

Abstract
Background
Stimulation of soluble guanylate cyclase (sGC) or inhibition of phosphodiesterase type-5 (PDE5) activates protein kinase G-1α (PKG1α) to counteract cardiac hypertrophy and failure. PKG1α acts within localized intracellular domains; however, its oxidation at cysteine-42, linking homo-monomers, alters this localization, impairing suppression of pathological cardiac stress. Since PDE5 and sGC reside in separate micro-domains, we speculated that PKG1α oxidation might also differentially influence the effects from their pharmacological modulation.
Methods and Results
Knock-in mice expressing a redox-dead PKG1α (PKG1αC42S) or littermate controls (PKG1αWT) were subjected to trans-aortic constriction (TAC) to induce pressure-overload, and treated with a PDE5 inhibitor (sildenafil, SIL), sGC activator (BAY-602770, BAY), or vehicle. In PKG1αWT controls, SIL and BAY similarly enhanced PKG activity and reduced pathological hypertrophy/fibrosis and cardiac dysfunction after TAC. However, SIL failed to protect the heart in PKG1αC42S, unlike BAY, which activated PKG and thereby facilitated protective effects. This corresponded with minimal PDE5 activation in PKG1αC42S TAC versus higher activity in controls, and little colocalization of PDE5 with PKG1αC42S (versus co-localization with PKG1αWT) in stressed myocytes.
Conclusions
In the stressed heart and myocytes, PKG1α C42 disulfide formation contributes to PDE5 activation. This augments the pathological role of PDE5 and so in turn enhances the therapeutic impact from its inhibition. PKG1α oxidation does not change the benefits from sGC activation. This finding favors the use of sGC activators regardless of PKG1α oxidation, and may help guide precision therapy leveraging the cGMP/PKG pathway to treat heart disease.
Keywords: heart failure, cyclic GMP, protein kinase G, hypertrophy, pharmacology, oxidative stress
Introduction
Despite many advances in therapy, heart failure (HF) continues to exact an enormous human, societal, and economic toll, and remains a leading cause of hospitalization, morbidity and mortality1. Among potential new therapies under active investigation are novel stimulators of the cyclic guanosine monophosphate (cGMP)-dependent protein kinase-1α (PKG1α) signaling system2-5. This pathway is long known for its vasodilatory role in pulmonary and systemic arterial beds, and has historically been leveraged to unload the right and/or left ventricles and reduce pulmonary congestion. PKG1α also confers direct myocardial effects6, where it serves as a counter-brake to blunt pathological hypertrophy and fibrotic pathways7-10 while also improving calcium homeostasis and reducing myocyte stiffness11, 12. As methods to stimulate PKG1α have expanded, leveraging the therapeutic potential of these myocardial effects has become more feasible.
Cyclic GMP is generated by soluble guanylate cyclase (sGC) activated by nitric oxide (NO) and by particulate GC downstream of natriuretic peptides. Inhibition of selective phosphodiesterases such as PDE5 can also elevate cGMP levels13 to engage PKG1α signaling. Acute studies in humans have employed both approaches and reported improvements in rest and exercise physiology in HF patients, including those with reduced (HFrEF) or preserved (HFpEF) ejection fraction5, 14, 15. Chronic PDE5 inhibition (PDE5-I) has benefited patients with dilated HF in some studies14, but not those with HFpEF4, 16. A Phase II trial with sGC stimulation by vericiguat yielded some potential results with HFrEF, less so in HFpEF2, 3. These findings motivated the current Phase III multicenter trial of this compound for HFrEF (NCT02861534).
A question raised by these variable responses to PDE5 inhibitors versus sGC stimulators is whether these methods of activating PKG1α are interchangeable, or if one method may be particularly more useful in the context of a certain underlying heart disease.
A major factor that impacts both cGMP synthesis and PKG1α signaling is oxidative stress. Oxidation of NO synthase reduces NO generation to favor synthesis of superoxide, and contributes to pathological cardiac remodeling, dysfunction, and vascular disease17, 18. Oxidation of sGC lessens its NO response to compromise cGMP synthesis19. PKG1α can be oxidized at cysteine 42 to form a disulfide bond between homo-dimer subunits20. In resistance arteries, this provides a vasodilatory mechanism in response to H2O221. However, in myocardium, C42-oxidation reduces the capacity of PKG1α activation to counter hormone, hemodynamic22, and cardiotoxic stress23. We found this was related to a plasma-membrane localization of PKG1α when the kinase is activated in a reduced form as opposed to a diffuse distribution when it becomes oxidized. Such membrane localization favors PKG1α targeting of critical proteins at the plasma membrane that are coupled to HF pathophysiology, enhancing its ameliorative impact22.
As the proteins synthesizing or hydrolyzing cGMP are also compartmentalized13, 24, redox-controlled relocalization of PKG1α might also alter its activation by different pharmacological methods to produce variable cardiac responses. The present study tests this hypothesis using intact mouse and cellular models, and compares the outcome and signaling of a selective PDE5 inhibitor versus sGC activator under each redox state of PKG1α. We reveal that the therapeutic impact of PDE5-I to block pathological stress is greatly hampered in myocytes and intact hearts expressing a redox-dead PKG1αC42S, whereas the efficacy of sGC stimulation is unaltered by the oxidative status of PKG1α.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure. This would be accomplished upon reasonable and direct request with the corresponding author, and data transmitted via a secured server by email.
Experimental Models
The study procedures were approved by the Johns Hopkins Animal Care and Use Committee in accordance with NIH guidelines. PKG1αC42S knock-in mice were generated on a C57BL6 background as previously reported21. Male mutant and littermate controls (WT) matched for body size were randomly assigned to experimental groups. Mice were subjected to 3-weeks of pressure overload from trans-aortic constriction (TAC, 7-0 prolene snare sized to a 27G needle)18. Surgical controls were subjected to a sham operation. Drug treatments were randomized between a PDE5-inhibitor (sildenafil; 200 mg/kg/day in Bio-Serv soft rodent chow), soluble guanylate cyclase activator (BAY602770; 0.3 mg/kg/day by oral gavage dissolved in Transcutol:Cremophor:water, 1:2:7 volume ratio), or to a corresponding vehicle. BAY602770 is an sGC heme-independent activator similar in action to cinaciguat25 but different from riociguat or vericiguat that are heme-dependent sGC stimulators26. The dose of BAY602770 was selected as the highest that had no impact on arterial blood pressure in chronically treated mice (Supplemental Figure 1). The sildenafil dose has been shown to be appropriate to achieve therapeutic free plasma concentrations in mice8, and also has no impact on arterial pressure. Serial echocardiography performed in conscious mice (Acuson Sequoia C256, 13-MHz; Siemens) provided indexes of LV chamber size and function18. In our mice, resting LV morphometry and function are nearly identical between both PKG genotypes at the age studied (3-4 months, Supplemental Figure 2) and even at one year (Supplemental Figure 3)22. Data analysis was performed blinded to treatment and genotype.
Isolated Myocyte Studies
Neonatal rat ventricular myocytes (NRVMs) were freshly isolated, and cultured for 24 hours before being infected with recombinant adenovirus (AdV) encoding human FLAG-tagged PKG1αWT or PKG1αC42S as described22. In other studies, NRVMs were co-infected with AdV expressing WT PDE5A, a PKG-phospho-mimetic PDE5 (S92D), or phospho-null PDE5 (S92A). These forms were generated from mouse lung cDNA subcloned into a pDsRed-C1 vector (Clontech)27. Twenty-four hours after infection, myocytes were stimulated with ET1 (10 nM) in serum-free DMEM media supplemented with 0.1% Insulin-Transferrin-Selenium (Life Technologies) for 48 hours. Adult myocytes were also studied as previously described27, isolated from PKG1αWT or PKG1αC42S hearts and exposed to 8-pCPT-cGMP (100 μM) ± sildenafil (1 μM) for 15 minutes with or without the selective PKG1α inhibitor DT3 (1 μM). Myocytes were washed in 1× PBS and flash-frozen as pellets. Lysates were probed by immunoblot for total or phosphorylated PDE5.
Myocardial Analysis
Details of tissue histological and molecular assays for gene and protein expression and phosphorylation are provided in an online supplement.
Statistical Analysis
Data in figures are presented as mean ± SD unless otherwise noted. The in vivo treatment protocols, were performed in separate cohorts, and therefore the analysis was performed within each genotype using a mixed factorial ANOVA with cohort and intervention serving as grouping variables, followed by post-hoc hypothesis testing between interventions using a Tukey test for multiple comparisons. Additional pair-wise testing between gene groups was performed using a Mann-Whitney test with a multiple comparisons correction (Bonferroni). For isolated tissue and cell culture analysis, 1- or 2-way ANOVA, or t-tests were used, or if groups failed normality testing or had significantly different variances (Brown-Forsythe test), a non-parametric Kruskal Wallis or Mann Whitney test was used. Sample size and statistical methods and results are provided in each figure and associated legends. Statistical analysis was performed using Systat Version 10 or GraphPad Ver. 7 software.
Results
PKG1α C42-oxidation is needed for PDE5-I but not sGC activation to counter TAC
PKG1αC42S and littermate PKG1αWT controls were subjected to trans-aortic constriction (TAC) and treated with the PDE5 inhibitor sildenafil (SIL), sGC activator (BAY), or vehicle. In PKG1αWT mice, 3-weeks of TAC doubled LV mass as measured by echocardiography and post-mortem analysis (Fig. 1A, 1B, Supplemental Table). Cardiac hypertrophy declined with either SIL or BAY treatment (e.g. for measured LV mass: -51% and –58% of the increase from TAC alone respectively, both p<0.0001, p=NS versus each other). TAC stimulated 45% less hypertrophy in PKG1αC42S mice (p<0.001), as previously observed22, though this still was significantly above sham control. In PKG1αC42S mice however, SIL did not reduce heart mass (+5%, p=NS), whereas it was diminished by BAY (-35%, p<0.03; red symbols, Fig 1A, 1B). The differential effects of the two drugs on LV mass between genotypes corresponded with molecular markers of pathological hypertrophy (B-type natriuretic peptide (Nppb), and regulator of calcineurin-1 (Rcan1), Supplemental Fig 4). We observed similar disparities on indexes of cardiac function (Fig 1C) and chamber dilation (Fig. 1D); SIL and BAY improved both in PKG1αWT after TAC, but only BAY did so in PKG1αC42S. TAC stimulated myocardial fibrosis significantly more in PKG1αWT than PKG1αC42S hearts, and here too, SIL suppressed fibrosis only in PKG1αWT, whereas BAY did so in both models (Fig. 1E, 1F, Supplemental Figure 5). Thus, PDE5-I counters cardiac pathophysiology induced by TAC so long as PKG1α oxidation at C42 can occur. By contrast, sGC activation is effective independent of this oxidative modification.
Figure 1. Maladaptive responses to pressure-overload are blunted by PDE5 inhibition in mice expressing PKG1αWT (WT) but not PKG1αC42S (KI).
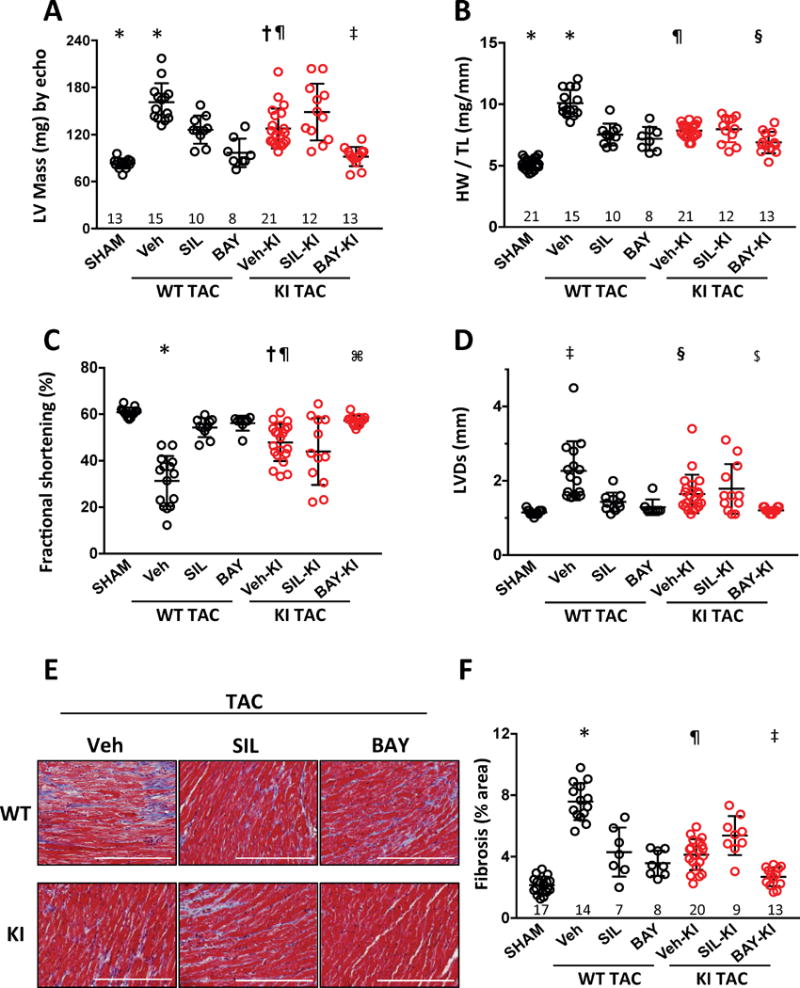
A, B) Effect of sildenafil (SIL), BAY602770 (BAY), or vehicle (Veh) on cardiac hypertrophy induced by TAC in mice expressing WT or C42S mutant PKG1α. Dot plots (mean ± SD). LV mass from echocardiography shown in Panel A; post-mortem heart mass normalized to tibia length in Panel B. Sample size for each group is shown above the x-axis. Statistical results shown are from mixed factorial ANOVA, with experimental cohort and treatment group as categories, and using a Tukey multiple comparisons test for between group significance testing (c.f. Supplemental Table): * p<0.001 vs other WT TAC groups and SHAM; † p=0.009 vs BAY-KI and <0.001 vs SHAM; ‡ p=0.002 vs SIL-KI, ¶ p<0.005 vs Veh-WT; § p=0.004 vs SIL-KI and p=0.001 vs Veh-KI. C, D) Left ventricular fraction shortening and end-systolic diameter from echocardiography analysis. * p<0.001 vs other WT groups; † p<0.001 vs SHAM, p=0.005 vs BAY-KI, p=0.31 vs SIL-KI, ¶ p<.001 vs Veh-WT; ⌘ p=0.001 vs SIL-KI; ‡ p<0.002 vs SHAM, BAY-WT, SIL-WT; § p<0.001 vs BAY-KI and vs SHAM, p=0.02 vs Veh-WT; $ p=0.05 vs SIL-KI. E) Example myocardial histology stained with Masson trichrome. Scale bar = 200 μm. F) Summary of myocardial interstitial fibrosis assessed by Masson’s trichrome staining. *p<0.002 vs SHAM, SIL-WT and BAY-WT; ¶ p<0.001 vs Veh-WT and BAY-KI; ‡ p<0.001 vs SIL-KI.
Preventing PKG1α C42-oxidation blunts PDE5 and corresponding PKG activation
A potential mechanism to explain why SIL is only effective in TAC hearts expressing WT but not PKG1αC42S relates to the impact of cGMP on the probability of forming the C42-dimer23. Binding of cyclic GMP to the regulatory domain of PKG1α leads to physical separation of the N-terminus monomers, reducing the probability of forming a disulfide between C42 residues28. As PDE5 inhibition increases cGMP by decreasing its hydrolysis, it could impede PKG1α oxidation and thereby benefit the heart against stress. However, this mechanism would not alter PKG1αC42S hearts since dimerization is already prevented by the mutation. If true, then SIL treatment should reduce the PKG1α dimer/monomer ratio in WT after TAC. We examined this and found no change in dimer/monomer ratio from SIL in TAC hearts (Fig. 2A). As previously shown22, there was no dimer present in PKG1αC42S.
Figure 2. SIL-induced PKG and TAC-stimulated PDE5 activation are suppressed in PKG1αC42S (KI) mice.
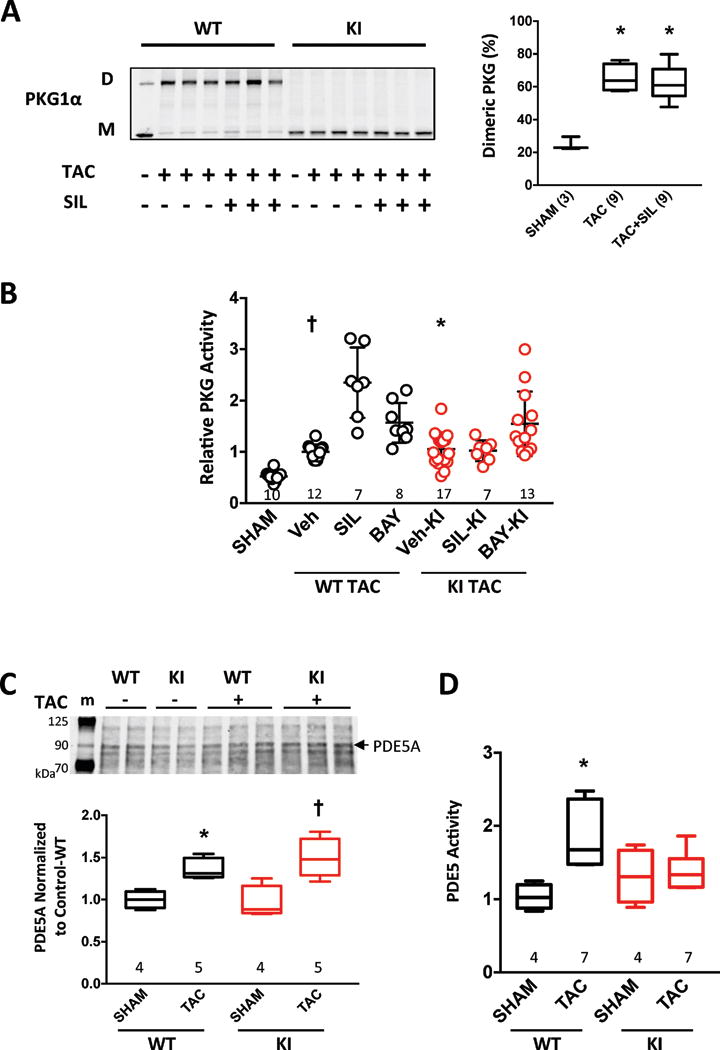
A) TAC stimulates PKG oxidation (D - disulfide dimer, M - monomer) in WT myocardium, and this is not altered by SIL co-treatment. Summary data to the right (Box plot, min-max whiskers; sample size shown, * p<0.0001 vs SHAM by one-way ANOVA. B) Myocardial PKG activity in both genotypes before and after TAC (mean ± SD). SIL increases this further only in WT mice subjected to TAC (p<0.0001 for interaction of genotype with TAC vs TAC+SIL), whereas BAY increases activity similarly in both genotypes (p=0.76 for interaction; p<0.0001 for BAY effect). Sample size provided in figure; results of Kruskal-Wallis analysis with multiple comparisons correction: † p<0.001 vs SHAM, SIL-WT and p=0.003 vs BAY-WT, * p=0.01 vs BAY-KI, p<0.001 vs SHAM. C) PDE5 protein expression in myocardium in control and TAC hearts from PKG1αWT or PKG1αC42S mice; top - example immunoblot (m-marker), bottom – summary data. Two-way ANOVA results: p<0.001 for TAC response, p=0.57 for genotype effect, p=0.32 for interaction of TAC and genotype. *p=0.01, † p<0.001 vs SHAM. D) PDE5 activation after TAC occurs in WT but not PKG1αC42S, p=0.021 for interaction of genotype and TAC. *p=0.001 vs Sham. Data are shown as box and whisker plots.
An alternative explanation for why SIL is ineffective in PKG1αC42S mice is that it does not augment PKG1α activity so long as C42-dimerization is prevented. In vitro myocardial PKG1α activity was similarly elevated in both genotypes after TAC (Fig. 2B). However, whereas TAC+SIL increased PKG1α activity further in PKG1αWT, it had no impact in PKG1αC42S mice. By contrast, BAY treatment increased PKG1α activity similarly in both genotypes (Fig 2B). We tested if the disparity in SIL activation of PKG1α might be due to less PDE5 expression in PKG1αC42S-TAC hearts. However, while TAC increased PDE5 expression modestly, it did so similarly in both models (Fig. 2C, supplemental Figure 6), so this could not explain the disparities. PDE5 activity is also regulated post-translationally, and activity increased after TAC only in PKG1αWT mice (Fig. 2D). This lack of PDE5 activation in PKG1αC42S would be anticipated to blunt the impact of PDE5-I therapy.
PKG1α C42-disulfide enhances PDE5 phosphorylation and thus activation
To further probe mechanisms underlying the lack of SIL responsiveness in PKG1αC42S-TAC, we turned to a cell-based model. Neonatal rat ventricular myocytes (NRVMs) were infected with adenovirus that expressed WT or C42S mutant PKG1α, and then were exposed to 48-hours of endothelin-1 (ET-1). This hypertrophic stress stimulated Nppb and Rcan1 gene expression (markers of pathological hypertrophy), but induction was less in PKG1αC42S versus PKG1αWT expressing cells (Fig. 3A, 3B). Both SIL and BAY suppressed gene expression when PKG1αWT was expressed, but only BAY did so when PKG1αC42S was expressed, recapitulating the in vivo disparities (Fig. 3C). Similar findings were obtained with phenylephrine as the hypertrophic agonist (Supplemental Figure 7). We then tested if SIL or BAY augmented cGMP to the same level independent of the form of PKG1α expressed. Both compounds similarly increased cGMP in myocytes expressing PKG1αWT; however, SIL was somewhat less effective than BAY in PKG1αC42S (Fig. 3D).
Figure 3. Disparity of drug response is observed in cells expressing PKG1αC42S.
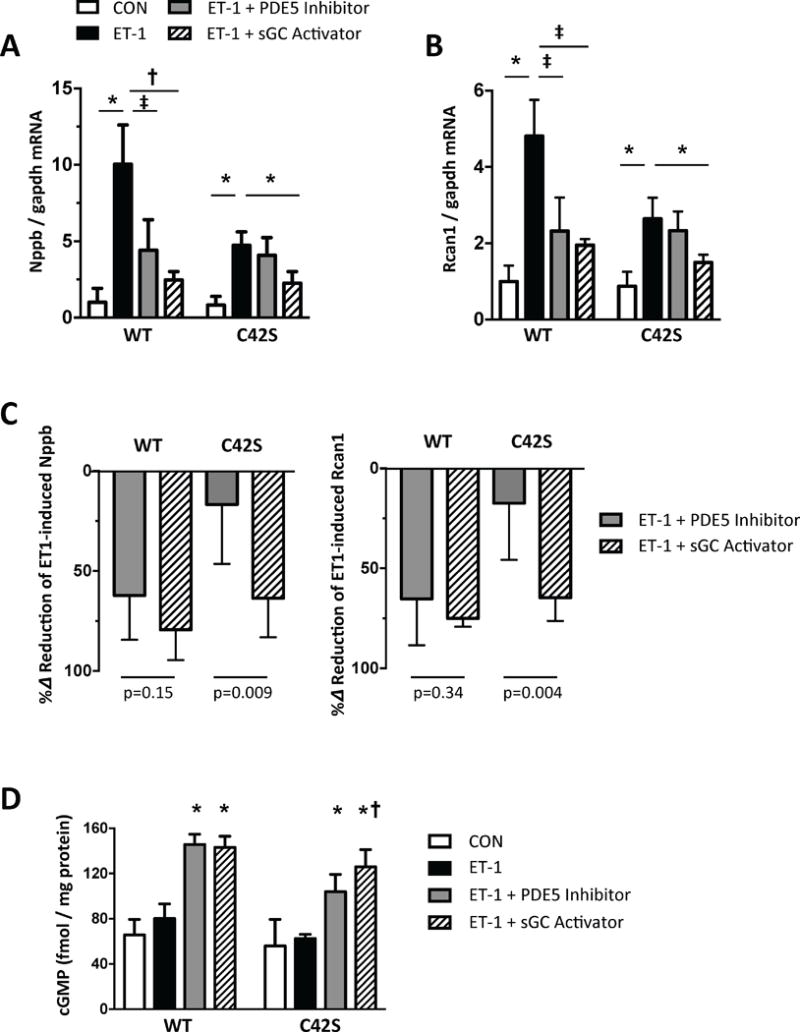
A, B) Nppb and Rcan1 mRNA expression normalized to Gapdh in NRVMs stimulated with endothelin-1 (ET1) ± SIL (1 μM) or BAY602770 (1 nM). N=6/group; * p<0.001, † p=0.003, ‡ p=0.03. C) Percent reduction of Nppb or Rcan1 expression induced by ET-1 due to each drug treatment. N=6/group; p values for Mann-Whitney-U test. D) Myocyte cGMP in NRVMs at rest, and with ET-1 exposure with or without SIL or BAY co-treatment. Analysis by 2 way ANOVA; Genotype p<0.0001; Drug Intervention p<0.0001; interaction p=0.043. Post-hoc Sidak multiple comparisons: * p<0.0001 vs CON and ET-1; † p=0.05 vs PDE5-I in C42S group.
Myocytes expressing either PKG1αWT or PKG1αC42S were next exposed to the cGMP-analog 8-pCPT-cGMP in combination with hydrogen peroxide (H2O2, 10 μM, for 20 minutes), the latter inducing C42-dimerization22. We again found PDE5 to be activated only in cells expressing PKG1αWT (Fig. 4A) despite similar PKG stimulation (Fig. 4B). Administering cGMP alone without H2O2 resulted in similar increases in PDE5 and PKG activity independent of the form of PKG1α expressed (Supplemental Figure 8). Thus, oxidation was required to observe the disparate response due to PKG1α redox genotype.
Figure 4. PKG1α C42 oxidation determines PDE5 activation and SIL efficacy in isolated myocytes.
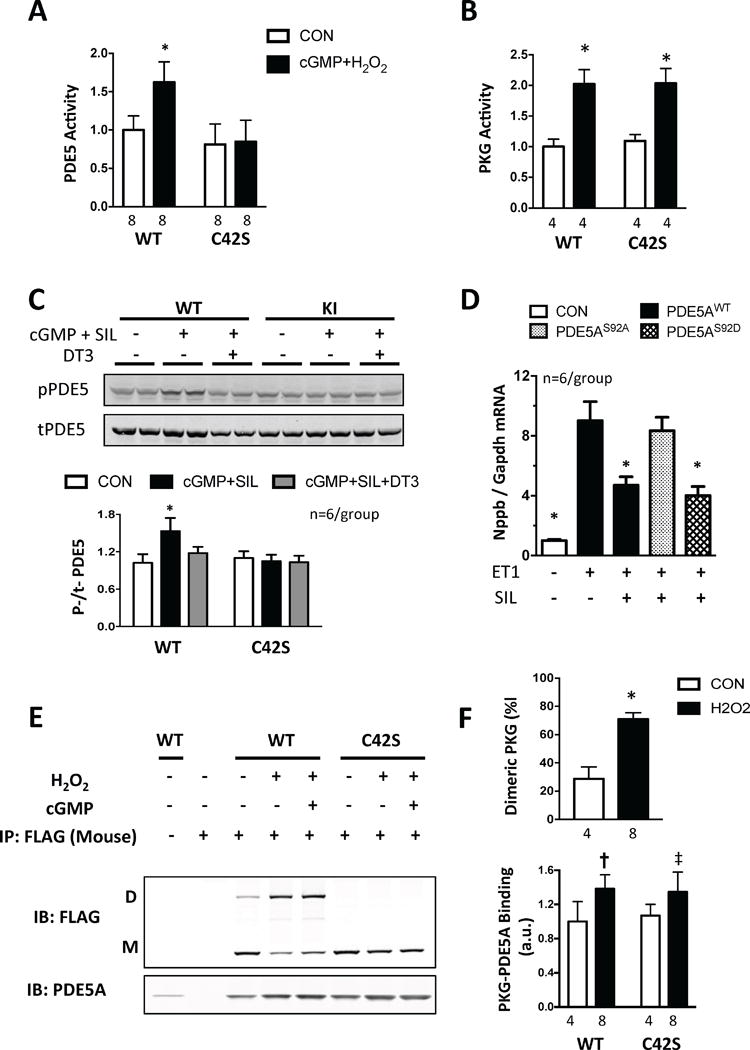
All bar graphs are shown as mean ± SD. A) PDE5 activity in NRVMs exposed to cGMP+ H2O2; p<0.001 for genotype effect; p=0.003 for interaction of cGMP+H2O2 × genotype by 2-way ANOVA; * p<0.001 vs CON. B) PKG activity before and after H2O2+cGMP; p<0.0001 for effect of cGMP+H2O2; p>0.5 for interaction of cGMP+H2O2 × genotype by 2-way ANOVA. C) PDE5 Ser92 phosphorylation and total protein level in adult mouse cardiomyocytes from WT and KI hearts. Example gel and summary data are shown. Cells were stimulated with cGMP+SIL with or without addition of PKG1α inhibitor DT3; * p<0.001 vs other groups. D) Nppb/Gapdh expression in response to ET-1 in NRVMs expressing PDE5WT, PDE5S92D or PDE5S92A. * P<0.001 vs WT and S92A responses, by 1-way ANOVA. E) Non-reducing gel for PKG-PDE5 interaction in NRVMs expressing FLAG-PKG1αWT or FLAG-PKG1αC42S. Pull-down used mouse monoclonal anti-FLAG antibody and gels probed for PDE5A or FLAG to detect dimer/monomer ratio of PKG1α forms. F) Summary data for studies as in Panel E. Upper) The percentage of dimeric PKG increases after H2O2 ± cGMP in WT expressing cells; * p<0.0001 vs CON. Lower) Co-precipitation of PDE5 and PKG1α: p<0.001 for effect of H2O2±cGMP, p=0.54 for stimulus genotype interaction. † p=0.009, ‡ p=0.06 vs CON.
PDE5 activity is increased by cGMP binding to a regulatory domain in the N-terminus (GAF domains) and by PKG-dependent phosphorylation at serine-9229, 30. This provides a negative feedback loop whereby cGMP/PKG activation results in greater PDE5 activity to counter PKG. SIL stimulated slightly less cGMP in PKG1αC42S expressing cells, so this might contribute to reduced PDE5 activity. Serine-92 phosphorylation could also play a role, and this was tested in adult myocytes, where we indeed found SIL increased S92 phosphorylation only in cells that expressed PKG1αWT (Fig. 4C). This change was prevented by co-incubation with the PKG inhibitor DT3, supporting involvement of the kinase. To test the functional impact of S92 phosphorylation on hypertrophic modulation by SIL, cardiomyocytes expressing either normal PDE5WT, or a mutated phospho-silenced (PDE5S92A) or phospho-mimetic (PDE5S92D) form, were exposed to 48-hours of ET1 with or without SIL. When PDE5WT or PDE5S92D was expressed, SIL blocked the hypertrophic response. However, this did not happen if PDE5S92A was expressed (Fig. 4D). Thus, the lack of S92 phosphorylation of PDE5 in cells expressing PKG1αC42S would be anticipated to lessen PDE5 activity, and thus reduce the impact of SIL on blunting hypertrophy.
PDE5 co-localizes with oxidized PKG1αWT more than with PKG1αC42S
To determine if PDE5 and PKG1α interact physically in a manner that hinges on PKG1α oxidation state, we performed immunoprecipitation from NRVMs, pulling down FLAG-tagged PKG1α (WT or C42S) and probing for PDE5. PDE5 co-precipitated with PKG1α, particularly with the addition of H2O2 or cGMP + H2O2. However, this increase was independent of C42-dimerization (i.e. oxidation; Fig. 4E, 4F), so changes in protein-protein interaction were unlikely to explain the results.
An alternative is a change in protein sub-cellular localization that can be missed in an immunoprecipitation assay. We previously reported that PKG1αC42S translocates to and remains primarily at the outer plasma membrane when cells or hearts are subjected to oxidant, hormone, or hemodynamic stress22. By contrast, oxidized WT PKG1α adapts a diffuse cytosolic distribution. Under normal conditions PDE5 is cytosolic, exhibiting a banding pattern coinciding with the Z-band protein α-actinin, but it becomes more diffuse when cells or hearts are stressed31. By contrast, sGC resides both in the cytosol and in the plasma membrane24, 32. Using confocal immunohistochemistry, we assessed the intracellular distribution of WT or C42S PKG1α with PDE5 (Fig. 5A). PDE5 was diffuse and remained so after ET-1 stimulation, and this corresponded to the distribution of PKG1αWT (Fig. 5B). However, PKG1αC42S was intensified at the outer plasma membrane upon exposure to ET1, and no longer localized with PDE5 (Fig. 5C). These findings support the notion that the spatial co-localization of PDE5 and PKG1α is diminished in myocytes subjected to stress so long as PKG1α oxidation at C42 is suppressed.
Figure 5. Oxidized WT but not C42S PKG1α co-localizes with PDE5 in NRVMs subjected to ET-1.
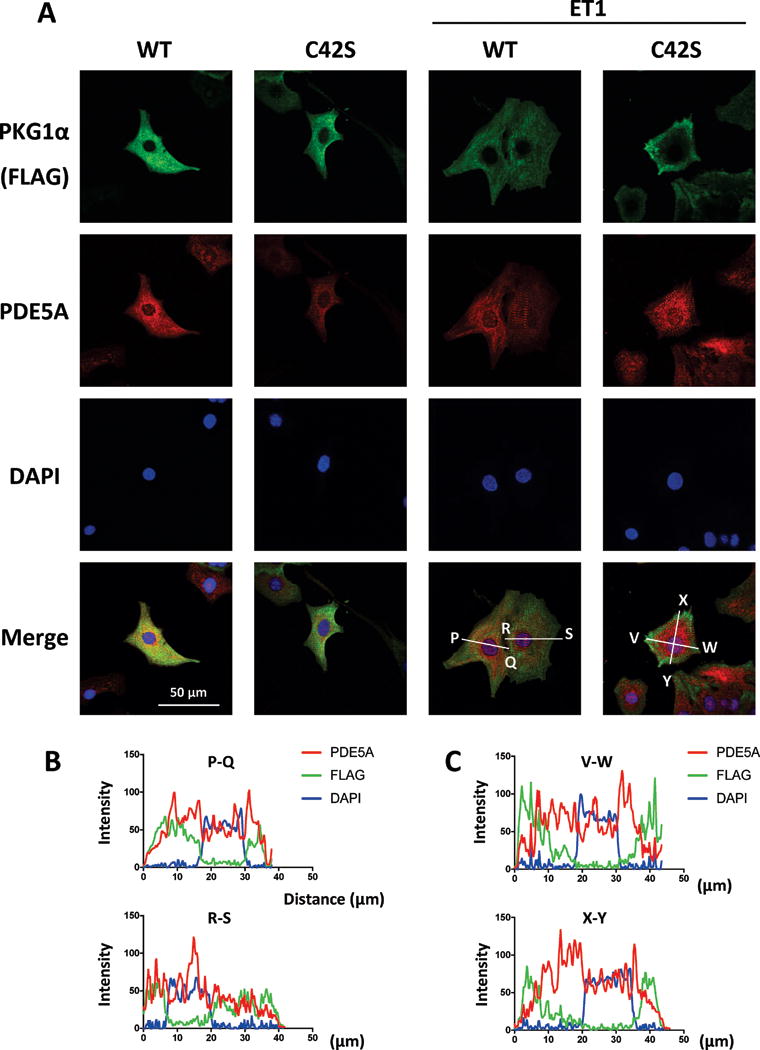
A) Representative confocal images showing subcellular localization of expressed PKG1αWT or PKG1αC42S (green), endogenous PDE5A (red), nuclei (DAPI, blue), and merged images (lower) in NRVMs with and without ET1 stimulation. Scale bar shows =50 μm. B, C) Densitometry line scan analysis shows correspondence of FLAG (PKG) and PDE5 in cells expressing PKG1αWT or PKG1αC42S. X-axis shows distance (μm) and Y-axis fluorescence intensity. In ET-1 exposed cells, PDE5 coincides with PKG1αWT but not PKG1αC42S outside the nucleus. Neither is nuclear in localization.
Discussion
In addition to its role in regulating vasomotor tone, cGMP-PKG1α signaling regulates molecular pathways that counter pathological stress. These include its suppression of Gq- coupled receptor signaling (e.g. angiotensin, endothelin) by activating regulator of G-protein coupled signaling RGS2 and RGS49, 33, and inhibition of the non-selective cation channel - transient receptor potential canonical channel 6 (TRPC6)34, 35; both of which reduce cardiac hypertrophy and fibrosis. Functional cardiac improvement is linked to PKG phosphorylation of phospholamban and troponin I to enhance calcium cycling and relaxation, and of titin to increase muscle compliance11, 12. PKG1α also provides protection against ischemic injury by improving mitochondrial function36, and it enhances protein quality control by increasing activity of the proteasome37.
The capacity of PKG1α to counter maladaptive stress in cardiomyocytes and the intact heart is further determined by the redox state of an intermolecular disulfide bond formed at Cys42 between its monomers. As previously reported22, 23, oxidation of PKG1α at this site diminishes its protective impact, whereas preventing oxidation by expressing a C42S mutation, enhances it. We now demonstrate that this post-translational modification is also an important determinant of the therapeutic efficacy of PDE5 inhibition, a common therapy used to stimulate this pathway. Our results are summarized in Figure 6, which shows that preventing such C42 oxidation decreases stress-induced activation of PDE5 due in part to reduced phosphorylation of PDE5 by PKG at serine 92. This means that despite PDE5-I, cGMP-mediated activation of PKG1α is diminished, and the cardioprotective effects that ensue are blunted. This does not apply to activation of sGC, which augments PKG1α activity and suppresses pathological stress regardless of the redox state of PKG1α. These data show relevance of myocardial oxidative stress to the efficacy of pharmacological methods used to stimulate PKG1α, and they likely have implications for precision therapy.
Figure 6. Summary scheme depicting impact of PKG1α oxidation on the efficacy of PDE5 inhibition or sGC stimulation to exert anti-hypertrophic regulation.
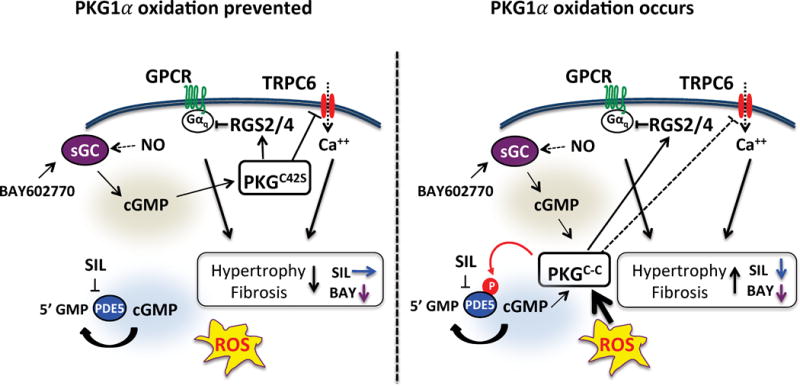
Left: By preventing PKG1α oxidation at C42, protection against hormone or hemodynamic stress is enhanced, with less hypertrophy and fibrosis as a result (black arrow). This involves PKG1αC42S translocation to the plasma membrane where it better inhibits RGS proteins and the cation channel TRPC6 (see text for details). This translocation, however, also moves PKG1αC42S away from PDE5, reducing the impact of PDE5-I (sildenafil, SIL), whereas sGC activation remains capable of activating PKG1αC42S in this location (blue/purple arrows). Right: PKG1α oxidation blunts its anti-hypertrophic/anti-fibrotic effects (black arrow) as it becomes less effective at targeting TRPC6 (as previously reported22 and depicted by a dashed line). However, the diffuse distribution of oxidized PKG1α means it can be impacted by cGMP enhanced following PDE5 inhibition, so now both BAY and SIL are effective as therapies to enhance anti-hypertrophic and fibrotic changes (blue/purple arrows). TRPC6, transient receptor potential canonical channel type 6; RGS, Regulator of G protein signaling; ROS, reactive oxygen species.
PKG1α must interact with specific substrates to achieve its multiple effects, and this is largely regulated by colocalization with the various proteins that generate cGMP or degrade it. An unusual feature of the PKG1α signaling system is that these components are mobile, so interactions and localization can be altered by physiological and pathological signaling inputs. An early example of this mobility was the discovery that inactivation of endothelial NOS (NOS3) in cardiomyocytes leads to migration of PDE5 away from its normal distribution at the z-disk to a more diffuse cytosolic distribution31, 38. A similar re-distribution was demonstrated in chronic heart disease models31, 39. Soluble GC shifts from cytosolic to plasma membrane compartments upon NO stimulation, and in the heart, the latter is further altered by chronic stress, leading to depressed sGC responsiveness to NO24, 32. PKG1α also translocates from the cytosol to the plasma membrane minutes after its stimulation by cGMP9, which is reversed upon its C42 oxidation22. Each of these studies has also shown how a change in location alters the function of a given component of the PKG1α signaling pathway to contribute to myocardial disease.
The current investigation supports the conclusion that the effective interaction between PDE5 and PKG1α, but not sGC and PKG1α, is sensitive to the latter’s redox state. An alternative explanation might have been that by blocking PKG1α oxidation at C42, the kinase became so effective at countering pathological stress that no further improvement was possible. However, TAC still stimulated pathophysiological changes in PKG1αC42S mice, and importantly these responses were diminished by sGC activation but not by SIL. Another alternative explanation is that SIL was not adequately dosed in PKG1αC42S-TAC. However, the doses we used were effective in cells and hearts expressing PKG1αWT. Moreover, even if a higher but still selective dose had more impact, this would still mean there is a substantial rightward shift in the dose response to SIL in PKG1αC42S versus WT controls, a finding with therapeutic implications. One prior study proposed that by elevating cGMP, SIL prevents C42 dimerization so as to mimic the protective effects from PKG1αC42S 23. However, we did not find evidence of such changes as the dimer/monomer ratio in WT-TAC hearts was increased similarly with or without SIL treatment. Furthermore, in the current study, we tested an alternative method to stimulate cGMP (sGC activation) and found it was effective, so the prior study’s explanation could not apply.
The PKG1αC42S redox-dead mutant was first shown to suppress resistance artery dilation in response to an oxidant, supporting prior evidence that PKG1α oxidation activates the kinase independent of cGMP20. However, Kalyanaraman et al40 have contested this, finding that PKG1α oxidation at C42 does not directly increase its activity but rather the C42S mutation reduces its activation by cGMP. In essence, they concluded that PKG1αC42S is a loss-of-function rather than oxidized PKG1α being a gain-of-function. This interpretation, however, cannot explain the behavior in myocardium, where PKG1αC42S is more protective under stress22, 23. We agree that PKG1α activity is probably not the primary altered property due to redox change, but believe its altered localization and consequent interactions are important factors. Kalyanaraman et al40 did not test this, as they used recombinant proteins and in vitro lysates that eliminate subcellular localization effects.
The current findings may have implications for clinical studies involving PDE5 inhibitors. Oxidative stress is a component of most forms of HF, and PKG1α oxidation increases in human failing myocardium22. However, the extent of such oxidation likely varies among patients, and this could impact the efficacy of PDE5 inhibitor strategies. In HFpEF, PDE5-I trials have included many patients with relatively mild cardiac disease, and often without ventricular hypertrophy4. Yet, prior experimental studies found that the milder the cardiac disease induced by loading stress, the less the therapeutic impact from PDE5-I41. The present data provides a new explanation for this finding, as oxidative stress is often greater with more severe disease. Biomarkers of oxidative stress or even oxidation of PKG1α itself may ultimately prove useful to help target PDE5-I therapy. Alternatively, the present findings endorse sGC activation (stimulation) strategies as they are effective regardless of the redox state of PKG1α.
In conclusion, the redox state of PKG1α defined by C42-dimerization determines the therapeutic efficacy of PDE5-I in hearts and cardiomyocytes under pathological stress. By contrast, sGC stimulation is effective regardless of this redox change. The most likely mechanism is that preventing the oxidation of PKG1α depresses its interaction with PDE5, whereas coordinated signaling between proteins is assisted when the kinase is oxidized. This may be specific to PDE5, and further studies assessing other strategies such as natriuretic peptide stimulation, organic nitrite or nitrate42, and PDE9 inhibitors43 are needed. Based on the present results, consideration of oxidative stress and specifically PKG1α oxidation should be made when contemplating the therapeutic use of PDE5 inhibitors.
Supplementary Material
A) What is new
Cyclic GMP-activated protein kinase G-1α conveys signaling linked to the nitric oxide and natriuretic peptide pathways. Its activation by phosphodiesterase type 5 (PDE5) inhibition or soluble guanylate cyclase (sGC) activation is being actively tested as a heart failure therapy.
Oxidation of PKG1α at cysteine-42 reduces its efficacy to counter pathological myocardial stress due to changes in its intracellular localization and thus protein interactions.
We now show that this oxidation is required in order for PDE5 inhibitors to protect against pathological hormone and mechanical stress, whereas sGC activation is protective independent of PKG1α redox state.
B) What are the clinical implications?
Post-translational changes in proteins – e.g. oxidation or phosphorylation - are well recognized, but rarely do they alter the efficacy of pharmaceuticals targeting the protein in an almost binary manner. Oxidative stress is common to most cardiovascular diseases, and PKG1α oxidation is found in failing human hearts.
In patients for whom activation of PKG1α is a desired therapy, those without sufficient myocardial stress to stimulate PKG1α-oxidation may be better served with sGC stimulators rather than PDE5 inhibitors.
When using PDE5 inhibitors in trials or individual patients, the status of myocardial redox maybe an important factor underlying subject response variability.
Acknowledgments
We thank Philip Eaton for providing the PKG1αWT and PKG1αC42S mice, as well as related DNA constructs. BAY 60-2770 was obtained under an MTA with Bayer HealthCare.
Sources of Funding: This work was supported by National Institute of Health Grants (R35 HL135827, RO1HL119012, T32-HL07227), AHA: 16SFRN28620000 Network Grant, Fondation Leducq TransAtlantic Network of Excellence (DAK), American Heart Association Mid-Atlantic Post-Doctoral Fellowship Grant (TN, MJR, DL), F31:HL134196 (KKS), and The Uehara Memorial Foundation Research Fellowship Grant Abroad (TN).
Footnotes
Disclosures: None
References
- 1.Ambrosy AP, Fonarow GC, Butler J, Chioncel O, Greene SJ, Vaduganathan M, Nodari S, Lam CS, Sato N, Shah AN, Gheorghiade M. The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol. 2014;63:1123–1133. doi: 10.1016/j.jacc.2013.11.053. [DOI] [PubMed] [Google Scholar]
- 2.Gheorghiade M, Greene SJ, Butler J, Filippatos G, Lam CS, Maggioni AP, Ponikowski P, Shah SJ, Solomon SD, Kraigher-Krainer E, Samano ET, Muller K, Roessig L, Pieske B, Investigators S-R, Coordinators Effect of Vericiguat, a Soluble Guanylate Cyclase Stimulator, on Natriuretic Peptide Levels in Patients With Worsening Chronic Heart Failure and Reduced Ejection Fraction: The SOCRATES-REDUCED Randomized Trial. JAMA. 2015;314:2251–2262. doi: 10.1001/jama.2015.15734. [DOI] [PubMed] [Google Scholar]
- 3.Pieske B, Maggioni AP, Lam CSP, Pieske-Kraigher E, Filippatos G, Butler J, Ponikowski P, Shah SJ, Solomon SD, Scalise AV, Mueller K, Roessig L, Gheorghiade M. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: results of the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED EF (SOCRATES-PRESERVED) study. Eur Heart J. 2017;38:1119–1127. doi: 10.1093/eurheartj/ehw593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O’Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E, Trial R. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309:1268–1277. doi: 10.1001/jama.2013.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zamani P, Rawat D, Shiva-Kumar P, Geraci S, Bhuva R, Konda P, Doulias PT, Ischiropoulos H, Townsend RR, Margulies KB, Cappola TP, Poole DC, Chirinos JA. Effect of inorganic nitrate on exercise capacity in heart failure with preserved ejection fraction. Circulation. 2015;131:371–380. doi: 10.1161/CIRCULATIONAHA.114.012957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong Q, Blanton RM. Protein kinase G I and heart failure: Shifting focus from vascular unloading to direct myocardial antiremodeling effects. Circ Heart Fail. 2013;6:1268–1283. doi: 10.1161/CIRCHEARTFAILURE.113.000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seo K, Rainer PP, Shalkey Hahn V, Lee DI, Jo SH, Andersen A, Liu T, Xu X, Willette RN, Lepore JJ, Marino JP, Jr, Birnbaumer L, Schnackenberg CG, Kass DA. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc Natl Acad Sci U S A. 2014;111:1551–1556. doi: 10.1073/pnas.1308963111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 9.Takimoto E, Koitabashi N, Hsu S, Ketner EA, Nagayama T, Bedja D, Gabrielson K, Blanton R, Siderovski DP, Mendelsohn ME, Kass DA. RGS2 mediates cardiac compensation to pressure-overload and anti-hypertrophic effects of PDE5 inhibition. J Clin Invest. 2009;119:408–420. doi: 10.1172/JCI35620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X, Eder P, Chang B, Molkentin JD. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci USA. 2010;107:7000–7005. doi: 10.1073/pnas.1001825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kruger M, Kotter S, Grutzner A, Lang P, Andresen C, Redfield MM, Butt E, dos Remedios CG, Linke WA. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res. 2009;104:87–94. doi: 10.1161/CIRCRESAHA.108.184408. [DOI] [PubMed] [Google Scholar]
- 12.Layland J, Li JM, Shah AM. Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol. 2002;540:457–467. doi: 10.1113/jphysiol.2001.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kokkonen K, Kass DA. Nanodomain Regulation of Cardiac Cyclic Nucleotide Signaling by Phosphodiesterases. Ann Rev Pharm Toxicol. 2017;57:455–479. doi: 10.1146/annurev-pharmtox-010716-104756. [DOI] [PubMed] [Google Scholar]
- 14.Guazzi M, Vicenzi M, Arena R, Guazzi MD. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: results of a 1-year, prospective, randomized, placebo-controlled study. Circ Heart Fail. 2011;4:8–17. doi: 10.1161/CIRCHEARTFAILURE.110.944694. [DOI] [PubMed] [Google Scholar]
- 15.Borlaug BA, Koepp KE, Melenovsky V. Sodium Nitrite Improves Exercise Hemodynamics and Ventricular Performance in Heart Failure With Preserved Ejection Fraction. J Am Coll Cardiol. 2015;66:1672–1682. doi: 10.1016/j.jacc.2015.07.067. [DOI] [PubMed] [Google Scholar]
- 16.Hoendermis ES, Liu LC, Hummel YM, van der Meer P, de Boer RA, Berger RM, van Veldhuisen DJ, Voors AA. Effects of sildenafil on invasive haemodynamics and exercise capacity in heart failure patients with preserved ejection fraction and pulmonary hypertension: a randomized controlled trial. Eur Heart J. 2015;36:2565–2573. doi: 10.1093/eurheartj/ehv336. [DOI] [PubMed] [Google Scholar]
- 17.Karbach S, Wenzel P, Waisman A, Munzel T, Daiber A. eNOS uncoupling in cardiovascular diseases–the role of oxidative stress and inflammation. Current Pharm Design. 2014;20:3579–3594. doi: 10.2174/13816128113196660748. [DOI] [PubMed] [Google Scholar]
- 18.Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–1231. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, Ak HS, Meurer S, Deile M, Taye A, Knorr A, Lapp H, Muller H, Turgay Y, Rothkegel C, Tersteegen A, Kemp-Harper B, Muller-Esterl W, Schmidt HH. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006;116:2552–2561. doi: 10.1172/JCI28371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schroder E, Browning DD, Eaton P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317:1393–1397. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- 21.Prysyazhna O, Rudyk O, Eaton P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat Med. 2012;18:286–290. doi: 10.1038/nm.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakamura T, Ranek MJ, Lee DI, Shalkey Hahn V, Kim C, Eaton P, Kass DA. Prevention of PKG1alpha oxidation augments cardioprotection in the stressed heart. J Clin Invest. 2015;125:2468–2472. doi: 10.1172/JCI80275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prysyazhna O, Burgoyne JR, Scotcher J, Grover S, Kass D, Eaton P. Phosphodiesterase 5 Inhibition Limits Doxorubicin-induced Heart Failure by Attenuating Protein Kinase G Ialpha Oxidation. J Biol Chem. 2016;291:17427–17436. doi: 10.1074/jbc.M116.724070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai EJ, Liu Y, Koitabashi N, Bedja D, Danner T, Jasmin JF, Lisanti MP, Friebe A, Takimoto E, Kass DA. Pressure-overload-induced subcellular relocalization/oxidation of soluble guanylyl cyclase in the heart modulates enzyme stimulation. Circ Res. 2012;110:295–303. doi: 10.1161/CIRCRESAHA.111.259242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knorr A, Hirth-Dietrich C, Alonso-Alija C, Harter M, Hahn M, Keim Y, Wunder F, Stasch JP. Nitric oxide-independent activation of soluble guanylate cyclase by BAY 60-2770 in experimental liver fibrosis. Arzneimittel-Forschung. 2008;58:71–80. doi: 10.1055/s-0031-1296471. [DOI] [PubMed] [Google Scholar]
- 26.Breitenstein S, Roessig L, Sandner P, Lewis KS. Novel sGC Stimulators and sGC Activators for the Treatment of Heart Failure. Handbook of Experimental Pharmacology. 2017;243:225–247. doi: 10.1007/164_2016_100. [DOI] [PubMed] [Google Scholar]
- 27.Zhang M, Takimoto E, Hsu S, Lee DI, Nagayama T, Danner T, Koitabashi N, Barth AS, Bedja D, Gabrielson KL, Wang Y, Kass DA. Myocardial remodeling is controlled by myocyte-targeted gene regulation of phosphodiesterase type 5. J Am Coll Cardiol. 2010;56:2021–2030. doi: 10.1016/j.jacc.2010.08.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burgoyne JR, Prysyazhna O, Rudyk O, Eaton P. cGMP-dependent activation of protein kinase G precludes disulfide activation: implications for blood pressure control. Hypertension. 2012;60:1301–1308. doi: 10.1161/HYPERTENSIONAHA.112.198754. [DOI] [PubMed] [Google Scholar]
- 29.Rybalkin SD, Rybalkina IG, Shimizu-Albergine M, Tang XB, Beavo JA. PDE5 is converted to an activated state upon cGMP binding to the GAF A domain. EMBO J. 2003;22:469–478. doi: 10.1093/emboj/cdg051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas MK, Francis SH, Corbin JD. Substrate- and kinase-directed regulation of phosphorylation of a cGMP-binding phosphodiesterase by cGMP. J Biol Chem. 1990;265:14971–14978. [PubMed] [Google Scholar]
- 31.Zhang M, Takimoto E, Lee DI, Santos CX, Nakamura T, Hsu S, Jiang A, Nagayama T, Bedja D, Yuan Y, Eaton P, Shah AM, Kass DA. Pathological cardiac hypertrophy alters intracellular targeting of phosphodiesterase type 5 from nitric oxide synthase-3 to natriuretic peptide signaling. Circulation. 2012;126:942–951. doi: 10.1161/CIRCULATIONAHA.112.090977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zabel U, Kleinschnitz C, Oh P, Nedvetsky P, Smolenski A, Muller H, Kronich P, Kugler P, Walter U, Schnitzer JE, Schmidt HH. Calcium-dependent membrane association sensitizes soluble guanylyl cyclase to nitric oxide. Nat Cell Biol. 2002;4:307–311. doi: 10.1038/ncb775. [DOI] [PubMed] [Google Scholar]
- 33.Tokudome T, Kishimoto I, Horio T, Arai Y, Schwenke DO, Hino J, Okano I, Kawano Y, Kohno M, Miyazato M, Nakao K, Kangawa K. Regulator of G-protein signaling subtype 4 mediates antihypertrophic effect of locally secreted natriuretic peptides in the heart. Circulation. 2008;117:2329–2339. doi: 10.1161/CIRCULATIONAHA.107.732990. [DOI] [PubMed] [Google Scholar]
- 34.Koitabashi N, Aiba T, Hesketh GG, Rowell J, Zhang M, Takimoto E, Tomaselli GF, Kass DA. Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition. J Mol Cell Cardiol. 2010;48:713–724. doi: 10.1016/j.yjmcc.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinoshita H, Kuwahara K, Nishida M, Jian Z, Rong X, Kiyonaka S, Kuwabara Y, Kurose H, Inoue R, Mori Y, Li Y, Nakagawa Y, Usami S, Fujiwara M, Yamada Y, Minami T, Ueshima K, Nakao K. Inhibition of TRPC6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A signaling in the heart. Circ Res. 2010;106:1849–1860. doi: 10.1161/CIRCRESAHA.109.208314. [DOI] [PubMed] [Google Scholar]
- 36.Costa AD, Pierre SV, Cohen MV, Downey JM, Garlid KD. cGMP signalling in pre- and post-conditioning: the role of mitochondria. Cardiovasc Res. 2008;77:344–352. doi: 10.1093/cvr/cvm050. [DOI] [PubMed] [Google Scholar]
- 37.Ranek MJ, Terpstra EJ, Li J, Kass DA, Wang X. Protein kinase g positively regulates proteasome-mediated degradation of misfolded proteins. Circulation. 2013;128:365–376. doi: 10.1161/CIRCULATIONAHA.113.001971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takimoto E, Champion HC, Belardi D, Moslehi J, Mongillo M, Mergia E, Montrose DC, Isoda T, Aufiero K, Zaccolo M, Dostmann WR, Smith CJ, Kass DA. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res. 2005;96:100–109. doi: 10.1161/01.RES.0000152262.22968.72. [DOI] [PubMed] [Google Scholar]
- 39.Senzaki H, Smith CJ, Juang GJ, Isoda T, Mayer SP, Ohler A, Paolocci N, Tomaselli GF, Hare JM, Kass DA. Cardiac phosphodiesterase 5 (cGMP-specific) modulates beta-adrenergic signaling in vivo and is down-regulated in heart failure. FASEB J. 2001;15:1718–1726. doi: 10.1096/fj.00-0538com. [DOI] [PubMed] [Google Scholar]
- 40.Kalyanaraman H, Zhuang S, Pilz RB, Casteel DE. The activity of cGMP-dependent protein kinase Ialpha is not directly regulated by oxidation-induced disulfide formation at cysteine 43. J Biol Chem. 2017;292:8262–8268. doi: 10.1074/jbc.C117.787358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagayama T, Hsu S, Zhang M, Koitabashi N, Bedja D, Gabrielson KL, Takimoto E, Kass DA. Pressure-overload magnitude-dependence of the anti-hypertrophic efficacy of PDE5A inhibition. J Mol Cell Cardiol. 2009;46:560–567. doi: 10.1016/j.yjmcc.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chirinos JA, Zamani P. The Nitrate-Nitrite-NO Pathway and Its Implications for Heart Failure and Preserved Ejection Fraction. Current Heart Failure Reports. 2016;13:47–59. doi: 10.1007/s11897-016-0277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, Bedja D, Kirk JA, Ranek MJ, Dostmann WR, Kwon C, Margulies KB, Van Eyk JE, Paulus WJ, Takimoto E, Kass DA. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature. 2015;519:472–476. doi: 10.1038/nature14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.