

Abstract
Background
Genome wide association studies have identified 23 loci for atrial fibrillation (AF), but the mechanisms responsible for these associations, as well as the causal genes and genetic variants, remain undefined.
Methods and Results
To identify the effect of common genetic variants on gene expression that might explain the mechanisms linking genome wide association loci with AF risk, we performed RNA sequencing of left atrial appendages from a biracial cohort of 265 subjects. Combining gene expression data with genome-wide single nucleotide polymorphism (SNP) data, we found that approximately two-thirds of the expressed genes were regulated in cis by common genetic variants at a false discovery rate of <0.05, defined as cis-expression quantitative trait loci (cis-eQTLs). 12 of 23 reported AF genome wide association loci displayed genome-wide significant cis-eQTLs, at PRRX1 (chromosome 1q24), SNRNP27 (1q24), CEP68 (2p14), FKBP7 (2q31), KCNN2 (5q22), FAM13B (5q31), CAV1 (7q31), ASAH1 (8p22), MYOZ1 (10q22), C11ORF45 (11q24), TBX5 (12q24), and SYNE2 (14q23), suggesting that altered expression of these genes plays a role in AF susceptibility. Allelic expression imbalance was employed as an independent method to characterize the cis-control of gene expression. 1248 of 5153 queried genes had cis-SNPs that significantly regulated allelic expression at a false discovery rate of <0.05.
Conclusions
We provide a genome wide catalog of the genetic control of gene expression in human left atrial appendage. These data can be used to confirm the relevance of genome wide association loci and to direct future functional studies to identify the genes and genetic variants responsible for complex diseases such as AF.
Journal Subject Terms: Arrhythmias, Functional Genomics, Gene Expression and Regulation
Keywords: genomics, right atrium, expression experiments, allelic expression imbalance cholesterol, RNA-sequencing, eQTL
Introduction
Atrial fibrillation (AF), the most common arrhythmia in the general population, is clearly heritable1. Genome-wide association studies (GWAS) have identified 23 loci associated with AF2–6, but the mechanisms for these associations and the causal genetic variants that underlie these associations have not yet been determined for any of these loci. The current study sought to identify the genes whose expression was altered by genetic variants associated with AF susceptibility and provide a genome-wide catalog of the genetic control of gene expression in human left atrial appendage tissue. The central hypothesis is that the mechanisms by which these GWAS variants are associated with AF is that they regulate the expression of nearby genes and that this altered expression can lead to changes in cell physiology and anatomy, which may predispose to atrial fibrillation.
Throughout life, thousands of genes must be expressed in a coordinated and regulated fashion for appropriate cardiac development and maintenance of physiological homeostasis. Much of this regulation is mediated by cis regulatory elements, such as enhancers, silencers, and promoters that bind proteins, alter histone methylation and acetylation, and regulate gene transcription. This regulation has been highlighted by the GTEx study7,8, where common genetic variants have been shown to be associated with the expression of nearby genes. These regulatory variants are called cis-expression quantitative trait loci (eQTLs) and have been identified across numerous human tissues. Some of these are tissue specific, while others function across many tissues. To increase our understanding of the AF GWAS loci we studied mRNA gene expression to determine the genetic control of gene expression in a primary AF target tissue, the human left atrium (LA). We previously performed transcriptome profiling via polyA+ RNA sequencing (RNAseq) in four paired human left and right atrial appendages and found 746 genes, including many long noncoding RNAs (lncRNAs), which were differentially expressed in these two chambers at a false discovery rate (FDR) of <0.0019.
In the current study we performed polyA+ RNAseq on LA appendage (LAA) tissue from 235 subjects of European descent and 30 subjects of African descent. We performed eQTL analyses to define common single nucleotide polymorphisms (SNPs) associated with LAA gene expression and found that ~2/3 of the expressed genes had genome-wide significant cis-eQTLs associated with their expression. This included 12 of the 23 reported AF GWAS loci top SNPs, with reassignment of the target candidate genes in several of these loci. We provide a public database for lookup of LAA eQTLs as a resource for future studies. We also performed allelic expression imbalance (AEI) analyses, as an independent method to identify genetic variants associated with gene expression.
Methods
The raw RNA-sequencing and genotyping will not be made available, but gene read counts are available in the GEO database (GSE69890). The statistical analytic methods are available on github.
Human Subjects
Left atrial appendage (LAA) tissues were collected from 251 patients who underwent elective cardiac surgery to treat AF, valve disease, or other cardiac disorders. AF history, type of AF, presence of structural heart disease, demographics, cardiac procedures, and other clinical data were collected in a research database. Atrial rhythm status was determined by review of electrocardiograms obtained prior to surgery. 14 additional LAA were obtained from non-failing, unused transplant donors. All surgical patients provided informed consent for research use of discarded atrial tissue. Prior to 2008 verbal consent was obtained and documented in the medical records in a process approved by the Cleveland Clinic Institutional Review Board (IRB). From 2008 onward, patients provided separate IRB-approved written informed consent. Tissue was also obtained from donor hearts not used for transplant with written consent for research use provided by the family. The IRB approved the studies included in this report.
Human Left Atrial Tissue Processing
Human LAA tissues were obtained from patients undergoing elective surgery to treat AF, valve disease, or other cardiac disorders. Specimens were snap frozen in liquid nitrogen and stored at -80°C until RNA extraction. LA tissue specimens were also obtained from non-failing donor hearts not used for transplant. These hearts were perfused with cardioplegia prior to explant and processed in the same manner as hearts used for organ transplant. As with the surgical specimens, donor tissue samples were snap frozen in liquid nitrogen and kept at -80°C until RNA extraction. Donor information was available for age, race, and sex.
Genomic DNA isolation and SNP microarray
DNA was extracted from 25–50 mg of LAA tissue. The tissue, in one mL of DNAzol® (Invitrogen), was homogenized (PowerGen700, Fisher Scientific) with sterile Omni Tip Disposable Generator Probes (Omni International). DNA was isolated from the homogenate following the DNAzol protocol. The DNA pellet was resuspended in 20 μl of 10 mM Tris buffer (pH 7.4) and the DNA concentration was optically evaluated (OD260 nm), diluted to 100 ng/μl and stored at −80°C until use. DNA was genotyped using Illumina Hap550v3 and Hap610-quad SNP microarrays. SNP data were imputed to 1000 Genomes Project phase 2 yielding ~19 million SNPs, using IMPUTE10 after filtering out variants falling below 0.5 on IMPUTE’s information statistic. For the eQTL analysis, we excluded SNPs with < 5% and <10% minor allele frequency in the European descent and African American cohorts resulting in roughly 6.8 and 6.6 million SNPs per cohort, respectively.
RNA isolation and sequencing
50–100 mg of LAA tissue was used to extract RNA. The tissue, in one mL of TRIzol® (Invitrogen), was homogenized with sterile Omni Tip Disposable Generator Probes. RNA was isolated from the homogenate following the TRIzol protocol. The RNA pellet was dried and re-suspended in 80 μl of RNase-free water, with concentration determined by OD260 nm and RNA stored at ‐80°C. Library generation for RNA-sequencing was done at the University of Chicago Genomics Facility using standard Illumina protocols (Part # 15015050, Rev A). Samples were filtered based on RNA quality as ascertained on an Agilent 2100 Bioanalyzer. Unstranded 100-bp paired-end sequencing was performed on the Illumina HiSeq 2000 platform and multiplexed to 6 samples across two lanes. Samples were de-multiplexed and aligned to hg19 using TopHat (v2.0.4)11 with the default options. Reads from exactly matched PCR duplicates were marked using picardtools (http://picard.sourceforge.net.) and excluded from further analysis. The sequence reads were mapped to the human genome to derive a digital count of the expression of genes, which were defined using the Ensembl (version 71) gene catalog.
Statistical analyses
RNAseq and eQTL analysis
Expression counts were obtained from aligned files using htseq12 counts against the human Ensembl gene annotation. On average, 26 million paired-end read fragments aligned to this annotation of the transcriptome across all of our samples. Reads were quantile normalized, and gene counts for eQTL analysis were variance-stabilized transformed using the R package Deseq213,14. Expression of each gene was adjusted by the following covariates: sex, genetic substructure based on four multidimensional scaling (MDS) factors, and 25 expression surrogate variable analysis (SVA) covariates. The SVA method is similar to principal component analysis, which uses unsupervised mathematical models to separate out the high variance components in high dimensional data. Thus, without manual normalization, the SVA method corrects for potential large effectors of gene expression such as read-depth, batch effects, and other technical variables, as well as environmental and disease effects such as AF status, history of structural heart disease, coronary artery disease, etc. Surrogate variables were calculated from the variance-stabilizing transformation (VST) data using the sva package15. eQTL analyses were performed using MatrixeQTL16 (2.1.0) to test associations between genotype and VST counts. These analyses were performed separately for each racial group with beta-coefficients calculated as the additive effect of one allelic difference on log2 gene expression. To correct for population substructure, genetic MDS was derived from SNP array genotyping. The top four MDS factors were uses as covariates for the regression analyses performed for eQTL calculations. The qvalue package was used to estimate false discovery rate (FDR) from the complete list of cis-eQTL SNP/genome wide expressed gene pairs p-values17. Linear regression and Q-Q plot comparison of the LAA eQTLs with selected tissues were performed using the version 6p analysis of GTEx project8.
Allele expression imbalance
Allelic expression imbalance (AEI) analysis was performed pooling the European and African descent subjects, using samtools18 and pysam packages, along with custom scripts (www.github.com/jeffhsu3/genda), which were used to obtain RNAseq allele-specific read counts at all genotyped SNPs residing in each Ensembl gene. These SNPs in the transcript are called “indicator SNPs”, and were used to determine the allelic ratio expression balance by counting RNAseq reads containing each allele. Indicator SNPs were then filtered on read counts (>20 heterozygotes at the indicator) for robust analysis. Outlier subjects were removed if their log2 allelic ratio was greater than 4.32, as these appeared to be due to indicator SNP genotyping error (subjects were in fact homozygous for the indicator SNP) or mapping errors. A non-parametric Mann-Whitney U-test was used to test the association of the absolute value of the log2 allelic ratio between homozygous and heterozygous individuals at common cis-SNPs (> 5% minor allele frequency with > 10 homozygotes of either allele in order to accurately capture the cis-SNP effect on AEI).
Results
Study participants
The demographics, relevant medical history, and surgical indications of the study population are shown in Table 1 and included 235 subjects with self-reported European ancestry and 30 self-reported African Americans. Using imputed SNP array data, the genetic ancestry was examined by principal component analysis, revealing that the self-identified African Americans were admixed or clustered closely with African reference samples, while the self-reported European ancestry samples clustered closely with European reference samples (Supplemental Figure 1).
Table 1.
Study Demographics
| Patient Characteristics | Total n = 265 |
European Descent n = 235 |
African Descent n=30 |
P-Value * |
|---|---|---|---|---|
| Age, median (IQR), y | 62.0 (53.0, 69.0) | 62.0 (54.5, 69.5) | 56.5 (50.2, 68.8) | 0.24 |
| Sex, Female, n (%) | 84 (31%) | 63 (26%) | 21 (70%) | 0.88 |
| BMI, median (IQR), (kg/m2)† | 27.2 (24.5, 30.3) | 27.2 (24.5, 30.3) | 26.2 (23.0, 31.6) | 0.63 |
| Diabetes, n (%)† | 35 (13%) | 28 (12%) | 7 (24%) | 0.07 |
| History of CAD, n (%)† | 160 (63%) | 143 (64%) | 17 (58%) | 0.69 |
| History of MVD, n (%)† | 136 (54%) | 114 (51%) | 22 (75%) | 0.02 |
| Hypertension, n (%)† | 137 (54%) | 115 (51%) | 22 (78%) | 0.01 |
| History of AF, n (%)† | 213 (84%) | 192 (86%) | 21 (72%) | 0.09 |
| AF Rhythm at surgery, n (%)† | 130 (51%) | 118 (53%) | 12 (41%) | 0.13 |
| Surgical Indication | ||||
| Donor, n (%) | 14 (5%) | 13 (5%) | 1 (3%) | 1.00 |
| Valve, n (%)‡ | 153 (57%) | 129 (54%) | 24 (80%) | 0.01 |
| CABG, n (%)‡ | 106 (40%) | 95 (40%) | 11 (36%) | 0.84 |
| Non-CABG Non-Valve, n (%) | 52 (19%) | 51 (21%) | 22 (73%) | <0.01 |
statistical significance between patients of European descent and African Descent, either by t-test, for quantitative traits, or chi-square, for categorical traits.
not including 1 African descent and 13 European descent donors, for which only age and sex were known.
patients could have an indication for both Valve and CABG surgeries
Abbreviations used: IQR, interquartile range; BMI, body mass index; CAD, coronary artery disease; MVD, mitral valve disease; CABG, coronary artery bypass graft.
Genotype effects on gene expression
The overall design of our analysis of the genetics of gene expression is shown in Supplemental Figure 2. We performed polyA+ selected RNAseq on LAA RNA to obtain expressed gene counts corresponding to the ~65,000 protein coding and non-coding genes contained in Ensembl gene annotation release 71. The average library size-adjusted reads for each gene for each sample gives a U-shaped frequency distribution (Supplemental Figure 3). Our definition of genes expressed in the LAA is those with a minimum of 1000 variance stabilized counts summed from all 235 European-descent samples which yielded 24,042 genes.
We examined the genetic effects on gene expression in LAAs in separate analyses of the European and African descent cohorts. 25 SVAs were determined to be optimal for eQTL identification, and these were used as co-variates in our eQTL analysis. We used a ±250 Kb window (500 Kb total) around the most distal 5′ transcription start site (TSS), as annotated by Ensembl, as our definition of a cis effect. We found 15,906 genes, representing 66% of the 24,042 expressed genes, including numerous long non-coding RNAs (lncRNAs), with at least one cis-eQTL that met our 0.05 q-value threshold. Supplemental Table 1 shows the top eQTL SNP for the 15,906 significant genes in European descent subjects and the associated eQTL q-value of the same SNP in African Americans. Only 713 significant cis-eQTLs (q<0.05) were found in the African-American subjects due to the small sample size, and a different SNP was often the most significant in these subjects vs. that of the European descent subjects (Supplemental Table 2). Nonetheless, there was a strong correlation (r=0.76, p=4.13E-278) of the effect size and direction between a set of 1464 cis-eQTLs in African Americans (meeting a more liberal q<0.5 threshold due to the small sample size) that the top cis-eQTL SNP for each gene (q<0.05) defined in European descent subjects (Figure 1). All significant SNP-gene pair cis-eQTL associations in the European descent subjects can be searched using our publicly available web application (http://afeqtls.lerner.ccf.org), which also displays the eQTL association values for the African American cohort.
Figure 1.
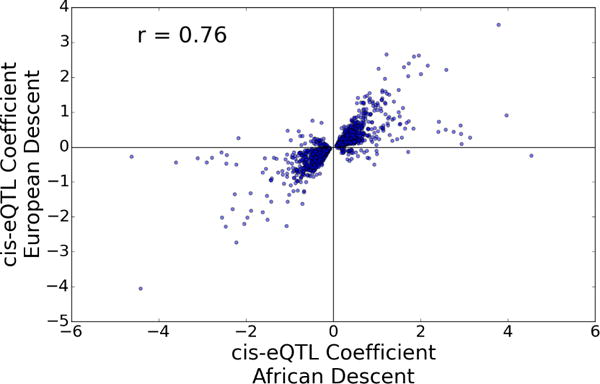
cis-eQTL replication between ethnic groups. The SNP with the most significant cis-eQTL (q<= 0.05) in European descent subjects for a given gene was filtered in the African-American subjects with a threshold of q <0.5, yielding a set of 1464 SNP-gene pairs in both populations. The eQTL beta-coefficients’ effect on target gene expression for that SNP for both the European-American (x-axis) and African-American (y-axis) subjects is plotted (r=0.76, p= 4.13E-278).
GTEx multiple tissue comparison
We examined the cross tissue replication of our 15,906 LAA eQTLs (SNP – gene pairs) by comparison against eQTL data from six tissues in the GTEx project7,8 including two heart tissues, the left ventricle (LV) and the right atrial appendage (RAA). There was a remarkable conservation of the LAA eQTLs with the six other tissues, as the effect and direction of the cis-SNPs on gene expression (beta coefficients) were highly correlated with r2 values ranging from 0.39 (coronary artery) to 0.67 (RAA) (Supplemental Figure 4). We found 586 LAA cis-eQTLs that were found not identified in LV and RAA tissues (Supplemental Table 3). The 23 AF GWAS SNPs identified in Table 2 were used to determine whether the LAA eQTLs were stronger than those found in other tissues. QQ plots were generated against a uniform distribution. When compared with three GTEx tissues (blood, LV, RAA), the LAA eQTLs showed greater significance, indicating that the LAA AF-GWAS eQTL set is the most relevant for AF (Supplemental Figure 5).
Table 2.
eQTLs and AEI in AF GWAS loci.
| AF GWAS locus | Nominal gene attribution | AF GWAS SNP* | Top eQTL Gene | Top eQTL Symbol | eQTL Beta† | eQTL p-value | eQTL q-value | GWAS SNP evidence AEI (p-value) | AEI indicator SNP |
|---|---|---|---|---|---|---|---|---|---|
| 1q21.3 | KCNN3, PMVK | rs6666258 | ENSG00000173207 | CKS1B | -0.05 | 4.53E-03 | 5.87E-02 | NA‡ | NA |
| 1q24.2 | METTL11B | rs12044963 | ENSG00000231437 | RP11-88H9.2 | 0.17 | 1.51E-02 | 7.36E-01 | NA | NA |
| 1q24.2 | PRRX1 | rs3903239 | ENSG00000116132 | PRRX1 | -0.16 | 2.86E-05 | 9.33E-04 | NA | NA |
| 2p13.3 | ANXA4, GMCL1 | rs3771537 | ENSG00000124380 | SNRNP27 | -0.05 | 6.22E-06 | 3.11E-04 | NA | NA |
| 2p14 | CEP68 | rs2540953 | ENSG00000011523 | CEP68 | -0.10 | 4.98E-13 | 7.13E-11 | NA | NA |
| 2q31.2 | TTN, TTN-AS1 | rs2288327 | ENSG00000079150 | FKBP7 | 0.10 | 2.40E-06 | 1.33E-04 | NA | NA |
| 3p25.1 | CAND2 | rs4642101 | ENSG00000144712 | CAND2 | -0.04 | 8.10E-03 | 8.85E-02 | NA | NA |
| 4q25 | PITX2 | rs6817105 | ENSG00000250103 | PANCR | 0.02 | 7.44E-01 | 7.76E-01 | NA | NA |
| 5q22.3 | KCNN2 | rs337711 | ENSG00000080709 | KCNN2 | -0.08 | 1.55E-03 | 3.35E-02 | NA | NA |
| 5q31.2 | WNT8A | rs2040862 | ENSG00000031003 | FAM13B | -0.24 | 6.91E-30 | 4.83E-27 | 2.10E-05 | rs3777118 |
| 6q22.31 | SLC35F1, PLN | rs4946333 | ENSG00000111860 | CEP85L | -0.04 | 7.70E-02 | 4.19E-01 | NA | NA |
| 6q22.31 | GJA1 | rs12664873§ | ENSG00000152661 | GJA1 | -0.07 | 1.62E-02 | ND§ | NA | NA |
| 7q31.2 | CAV1 | rs3807989 | ENSG00000105974 | CAV1 | -0.10 | 1.55E-07 | 8.96E-06 | 1.98E-9 | rs1049337 |
| 8p22 | ASAH1, PCM1 | rs7508 | ENSG00000104763 | ASAH1 | 0.08 | 1.69E-07 | 1.22E-05 | 5.60E-08 | rs3810 |
| 9q22.32 | C9ORF3 | rs10821415 | ENSG00000158169 | FANCC | 0.04 | 1.84E-02 | 1.54E-01 | NA | NA |
| 10q22.2 | SYNPO2L | rs10824026 | ENSG00000177791 | MYOZ1 | -1.87 | 2.90E-38 | 3.65E-35 | NA | NA |
| 10q24.33 | NEURL1 | rs6584555 | ENSG00000173915 | USMG5 | 0.03 | 4.13E-02 | 2.52E-01 | NA | NA |
| 10q24.33 | SH3PXD2A | rs2047036 | ENSG00000107957 | SH3PXD2A | -0.01 | 3.80E-01 | 6.53E-01 | NA | NA |
| 11q24 | KCNJ5 | rs75190942 | ENSG00000174370 | C11orf45 | 0.29 | 1.59E-06 | 9.20E-05 | NA | NA |
| 12q24.21 | TBX5 | rs10507248 | ENSG00000089225 | TBX5 | 0.07 | 4.87E-04 | 1.03E-02 | NA | NA |
| 14q23.2 | SYNE2 | rs1152591 | ENSG00000054654 | SYNE2 | -0.17 | 2.31E-19 | 6.73E-17 | 3.02E-09 | rs35648226 |
| 15q24.1 | HCN4 | rs7164883 | ENSG00000183324 | REC114 | -0.15 | 6.71E-02 | 3.27E-01 | NA | NA |
| 16q22.3 | ZFHX3 | rs2106261 | ENSG00000259768 | antisense-ZFHX3 | -0.09 | 1.57E-01 | 4.80E-01 | NA | NA |
Trans-eQTL analysis
We identified 58 trans-eQTLs in the subjects of European descent using the conservative definition of the trans-SNP and its regulated transcript being on different chromosomes. We used a stringent Bonferroni adjusted p-value threshold of <1E-13 (Supplemental Table 4) as trans-eQTLs show modest to low replication between studies19,20. We examined the effect size of these 58 trans-eQTLs in the African American subjects, and there was significant correlation (Supplemental Table 4 and Supplemental Figure 6, r=0.35, p=1.91E-3), indicating replication for most of these trans-eQTLs, with several outliers.
cis-eQTLS at AF GWAS loci to identify candidate causal variants
Among the 23 top independent AF GWAS SNPs (Table 2), we found 12 SNPs that had one or more genome-wide significant cis-eQTLs (q<0.05, using all eQTL SNP-gene pairs in 500 Kb intervals that overlap with the 23 GWAS SNPs). Another 6 AF GWAS SNPs had cis-eQTLs that met the more liberal threshold of p<0.05. RNAseq confirmed our prior finding that the AF associated SNPs at chr 4q25 are not associated with LAA expression of PITX2c21. The top eQTL gene in each GWAS locus was not always the nominally attributed closest candidate gene. For example at the chr. 5q31 locus the most significant eQTL was for FAM13B, while this locus had previously been putatively associated with WNT8A. Similarly, the chr. 10q22 locus was most strongly associated with MYOZ1, whereas SYNPO2L was the closest gene. Supplemental Table 5 provides a complete list of all genes in each GWAS locus with their eQTLs values for the corresponding GWAS SNP. Supplemental Table 5 additionally provides the top cis-eQTL SNP for each gene in these GWAS loci. This analysis demonstrates that the GWAS SNP can be a significant cis-eQTL for two or more genes. For example the chr. 10q22 AF GWAS SNP has genome-wide significant cis-eQTLs for both SYNPO2L and MYOZ1. We found that the GWAS SNP was often not the strongest cis-eQTL SNP. For example, in the chr. 11q24 locus a significant eQTL for C11ORF45 was found, but the strongest eQTL SNP (rs4937390) was not the GWAS SNP (rs75190942) and was not in strong linkage disequilibrium (LD) with the GWAS SNP (r2=0.21).
The most significant AF GWAS SNP cis-eQTL was for rs10824026 on chr. 10q22 for MYOZ1 (q = 3.65E-35, Table 2, Figure 2), as previously reported in a microarray study of 53 LA samples22. In addition, the SYNPO2L gene has the second strongest eQTL at this locus (q=1.63E-10, Figure 2B, Supplemental Table 5). As shown in Figure 2, there are additional SNPs in LD with rs10824026 that have stronger cis-eQTLs than the GWAS SNP rs10824026, indicating that the AF GWAS SNP may not be the causal SNP at this locus. The AF risk allele of rs10824026 was associated with decreased expression of the MYOZ1 gene but increased expression of the SYNPO2L gene (Figure 2), indicating that altered expression of either or both of these genes might contribute to AF susceptibility.
Figure 2.
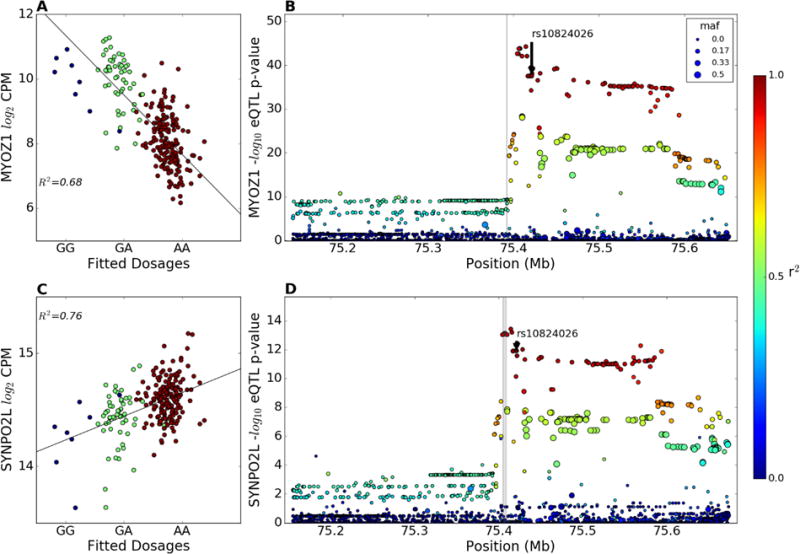
MYOZ1 and SYNPO2L cis-eQTLs at the chr 10q22 AF risk locus. (A) Partial regression plot showing the effect of the AF GWAS SNP rs10824026 on the LA expression of the MYOZ1 gene in European descent subjects after adjusting for genetic MDS, transcription SVAs, and gender. Blue dots are subjects homozygous for the minor allele, green dots are heterozygotes, and red dots are homozygous for the major (risk) allele (r2 = 0.68, q-value = 3.65E-35). (B) eQTL p-values and LD relationship (color scale) with rs10824026 in the 0.5 Mb region around the MYOZ1 TSS in European subjects, with the gray bar indicating the gene boundary. (C) Effect of rs10824026 on the LA expression of the SYNPO2L gene in European subjects (r2 = 0.76, q-value = 8.31E-11). (D) eQTL p-values and LD relationship with rs10824026 in the 0.5 Mb region around the SYNPO2L TSS European subjects.
The second strongest AF GWAS SNP cis-eQTL (Table 2) was found at chr. 5q31 for the FAM13B gene (q=4.83E-27, Figure 3A, B). Although this GWAS SNP was nominally attributed by proximity to the WNT8A gene, WNT8A is not expressed in our LA samples and our eQTL analysis suggests that the gene at this locus responsible for AF susceptibility is FAM13B, encoding a poorly characterized member of the Rho GTPase activation domain protein (RhoGAP) gene family. The AF risk allele is associated with lower LAA FAM13B expression. The cis-eQTL plot for left atrial FAM13B expression in European descent subjects shows many equally significant eQTL SNPs in the same LD block stretching over ~150 kb, including the AF GWAS SNP rs2040862 (Figure 3B).
Figure 3.
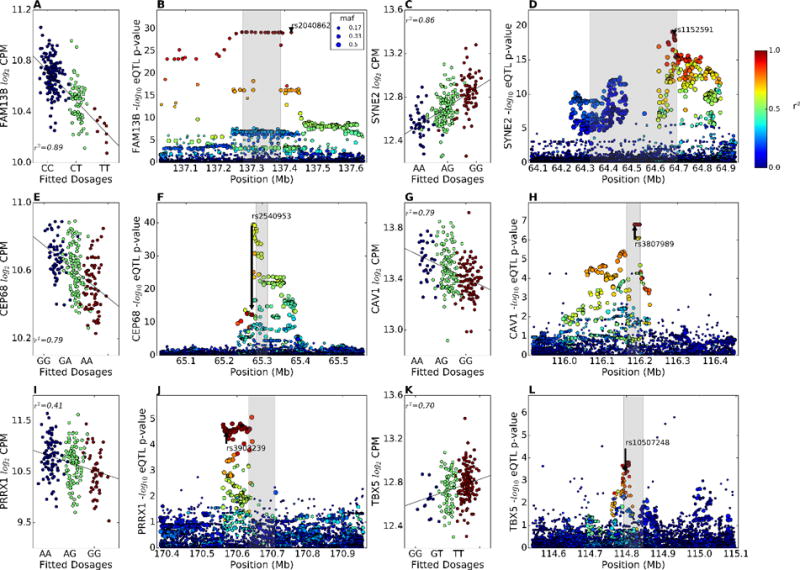
Cis-eQTLs for GWAS genes. (A, C, E, G, I, K) Partial regression plot showing the effect of the AF GWAS SNPs rs2040862, rs1152591, rs2540953, rs3807989, rs3903239, and rs10507248 on the LA expression of the FAM13B, SYNE2, CEP68, CAV1, PRRX1, and TBX5 genes in European descent subjects after adjusting for genetic MDS, transcription SVAs, and gender. Blue dots are subjects homozygous for the reference allele (GRCh37 build), green dots are heterozygotes, and red dots are homozygous for the variant allele. (B, D, F, H, J, L) eQTL p-values and LD relationship (color scale) with the GWAS SNP (arrow) in the 0.5 Mb region around the TSS of the FAM13B, SYNE2, CEP68, CAV1, PRRX1, and TBX5 genes in European subjects.
The third strongest GWAS SNP eQTL was for the SYNE2 gene (q=6.72E-17, Figures 3C, D). SYNE2 encodes the nuclear envelope protein nesprin2, which links the nuclear envelope to the cytoskeleton and controls subcellular nuclear localization23,24. The GWAS SNP (rs1152591) risk allele is also associated with lower LAA expression of SYNE2.
GWAS SNPs at nine other loci were also significantly associated in cis with the expression of PRRX1, SNRNP27, CEP68, FKBP7, KCNN2, CAV1, AHAH1, C11ORF45, and TBX5 (Figures 3E–L, Supplemental Figure 7). There were six more GWAS SNPs with cis-eQTLs that met the more liberal p<0.05 threshold; these SNPs were associated with the expression of CKS1B, RP11-88H9.2, CAND2, GJA1, FANCC, and USMG5 (Table 2, Supplemental Table 5).
Allelic expression imbalance
We examined the effects of genetic variation on gene expression using AEI, an analysis method that eliminates the most common sources of variation among our human left atrial samples. In this method we count expression of the two alleles in the RNAseq data within each individual that is heterozygous for an exonic “indicator SNP”. These indicator SNPs were identified by DNA SNP array genotyping. Combining our subjects of European and African American descent, we examined all expressed genes with at least 20 subjects heterozygous for one or more indicator SNPs. To find the top cis-SNP associated with AEI, which we call the “cis-AEI SNP”, we used a non-parametric two-tailed t-test, as previously described25, to calculate the p-value for association of each cis-SNP within ±250 Kb of the TSS with AEI for each indicator SNP. For example, the SYNE2 transcript contains the indicator SNP rs35648226 that was heterozygous in 116 subjects, and we ranked the log2 expression of the minor/major allele counts for each of these subjects (Figure 4A). Each bar represents one subject that was color-coded as homozygous (green) or heterozygous (orange) for the cis-AEI SNP rs2738413, which is in strong LD with the SYNE2 locus AF GWAS SNP rs1152591. The top cis-AEI SNP, rs2738413 (p=3.02E-9), and all other SNPs in this locus are shown in an AEI Manhattan plot (Figure 4B). It is evident that the cis-AEI SNP was not in perfect LD with the indicator SNP (LD between indicator and cis-AEI SNP r2 = 0.39 in our dataset), as heterozygotes for the cis-AEI SNP were found at both ends of the allelic ratio plot (Figure 4A). We compared the Manhattan plots for the cis-AEI and cis-eQTLs in this locus (Figures 4B, C) and found excellent concordance, although a different variant (chr14:64673560:D) in strong LD with rs1152591 had the strongest eQTL.
Figure 4.
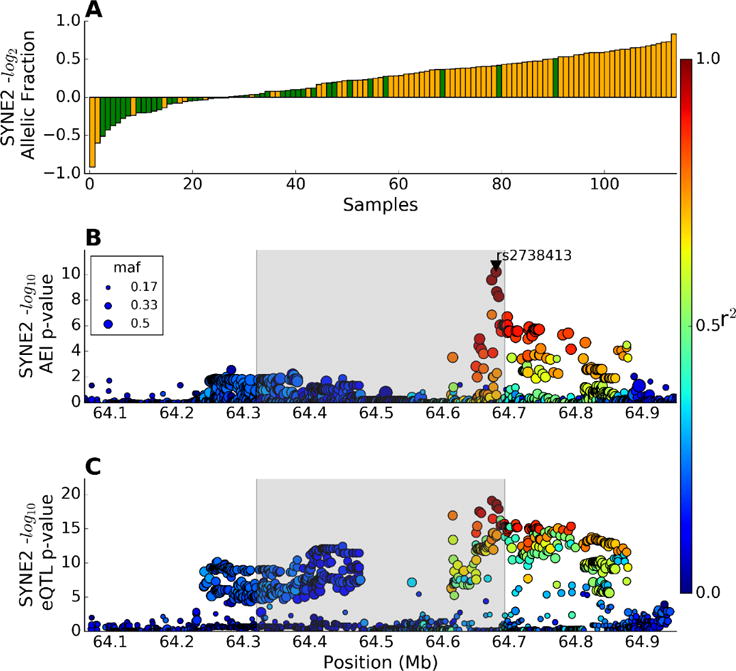
Allelic expression imbalance and cis-eQTLs for the SYNE2 gene. (A) The log2 allelic expression ratio is plotted in ascending order for the 116 subjects who were heterozygous for the exonic indicator SNP rs35648226. The green and orange bars represent subjects homozygous and heterozygous for the top cis-AEI SNP rs2738413, respectively. (B) cis-AEI SNP Manhattan plot of the –log10 p-values for association of each cis-SNP in this locus with AEI, revealing that the GWAS SNP rs1152591 was not the top cis-AEI SNP. LD with the top cis-AEI SNP rs2738413 is indicated by the color scale and the gray shading shows the SYNE2 gene boundaries. (C) cis-eQTL Manhattan plot of the –log10 p-values for association of each cis-SNP in this locus with SYNE2 expression, showing that there is a different top eQTL SNP, but this SNP is in high LD with both the cis-AEI and GWAS SNPs
Global AEI analysis was performed for 5153 genes that met our genotype and expression criteria. We found an AEI association for 1248 genes (24% of queried genes) using a q-value threshold of 0.05 (Supplemental Table 6). The major reason that only a fraction of the analyzed genes had significant AEI was that for many of these genes the number of subjects heterozygous for the indicator SNP was underpowered. In many cases, the top AEI SNP differed from the top eQTL SNP. AEI analysis may provide insight into the functional SNP, as the AEI method controls for expression within each subject and eliminates many variables in the eQTL analysis. AEI within the AF-GWAS loci is shown in Supplemental Table 5.
Discussion
We found that most of the LAA expressed genes (66% in the European descent subjects) were controlled by nearby common genetic variants (cis-eQTLs). This is concordant with other large human transcriptomic studies, as ~69% of the genes expressed in whole blood had cis-eQTLs in 5626 subjects26. Thus, common variants have large cis-effects on global gene expression and robust eQTL identification is detectable for sample sizes in the low hundreds.
GWAS studies have been invaluable in identifying common genetic variants associated with complex diseases such as AF. However, it is still an arduous task to identify causal SNPs and the mechanisms by which they alter disease susceptibility, particularly when the disease-associated genetic variants are intergenic. Here, we use an intermediate phenotype, gene expression, to identify potential causal genetic variants, based on the hypothesis that these SNPs work by regulating gene expression. To gain deeper insight into the functional role of the AF GWAS SNPs we evaluated their roles as regulatory SNPs by ascertaining their effects on the expression of nearby genes via RNAseq of a large biracial set of human left atrial appendages.
Overall, genome-wide significant cis-eQTLs were discovered for 12 of the 23 AF GWAS SNPs, and another 6 GWAS SNPs displayed uncorrected cis-eQTL p values <0.05. One reason for the incomplete representation of genome-wide cis-eQTLs for the GWAS SNPs is that the remaining SNPs may control gene expression at a different time (during heart development or maturation) and/or place (e.g., left atrial pulmonary veins). This may explain the absence of an identifiable eQTL at the chr. 4q25 locus for PITX2c in adult left atrial tissue, despite this locus harboring the strongest AF GWAS SNP21.
The AF-GWAS SNP with the strongest eQTL was rs10824026, associated with expression of both the MYOZ1 and SYNPO2L gene, although in opposite directions. While a previously reported study identified rs3740293, a SNP in LD rs10824026, as a cis-eQTL for MYOZ1 in LA and RA tissue, it did not identify the SNP as an eQTL for SYNPO2L22. MYOZ1 encodes the myozenin1 protein, which interacts with calcineurin and colocalizes with the Z-disc protein α-actinin. SYNPO2L encodes synaptopodin 2-like protein, initially described in cardiomyocytes derived from pluripotent stem cells; this protein is also highly expressed in the Z-disc and, like myozenin-1, it interacts with α-actinin. Also called cytoskeletal heart-enriched actin-associated protein (CHAP), its knockdown in zebrafish leads to aberrant heart and skeletal muscle development, disorganized sarcomeres, and diminished cardiac contractility27. The inverse regulation of the expression of these two adjacent genes suggests that the ratio of the encoded proteins that share a similar subcellular localization may play a role in AF susceptibility.
The second strongest GWAS SNP eQTL was for FAM13B, at a locus previously attributed to WNT8A. At this locus, the minor allele of rs2040862 is the AF risk allele, and this allele is associated with decreased expression of FAM13B. The FAM13B gene encodes an uncharacterized protein containing a Rho GTPase activation protein (RhoGAP) domain. A different SNP at chr. 5q31 (rs1004989) that was previously associated with the electrocardiographic QT-interval was found to have a significant eQTL for FAM13B expression in left ventricle tissue samples28; however, rs1004989 is not in LD with the AF GWAS SNP (rs2040862) or the left atrial FAM13B top eQTL SNP (rs17171731), perhaps illuminating tissue-specific regulatory variants controlling FAM13B gene expression.
Recently an AF candidate gene and GWAS region SNP eQTL study was reported using 122 RAA samples29. Cis-eQTLs were identified for PITX2a in RAA, but the LAA-expressed PITX2c transcript isoform was not expressed in RAA. We found no comparable cis-eQTL for PITX2c in LAA tissue. Additional RAA cis-eQTLs were identified for CAV1, MOYZ1, C9ORF3, and FANCC29, genes for which we also identified LAA cis-eQTLs.
Limitations of our study include statistical power; thus, absence of an eQTL does not guarantee that there is no genetic control of gene expression at any given locus30. We also cannot detect the effects of rare coding mutations. Exome sequencing studies have recently found short truncating mutations at the 10q22 AF locus in SYNPOL2 that may potentially play a role in AF31. In addition, gene expression is most likely controlled by multiple variants, both local and distant. Our combination of eQTL and AEI analysis may, in some cases, help to identify causal candidate SNPs that are stronger than the top AF GWAS SNPs, which can then be tested in functional genomic studies. The effect of heart explanting on gene expression is unknown, but this covariate and all other phenotypic and environmental effects on gene expression are potentially corrected for by our SVA method. Another limitation is that our study was performed on LAA, often surgically removed to reduce stroke risk; although, other regions such as those in and nearer the pulmonary vein may be more relevant to AF pathogenesis. Finally, our study provides a valuable resource to the community with a LA-specific eQTL browser, which may be particularly useful for genetic studies of AF, and more broadly for the identification of functional SNPs for other cardiovascular traits and diseases.
Supplementary Material
Clinical Perspective.
Atrial fibrillation is the most common arrhythmia, and it is associated with increased risk of stroke and death. Family studies as well as genome-wide association studies have shown that genetics play a strong role in susceptibility to atrial fibrillation. We performed RNA sequencing along with genome-wide genotyping on left atrial appendages from a biracial cohort of 265 subjects. This allowed us to identify common genetic variants associated with the expression of nearby genes in the left atrium. We combined our data with results from the most recent atrial fibrillation genome-wide association study, allowing us to suggest candidate causal genes and candidate causal genetic variants, with the hypothesis that most common genetic variants resulting in increased susceptibility to atrial fibrillation act via regulating the expression of nearby genes. Our studies pave the way for future functional studies to confirm the causal genes and genetic variants, which can yield novel insight into atrial fibrillation pathogenesis and suggest novel therapeutic strategies to prevent or treat this disease.
Acknowledgments
Data Access: Variance normalized gene expression levels for each subject are available in the GEO database, accession number GSE69890.
Sources of Funding: This work was supported by the NIH grants R01 HL 090620 and R01 HL 111314 to MKC, DVW, JB, and JDS, the NIH National Center for Research Resources for Case Western Reserve University and Cleveland Clinic Clinical and Translational Science Award UL1-RR024989, the Cleveland Clinic Department of Cardiovascular Medicine philanthropy research funds, and the Tomsich Atrial Fibrillation Research Fund. JH was supported by the NIHtraining grant T32 GM 088088. JDS was supported by the Geoffrey Gund Endowed Chair for Cardiovascular Research.
Footnotes
Disclosures: None
References
- 1.Ellinor PT, Yoerger DM, Ruskin JN, MacRae CA. Familial aggregation in lone atrial fibrillation. Hum Genet. 2005;118:179–184. doi: 10.1007/s00439-005-0034-8. [DOI] [PubMed] [Google Scholar]
- 2.Ellinor PT, Lunetta KL, Glazer NL, Pfeufer A, Alonso A, Chung MK, et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet. 2010;42:240–4. doi: 10.1038/ng.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44:670–5. doi: 10.1038/ng.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sinner MF, Tucker NR, Lunetta KL, Ozaki K, Smith JG, Trompet S, et al. Integrating genetic, transcriptional, and functional analyses to identify five novel genes for atrial fibrillation. Circulation. 2014 doi: 10.1161/CIRCULATIONAHA.114.009892. CIRCULATIONAHA–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Low S-K, Takahashi A, Ebana Y, Ozaki K, Christophersen IE, Ellinor PT, et al. Identification of six new genetic loci associated with atrial fibrillation in the Japanese population. Nat Genet. 2017;49:953–958. doi: 10.1038/ng.3842. [DOI] [PubMed] [Google Scholar]
- 6.Christophersen IE, Rienstra M, Roselli C, Yin X, Geelhoed B, Barnard J, et al. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat Genet. 2017;49:946–952. doi: 10.1038/ng.3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ardlie KG, Deluca DS, Segrè AV, Sullivan TJ, Young TR, Gelfand ET, et al. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science (80-) 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu J, Hanna P, Van Wagoner DR, Barnard J, Serre D, Chung MK, et al. Whole genome expression differences in human left and right atria ascertained by RNA sequencing. Circ Cardiovasc Genet. 2012;5:327–35. doi: 10.1161/CIRCGENETICS.111.961631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–13. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 11.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anders S, Pyl PT, Huber W. HTSeq–A Python framework to work with high-throughput sequencing data. Bioinformatics. 2014:btu638. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:e161. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–8. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Nas A, Ingram-Drake L, Sinsheimer JS, Wang SS, Schadt EE, Drake T, et al. Expression Quantitative Trait Loci: Replication, Tissue- and Sex-Specificity in Mice. Genetics. 2010;185:1059–1068. doi: 10.1534/genetics.110.116087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grundberg E, Small KS, Hedman ÅK, Nica AC, Buil A, Keildson S, et al. Mapping cis-and trans-regulatory effects across multiple tissues in twins. Nat Genet. 2012;44:1084–1089. doi: 10.1038/ng.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gore-Panter SR, Hsu J, Hanna P, Gillinov AM, Pettersson G, Newton DW, et al. Atrial Fibrillation associated chromosome 4q25 variants are not associated with PITX2c expression in human adult left atrial appendages. PLoS One. 2014;9:e86245. doi: 10.1371/journal.pone.0086245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin H, Dolmatova EV, Morley MP, Lunetta KL, McManus DD, Magnani JW, et al. Gene expression and genetic variation in human atria. Heart Rhythm. 2014;11:266–271. doi: 10.1016/j.hrthm.2013.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A, et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. 2007;16:2816–33. doi: 10.1093/hmg/ddm238. [DOI] [PubMed] [Google Scholar]
- 24.Lüke Y, Zaim H, Karakesisoglou I, Jaeger VM, Sellin L, Lu W, et al. Nesprin-2 Giant (NUANCE) maintains nuclear envelope architecture and composition in skin. J Cell Sci. 2008;121:1887–98. doi: 10.1242/jcs.019075. [DOI] [PubMed] [Google Scholar]
- 25.Xiao R, Scott LJ. Detection of cis-acting regulatory SNPs using allelic expression data. Genet Epidemiol. 2011;35:515–525. doi: 10.1002/gepi.20601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huan T, Liu C, Joehanes R, Zhang X, Chen BH, Johnson AD, et al. A systematic heritability analysis of the human whole blood transcriptome. Hum Genet. 2015:1–16. doi: 10.1007/s00439-014-1524-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beqqali A, Monshouwer-Kloots J, Monteiro R, Welling M, Bakkers J, Ehler E, et al. CHAP is a newly identified Z-disc protein essential for heart and skeletal muscle function. J Cell Sci. 2010;123:1141–1150. doi: 10.1242/jcs.063859. [DOI] [PubMed] [Google Scholar]
- 28.Arking DE, Pulit SL, Crotti L, der Harst P, Munroe PB, Koopmann TT, et al. Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat Genet. 2014;46:826–836. doi: 10.1038/ng.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin RIR, Babaei MS, Choy M-K, Owens WA, Chico TJA, Keenan D, et al. Genetic variants associated with risk of atrial fibrillation regulate expression of PITX2, CAV1, MYOZ1, C9orf3 and FANCC. J Mol Cell Cardiol. 2015;85:207–214. doi: 10.1016/j.yjmcc.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Nica AC, Parts L, Glass D, Nisbet J, Barrett A, Sekowska M, et al. The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet. 2011;7:e1002003. doi: 10.1371/journal.pgen.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lubitz SA, Brody JA, Bihlmeyer NA, Roselli C, Weng L-C, Christophersen IE, et al. Whole Exome Sequencing in Atrial Fibrillation. PLoS Genet. 2016;12:e1006284. doi: 10.1371/journal.pgen.1006284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.