomine™ suggest that CSGlcA-T is specifically upregulated in melanoma tissues compared to control peri-tumor tissues among many human cancer types included in the analysis (Figure S3A, lower). These data together suggest a cancer type-specific upregulation of CSGlcA-T in human melanoma.
Figure 3. CSGlcA-T knockdown attenuates CHSA production and selectively inhibits BRAF V600E-dependent melanoma cell transformation in the presence of PTEN.

(A) Real time PCR and Western blot results showing the mRNA and protein level of CSGlcA-T (left and middle, respectively) and Western blot results showing CHSA level in multiple melanoma cell lines comparing with normal melanocyte Mel-ST cells (right).
(B) Effect of CSGlcA-T knockdown on proliferation rates of melanoma HMCB cells harboring NRAS Q61K (left), A375 cells harboring BRAF V600E (middle), or BRAF V600E SK-MEL-24 cells with PTEN deletion (right). Insets: Western blots showing the CSGlcA-T knockdown efficiency in each cell line.
(C) Effect of CSGlcA-T knockdown on monolayer colony formation of diverse melanoma cells including PTEN-expressing NRAS Q61K HMCB cells, BRAF V600E A375 cells, and BRAF V600E SK-MEL-24 cells with PTEN deletion.
(D) Rescue effect of CSGlcA-T knockdown with overexpression of FLAG-tagged wild type CSGlcA-T or mutant enzyme-dead D184A on cell proliferation rates of BRAF V600E melanoma A375 cells.
(E) Effect of CSGlcA-T knockdown on cell proliferation rates of BRAF V600E-positive A375 cell line with PTEN knockout.
(F) Western blot showing effects of CSGlcA-T knockdown on AKT, MEK1, and EKR1/2 phosphorylation in HMCB, A375, SK-MEL-24, and PTEN knockout A375 cells (left to right panels, respectively).
(G) Effect of CSGlcA-T knockdown on xenograft tumor growth (left), tumor weight (middle), and tumor CHSA levels (right) in nude mice inoculated with melanoma BRAF V600E-positive A375 and NRAS Q61K HMCB cells (upper panels), or BRAF V600E-positive SK-MEL-24 cells with PTEN deletion and A375 PTEN knockout cells (lower panels).
(H) Western blot showing AKT, MEK1, and ERK1/2 phosphorylation in tumor lysates obtained from xenograft tumors with CSGlcA-T knockdown.
Data are representative of two experimental replications for (A) and (D), and three experimental replications for (B, C, E, F and H). For (G), data reflect a single cohort experiment (A375 xenograft: n=10 tumors from 12 mice; HMCB xenograft: n=12 tumors from 12 mice; SK-MEL-24 xenograft: n=8 tumors from 12 mice; A375 PTEN-KO xenograft: n=10 tumors from 12 mice.) Data are mean ± SEM for tumor growth and mean ± s.d. for tumor weight; p values were obtained by a two-way ANOVA test for tumor growth rates and a two-tailed Student's test for tumor masses (*0.01 < p < 0.05; **0.001 < p < 0.01; ***p<0.001; ns, not significant). For RT-PCR, cell proliferation and colony formation, error bars indicate ± s.d. of three technical replicates.
See also Figures S3-S4.
Stable knockdown (KD) of CSGlcA-T resulted in reduced cell proliferation rates in A375 and SK-MEL-5 cells expressing BRAF V600E and PTEN, but not in control cells including HMCB and SK-MEL-2 cells expressing mutant NRAS (NRAS Q61K and Q61R, respectively) or SK-MEL-24 and C32 cells expressing BRAF V600E with PTEN deletion (Figures 3B and S3B). In addition, CSGlcA-T knockdown attenuated colony formation potential of A375 cells but not control HMCB or SK-MEL-24 cells (Figure 3C). Consistent with these findings, silencing CSGlcA-T resulted in significantly decreased cell proliferation and decreased AKT phosphorylation in immortalized melanocyte Mel-ST cells expressing BRAF V600E but not in cells expressing BRAF WT (Figure S3C).
We next generated “rescue” A375 cell lines with stable knockdown of endogenous CSGlcA-T and rescue expression of wild-type (WT) or an enzyme-dead D184A mutant (Izumikawa et al., 2008) of an shRNA-resistant, FLAG-tagged human CSGlcA-T form (Figure 3D). Rescue expression of FLAG-CSGlcA-T WT, but not D184A mutant, significantly reversed the reduced cell proliferation upon CSGlcA-T knockdown in A375 cells (Figure 3D). In addition, stable knockout of PTEN also reversed the inhibitory effect of CSGlcA-T knockdown on A375 cell proliferation (Figure 3E). Moreover, PTEN re-expression restored the sensitivity of BRAF V600E/PTEN-null SK-MEL-24 cells to CSGlcA-T knockdown with reduced cell proliferation (Figure S3D). Consistently, CSGlcA-T knockdown resulted in decreased phosphorylation levels of AKT but not MEK1 or ERK1/2 in A375 and SK-MEL-5 cells expressing BRAF V600E and PTEN, as well as in BRAF V600E/PTEN-null SK-MEL-24 cells with PTEN re-expression, but not in control mutant NRAS-expressing HMCB and SK-MEL-2 cells, or BRAF V600E-expressing SK-MEL-24 and C32 cells with PTEN deletion or A375 cells with PTEN KO (Figures 3F, S4A and S4B). Similar results were obtained using a xenograft mouse model, where CSGlcA-T knockdown resulted in decreased intracellular CHSA levels, leading to reduced growth rates, masses, and sizes of tumors (Figure 3G and S4C) as well as decreased AKT phosphorylation (Figure 3H) in nude mice with A375 xenograft tumors but not in control mice with HMCB (NRAS Q61R), SK-MEL-24 (BRAF V600E/PTEN-del), or A375 PTEN KO tumors.
CSGlcA-T inhibits PTEN, which is essential to maintain PIP3 levels and subsequently sustain AKT activation in BRAF V600E-expressing cells but dispensable in cells expressing mutant NRAS or PI3KCA
We next sought to explore the underlying molecular mechanism. We found that CSGlcA-T knockdown resulted in decreased phosphatidylinositol (3,4,5)-trisphosphate (PIP3) levels in A375 and SK-MEL-5 cells (BRAF V600E/PTEN) but not in control mutant NRAS-expressing HMCB and SK-MEL-2 cells or in SK-MEL-24 and C32 cells expressing BRAF V600E with PTEN deletion (Figure 4A). Surprisingly, we found that CSGlcA-T knockdown commonly led to increased PTEN activity with reduced inhibitory phosphorylation at Ser380/Thr382/Thr383 of PTEN in all of the tested melanoma cells despite their different oncogenic mutational backgrounds (Figure 4B, left and right, respectively). However, we found that, although CSGlcA-T knockdown did not alter PI3K kinase activity in these melanoma cells (Figure 4C), the PI3K kinase activity levels in HMCB and SK-MEL-2 cells expressing mutant NRAS were significantly higher than in BRAF V600E expressing melanoma cells including A375, SK-MEL-5, SK-MEL-24 and C32 (Figure 4D). Consistent with these findings, transient transfection of mutant NRAS Q61K resulted in significantly higher PI3K kinase activity in NIH/3T3 cells compared to control cells expressing NRAS, BRAF, or BRAF V600E (Figure S5A). Moreover, treatment with PI3K inhibitor LY294002 resulted in further decreased AKT phosphorylation levels in A375 cells with stable knockdown of CSGlcA-T but not in CSGlcA-T knockdown HMCB or A375 PTEN-KO cells (Figure 4E), suggesting a PI3K-independent mechanism in A375 cells that contributes to AKT phosphorylation, which is likely mediated through PTEN.
Figure 4. CSGlcA-T inhibits PTEN to sustain AKT activation by maintaining PIP3 levels in BRAF V600E-expressing cells but not in cells expressing mutant NRAS or PI3KCA.

(A) Effect of CSGlcA-T knockdown on PI(3,4,5)P3 (PIP3) mass in NRAS mutant HMCB and SK-MEL-2 cells, BRAF V600E A375 and SK-MEL-5 cells, BRAF V600E SK-MEL-24 and C32 cells with PTEN deletion and PTEN knockout A375 cells.
(B) Effect of CSGlcA-T knockdown on PTEN activity (left) and PTEN phosphorylation (right) in NRAS mutant HMCB and SK-MEL-2 cells and BRAF V600E A375 and SK-MEL-5 cells.
(C) Effect of CSGlcA-T knockdown on PI3-kinase activity in NRAS mutant HMCB and SK-MEL-2 cells, BRAF V600E A375 and SK-MEL-5 cells, BRAF V600E SK-MEL-24 and C32 cells with PTEN deletion.
(D) Comparison of PI3-kniase activity in diverse melanoma cell lines with NRAS mutant or BRAF V600E.
(E) Western blot results showing the effect of CSGlcA-T knockdown on AKT phosphorylation of BRAF V600E A375 cells, NRAS Q61R HMCB cells, and A375 PTEN knockout cells in the presence or absence of PI3K inhibitor LY294002.
(F) Rescue effect on cell proliferation rate (left) and AKT phosphorylation (right) by overexpression of PI3KCA H1047R mutant in A375 cells with or without CSGlcA-T knockdown.
(G-H) Effects of stable knockdown of CSGlcA-T on AKT and PTEN phosphorylation (G) and cell proliferation rates (H) in human colorectal WiDr cells (G, left and H, upper) expressing BRAF V600E and PTEN and HT-29 cells expressing BRAF V600E, PTEN and an active PI3KCA P449T mutant (G, right and H, lower).
Data are representative of three experimental replications for (A-C) and two experimental replications for (D-H). Error bars indicate ± s.d. of two technical replications for (A, C, D) and three technical replications for (B, F, H); p values were obtained by a two-tailed Student's test except a two-way ANOVA test for (F and H) (*0.01 < p < 0.05; **0.001 < p < 0.01; ***p<0.001 ns, not significant).
Also see Figure S5.
Consistent with these results, transient transfection with a HA-tagged, constitutively active form of AKT (T308D/S473D; (Scheid et al., 2002)) but not a kinase-dead form of AKT (K179A/T308A/S473A; (Wang et al., 1999)) significantly rescued the decreased cell proliferation rates in A375 cells with stable knockdown of CSGlcA-T (Figure S5B). In addition, expression of a constitutively active form of PI3KCA (H1047R; (Zhao et al., 2005)) also reversed the inhibitory effect of CSGlcA-T knockdown on cell proliferation (Figure 4F, left) as well as AKT phosphorylation (Figure 4F, right) in A375 cells. Moreover, stable knockdown of CSGlcA-T resulted in decreased AKT phosphorylation (Figure 4G, left) and reduced cell proliferation rates (Figure 4H, upper) in human colorectal WiDr cells expressing BRAF V600E and PTEN, but not in control colorectal HT-29 cells expressing BRAF V600E, PTEN and an active PI3KCA P449T mutant (Figure 4G, right and Figure 4H, lower, respectively). These data together suggest that CSGlcA-T-dependent PTEN inhibition represents a common mechanism that contributes to AKT activation in cancer cells, which is essential to sustain AKT activation in BRAF V600E-expressing cells but dispensable in cells expressing mutant NRAS, which directly activates PI3K.
CSGlcA-T signals through CHSA to mediate PTEN inhibition
Consistently, oral gavage with CHSA resulted in decreased PTEN activity in tumors derived from PDX mouse models of melanoma, whereas stable knockdown of CSGlcA-T led to increased PTEN activity in melanoma cell xenograft tumors (Figure 5A, left and right panels, respectively). The decreased and increased PTEN activity correlated with increased and decreased inhibitory phosphorylation levels of PTEN, respectively, in these tumor samples (Figure 5B). We also found that treatment with increasing concentrations of CHSA resulted in increased intracellular CHSA levels in A375 cells in a dose dependent manner (Figure 5C). Confocal imaging revealed that chondroitin sulfate fluorescein, when added to culture media, was taken up into the cytosol of A375 melanoma cells (Figure 5D), likely through endocytosis as reported (Smedsrod et al., 1985). We determined the intracellular concentrations of CHSA in diverse melanoma cells by Western blot using purified CHSA as a standard (Figure S6A), and found intracellular CHSA levels of approximately 99.2, 77.5, 116.7, and 198.0 μg/mL in A375, SK-MEL-5, SK-MEL-2, and HMCB melanoma cells, respectively. Consistently, treatment with increasing concentrations of CHSA resulted in dose dependent increases in cell proliferation rates (Figures 5E, 5G and 5H, left panels) and increased phosphorylation levels of AKT and PTEN (Figures 5E, 5G and 5H, right panels) in A375 cells with endogenous CHSA, whereas CHSA treatment did not affect cell proliferation in A375 PTEN-KO cells with stable knockout of PTEN (Figure 5F) or control cells including PMWK cells expressing wild type BRAF and HMCB cells harboring NRAS Q61R mutation (Figure S6B, left and right, respectively). In addition, CHSA treatment resulted in increased cell proliferation rates (Figures S6C, left) and increased phosphorylation levels of AKT and PTEN (Figures 6C right) in SK-MEL-24 cells with PTEN re-expression. Moreover, CHSA treatment effectively rescued not only the reduced cell proliferation rates but also decreased phosphorylation levels of AKT and PTEN in A375 and SK-MEL-5 cells with stable knockdown of CSGlcA-T (Figure 5G and Figure S6D), and A375 cells treated with BRAF inhibitors including PLX4032 (Figure 5H) or dabrafenib (Figure S6E).
Figure 5. CSGlcA-T signals through CHSA to inhibit PTEN.

(A) Effect of CHSA treatment on PTEN activity in engrafted PDX tumor tissues from NSG mice fed with PBS or CHSA (left) and xenograft tumors of HMCB and A375 cells with or without CSGlcA-T knockdown (right).
(B) Western blot showing the effect on PTEN phosphorylation in PDX tumor tissues from NSG mice fed with PBS or CHSA (left 3 panels) and xenograft tumors of HMCB and A375 cells with CSGlcA-T knockdown (right 2 panels).
(C) Western blot showing the intracellular CHSA level following treatment with increasing concentrations of CHSA in A375 cells (upper). Lower panel shows quantification of the Western blotting.
(D) Confocal imaging results showing the uptake of fluorescein-labelled chondroitin sulfate into the cytosol of HMCB and A375 melanoma cells. Scale bars represent 10 μM.
(E) Effect of increasing treatment dosage of CHSA on cell proliferation rate (left) and phosphorylation level of AKT and PTEN (right) in BRAF V600E A375 melanoma cells.
(F) Effect of CHSA treatment on cell proliferation rate in A375 melanoma cells with PTEN knockout. Inset: Western blot showing the knockout efficiency of PTEN in A375 cells.
(G) Rescue effect of CHSA treatment on cell proliferation rate (left) and phosphorylation level of AKT and PTEN (right) in A375 cells with or without stable knockdown of CSGlcA-T.
(H) Rescue effect of CHSA treatment on cell proliferation rate (left) and phosphorylation level of AKT and PTEN (right) in A375 cells in the presence or absence of BRAF V600E inhibitor PLX4032.
Data are representative of three experimental replications for (A) and two experimental replications for (B-H). Error bars indicate ± s.d. of three technical replications, p values were obtained by a two-tailed Student's test for (A) and a two-way ANOVA test for (E-H) (***p<0.001; ns, not significant).
Also see Figure S6.
Figure 6. The chain form of CHSA is essential for PTEN phosphorylation and inhibition.

(A-B) Effect of CHSA (left), digested CHSA (middle) and disaccharide (right) treatment on AKT and PTEN phosphorylation (A) and PTEN activity (B) in BRAF V600E A375 melanoma cells in a cell free assay.
(C-D) Effect of CHSA (left), digested CHSA (middle) and chondroitin disaccharide (right) treatment on AKT and PTEN phosphorylation (C) and PTEN activity (D) in BRAF V600E SK-MEL-5 melanoma cells in a cell free assay.
(E-F) Effect of CHSA (left), digested CHSA (middle) and chondroitin disaccharide (right) treatment on AKT and PTEN phosphorylation (E) and PTEN activity (F) in control NRAS Q61K expressing HMCB melanoma cells in a cell free assay.
Data are representative of three experimental replications. Error bars indicate ± s.d. of three technical replicates. p values were obtained by a two-tailed Student's test (***p<0.001; ns, not significant).
Also see Figure S7.
CHSA requires its chain form to specifically promote CK2-PTEN binding and consequent phosphorylation and inhibition of PTEN
We next sought to elucidate the molecular mechanism by which CHSA inhibits PTEN. We found that treatment with CHSA in the chain form resulted in increased phosphorylation levels of PTEN and AKT (Figure 6A, left) accompanied by reduced PTEN activity in A375 cells expressing BRAF V600E and PTEN (Figure 6B, left), but not in cells treated with CHSA digested with chondroitinase ABC (Yamagata et al., 1968) that breaks down long chains of chondroitin sulfate to short chains and single disaccharide units (Figures 6A-6B, middle panels) or purified single units of disaccharide (Figures 6A-6B, right panels). Similar results were obtained using BRAF V600E and PTEN-expressing SK-MEL-5 melanoma cells (Figures 6C-6D). In contrast, although treatment with CHSA chains also resulted in increased phosphorylation levels of PTEN (Figure 6E, left) with reduced PTEN activity (Figure 6F, left) in HMCB cells expressing NRAS Q61R mutant, phosphorylation levels of AKT were not altered (Figure 6E, left), whereas treatment with CHSA digested by chondroitinase ABC or purified disaccharide units did not affect phosphorylation levels of AKT and PTEN or PTEN activity in HMCB cells (Figures 6E-6F, middle and right panels, respectively). Consistent with these findings, unlike CHSA chains, treatment with digested CHSA or purified disaccharide did not alter cell proliferation rates of A375 cells (Figure S7A, left and right, respectively).
CK2 was reported to phosphorylate and inhibit PTEN (Song et al., 2012). Since CHSA did not affect PTEN activity when incubated with recombinant PTEN (Figure S7B) or CK2 kinase activity in an in vitro kinase assay where purified CK2 was incubated with purified myelin basic protein (MBP) as a nonspecific exogenous substrate (Figure S7C), we next sought to determine whether CHSA affects CK2-dependent phosphorylation of PTEN. We found that CHSA enhanced CK2-dependent phosphorylation of PTEN in a dose-dependent manner in an in vitro kinase assay where immunoprecipitated CK2 was incubated with recombinant PTEN (rPTEN) in the presence of increasing concentrations of CHSA (Figure 7A, left). In contrast, neither chondroitinase ABC-digested CHSA nor purified disaccharide units were able to affect CK2-dependent PTEN phosphorylation (Figure 7A, middle and right, respectively).
Figure 7. The chain form of CHSA is required for PTEN-CK2 interaction.

(A) CK2 dependent phosphorylation of PTEN in an in vitro kinase assay using endogenous CK2 from A375 melanoma cells treated with CHSA (left), digested CHSA (middle) and disaccharide (right) incubated with recombinant PTEN as substrate.
(B) Effect of CHSA (left) and disaccharide (right) treatment on endogenous CK2-PTEN interaction in BRAF V600E A375 melanoma cells.
(C) Western blot showing the binding ability of purified recombinant PTEN pre-treated with increasing concentration of CHSA with purified CK2 protein (left), as well as the binding ability of purified recombinant CK2 pre-treated with increasing concentration of CHSA with purified PTEN protein (right).
(D) Western blot results showing disaccharide pre-treatment of recombinant PTEN (left) or recombinant CK2 (right) does not affect the binding ability of rPTEN to CK2 or rCK2 to rPTEN, respectively.
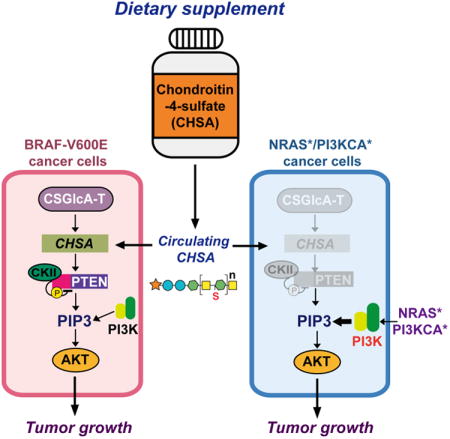
(E) Proposed working model: Dietary supplement CHSA results in increased circulating and intracellular CHSA levels, leading to PTEN inhibition and subsequent AKT activation, and consequently promotes BRAF V600E melanoma tumor growth (left; pink box). CHSA-dependent inhibition of PTEN is dispensable in melanoma cells expressing mutant NRAS or PI3KCA (right; in gray color), which can directly activate PI3K-AKT pathway (right; blue box).
Data are representative of three experimental replications.
Also see Figure S7.
Moreover, we found that CHSA treatment resulted in enhanced CK2-PTEN interaction assessed by increased CK2 co-immunoprecipitated with PTEN in A375 cells (Figure 7B, left) but not in cells treated with purified disaccharide (Figure 7B, right). To determine whether CHSA directly or indirectly affects the CK2-PTEN complex, we performed cell-free, in vitro binding assays using purified recombinant CK2 and PTEN in the presence of increasing amounts of CHSA. We found that purified PTEN pre-treated with increasing concentrations of CHSA showed increased binding ability to CK2 (Figure 7C, left), whereas CHSA-pre-treated CK2 did not demonstrate altered binding ability to PTEN (Figure 7C, right). In contrast, pre-treatment with purified disaccharide did not alter the binding ability of rCK2 or rPTEN to each other (Figure 7D). Together, these data suggest an important role of CHSA in CSGlcA-T mediated inhibition of PTEN, which is specifically essential to sustain AKT activation in BRAF V600E-dependent melanoma transformation.
Discussion
Our results reveal a pathologic link between dietary supplements and specific oncogenic mutations, such that CHSA treatment results in increased circulating and consequently intracellular CHSA levels that promote BRAF V600E tumor growth (Figure 7E, left). Our findings also suggest that, although the majority of widely used dietary supplements are generally safe, some may exhibit pro-tumor effects in populations with specific genetic backgrounds. Thus, deciphering the molecular mechanisms by which certain dietary supplements may increase cancer risk and contribute to tumorigenesis and disease development in patients with particular genetic alterations has highly impactful clinical significance. For example, such information will allow physicians or pharmacists to take an individual's specific genetic background into consideration when providing reliable advice for dietary supplement choices with low cancer risk. Moreover, the availability of such information to the public will educate people to consider dietary supplement selection more specifically based on their genetic background. This may drive individuals to seek advice regarding dietary supplements from their physician or other informed medical sources; unlike drugs, many people currently self-prescribe dietary supplements without consulting a doctor (Brown, 2017). Lastly, these findings further support our previous suggestion to develop the concept of a “precision diet” (Xia et al., 2017), which should be designed based on individual genetic background to include dietary components and supplements that will lower cancer risk and provide cancer prevention.
Our findings have additional clinical impact, suggesting that taking chondroitin sulfate with CHSA as a primary component as a dietary supplement for osteoarthritis or other health problems may not only worsen the disease burden in patients with BRAF V600E melanoma or other related cancers but also increase cancer risk in individuals with BRAF V600E-positive premalignant lesions. In addition, our data also demonstrate that treatment with CHSA confers drug resistance to BRAF V600E-expressing melanoma cells in vitro and in vivo when treated with BRAF inhibitors. Taken together, these findings suggest that CHSA as a dietary supplement may commonly increase cancer risk and/or confer drug resistance through PTEN inhibition, while patients with BRAF V600E melanoma in particular should avoid using chondroitin sulfate as a dietary supplement. Furthermore, data analysis using Oncomine™ and diverse human cancer cells suggest that CSGlcA-T is specifically upregulated in melanoma cells. Therefore, CSGlcA-T may serve as a biomarker for melanoma. Further mechanistic studies are required to elucidate the molecular basis underlying the cancer type-specific upregulation of CSGlcA-T in melanoma.
Our data further suggest that the common upregulation of CSGlcA-T in melanoma cells results in relatively high levels of CHSA, leading to inhibition of PTEN, which is specifically essential to sustain AKT activation in BRAF V600E-expressing melanoma cells (Figure 7E, left). However, this CHSA-dependent inhibition of PTEN is dispensable in melanoma cells expressing mutant NRAS or PI3KCA, which directly activate the PI3K-AKT pathway (Figure 7E, right) (Rodriguez-Viciana et al., 1994). Therefore, these data not only link the CSGlcA-T-CHSA glycosaminoglycan biosynthesis pathway to the CK2-PTEN signaling cascade, suggesting an intracellular signaling function of chondroitin-4-sulfate that is independent of its role as an extracellular matrix component, but also reveal distinct mechanisms to sustain AKT activation required in cancer cells transformed by different oncogenes. This is consistent with previous reports describing a reciprocal mutational status for PTEN and NRAS in human melanoma cells (Tsao et al., 2000; Wu et al., 2003). These findings also suggest that melanoma-specific upregulation of CSGlcA-T may provide a cancer type-specific “evolutionary advantage” that may explain why BRAF mutations are relatively more frequent in melanoma compared with other human malignancies. Since PTEN loss is mostly observed as a late event in melanoma (Wu et al., 2003), it is possible that the CSGlcA-T-CHSA axis-dependent PTEN inhibition may function as an early event and contribute to BRAF V600E-dependent melanoma transformation, while PTEN loss may be a late event as the disease progresses to eventually achieve further PTEN inhibition.
We demonstrated that enzymatic activity is required for CSGlcA-T to contribute to cell proliferation of melanoma cells expressing BRAF V600E, suggesting that the CSGlcA-T-CHSA axis may represent a therapeutic target to treat BRAF V600E positive melanoma. Currently, specific CSGlcA-T inhibitors are not available. Future anti-chondroitin sulfate drugs such as small molecule inhibitors of CSGlcA-T or inhibitory derivatives of chondroitin-4-sulfate that can block chondroitin-4-sulfate-PTEN binding may represent alternative clinical treatments or supplements for BRAF V600E-positive melanoma patients. However, inhibition of the CSGlcA-T-CHSA axis at the whole organism level may cause health problems due to chondroitin sulfate deficiency, such as, but not limited to, osteoarthritis and joint pain; symptoms of CSGlcA-T-deficiency, which is associated with mild skeletal dysplasia and joint laxity (Vodopiutz et al., 2017); or symptoms of Morquio syndrome, which is an autosomal recessive and progressive disease resulting from a deficiency of N-acetylhexosamine sulfate sulfatase that is characterized by abnormal heart and skeletal development, hypermobile joints, and large fingers because of the excretion of keratan sulfate and chondroitin sulfate (Matalon et al., 1974; Mikles and Stanton, 1997). These concerns warrant further detailed toxicity and pharmacokinetics studies to evaluate potential anti-chondroitin sulfate therapies in clinical BRAF V600E-positive cancer treatment.
Our study also raises additional questions. It is not clear why only the chain form of chondroitin-4-sulfate functions to promote CK2-PTEN interaction and consequent phosphorylation and inhibition of PTEN, whereas disaccharide single units or digested CHSA fail to do so. Future molecular and structural studies are warranted. However, this observation is consistent with the previous report that CSGlcA-T is crucial for chondroitin polymerization and elongation (Izumikawa et al., 2008), suggesting the importance of the chain formation in CHSA-dependent PTEN inhibition. Future studies are also warranted to determine whether CHSA specifically binds to PTEN and consequently enhances PTEN recruitment to CK2, and/or stabilizes CK2-PTEN interaction, and whether other types of chondroitin sulfate, in particular chondroitin-6-sulfate, are able to inhibit PTEN in a similar way and consequently promote BRAF V600E tumor growth. Lastly, the limitations of the experimental platforms employed in this study, such that xenograft mouse models of human melanoma cannot represent tumorigenesis and progression of skin cancer, micronutrient delivery to skin tissue, or a relevant immune response, warrant future studies using more informative models such as BRAF V600E-positive genetically engineered mouse (GEM) models of PTEN proficient and deficient melanomas.
Detailed methods are provided in the online version of this paper and include the following:
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jing Chen (jchen@emory.edu).
Experimental Model and Subject Details
Animals
Approval of the use of mice and designed experiments was given by the Institutional Animal Care and Use Committee of Emory University. Nude mice (nu/nu, female 6-week old, Envigo) were used for xenograft studies and NSG mice (NOD scid gamma, female 6-week old, The Jackson Lab) were used for Patient derived xenograft mouse model studies. Animals are drug and test naïve.
Human studies
Approval of the use of human specimens was given by the Institutional Review Board of Emory University School of Medicine. Human melanoma PDX models including TM01612 (SEX: Male; AGE: 50; STAGE/GRADE: IV), TM01386 (SEX: Male; AGE: 60; STAGE/GRADE: not specified), TM01149 (SEX: Male; AGE: 77; STAGE/GRADE: IV), and TM00943 (SEX: Male; AGE: 60; STAGE/GRADE: not specified) were obtained from The Jackson Laboratory. The BRAF gene mutation, disease developmental stage, patient age/sex, and gender identity information were retrieved from the Mouse Tumor Biology Database (MTB), Mouse Genome Informatics, The Jackson Laboratory, Bar Harbor, Maine.
Cell lines
A375, SK-MEL-5, PMWK, WM-266-4, CHL-1, C32, A2058, Mel-ST, and HEK293T were cultured in Dulbecco Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS). HMCB and SK-MEL-2 cells were cultured in Eagle's Minimum Essential Medium (EMEM) with 10% FBS. SK-MEL-24 cells were cultured in EMEM with 15% FBS. NIH-3T3 cells were cultured in DMEM with 10% bovine calf serum (BCS). EL-1 cells were cultured in Iscove's modified Dulbecco's medium (IMDM) supplemented with 0.05mM 2-mercaptoethanol, 0.1mM hypoxanthine, 0.016mM thymidine and 10% bovine serum (FBS). KG-1a, K562, Molm14, NB4, THP-1, NOMO-1, H1299, A549, H157, NCI-H596, EKVX, HCC827, NCI-H358 and H1975 cells were cultured in RPMI 1640 medium with 10% FBS. HOK cell were cultured in Oral Keratinocyte Medium (OKM) with oral keratinocyte supplement (OKGS). Tu212, 212LN, Tu686, Tu167, FaDu, UDSCC22B and UMSCC47 cells were cultured in DMEM/F12 medium with 10% FBS. BEAS-2B cells were cultured using BEGM™ Bronchial Epithelial Cell Growth Medium (LONZA). All the cells were cultured at 37°C and 5% CO2. Cells purchased commercially are authenticated by STR DNA typing. Cells from other sources were not authenticated. All cell lines are negative for mycoplasma contamination. Please also see detailed information of each cell line in KEY RESOURCES TABLE.
Key Resources Table.
| Reagent Or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal AKT1 antibody | Cell Signaling Technology | Cat# 2938S; Clone# C73H10; RRID: AB_915788. |
| Rabbit monoclonal P-AKT (S473) antibody | Cell Signaling Technology | Cat# 4060S; Clone# D9E; RRID: AB_2315049. |
| Rabbit monoclonal P-AKT (T308) antibody | Cell Signaling Technology | Cat# 2965S; Clone# C31E5E; RRID: AB_2255933. |
| Rabbit monoclonal P-MEK1/2 (S217/221) antibody | Cell Signaling Technology | Cat# 9154S; Clone# 41G9; RRID: AB_2138017. |
| Mouse monoclonal MEK1 antibody | Cell Signaling Technology | Cat# 2352S; Clone# 61B12; RRID: AB_10693788. |
| Rabbit polyclonal p44/42 MAP kinase (phosphorylated Erk1/2) antibody | Cell Signaling Technology | Cat# 9101L; Clone# N/A; RRID: AB_331646. |
| Rabbit polyclonal p44/42 MAPK (Erk1/2) antibody | Cell Signaling Technology | Cat# 9102S; Clone# N/A; RRID: AB_330744. |
| Rabbit monoclonal PTEN, P-Ser380/Thr382/Thr383 antibody | Cell Signaling Technology | Cat# 9549S; Clone# 44A7; RRID: AB_659891. |
| Rabbit monoclonal PI3 Kinase p85 antibody | Cell Signaling Technology | Cat# 4257S; Clone# 19H8; RRID: AB_10695255. |
| Rabbit monoclonal P-Bad (S136) antibody | Cell Signaling Technology | Cat# 4366S; Clone# D25H8; RRID: AB_10547878. |
| Rabbit monoclonal Bad antibody | Cell Signaling Technology | Cat# 9239S; Clone# D24A9; RRID: AB_2062127. |
| Rabbit monoclonal P-GSK-3-beta (S9) antibody | Cell Signaling Technology | Cat# 9322S; Clone# D3A4; RRID:AB_2115196. |
| Rabbit monoclonal GSK-3-beta XP antibody | Cell Signaling Technology | Cat# 12456S; Clone# D5C5Z; RRID: N/A. |
| Rabbit polyclonal CSGLCAT antibody | Abcam | Cat# ab75050; Clone# N/A; RRID: AB_1523416. |
| Rabbit polyclonal Ki67 antibody | Abcam | Cat# ab15580; Clone#N/A; RRID: AB_443209. |
| Rabbit polyclonal Anti-Phospho - (Ser/Thr) Phe antibody | Abcam | Cat# ab17464; Clone# N/A; RRID: AB_443891. |
| Rat monoclonal anti-mouse IgG VeriBlot for IP secondary antibody (HRP) | Abcam | Cat# ab131368; Clone# N/A; RRID: N/A. |
| Mouse monoclonal PTEN antibody | Santa Cruz Biotechnology | Cat# sc-7974; Clone# A2B1; RRID: AB_628187. |
| Mouse monoclonal casein kinase II alpha antibody | Santa Cruz Biotechnology | Cat# sc-373894; Clone# E-7; RRID: AB_10947405. |
| Rabbit monoclonal Anti-HA-probe Polyclonal antibody, Unconjugated | Santa Cruz Biotechnology | Cat# sc-805; Clone# Y-11; RRID: AB_631618. |
| Mouse monoclonal anti-Chondroitin 4 Sulfate antibody | Millipore | Cat# MAB2030; Clone# be-123; RRID: AB_94510. |
| Mouse monoclonal ANTI-FLAG M2 antibody | Sigma-Aldrich | Cat# F3165; Clone# M2; RRID: AB_259529. |
| Mouse monoclonal Anti-beta-Actin antibody | Sigma-Aldrich | Cat# A1978; Clone# AC-15; RRID: AB_476692. |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| Patient-derived xenografts (PDX) TM00943; TM01612; TM01386; TM01149. |
The Jackson Laboratory | Mouse Tumor Biology Database (MTB), Mouse Genome Informatics, The Jackson Laboratory, Bar Harbor, Maine. World Wide Web (URL: http://www.informatics.jax.org/) |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Myelin Basic Protein bovine | Sigma-Aldrich | Cat# M1891 |
| Vemurafenib (PLX4032, RG7204) | Selleck Chemicals | Cat# S1267 CAS: 918504-65-1 |
| Dabrafenib (GSK2118436) | Selleck Chemicals | Cat# S2807 CAS: 1195765-45-7 |
| PTEN phospholipid phosphatase | Echelon Biosciences | Cat# S1105 |
| LY294002 | Selleck Chemicals | Cat# E3000 |
| Casein Kinase 2 Protein | Millipore | Cat# 14-197 Product# 6QQ199 |
| Chondroitin sulfate A sodium salt from bovine trachea | Sigma-Aldrich | Cat# C9819 CAS: 39455-18-0 |
| Chondroitinase ABC from Proteus vulgaris | Sigma-Aldrich | Cat# C3367 CAS: 9024-13-9 |
| Chondroitin disaccharide Δdi-4S sodium salt | Sigma-Aldrich | Cat# C4045 CAS: 136144-56-4 |
| Phosphatidylinositol 3,4,5-trisphosphate diC8 (PI(3,4,5)P3 diC8) | Echelon Biosciences | Cat# P-3908 CAS: 163563-77-7 |
| Chondroitin Sulfate Fluorescein | Creatvie PEGWorks | Cat# CS-101 |
| ProLong™ Gold AntifadeMountant with DAPI | Invitrogen | Cat# P36931 |
| Adenosine 5′-triphosphate (ATP) disodium salt hydrate | Sigma-Aldrich | Cat# A1852 CAS: 34369-07-8 |
| Hygromycin B | Gibco | Cat# 10687010 CAS: 31282-04-9 |
| Puromycin dihydrochloride from Streptomyces alboniger | Sigma-Aldrich | Cat# P8833 CAS: 58-58-2 |
| 3,3′-Diaminobenzidine | Sigma-Aldrich | Cat# D8001 CAS: 91-95-2 |
| Hematoxylin | Sigma-Aldrich | Cat# H3136 CAS: 517-28-2 |
| Crystal Violet | Sigma-Aldrich | Cat# C3886 CAS: 548-62-9 |
| Critical Commercial Assays | ||
| PfuTurbo DNA polymerase | Agilent Technologies | Cat# 600250 |
| Two-Step RT-PCR kit | TaKaRa/Clontech | Cat# RR019A |
| BEGM™ Bronchial Epithelial Cell Growth Medium | LONZA | Cat# CC-3170 |
| TRIzol® Reagent RNA extraction | Invitrogen | Cat# 15596026 |
| FuGENE® 6 Transfection Reagent | Promega | Cat# E2691 |
| TransIT®-LT1 Transfection Reagent | Mirus | Cat# MIR 2305 |
| Lipofectamine™ 3000 Transfection Reagent | Invitrogen | Cat# L3000008 |
| PowerUp™ SYBR™ Green Master Mix | Applied Biosystems | Cat# A25742 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | ROCHE | Cat# 4693159001 |
| RIPA Buffer | Sigma-Aldrich | Cat# R0278 |
| PI(3)P Mass ELISA | Echelon Biosciences | Cat# K-3300 |
| Malachite Green Assay Kit | Echelon Biosciences | Cat# K-1500 |
| PI3-Kinase Activity ELISA: Pico | Echelon Biosciences | Cat# K-1000s |
| DakoEnVision+ System, HRP | Dako/Agilent | Cat# K4002 |
| Deposited Data | ||
| Original images were deposited to Mendeley data | This paper | https://doi:10.17632/hmg3v5s7ty.1 |
| Experimental Models: Cell Lines | ||
| Human: A375 cells | ATCC | Cat# CRL-1619; RRID: CVCL_0132 |
| Human: SK-MEL-5 cells | ATCC | Cat# HTB-70; RRID: CVCL_0527 |
| Human: PMWK cells | ATCC | Cat# CRL-2624; RRID: CVCL_A665 |
| Human: WM-266-4 cells | ATCC | Cat# CRL-1676; RRID: CVCL_2765 |
| Human: CHL-1 cells | ATCC | Cat# CRL-9446; RRID: CVCL_1122 |
| Human: A2058 cells | ATCC | Cat# CRL-11147; RRID: CVCL_1059 |
| Human: Mel-ST cells | (Kang et al., 2015) | Cat# N/A; RRID: N/A |
| Human: HEK293T cells | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| Human: HMCB cells | ATCC | Cat# CRL-9607; RRID: CVCL_3317 |
| Human: SK-MEL-2 cells | ATCC | Cat# HTB-68; RRID: CVCL_0069 |
| Human: SK-MEL-24 cells | ATCC | Cat# HTB-71; RRID: CVCL_0599 |
| Human: WiDr cells | ATCC | Cat# CCL-218 RRID: CVCL_2760 |
| Human: HT-29 cells | ATCC | Cat# HTB-38 RRID:CVCL_0320 |
| Mouse: NIH/3T3 cells | ATCC | Cat# CRL-1658; RRID: CVCL_0594 |
| Human: EL 1 | ATCC | Cat# CRL-9854; RRID: CVCL_3680 |
| Human: KG-1α | ATCC | Cat# CRL-246.1; RRID: CVCL_3602 |
| Human: K-562 | ATCC | Cat# CCL-243; RRID: CVCL_0004 |
| Human: Molm14 | (Fan et al., 2016) | Cat# N/A; RRID: N/A |
| Human: NB4 | CLS | Cat# 300299/p638_NB-4; RRID: CVCL_0005 |
| Human: THP-1 | ATCC | Cat# TIB-202; RRID: CVCL_0006 |
| SKM-1 | DSMZ | Cat# ACC-547; RRID: CVCL_0098 |
| Human: NOMO-1 | DSMZ | Cat# ACC-542; RRID: CVCL_1609 |
| Human: BEAS-2B | ATCC | Cat# CRL-9609; RRID: CVCL_0168 |
| Human: H1299 | ATCC | Cat# CRL-5803; RRID: CVCL_0060 |
| Human: A549 | ATCC | Cat# CCL-185; RRID: CVCL_0023 |
| Human: H157 | ATCC | Cat# CRL-5802; RRID: CVCL_0463 |
| Human: NCI-H596 | ATCC | Cat# HTB-178; RRID: CVCL_1571 |
| Human: EKVX | (Fan et al., 2016) | N/A |
| Human: HCC827 | ATCC | Cat# CRL-2868; RRID: CVCL_2063 |
| Human: NCI-H358 | ATCC | Cat# CRL-5807; RRID: CVCL_1559 |
| Human: H1975 | ATCC | Cat# CRL-5908; RRID: CVCL_1511 |
| Human: HOK | (Fan et al., 2016) | N/A |
| Human: Tu212 | (Kang et al., 2010) | N/A |
| Human: 212LN | (Kang et al., 2010) | N/A |
| Human: Tu686 | (Kang et al., 2010) | N/A |
| Human: Tu167 | provided by Dr. Sumin Kang, EMORY University, Atlanta, GA | N/A |
| Human: FaDu | ATCC | Cat# HTB-43; RRID: CVCL_1218 |
| Human: UMSCC47 | (Fan et al., 2016) | N/A |
| Human: UMSCC22B | provided by Dr. Georgia Chen, EMORY University, Atlanta, GA | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Heterozygous Hsd:Athymic Nude-Foxn1nu/Foxn1+ | Envigo/Harlan | Cat# 069(nu)/070(nu/+) |
| Mouse: NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ | The Jackson Laboratory | Stock# 005557 RRID: IMSR_JAX:005557 |
| PDX tumor carrying mouse: NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ | The Jackson Laboratory | N/A |
| Oligonucleotides | ||
| shRNA targeting CSGlcA-T sequence #1: 5′-CGCTCATTTGAACTGGCCAAA-3′ | BROAD Institute/Open Biosystems | N/A |
| shRNA targeting CSGlcA-T sequence #2:5′-CGGCTAGACCAAAGTGATGAA-3′ | BROAD Institute/Open Biosystems | N/A |
| RT-PCR primers for CHPF2, Forward: 5′-GTCACGGAGTCTCCTGCTTC-3′ | This paper | N/A |
| RT-PCR primers for CHPF2, Reverse: 5′-GGTCCCTATTTTTGGCCAGT-3′ | This paper | N/A |
| RT-PCR primers for GAPDH, Forward: 5′-CTGGGCTACACTGAGCACC-3′ | This paper | N/A |
| RT-PCR primers for GAPDH, Reverse: 5′-AAGTGGTCGTTGAGGGCAATG-3′ | This paper | N/A |
| Primers: CSGlcA-T D184A mutation, Forward: 5′-CTTCATGCAGGATGCAACATATGTGCAGGC-3′ | This paper | N/A |
| Primers: CSGlcA-T D184A mutation, Reverse: 5′-GCCTGCACATATGTTGCATCCTGCATGATGAAG-3′ | This paper | N/A |
| Recombinant DNA | ||
| hPTEN CRISPR/Cas9 KO plasmid | Santa Cruz Biotechnology | sc-422475 |
| hPTEN CRISPR/Cas9 HDR plasmid | Santa Cruz Biotechnology | sc-422475-HDR |
| pBabe puro HA PIK3CA H1047R | pBabe puro HA PIK3CA H1047R(Zhao et al., 2005) | Addgene plasmid # 12522 |
| pcDNA3 HA PKB AAA | pcDNA3 HA PKB AAA was a gift from Morris Birnbaum | Addgene plasmid # 16001 |
| HA PKB T308D S473D pcDNA3 | HA PKB T308D S473D pcDNA3 (Scheid et al., 2002) | Addgene plasmid # 14751 |
| pcDNA3 GFP PTEN | 809 pcDNA3 GFP PTEN was a gift from William Sellers (Vazquez et al., 2001) | Addgene plasmid # 10759 |
| pLHCX-FLAG-CSGlcA-T | This paper | N/A |
| pLHCX-FLAG-CSGlcA-T D184A | This paper | N/A |
| pMSCV-FLAG-BRAF | (Kang et al., 2015) | N/A |
| pMSCV-FLAG-BRAF V600E | (Kang et al., 2015) | N/A |
| Software and Algorithms | ||
| Patient-derived xenografts (PDX) | The Jackson Laboratory | http://www.informatics.jax.org/ |
| Human pLKO.1 the RNAi consortium (TRC) Library | BROAD Institute/ Open Biosystems | https://www.broadinstitute.org/rnai-consortium/rnai-consortium-shrna-library |
| CSGlcA-T mRNA expression | ONCOMINE™ | https://www.oncomine.org/resource |
| GraphPad Prism 7 software | GraphPad Software | https://www.graphpad.com/ |
Method Details
Xenograft studies
Nude mice (Heterozygous Hsd:Athymic Nude-Foxn1nu/Foxn1+, Envigo) were subcutaneously injected with 1×106 melanoma cells on the flank. For chondroitin-4-sulfate (CHSA) and BRAF-V600E inhibitor treatment experiments, mouse littermates were randomly assigned to 4 experimental groups when the size of the tumor sizes reached around 50-100 mm3. 100mg/kg/day CHSA and 10mg/kg/day of BRAF-V600E inhibitor PLX4032 were given to mice by oral gavage until the experiment endpoint. PBS was a diluent control. Tumor growth was recorded every two days from 7-12 days after inoculation by measurement of two perpendicular diameters using the formula 4π/3 × (width/2)2 × (length/2). The masses of tumors (mg) derived from treatments were analyzed. For patient-derived xenografts (PDX), NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, The Jackson Lab) carrying patient tumor specimens were sacrificed once the PDX tumor size reached around 1500mm3. The tumor was then excised and small evenly cut pieces were implanted subcutaneously into the flank of 6-week old female NOD.scid mice (The Jackson Laboratory). The mice were evenly divided into 2 groups when tumor sizes reached around 50-100 mm3 before treatment. For CHSA treatment, the dosage of CHSA (Chondroitin sulfate A sodium salt from bovine trachea, Sigma) that given to mice daily was calculated based on that chondroitin sulfate is mostly administered orally at doses ranging from 800 to 2000 mg/day to human (http://www.mayoclinic.org/drugs-supplements/chondroitin-sulfate/dosing/hrb-20058926). Given the average human body weight is around 60kg (Walpole et al., 2012), the suggested dosage is approximately 33mg/kg/day, while several doses can be administered daily to patients with osteoarthritis (Henrotin et al., 2010). Considering all the factors above, 100mg/kg/day CHSA was given to mice by oral gavage for around 30 days until the experiment endpoint. PBS was a diluent control. Tumor growth was recorded every two days from one week after inoculation by measurement of two perpendicular diameters using the formula 4π/3 × (width/2)2 × (length/2). The masses of tumors (mg) derived from treatments were analyzed. Please also see detailed mice PDX model information in KEY RESOURCES TABLE.
Site specific mutagenesis, stable knockdown, and protein overexpression
pLHCX-FLAG-CSGLcA-T D184A plasmid DNA was generated from pLHCX-FLAG-CSGLcA-T through site specific mutagenesis using PfuTurbo DNA polymerase (Agilent Technologies) and PCR. Primers for CSGLcA-T D184A mutagenesis are 5′-CTTCATGCAGGATGCAACATATGTGCAGGC-3′ (Forward) and 5′-GCCTGCACATATGTTGCATCCTGCATGATGAAG-3′ (Reverse). Stable knockdown of endogenous CSGlcA-T was achieved using lentiviral vectors harboring shRNA constructs (Human pLKO.1 the RNAi consortium (TRC) Library, Open Biosystems). shRNA targeting CSGlcA-T sequences are 5′-CGCTCATTTGAACTGGCCAAA-3′ and 5′-CGGCTAGACCAAAGTGATGAA-3′. To produce lentivirus, each construct was co-transfected with psPAX2 packaging plasmid and pMD2.G envelope plasmid (Addgene) into HEK293T cells using TransIT®-LT1 Transfection Reagent (Mirus Bio) according to the manufacturer's instructions. Lentivirus-containing supernatant medium was collected 48 hours after transfection, filtered before adding to the indicated host cell lines. Target cells transduced with lentivirus-containing supernatant were subjected to puromycin (Puromycin dihydrochloride from Streptomyces alboniger, Sigma-Aldrich) selection. Knockdown of endogenous proteins was confirmed by Western blot. Stable overexpression in A375 cells was conducted by using retroviral vectors harboring FLAG-tagged CSGlcA-T wild type, FLAG-tagged CSGlcA-T D184A mutant (pLHCX-FLAG-CSGLcA-T D184A) and HA-tagged PI3KCA active form H1047R (pBabe puro HA PIK3CA H1047R, (Zhao et al., 2005), Addgene). Stable overexpression in Mel-ST cells was conducted by using retroviral vectors harboring FLAG-tagged BRAF and FLAG-tagged BRAF V600E (pMSCV-FLAG-BRAF/FLAG-BRAF V600E) (Kang et al., 2015). Briefly, to produce retrovirus, each construct was co-transfected with VSVG, EcoPak packaging plasmid and envelope plasmid (Addgene) into HEK293T cells using FuGENE™ reagent Transfection Reagent (Promega) according to the manufacturer's instructions. Retrovirus-containing supernatant medium was collected 48 hours after transfection, filtered before adding to the indicated host cell lines with HEPES to adjust the pH. Target cells were subjected to hygromycin (Hygromycin B, Gibco) or puromycin selection. The overexpression of proteins was confirmed by Western blot using antibodies against FLAG or HA. PTEN CRISPR/Cas9 knockout A375 stable cell line was generated by using hPTEN CRISPR/Cas9 KO Plasmid and hPTEN HDR Plasmid (Sand Cruz Biotechnology) followed by manufacture instruction. Briefly, both plasmids were co-transfected into A375 cells using Lipofectamine™ 3000 Transfection Reagent (Invitrogen). Cells were exposed in puromycin 48 hours after transfection for selecting for 24 hours, then the puromycin was removed from the cells and the cells were cultured to reach sufficient number for further experiments. Knockout of PTEN protein was confirmed by Western blot. Please also see detailed information related to shRNA sequence, plasmids name, and reagents information in KEY RESOURCES TABLE.
Antibodies
Antibodies against AKT1, p-AKT (S473), p-AKT (T308), p-MEK1/2 (S217/221), MEK1, p-ERK1/2 (p44/42 MAP kinase (phosphorylated Erk1/2)), ERK1/2 (p44/42 MAPK (Erk1/2)), PI3 Kinase p85, p-PTEN (S380/T382/T383), p-Bad (S136), Bad, p-GSK-3-beta (S9) and GSK-3-beta XP were obtained from Cell Signaling Technology; Antibodies against CSGlcA-T, Ki67, pan Phospho-(Ser/Thr) and Heavy chain-free anti-mouse IgG (anti-mouse IgG VeriBlot for IP secondary antibody (HRP)) were obtained from Abcam; Antibodies against PTEN, CK2 and HA-probe were from Santa Cruz Biotechnology; Antibody against Chondroitin-4-sulfate was from Millipore; Antibodies against FLAG-probe and beta-action were from Sigma-Aldrich.
Transient transfection
HA-tagged pcDNA-AKT-AAA (pcDNA3 HA PKB AAA, kinase dead, Addgene) and HA-tagged pcDNA-AKT-DD (HA PKB T308D S473D pcDNA3, constitutively active, Addgene) (Scheid et al., 2002) were transfected into A375 cells using TransIT®-LT1 Transfection Reagent (Mirus Bio) according the manufacturer's instructions. GFP-tagged pcDNA-PTEN (pcDNA3 GFP PTEN, Addgene) (Vazquez et al., 2001) was transfected into SK-MEL-24 cells using Lipofectamine 3000 Reagent (Invitrogen) according to the manufacturer's instructions. Cells were seeded for cell proliferation assay 24 hours after transfection. Cells were harvested for Western blot 48 hours after transfection.
Cell proliferation assay
Cell proliferation assays were performed by seeding 5 × 104 cells in a 6-well plate with daily counting of cell number. Cell proliferation was determined by cell numbers recorded at 1 to 6 days after being seeded and normalized to that of each of the cell lines at the starting time (T=0 hr) by trypan blue exclusion using TC20 Automated Cell Counter (BioRad).
Quantitative RT-PCR
Total RNA of diverse human melanoma cell lines was extracted by using TRIzol® Reagent RNA extraction (Invitrogen). The total cDNA was prepared through Two-Step RT-PCR kit (TaKaRa/Clontech). Primers for real-time PCR that targeting CHPF2 and GAPDH were obtained from Integrated DNA Technologies (IDT). The real time PCR was performed by using PowerUp™ SYBR™ Green Master Mix and fast 7500 systems (Applied Biosystems™). Primers sequences for gene-specific amplicons are: 5′-GTCACGGAGTCTCCTGCTTC -3′ (CHPF2/forward); 5′- GGTCCCTATTTTTGGCCAGT -3′ (CHPF2/reverse); 5′- CTGGGCTACACTGAGCACC -3′ (GAPDH/forward); 5′-AAGTGGTCGTTGAGGGCAATG -3′ (GAPDH/reverse). Please also see detailed reagent information in KEY RESOURCES TABLE.
Colony formation assay
Briefly, around 500 cells of each melanoma cell lines harboring either pLKO.1 or shCSGLcA-T were seeded per well of 6-well plates for monolayer colony formation. Colonies were stained and analyzed with 0.5% crystal violet (Sigma-Aldrich) 15-20 days after seeding.
Cell culture treatment
CHSA treatment was performed by incubating cells with 20-100μg/ml or indicated concentration for 24-48 hours. BRAF-V600E inhibitor treatment was performed by incubating cells with 10μM Vemurafenib (PLX4032, RG7204, Selleck Chemicals) or Dabrafenib (GSK2118436, Selleck Chemicals) for 12 hours. LY294002 treatment was performed by incubating cells with 10mM LY294002 (Selleck Chemicals) for 8-10 hours.
Sample preparation
For mice serum collection, mice peripheral blood was collected from mice retro-orbitally on the day of the experimental end point. Samples were then incubated on ice for 2 hours and centrifuged at 3000 rpm for 10 minutes (4°C), then immediately applied to the indicated assays. For chondroitin-4-sulfate detection by Western blot, lysates were pre-treated with 0.1-0.5 unit of chondroitinase ABC enzyme (Chondroitinase ABC from Proteus vulgaris, Sigma-Aldrich) per 1mg of cell and serum sample and incubated at room temperature for 1 hour, followed by Western blot. RIPA buffer (Sigma-Aldrich) was used for cell lysate preparation for membrane protein release purpose. For preparation of digested CHSA, in a 50μl reaction, add 10μg CHSA, 0.5 μl (1U) Chondroitinase ABC from Proteus vulgaris (Sigma-Aldrich) in 40mMTris-HCl (adjust pH to 8.0 with 200 mM sodium acetate). Samples were incubated at 37°C for 8-12 hours with gentle shaking. Reaction mixtures were then filtered through 10kD Spin Column (abcam) to remove Chondroitinase ABC.
Detection and quantification of PI(3,4,5)P3 from cells
The PI(3,4,5)P3 (PIP3) level in melanoma cell lines with CSGLcA-T knockdown was assessed using the PI(3)P Mass ELISA Kit (Echelon) following the manufacturer's instructions. PIP3 mass was normalized by cell number of each sample.
PI3-kinase activity assay
Class I phosphoinositide 3-kinase (PI3-K) activity in melanoma cell lines was measured using the PI3-Kinase Activity ELISA Kit (PI3-Kinase Activity ELISA: Pico) from Echelon following the manufacturer's instructions. PI3-Kinase activity was normalized by cell number of each sample. The antibody used for endogenous PI3-Kinase immunoprecipitation was obtained from Cell Signaling Technology.
Confocal fluorescence analysis
A375 and HMCB melanoma cells were seeded in 24 well plates with autoclaved coverslip (Fisher) one day before treatment to let the cells grow and attach onto the surface of the coverslip. 100μg/ml Chondroitin Sulfate Fluorescein (Creatvie PEGWorks) or vehicle control PBS was then added into the culture media for around 24 hours. After treatment, cells were washed with PBS several times to remove the superfluous chondroitin sulfate that may generate background signal. The coverslip with cells was then fixed using 10% paraformaldehyde at room temperature for 10 minutes, followed by washing 3 times with PBS. 7-10μl of ProLong Gold Antifade Mountant solution was placed onto a glass slide with DAPI (ProLong™ Gold Antifade Mountant with DAPI, Invitrogen) and the coverslip placed on the glass slide with the cells facedown. The edges were sealed with transparent nail polish, air dried in the dark, and the cells subjected to confocal fluorescence imaging and data analysis.
Cell-free system assays
Cell free assays were performed as previously described (Lin et. al 2015). Briefly, A375, SK-MEL-5 and HMCB melanoma cells were harvested once confluent, pelleted and re suspended in PBS (final volume approximately 100μl), followed by sonication. The cell mixture was then diluted 4 times, followed by adding different doses of CHSA, digested CHSA or disaccharide unit (Chondroitin disaccharide Δdi-4S sodium salt, Sigma-Aldrich) to reach the final concentrations of 5, 12.5, 25, 50μg/ml (equals to final concentrations of 20, 50, 100, 200μg/ml, respectively, in cells) and incubated for around 1 hour at 37°C with gentle shaking every 10 minutes. After incubation, equal volume of PBS containing 2% NP40 and inhibitor cocktails (cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail, ROCHE) was added into the mixture. The samples were then centrifuged and the supernatant was retained for determination of endogenous PTEN activity as described or for Western blot of PTEN and AKT phosphorylation as well as PTEN-CK2 binding assay.
PTEN activity assay
PTEN activity in the indicated cell lines and tumor tissues was assessed using the PTEN Malachite Green Assay Kit from Echelon. To determine the PTEN activity in cells, endogenous PTEN from 0.5-1×107 cells was bound to protein G-Sepharose beads using PTEN antibody by immunoprecipitation. For PTEN activity in xenograft tumor tissues, around 2mg total tumor lysates were used. The PTEN-bound beads were washed 3 times with 1×TBS and 2 times with PTEN activity assay buffer (TBS plus 10mM DTT). 3000 pmol purified PIP3 was then added into the reaction buffer as reaction substrate. For assays using recombinant PTEN, 100ng purified recombinant PTEN phospholipid phosphatase (Echelon) was added for each reaction. PTEN activity was measured by detecting the free phosphate generated by the conversion of PIP3 (Phosphatidylinositol 3,4,5-trisphosphate diC8 (PI(3,4,5)P3 diC8), Echelon) to PIP2.
Kinase assays
Both endogenous CK2 immunoprecipitated from A375 cells (CK2-bound beads using CK2 antibody from Santa Cruz) and purified recombinant Casein Kinase 2 Protein (Millipore) were used in this study. CK2 in vitro kinase assay was performed by adding CK2 enzyme to 1×CK2 reaction buffer (20 mM Tris-HCl, 50 mM KCl, 10 mM MgCl2, pH 7.5) in the absence of 200μM ATP. For endogenous CK2, beads were washed three times with 1×TBS and twice in 1×CK2 reaction buffer plus ATP (Adenosine 5′-triphosphate (ATP) disodium salt hydrate, Sigma-Aldrich) before use. 100ng recombinant PTEN or 10μg MBP (Myelin Basic Protein bovine, Sigma-Aldrich) was used as substrate per reaction, in the presence or absence of CHSA as indicated. Kinase assay was carried out for 30 minutes at 30°C, followed by SDS-PAGE and Western blot detection.
PTEN and CK2 sequential binding assay
Per reaction, 50ng of purified recombinant PTEN (Echelon) or CK2 (Millipore) was suspended in 500μl 1×TBS, followed by the addition of PTEN or CK2 antibody, respectively. Samples were incubated for about 3 hours at 4°C, then 20μl protein G beads were added into the mixture for further incubation at 4°C for about 1 hour. The beads were then washed 3 times with 1×TBS, followed by incubating in 1×TBS buffer in the presence 0, 20, 50, 100, 200mg/ml CHSA or disaccharide for around 1 hour at 25°C. The protein-bound beads were then washed 3 times with 1×TBS to remove the superfluous CHSA or disaccharide. Next, an equal amount of recombinant CK2 or PTEN was added to the PTEN-bound or CK2-bound beads, respectively. After immunoprecipitation at 4°C overnight, the beads were then washed 3 times with 1×TBS and eluted with SDS buffer for Western blotting.
Immunoprecipitation
For endogenous PTEN and CK2 interaction testing, 1-2mg of total cell lysates were incubated with anti-PTEN or anti-CK2 antibody overnight at 4 °C. After incubation, protein G-Sepharose was used for precipitation for 2 hours. The beads were then washed with 1×TBS and eluted with SDS sample buffer for the Western blotting analysis.
Immunohistochemical staining
Ki67 staining was performed as previously described (Kang et al., 2015). Briefly, resected tumors from PDX and cell line xenograft mice were fixed in 10% buffered formalin, embedded in paraffin and mounted on slides. After deparaffinization and rehydration, mouse tumor sections were then incubated in 3% hydrogen peroxide to suppress endogenous peroxidase activity. Antigen retrieval was achieved by microwaving the sections in 10mM sodium citrate (pH6.0). 10% goat serum was then used to block the sections. Human Ki67 antibody (Abcam) was applied to the mouse tumor sections for overnight at 4°C, respectively. Detection was achieved with the Dako IHC kit (Dako EnVision+ System, HRP, Agilent technologies). Slides were stained with 3,3′-diaminobenzidine (Sigma-Aldrich), washed, counterstained with hematoxylin (Sigma-Aldrich), dehydrated, and mounted. Images of each slide were taken using an inverted microscope for data analysis.
Quantification and Statistical analysis
In studies in which statistical analyses were performed, a 2-tailed Student's t test was used to generate p values, except a two-way ANOVA was used for cell proliferation assay, tumor growth and tumor weight analysis. “n” represents number of animals in each group and the values are included in the main and supplemental figures. p values less than or equal to 0.05 were considered significant. Data shown are from one representative result of multiple experimental replications. Data with error bars represent mean ± s.d., except for tumor growth curves in Figures 1A, 1B, 1C, 2A, 2B, 2C and 3G, which represent mean ± SEM. There is no estimate of variation in each group of data and the variance is similar between the groups. No statistical method was used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment. For animal studies, animals were randomly chosen and concealed allocation and outcome assessment was blinded. All data are expected to have normal distribution. Statistical analysis and graphical presentation was performed using Prism 6.0 (GraphPad).
Data and Software Availability
All software used in this study is listed in the Key Resource Table. Original imaging data have been deposited to Mendeley Data and are available at https://doi:10.17632/hmg3v5s7ty.1.
Supplementary Material
Research Highlight.
Chondroitin-4-sulfate selectively promotes BRAF V600E/PTEN tumor growth
The CSGlcA-T-CHSA axis enhances CK2-dependent inhibition of PTEN
PTEN inhibition by CHSA is dispensable in mutant NRAS or PI3KCA cancer cells
Dietary supplements may exhibit oncogene-dependent pro-tumor effects
Acknowledgments
We thank Dr. Anthea Hammond for critical reading and editing of the manuscript. This work was supported in part by NIH grants CA140515, CA183594, CA174786 (J.C.), Joel A. Katz Music Medicine Fund supported by the T.J. Martell Foundation/Winship Cancer Institute (J.C. and R.L.). R.L. is a Special Fellow of The Leukemia & Lymphoma Society. J.C. is a Winship 5K Scholar.
Footnotes
Supplemental Information: Supplemental information includes seven figures.
Author Contributions: R.L. designed and performed experiments, analyzed data, and wrote the manuscript. S.X., and C.S. designed and performed experiments, and analyzed data. Y.L., X.G., D.C., M.W., H.-B.K., Y.P., and S. Liu performed experiments, and analyzed data. Y.R.C., O.A.-W., T.M., R.R.K., D.H.L. provided crucial study materials and reagents. M.R. analyzed data. F.R.K., S. Lonial, and J.C. supervised studies and edited the manuscript. J.C. designed experiments, analyzed data, and wrote the manuscript.
Declaration of Interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, Hirth P. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nature reviews Drug discovery. 2012;11:873–886. doi: 10.1038/nrd3847. [DOI] [PubMed] [Google Scholar]
- Brasky TM, Kristal AR, Navarro SL, Lampe JW, Peters U, Patterson RE, White E. Specialty supplements and prostate cancer risk in the VITamins and Lifestyle (VITAL) cohort. Nutr Cancer. 2011;63:573–582. doi: 10.1080/01635581.2011.553022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AC. An overview of herb and dietary supplement efficacy, safety and government regulations in the United States with suggested improvements. Part 1 of 5 series. Food Chem Toxicol. 2017;107:449–471. doi: 10.1016/j.fct.2016.11.001. [DOI] [PubMed] [Google Scholar]
- Carney SL, Muir H. The structure and function of cartilage proteoglycans. Physiol Rev. 1988;68:858–910. doi: 10.1152/physrev.1988.68.3.858. [DOI] [PubMed] [Google Scholar]
- Clegg DO, Reda DJ, Harris CL, Klein MA, O'Dell JR, Hooper MM, Bradley JD, Bingham CO, 3rd, Weisman MH, Jackson CG, et al. Glucosamine, chondroitin sulfate and the two in combination for painful knee osteoarthritis. N Engl J Med. 2006;354:795–808. doi: 10.1056/NEJMoa052771. [DOI] [PubMed] [Google Scholar]
- Conte A, Volpi N, Palmieri L, Bahous I, Ronca G. Biochemical and pharmacokinetic aspects of oral treatment with chondroitin sulfate. Arzneimittelforschung. 1995;45:918–925. [PubMed] [Google Scholar]
- Delmas A, Cherier J, Pohorecka M, Medale-Giamarchi C, Meyer N, Casanova A, Sordet O, Lamant L, Savina A, Pradines A, et al. The c-Jun/RHOB/AKT pathway confers resistance of BRAF-mutant melanoma cells to MAPK inhibitors. Oncotarget. 2015;6:15250–15264. doi: 10.18632/oncotarget.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Lin R, Xia S, Chen D, Elf SE, Liu S, Pan Y, Xu H, Qian Z, Wang M, et al. Tetrameric Acetyl-CoA Acetyltransferase 1 Is Important for Tumor Growth. Mol Cell. 2016;64:859–874. doi: 10.1016/j.molcel.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney GT, Messina JL, Fedorenko IV, Sondak VK, Smalley KS. Paradoxical oncogenesis--the long-term effects of BRAF inhibition in melanoma. Nat Rev Clin Oncol. 2013;10:390–399. doi: 10.1038/nrclinonc.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlee H, Crew KD, Shao T, Kranwinkel G, Kalinsky K, Maurer M, Brafman L, Insel B, Tsai WY, Hershman DL. Phase II study of glucosamine with chondroitin on aromatase inhibitor-associated joint symptoms in women with breast cancer. Support Care Cancer. 2013;21:1077–1087. doi: 10.1007/s00520-012-1628-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hathcock JN, Shao A. Risk assessment for glucosamine and chondroitin sulfate. Regul Toxicol Pharmacol. 2007;47:78–83. doi: 10.1016/j.yrtph.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Henrotin Y, Mathy M, Sanchez C, Lambert C. Chondroitin sulfate in the treatment of osteoarthritis: from in vitro studies to clinical recommendations. Ther Adv Musculoskelet Dis. 2010;2:335–348. doi: 10.1177/1759720X10383076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Jin L, Jiang CC, Long GV, Scolyer RA, Wu Q, Zhang XD, Mei Y, Wu M. AEBP1 upregulation confers acquired resistance to BRAF (V600E) inhibition in melanoma. Cell Death Dis. 2013;4:e914. doi: 10.1038/cddis.2013.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumikawa T, Koike T, Shiozawa S, Sugahara K, Tamura J, Kitagawa H. Identification of chondroitin sulfate glucuronyltransferase as chondroitin synthase-3 involved in chondroitin polymerization: chondroitin polymerization is achieved by multiple enzyme complexes consisting of chondroitin synthase family members. J Biol Chem. 2008;283:11396–11406. doi: 10.1074/jbc.M707549200. [DOI] [PubMed] [Google Scholar]
- Johnson DB, Sosman JA. Update on the targeted therapy of melanoma. Current treatment options in oncology. 2013;14:280–292. doi: 10.1007/s11864-013-0226-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan KM, Arden NK, Doherty M, Bannwarth B, Bijlsma JW, Dieppe P, Gunther K, Hauselmann H, Herrero-Beaumont G, Kaklamanis P, et al. EULAR Recommendations 2003: an evidence based approach to the management of knee osteoarthritis: Report of a Task Force of the Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT) Ann Rheum Dis. 2003;62:1145–1155. doi: 10.1136/ard.2003.011742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HB, Fan J, Lin R, Elf S, Ji Q, Zhao L, Jin L, Seo JH, Shan C, Arbiser JL, et al. Metabolic Rewiring by Oncogenic BRAF V600E Links Ketogenesis Pathway to BRAF-MEK1 Signaling. Molecular cell. 2015;59:345–358. doi: 10.1016/j.molcel.2015.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Elf S, Lythgoe K, Hitosugi T, Taunton J, Zhou W, Xiong L, Wang D, Muller S, Fan S, et al. p90 ribosomal S6 kinase 2 promotes invasion and metastasis of human head and neck squamous cell carcinoma cells. J Clin Invest. 2010;120:1165–1177. doi: 10.1172/JCI40582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor ED, Rehm CD, Du M, White E, Giovannucci EL. Trends in Dietary Supplement Use Among US Adults From 1999-2012. JAMA. 2016;316:1464–1474. doi: 10.1001/jama.2016.14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapik JJ, R TJ, Austin KG, Steelman RA, Gannon J, Farina EK, Lieberman HR. Temporal trends in dietary supplement prescriptions of United States military service members suggest a decrease in pyridoxine and increase in vitamin D supplements from 2005 to 2013. Nutr Res. 2016;36:1140–1152. doi: 10.1016/j.nutres.2016.09.001. [DOI] [PubMed] [Google Scholar]
- Lamari FN, Karamanos NK. Structure of chondroitin sulfate. Adv Pharmacol. 2006;53:33–48. doi: 10.1016/S1054-3589(05)53003-5. [DOI] [PubMed] [Google Scholar]
- Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, Dalin MG, Akyurek LM, Lindahl P, Nilsson J, et al. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med. 2015;7:308re308. doi: 10.1126/scitranslmed.aad3740. [DOI] [PubMed] [Google Scholar]
- Matalon R, Arbogast B, Justice P, Brandt IK, Dorfman A. Morquio's syndrome: deficiency of a chondroitin sulfate N-acetylhexosamine sulfate sulfatase. Biochem Biophys Res Commun. 1974;61:759–765. doi: 10.1016/0006-291x(74)91022-5. [DOI] [PubMed] [Google Scholar]
- Mikles M, Stanton RP. A review of Morquio syndrome. Am J Orthop (Belle Mead NJ) 1997;26:533–540. [PubMed] [Google Scholar]
- Monfort J, Pelletier JP, Garcia-Giralt N, Martel-Pelletier J. Biochemical basis of the effect of chondroitin sulphate on osteoarthritis articular tissues. Ann Rheum Dis. 2008;67:735–740. doi: 10.1136/ard.2006.068882. [DOI] [PubMed] [Google Scholar]
- Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson AR, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–2760. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna D, Karreth FA, Rust AG, Perez-Mancera PA, Rashid M, Iorio F, Alifrangis C, Arends MJ, Bosenberg MW, Bollag G, et al. BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc Natl Acad Sci U S A. 2015;112:E536–545. doi: 10.1073/pnas.1418163112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautiainen S, Manson JE, Lichtenstein AH, Sesso HD. Dietary supplements and disease prevention - a global overview. Nat Rev Endocrinol. 2016;12:407–420. doi: 10.1038/nrendo.2016.54. [DOI] [PubMed] [Google Scholar]
- Ricciardelli C, Mayne K, Sykes PJ, Raymond WA, McCaul K, Marshall VR, Tilley WD, Skinner JM, Horsfall DJ. Elevated stromal chondroitin sulfate glycosaminoglycan predicts progression in early-stage prostate cancer. Clin Cancer Res. 1997;3:983–992. [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- Rohrmann K, Niemann R, Buddecke E. Two N-acetylgalactosaminyltransferase are involved in the biosynthesis of chondroitin sulfate. Eur J Biochem. 1985;148:463–469. doi: 10.1111/j.1432-1033.1985.tb08862.x. [DOI] [PubMed] [Google Scholar]
- Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med. 2014;6:221ra215. doi: 10.1126/scitranslmed.3007653. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Marignani PA, Woodgett JR. Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol Cell Biol. 2002;22:6247–6260. doi: 10.1128/MCB.22.17.6247-6260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwingshackl L, Boeing H, Stelmach-Mardas M, Gottschald M, Dietrich S, Hoffmann G, Chaimani A. Dietary Supplements and Risk of Cause-Specific Death, Cardiovascular Disease, and Cancer: A Systematic Review and Meta-Analysis of Primary Prevention Trials. Adv Nutr. 2017;8:27–39. doi: 10.3945/an.116.013516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbert JE, Sugumaran G. Biosynthesis of chondroitin/dermatan sulfate. IUBMB life. 2002;54:177–186. doi: 10.1080/15216540214923. [DOI] [PubMed] [Google Scholar]
- Smedsrod B, Kjellen L, Pertoft H. Endocytosis and degradation of chondroitin sulphate by liver endothelial cells. Biochem J. 1985;229:63–71. doi: 10.1042/bj2290063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- Sprouse AA, van Breemen RB. Pharmacokinetic Interactions between Drugs and Botanical Dietary Supplements. Drug Metab Dispos. 2016;44:162–171. doi: 10.1124/dmd.115.066902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi R, Salvo MC. Utilization and Safety of Common Over-the-Counter Dietary/Nutritional Supplements, Herbal Agents, and Homeopathic Compounds for Disease Prevention. Med Clin North Am. 2016;100:1089–1099. doi: 10.1016/j.mcna.2016.04.017. [DOI] [PubMed] [Google Scholar]
- Tsao H, Zhang X, Fowlkes K, Haluska FG. Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res. 2000;60:1800–1804. [PubMed] [Google Scholar]
- Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem. 2001;276:48627–48630. doi: 10.1074/jbc.C100556200. [DOI] [PubMed] [Google Scholar]
- Vodopiutz J, Mizumoto S, Lausch E, Rossi A, Unger S, Janocha N, Costantini R, Seidl R, Greber-Platzer S, Yamada S, et al. Chondroitin Sulfate N-acetylgalactosaminyltransferase-1 (CSGalNAcT-1) Deficiency Results in a Mild Skeletal Dysplasia and Joint Laxity. Hum Mutat. 2017;38:34–38. doi: 10.1002/humu.23070. [DOI] [PubMed] [Google Scholar]
- Walpole SC, Prieto-Merino D, Edwards P, Cleland J, Stevens G, Roberts I. The weight of nations: an estimation of adult human biomass. BMC Public Health. 2012;12:439. doi: 10.1186/1471-2458-12-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Somwar R, Bilan PJ, Liu Z, Jin J, Woodgett JR, Klip A. Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol Cell Biol. 1999;19:4008–4018. doi: 10.1128/mcb.19.6.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Goel V, Haluska FG. PTEN signaling pathways in melanoma. Oncogene. 2003;22:3113–3122. doi: 10.1038/sj.onc.1206451. [DOI] [PubMed] [Google Scholar]
- Xia S, Lin R, Jin L, Zhao L, Kang HB, Pan Y, Liu S, Qian G, Qian Z, Konstantakou E, et al. Prevention of Dietary-Fat-Fueled Ketogenesis Attenuates BRAF V600E Tumor Growth. Cell metabolism. 2017;25:358–373. doi: 10.1016/j.cmet.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata T, Saito H, Habuchi O, Suzuki S. Purification and properties of bacterial chondroitinases and chondrosulfatases. J Biol Chem. 1968;243:1523–1535. [PubMed] [Google Scholar]
- Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A. 2005;102:18443–18448. doi: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.