

Key Points
Dock8 regulates the expression of CD19 and WASP.
BCR clustering and B-cell spreading are decreased in memory B cells of Dock8 patients.
Abstract
Dock8 deficiency leads to immunodeficiency, and the role of Dock8 in B-cell development and function has been revealed; however, the role of DocK8 on B-cell receptor (BCR) signaling and function of memory B cells remains elusive. In this study, we generated a Dock8 knockout mouse model and collected peripheral blood mononuclear cells from Dock8 patients to study the effect of Dock8 deficiency on the BCR signaling and activation of memory B cells with confocal microscopy and total internal reflection fluorescence microscopy. The activation of key, positive upstream BCR signaling molecules, pCD19 and phosphorylated Brutons tyrosine kinase (pBtk), is reduced. Interestingly, the total protein and activated levels of Wiskott–Aldrich syndrome protein (WASP) are decreased in Dock8-deficient mouse B cells. Our previous research has shown that WASP positively regulates cd19 transcription; furthermore, we found that Dock8 regulates cd19 transcription. What we found in Dock8 patients can be a phenotype copied from Dock8 mice. The early activation of memory B cells from Dock8 patients is disrupted with reduced BCR clustering, B-cell spreading, and signalosome recruitment into the degree of naïve B cells, as well as the transition from naïve B cells to unswitched memory B cells. Overall, our study provides a novel mechanism for Dock8 regulation of BCR signaling by regulating cd19 transcription, as well as the underlying mechanism of noncompetence of memory B cells in Dock8 patients.
Visual Abstract
Introduction
DOCK8 belongs to a class of guanine nucleotide exchange factors that modulate the activity of ρ guanosine triphosphate hydrolase enzymes (GTPases).1 DOCK8 deficiency is the primary cause of autosomal recessive hyperimmunoglobulin E (hyper-IgE) syndrome (AR-HIES), which was first described in 2004,2 and mutations within the DOCK8 gene were subsequently demonstrated as causing AR-HIES.3 AR-HIES patients develop severe allergies, bearing elevated serum IgE levels and high peripheral eosinophil counts.4,5 For previous research, we screened 7 Chinese candidate patients for mutations within the DOCK8 gene and identified 3 large novel homozygous deletions and 4 novel point mutations with targeted deep sequencing.6
In Dock8 mutant mice, the longevity and affinity maturation of T-dependent antibody responses is severely disrupted.7 The germinal center formation is crippled, and the formation of an immunological synapse is defective. The deficiency of Dock8 has no effect, however, on some other B-cell receptor (BCR) signaling upon stimulation with anti-IgM, such as calcium flux and Erk signaling or the activation of classical molecules including CD69, CD86, and CD25.8 Dock8 patients have a breakdown of the peripheral B-cell tolerance because of the defective suppression of Tregs.9 Circulating CD19+CD27+ memory B cells are severely decreased in Dock8 patients, and circulating IgD+CD27+ marginal-zone-like (MZ-like) B cells are also decreased, which can similarly be seen in Dock8-deficient mice.7,8 Peripheral B cells of DOCK8-deficient patients are almost all naïve and fail to proliferate and secrete IgM and IgG in response to cytosine guanine dinucleotide (CpG).7 Additionally, CpG-driven phosphorylation of Syk and Stat3 is reduced in peripheral blood mononuclear cells (PBMCs) of Dock8 patients, which is dependent on the TLR9-MyD88.7 However, the role of Dock8 on BCR signaling still remains elusive. Additionally, most of the DOCK8-deficient patients are caused by the frame shift or gene deletion in different exons of the dock8 gene instead of by point mutation, so it would be interesting to generate an optimal mouse model to mirror human diseases, and we generated a Dock8-deficient mouse strain with a frame shift in the first exon of dock8. Because the frame shift occurs in the first exon of dock8, the possibility of generating a functional peptide of Dock8 is reduced because of the limited size. The difference is that the frame shift may occur in other exons in human disease.
Considering CD19 as the hallmark of B-cell development, the role of CD19 has been studied extensively in B-cell function. CD19 knockout (KO) B cells have defects in BCR signaling, cell spreading, and BCR microcluster formation.10 Deficiency of CD19 causes a collapse in the formation of germinal-center B cells and differentiation of marginal-zone B cells as well as in the T-dependent antibody response.11-13 CD19-mediated Btk signaling is critical for the maintenance and generation of MZ B cells.14 In Dock8-deficient mice, the frequency of marginal-zone B cells is also severely reduced.8 The phenotype of Dock8 KO mice recapitulates that of CD19 KO mice in the differentiation of peripheral B cells as well as in the BCR signaling, which indicates that Dock8 might regulate CD19 to control the differentiation of marginal-zone B cells.
Dock8 is involved in the actin cytoskeleton reorganization, and the function of ρGTPase in lymphocyte signaling is important. The absence of Rac1 and Rac2 leads to the disruption of actin polymerization, T-cell receptor (TCR) clustering, and calcium flux and of P38 activation.15-17 The absence of Rac2 causes the reduction of B1a and marginal-zone B cells, calcium flux, and proliferation.17 Wiskott–Aldrich syndrome protein (WASP) is a well-known critical downstream protein of ρGTPase and its deficiency has a similar phenotype as that of Dock8 deficiency, although it is still unknown whether Dock8 can regulate the activation of WASP in B cells.
In this study, we have generated Dock8-deficient mice with the TALEN technique to knock out the 45 nucleotides in exon1 and used the PBMCs from the 3 Dock8 patients screened before (Table 1). We found that Dock8 and WASP are coordinated during BCR activation and that the absence of Dock8 leads to downregulation of BCR signaling of the phosphorylated Brutons tyrosine kinase (pBtk) and pCD19 as well as of the expression and activation of WASP. Mechanistically, Dock8 can regulate the expression of CD19 and WASP. By using total internal reflection fluorescence microscopy (TIRFM), we found that the magnitude of BCR clustering, B-cell spreading, and signalosome recruitment is drastically reduced in the memory B cells of Dock8 patients and close to the level of naïve B cells. Our study has provided a new pathway demonstrating that Dock8 regulates the expression of CD19 and WASP and consequent BCR signaling and a new underlying mechanism for the defective activation of memory B cells in Dock8 patients.
Table 1.
The mutations of amino acids in Dock8 protein with TALEN technique
| Mouse type | Amino acid |
|---|---|
| WT | MATLPSAERRAFALKINRYSSSEIRKQFTLPPNLGQYHRHSISTSGFPSLQLPQLYEPVE |
| MT | MATLPSAERLRAQDQQVFIVRNKEAVYAPTQPRTVPSAQYQYIWFPLSSATSAL |
MT, mutant type; WT, wild type.
Materials and methods
Patients and control subjects
From 2014 to 2015, a total of 3 Chinese patients with Dock8 mutations from 3 unrelated Chinese families were enrolled in the present study. The diagnosis of Dock8 patients was as previously described.6 Healthy control subjects consisted of 3 age-matched subjects (average age). Signed consent from all the children’s parents was obtained with the approval of the ethics committee of the Children’s Hospital of Chongqing Medical University.
Statistical analysis
Statistical significance was assessed using the Mann-Whitney U test by Prism software (GraphPad Software). The P values were determined in comparison with the naïve or memory B cells of heathy controls (HCs) (P < .01).
Results
Dock8 is involved in BCR activation
To determine whether or not Dock8 is involved in the BCR activation, we examined the spatiotemporal relationship between BCR and Dock8 by using an antibody specific to Dock8 and confocal microscopy. At 0 minutes, Dock8 was distributed between both the plasma membrane and cytoplasm (Figure 1A). At 5 and 10 minutes, Dock8 was redistributed and cocapped with the BCR cluster (Figure 1A). At 30 minutes, Dock8 underwent endocytosis, together with the BCR (Figure 1A). As was expected, we almost could not detect Dock8 staining in B cells from Dock8-deficient mice (Figure 1C), and we found that the BCRs were located on the membrane for all the time points examined (Figure 1B). Therefore, we used a BCR internalization assay to examine the effect of Dock8 deficiency on BCR endocytosis and found that the percentage of BCRs remaining on the cell surface was significantly increased in Dock8-deficient B cells in comparison with that of WT B cells (Figure 1E). We used the correlation coefficient to determine the colocalization of BCR and Dock8 quantitatively. The colocalization between BCR and Dock8 was increased over 30 minutes and increased significantly at 10 and 30 minutes in comparison with 0 minutes (Figure 1D). To rule out the effect of BCR intensity on the internalization, we examined the BCR intensity by staining CD79α, and no difference was observed between WT and KO B cells (Figure 1F). This result suggests that Dock8 is involved in BCR activation and internalization. To confirm the deletion efficiency of Dock8 on the protein level, we did a Western blot to exam the Dock8 expression in the spleen of KO mice and found the absence of Dock8 in Dock8 KO mice, which indicates the successful deletion of Dock8 in KO mice (Figure 1G). To observe the effect of Dock8 deficiency on the impact of spleen architecture, we did a hematoxylin and eosin staining of spleen sections from WT and KO mice. We found that the germinal center in the spleen of Dock8 KO mice did not have a defined structure and had scattered lymphocytes (Figure 1H). This result indicates that Dock8 is critical to maintain the structure of the germinal center in the spleen.
Figure 1.

Dock8 is involved in BCR activation and internalization. We incubated murine splenic WT or Dock8 KO B cells with AF546–mB-Fab′–anti-Ig, to mimic sAg, for 10 minutes at 4°C to label the BCR. Then, the cells were incubated either with streptavidin or with the medium alone (0 minutes) as a control at 37°C for varying lengths of time. (A-B) After fixation and permeabilization, the cells were stained for Dock8 and analyzed using confocal microscopy (CFm). (C) The expression levels of Dock8 in WT and KO B cells quantified using NIS-Elements AR 3.2 software. (D) The correlation coefficient between BCR and Dock8 was quantified using NIS-Elements AR 3.2 software. Shown are representative images in which more than 50 cells were individually analyzed using NIS-Elements AR 3.2 software; mean values (±SD) are from 3 independent experiments. (E) Flow cytometry analysis of BCR internalization by quantifying the percentage of biotin-F(ab′)2–anti-Ig–labeled BCR remaining on the cell surface after the 37°C chase. Shown are the average percentages (±SD) from 3 independent experiments. (F) Flow cytometry analysis of BCR intensity of splenic WT or Dock8 KO B cells. (G) Western blot of Dock8 from the splenocytes of WT and Dock8 mice; glyceraldehyde-3-phosphate dehydrogenase was used as a loading control. (H) Hematoxylin and eosin staining of spleen from WT and Dock8 KO mice (19-23 weeks old). Scale bars, 2.5 μm. *P < .01; **P < .001; ***P < .0001. NS, not significant.
The absence of Dock8 downregulates BCR signaling and the expression of WASP
To determine the effect of Dock8 deficiency on BCR signaling, we examined the levels of pBtk, the key positive molecule of upstream BCR signaling, as well as protein tyrosine phosphorylation (pY), the total level of BCR signaling by using confocal microscopy and flow cytometry. The colocalization of pY and pBtk with BCR was increased over 10 minutes and decreased at 30 minutes in WT B cells, but it was dramatically decreased in DOCK8 KO total and follicular (FO) B cells (Figure 2A-C; supplemental Figure 1). The levels of pBtk and pY in both WT and KO B cells were increased over 5 minutes and then decreased for 30 minutes by using flow cytometry (Figure 2D-E). However, the levels of pY and pBtk in DOCK8 KO B cells were significantly lower than those of WT B cells upon soluble antigen (sAg) stimulation (Figure 2D-E). To normalize the signaling level, we measured the levels of total Btk by flow cytometry, and no difference was detected between WT and Dock8 KO B cells (supplemental Figure 2A). Additionally, Dock8 was reported to affect the differentiation of MZ B cells severely,11,14 and therefore the signaling levels were examined exclusively for FO B cells. Similarly, the levels of pY, pBtk, and phosphorylated extracellular regulated protein kinases (pERK) were both decreased in FO B cells of KO mice (supplemental Figure 2B-D). These results imply that Dock8 regulates BCR signaling positively. WASP has been reported as a downstream effector of Dock8, so we examined the spatiotemporal relationship between Dock8 and phosphorylated WASP (pWASP) during BCR activation by using confocal microscopy, as well as the messenger RNA (mRNA) levels of wasp and protein-expression levels of WASP in Dock8-deficient B cells by using flow cytometry. We found that the colocalization between Dock8 and pWASP in B cells peaked at 5 minutes and was significantly higher than that at 0 minutes (Figure 2F-G). The mRNA levels of wasp and protein-expression levels of WASP were significantly reduced in Dock8-deficient B cells (Figure 2H-I). We further examined the mRNA levels of Dock8 KO FO B cells because MZ B cells with higher expression of WASP were reduced in Dock8 mice, and the mRNA levels of wasp do not have changes in FO B cells (supplemental Figure 1D). This indicates that the reduction of mRNA levels of wasp in total B cells is due to the decrease of MZ B cells. WASP-interacting protein (WIP) promotes the expression of WASP.18-20 The protein and mRNA levels of WIP were examined by Western blot and reverse transcription polymerase chain reaction (RT-PCR), and both of them were significantly decreased in Dock8- deficient B cells (supplemental Figure 3A-B). These results suggest that Dock8 positively regulates the transcription of wip.
Figure 2.

The levels of tyrosine and Btk phosphorylation in BCR clusters in response to sAg is reduced in Dock8 KO B cells. Murine splenic B cells were incubated with AF546–mB-Fab′–anti-Ig without or with streptavidin (sAg) at 4°C, washed, and warmed to 37°C for varying lengths of time. After fixation and permeabilization, the cells were stained for pY, pBtk, Dock8, WASP, and pWASP and were analyzed using CFm (A-B,F) or flow cytometry (D-E,I). The Pearson’s correlation coefficients between BCR and pY/pBtk (C) or between BCR and Dock8/pWASP (G) staining in sAg-stimulated cells were determined using NIS-Elements AR 3.2 software. (H) Real-time PCR to examine the mRNA expression levels of wasp in sorted Dock8 B cells. Flow cytometry analysis of the MFI of pY and pBtk in splenic B cells after stimulation with sAgs (D-E) or the MFI of WASP in splenic B cells without stimulation (I). Shown are representative images in which more than 50 cells were individually analyzed using NIS-Elements AR 3.2 software, and mean values (±SD) are from 3 independent experiments. Scale bars, 2.5 μm. *P < .01; **P < .001. ISO, isotype control.
The absence of Dock8 leads to the reduced activation of WASP and CD19
Because Dock8 regulates the expression levels of WASP, we asked whether Dock8 deficiency also affects the activation of WASP in B cells. To determine this, we stained for activated WASP by using specific antibodies for pWASP and examined this using confocal microscopy and flow cytometry. We used the correlation coefficient to determine the colocalization of BCR and pWASP quantitatively. The colocalization between BCR and pWASP in WT B cells was increased for 10 minutes and decreased for 30 minutes afterward, but the colocalization of BCR with pWASP in DOCK8-deficient B cells was significantly lower than that of WT B cells (Figure 3A-C). In both WT and KO B cells, the levels of pWASP were increased over the first 10 minutes and then decreased over 30 minutes, but the levels of pWASP in DOCK8 KO B cells were significantly lower than those in WT B cells upon sAg stimulation by using flow cytometry (Figure 3D). After normalization, the levels of pWASP in Dock8 KO B cells were still lower, although the total levels of WASP were reduced (supplemental Figure 4). CD19 has been reported to regulate Btk upstream, and therefore we examined the effect of Dock8 deficiency on the activation of CD19 by using specific antibodies for phosphorylated CD19 (pCD19) with confocal microscopy and flow cytometry. The colocalization between pCD19 and BCR was increased over the first 10 minutes and decreased afterward in WT B cells, and the deficiency of Dock 8 significantly decreased the colocalization between pCD19 and BCR (Figure 3E-G). In both WT and DOCK8 KO B cells, the levels of pCD19 were increased over the first 10 minutes and then decreased for 30 minutes, but the levels of pCD19 in KO B cells were significantly lower than those in WT B cells upon sAg stimulation by using flow cytometry (Figure 3H). To determine at which step Dock8 regulates BCR signaling on a transcriptional level, we used RT-PCR to examine the mRNA levels of cd19, btk, cd21, and cd81 and found that only the mRNA levels of cd19 were decreased in Dock8-deficient B cells (Figure 3I-L). These results suggest that Dock8 might positively regulate the activation of WASP, CD19, and the transcription of cd19.
Figure 3.

The recruitment of pWASP and pCD19 to BCR clusters in B cells stimulated by sAg is decreased in Dock8 KO B cells. Murine splenic B cells were incubated with AF546–mB-Fab′–anti-Ig without or with streptavidin (sAg) at 4°C, washed, and warmed to 37°C for varying lengths of time. After fixation and permeabilization, the cells were stained for pWASP and pCD19 and analyzed using CFm (A-C,E-G) or flow cytometry (D,H). Flow cytometry analysis of the MFI of pWASP (D) and pCD19 (H) after stimulation with sAgs. The Pearson’s correlation coefficients between BCR and pWASP (C) or between BCR and pCD19 (G) staining in sAg-stimulated cells were determined using NIS-Elements AR 3.2 software. The relative mRNA levels of cd19, btk, cd21, and cd81 in splenic B cells examined by RT-PCR (I-L). Shown are representative images in which more than 50 cells were individually analyzed using NIS-Elements AR 3.2 software; shown are mean values (±SD) from 3 independent experiments. Scale bars, 2.5 μm. *P < .01; **P < .001.
The absence of Dock8 disrupts the differentiation of marginal zone and GC B cells
The CD19-mediated Btk signaling has been shown to control the generation of MZ B cells as well as the formation of germinal center (GC) B cells.11,14 We examined the effect of Dock8 deficiency on the generation of FO and MZ B cells by using flow cytometry. We did not observe any changes for the follicular B cells between WT and KO mice, either for the frequency or for the number (Figure 4A-C). However, we found that the percentage of MZ B cells (CD21highCD23low) was dramatically decreased in Dock8 KO mice, as well as the total numbers (Figure 4D-F). We also found a decreased frequency and number of GC B cells in nonimmunized KO mice, and the frequency of GC B cells in nonimmunized WT mice was around 7 times lower than that of immunized mice (Figure 4G-I). To clearly detect whether the GC formation has defects in Dock8 KO mice, we immunized the Dock8 KO mice and the control littermates with NP14-OVA for 2 weeks. Similarly, we found decreased frequency and number of GC B cells in Dock8 KO mice (Figure 4J-L). These results indicate that Dock8 is indispensable for the generation of peripheral MZ and GC B cells, which is consistent with a previous report.8
Figure 4.

The differentiation of marginal-zone and germinal-center B cells is reduced in Dock8 KO mice. B cells from nonimmunized and immunized WT (n = 5) and Dock8 KO (n = 5) mice were stained with labeled Abs specific for surface markers of FO, MZ, and GC B cells. Gating strategy was as follows: FO B cells (IgM-IgD+), MZ B cells (CD23lowCD21high). Anti-mouse Abs and reagents used to stain splenic MZ B cells included allophycocyanin (APC) anti-CD21 (BioLegend), fluorescein isothiocyanate (FITC) anti-B220 (BioLegend), and phycoerythrin (PE) anti-CD23 (BD Biosciences). Anti-mouse Abs and reagents to stain splenic GC B cells included FITC-anti-CD95 (BD Biosciences), APC-anti-GL7, and PerCP-Cy5.5-anti-B220 (BD Biosciences). Anti-mouse Abs and reagents used to stain FO B cells included FITC-anti-B220, Percp-Cy5.5-anti-IgD (BioLegend), and Efluor450-anti-IgM (eBioscience) at 4°C. Then, samples were analyzed by flow cytometry. Shown are representative dot plots (A,D,G,J) and the average percentages (+SD) and numbers of cells extracted from the spleen (B-C,E-F,H-I,K-L) of 3 independent experiments. *P < .01; **P < .001.
B cells from Dock8 patients have reduced BCR signaling and altered actin organization
To determine the effect of Dock8 deficiency physiologically, we used PBMCs from 3 Dock8 patients between the ages of 9 and 17 years with a clinical diagnosis showing that expression of Dock8 is absent and autoantibody is not present (Table 2). Sorted human B cells from age-matched HCs and patients were stimulated with sAg and stained with pY, pBtk, pWASP, and actin and then examined by confocal microscopy and flow cytometry. We found that the colocalization between BCR and pY/pBtk or pWASP/actin was profoundly reduced in human B cells from Dock8 patients in comparison with those from HCs (Figure 5A-F). The levels of pY and pBtk were increased for the first 5 minutes and then decreased for 30 minutes in B cells of HCs; the levels of pY and pBtk peaked at 10 minutes in the B cells of Dock8 patients and were significantly lower than those of HCs, as seen with flow cytometry (Figure 5G-H). Interestingly, the staining of pBtk was distributed throughout the whole of B cells of Dock8 patients without stimulation (0 minutes). For pWASP and F-actin, the basal levels of pWASP and F-actin were lower in the B cells of Dock8 patients than in those of HCs (Figure 5I-J). The levels of pWASP peaked at 10 minutes both in HCs and in patients, yet the levels of pWASP were significantly lower in the B cells of Dock8 patients (Figure 5I). The levels of F-actin were decreased for the first 5 minutes and then increased afterward for 30 minutes in both HCs and Dock8 patients but were lower in Dock8 patients than in HCs (Figure 5J). We also examined the total levels of CD19 and pCD19 in the B cells of HCs and Dock8 patients by confocal microscopy and flow cytometry. The colocalization between pCD19 and BCR was also significantly decreased in the B cells of Dock8 patients in comparison with that of HCs upon sAg stimulation (Figure 6A-C). The mean fluorescence intensity (MFI) of CD19 was significantly lower in patients than in HCs, quantified by NIS-Elements AR 3.2 software (Figure 6D). The levels of pCD19 peaked at 5 minutes and then decreased at 30 minutes in the B cells of HCs; it also peaked at 10 minutes in Dock8 patients but was significantly lower than that of HCs, as indicated by flow cytometry (Figure 6E). Our previous research has shown that WASP regulates the transcription of cd19.21 Dock8 positively regulates wasp upstream, and therefore we used a luciferase assay to determine whether Dock8 regulates the transcription of cd19. We transfected 293 cells with a pGL3 vector carrying cd19 promoter together with pcDNA3.1 or pcDNA3.1-Dock8, as well as internal-control renilla luciferase-thymidine kinase reporter vector (pRL-TKB). The fluorescence signal of 293 cells transfected with pGL3-CD19 and pcDNA3.1-Dock8 (pGL3-CD19+Dock8) was significantly higher than that of pGL3-CD19 and pcDNA3.1 (pGL3-CD19) (Figure 6F). These results collectively suggest that BCR signaling and actin organization are altered in Dock8 patients and further confirmed that Dock8 regulates the transcription of cd19.
Table 2.
Clinical characteristics of 3 DOCK patients
| Patient ID | Age at sample collection, y | Clinical score | Protein expression | Exon location | Auto antibody |
|---|---|---|---|---|---|
| P1 | 17 | 75 | Absent | 19-48 exon het del, | None |
| c.5842 delG,c.5843C>A | |||||
| p.A1948fsX1953 | |||||
| P2 | 17 | 62 | Absent | Exon 11 hom del. Exon 12-33 het del | None |
| P3 | 9 | 45 | Absent | c.1278-1279 delTG p.V427fsX435 | None |
Figure 5.

The recruitment of BCR signalosomes and actin to BCR clusters is decreased in Dock8 patients. Human B cells from HCs and Dock8 patients were incubated with AF546–mB-Fab′–anti-Ig without or with streptavidin (sAg) at 4°C, washed, and warmed to 37°C for varying lengths of time. After fixation and permeabilization, the cells were stained for pY, pBtk, pWASP, and F-actin and were analyzed using CFm (A-D) or flow cytometry (G-J). The Pearson’s correlation coefficients between BCR and pY/pBtk (E) or between BCR and pWASP/actin (F) staining in sAg-stimulated cells were determined using NIS-Elements AR 3.2 software. Flow cytometry analysis of the MFI of pY (G), pBtk (H), pWASP (I), and F-actin (J) after stimulation with sAgs. Shown are representative images in which more than 50 cells were individually analyzed using NIS-Elements AR 3.2 software, and mean values (±SD) are from 3 independent experiments. All 3 patients were included, and each patient was an independent experiment. Scale bars, 2.5 μm. *P < .01. P, probability.
Figure 6.

Dock8 deficiency disrupts the activation and transcription of CD19 as well as the early activation of memory B cells. Human B cells from HCs and Dock8 patients were incubated with AF546–mB-Fab′–anti-Ig without or with streptavidin (sAg) at 4°C, washed, and warmed to 37°C for varying lengths of time. (A-E) After fixation and permeabilization, the cells were stained for pCD19 and CD19 and analyzed using CFm (A-D) or flow cytometry (E). (C) The Pearson’s correlation coefficients between BCR and pCD19 staining in sAg-stimulated cells were determined using NIS-Elements AR 3.2 software. (D) The MFI of CD19 in B cells from HC and Dock8 patients was measured by NIS-Elements AR 3.2 software. (E) The MFI of pCD19 in sAg-stimulated cells were determined by flow cytometry. (F) Two hundred ninety-three cells in 24-well plates were transfected with 0.45 μg pGL3-CD19, 0.45 μg pcDNA3.1, or pcDNA3.1-Dock8 and 0.045 μg pRL-TKB (internal control) for 24 hours, followed by luceferase reporter assay; pGL3-promoter and pGL3-basic were used as positive and negative controls. TIRFM and interference reflection microscopy analysis of pY and pBtk staining in the contact zone of naïve and memory B cells from HCs and Dock8 patients incubated with membrane-tethered Fab′–anti-Ig. Shown are representative images (G-J) and the B-cell contact area (K), the MFI of Fab′–anti-Ig in the B-cell contact zone (L), MFI (±SD) of pY (M), and pBtk (N) in the B-cell contact zone from 3 independent experiments. All 3 patients were included, and each patient was an independent experiment. Scale bars, 2.5 µm. *P < .01; **P < .001, in comparison with naïve B cells from Dock8 patients. M, memory B cell; N, naïve B cell.
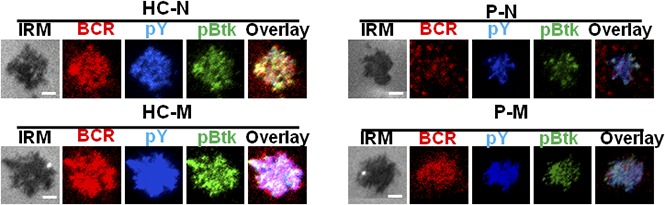
Memory B cells from Dock8 patients have disrupted early activation
CD19 is critical for the early activation of B cells10; to determine the effect of Dock8 deficiency on the early activation of human B cells, we examined the BCR clustering and B- cell spreading in HCs and Dock8 patients’ B cells upon stimulation with membrane-associated antigens by TIRFM. Memory B cells were distinguished from naïve B cells by CD27 staining. In HCs, the contact area between naïve B cells and the antigen-containing lipid bilayer increased for the first 5 minutes and started to contract afterward (Figure 6G,K). In HCs memory B cells, the contact area was dramatically increased in comparison with that of HCs naïve B cells (Figure 6G,I,K). The contact area dynamics of naïve B cells from Dock8 patients was similar to those of HCs’ naïve B cells (Figure 6G-H,K). However, the contact area of memory B cells from Dock8 patients was drastically decreased in comparison with that of HCs’ memory B cells, showing no increase in the contact area in comparison with naïve B cells from Dock8 patients (Figure 6G-K). The MFI used to gauge the BCR clustering in the contact area of HCs’ naïve B cells increased to 7 minutes (Figure 6G,L), and the MFI in the contact zone of HCs’ memory B cells was significantly increased in comparison with that of naïve B cells in HCs (Figure 6G,I,L). But the MFI in the contact zone of memory B cells from Dock8 patients was significantly reduced in comparison with that of HCs’ memory B cells, and even similar to the level of naïve B cells from Dock8 patients (Figure 6G-J,L). To obviate the effect of Dock8 deficiency on the BCR intensity, we determined the BCR intensity with CD79α antibody by using flow cytometry, and no difference was observed between HCs and Dock8 patients (data not shown). Together, these results imply that Dock8 deficiency fails to promote the increase of BCR clustering and B-cell spreading from naïve B cells to memory B cells. To further determine the effect of Dock8 deficiency on the signalosome recruitment in human B cells upon mAg stimulation, we examined the total level of BCR signaling pY, and the phosphorylation of the key positive upstream signaling molecule Btk (pBtk). In HCs’ naïve B cells, the recruitment of pY peaked at 3 minutes and decreased at 7 minutes (Figure 6G,M). The level of pY in the contact zone of HCs’ memory B cells was profoundly increased in comparison with that of HCs naïve B cells (Figure 6G,I,M). However, the level of pY in the contact zone of naïve and memory B cells in Dock8 patients was of the same level with each other and significantly reduced in comparison with that of HC naïve and memory B cells, respectively (Figure 6G-M). Similar to the behavior of the activation of pY, the activation of Btk in HCs’ naïve or memory B cells was also significantly higher than that of pBtk in naïve or memory B cells from Dock8 patients (Figure 6G-J,N). To confirm the effect of Dock8 deficiency on the BCR signaling, we determined the phosphorylated levels of Erk and Btk in B cells upon stimulation with sAgs by using the BD Phosflow method. The levels of pERK and pBtk were increased in HCs’ memory B cells and reduced in naïve or memory B cells of Dock8 patients (Figure 7A-B). Interestingly, we found that the transition from naïve B cells to unswitched memory B cells (IgD+CD27+) was abolished for Dock8 patients in comparison with that of HCs (Figure 7C) after in vitro stimulation. To further elucidate the Ig levels in memory B cells, IgM, IgG, and IgE were stained. The levels of IgE were enhanced in Dock8 patients, which is consistent with previous reports. Surprisingly, it was hard to detect IgM memory B cells, and the levels of IgG memory B cells were reduced in Dock8 patients (supplemental Figure 5A). We also found a decreased expression of CD19 in total B cells as well as naïve and memory B cells from Dock8 patients in comparison with that of HCs (Figure 7D-E; supplemental Figure 5B) but no difference for the expression of CD81 that is associated with CD19 (Figure 7F). These results suggest that Dock8 deficiency blocks the boost of signaling from naïve cells to memory B cells.
Figure 7.

Dock8 deficiency cripples the transition from naïve B cells to unswitched memory B cells. Enriched B cells from PBMCs of HCs and Dock8 patients were stimulated with sAgs for varying lengths of time and fixed and stained with anti-pERK and pBtk antibody for flow cytometry. (A-B) The quantification of MFI of pERK (A) and pBtk (B) in naïve or memory B cells from HCs or Dock8 patients. (C) Flow cytometry analysis of naïve and memory B cells from HCs and Dock8 patients distinguished by IgD and CD27 staining. (D) The flow plot of CD19 expression in total B cells of HCs and Dock8 patients. (E-F) The quantification of MFI of CD19 and CD81 in naïve or memory B cells from HCs or Dock8 patients with flow cytometry (patients 1-3). Shown are the mean values (±SD) from 3 independent experiments. *P < .01; **P < .001.
Discussion
In this study, we investigated the role of Dock8 deficiency on BCR signaling by using germline Dock8-deletion mice and the PBMCs from Dock8-deficient patients. We found that Dock8 is involved in the BCR activation and that the absence of Dock8 leads to the diminishment of the key positive BCR signaling molecule Btk. Furthermore, the upstream molecule of Btk-CD19’s activation is severely disrupted. By analyzing the mRNA levels of cd19 and wasp, we found that Dock8 deficiency downregulates the transcription of cd19 in mouse B cells. We found that the magnitudes of BCR clustering, B-cell spreading, and BCR signalosome recruitment are severely disrupted in memory B cells of Dock8 patients in comparison with that of HC memory B cells and close to the levels of naïve B cells. The expression levels of CD19 are also dramatically reduced in B cells of Dock8 patients, which is similar to the results from Dock8 KO mice. We also revealed that Dock8-deficient human B cells have a reduced activation of WASP and actin polymerization. Overall, this is the first report, to our knowledge, that Dock8 positively regulates the transcription of cd19 and that there is a defect in early activation of memory B cells in Dock8 patients.
One interesting future direction would be to determine how Dock8 regulates WASP in B cells. Recently, the Dock8-WIP-WASP axis has been reported to link TCR and actin.22 The phenotypes of Dock8 mice and patients almost copy those of WASP KO mice or WASP patients.21 As a guanine nucleotide exchange factor, Dock8 has been involved in the activation of Rac1 and cdc42.23,24 Interestingly, Dock8 deficiency does not affect the total Cdc42 activity in dendritic cells, and it is disrupted only at the leading edge of spreading membrane, causing a severe defect in cell polarization and migration.
Therefore, DOCK8 regulates DC migration by controlling Cdc42 activity spatially.24 Cdc42 is a critical activator upstream of WASP by interacting with its GTPase-binding domain.25-27 In our studies, we found that the global activity of WASP is reduced. It would be of interest to explore whether Dock8 regulates the activation of WASP via Cdc42. What also requires confirmation is whether Dock8 regulates the transcription of cd19 via WASP. Two hundred ninety-three cells lack the WASP expression, and it is possible that Dock8 regulates the CD19 transcription via WAVE in 293 cells. Therefore we need to cross the Dock8 KO mice with WASP transgenic mice to examine whether the defect of cd19 in Dock8 KO mice can be rescued.
To be consistent with previous reports, our data support the notion of Dock8-deficient B cells having reduced activation of pSyk and pSTAT-3 upon stimulation with CpG.7 But it is also contradictory with the report that the activation of Erk and calcium flux is not affected in Dock8-mutant mice determined from a genetic screen in Goodnow’s group.8 The discrepancy could be due to the difference of Dock8 deficiency, the background of the mice, or the dose and type of stimulants. Goodnow's group used MD4 transgenic mice and they used IgM or CD40 ligand to stimulate the B cells.
In summary, our study reveals a new pathway whereby Dock8 regulates BCR signaling through regulating CD19 and WASP expression. Furthermore, the underlying cellular molecular mechanism of defective memory response in Dock8 patients is due to the inefficient early activation events of their memory B cells. Overall, it will provide a therapeutic design for the treatment of Dock8 patients.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Katrina Randall for the helpful discussions regarding this manuscript.
This work was supported by the Natural Science Foundation of China (81701628, 81722002, and 31500709) and the Public Welfare Scientific Research Project of China (201402012).
Authorship
Contribution: X.S. and J.W. carried out the initial analyses and drafted the initial manuscript; T.Q., Y.Z., L.H., L.N., X.B., Y.J., and X.X. performed the microscopic and flow cytometry assay; H.M., Y.Z., W.S., X.T., and Z.Z. reviewed and revised the manuscript; X.Z. and C.L. conceptualized and designed the study and reviewed and revised the manuscript; and all authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Chaohong Liu, Department of Pathogen Biology, School of Basic Medicine, Huazhong University of Science and Technology, No. 13, Hangkong Rd, Wuhan 430030, China; e-mail: chaohongliu80@126.com; Xiaodong Zhao, Chongqing City Key Laboratory of Child Infection and Immunity, Children's Hospital of Chongqing Medical University, No. 136, Zhongshan 2nd Rd, Yuzhong District, Chongqing 400014, China; e-mail: zhaoxd530@aliyun.com; and Zhiyong Zhang, Chongqing City Key Laboratory of Child Infection and Immunity, Children's Hospital of Chongqing Medical University, No. 136, Zhongshan 2nd Rd, Yuzhong District, Chongqing 400014, China; e-mail: zzy510214@126.com.
References
- 1.Miyamoto Y, Yamauchi J. Cellular signaling of Dock family proteins in neural function. Cell Signal. 2010;22(2):175-182. [DOI] [PubMed] [Google Scholar]
- 2.Ruusala A, Aspenström P. Isolation and characterisation of DOCK8, a member of the DOCK180-related regulators of cell morphology. FEBS Lett. 2004;572(1-3):159-166. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Q, Davis JC, Lamborn IT, et al. . Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361(21):2046-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grimbacher B, Holland SM, Puck JM. Hyper-IgE syndromes. Immunol Rev. 2005;203(1):244-250. [DOI] [PubMed] [Google Scholar]
- 5.Buckley RH, Wray BB, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics. 1972;49(1):59-70. [PubMed] [Google Scholar]
- 6.Qin T, An Y, Liu C, et al. . Novel DOCK8 gene mutations lead to absence of protein expression in patients with hyper-IgE syndrome. Immunol Res. 2016;64(1):260-271. [DOI] [PubMed] [Google Scholar]
- 7.Jabara HH, McDonald DR, Janssen E, et al. . DOCK8 functions as an adaptor that links TLR-MyD88 signaling to B cell activation. Nat Immunol. 2012;13(6):612-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Randall KL, Lambe T, Johnson AL, et al. . Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nat Immunol. 2009;10(12):1283-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janssen E, Morbach H, Ullas S, et al. . Dedicator of cytokinesis 8-deficient patients have a breakdown in peripheral B-cell tolerance and defective regulatory T cells. J Allergy Clin Immunol. 2014;134(6):1365-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Depoil D, Fleire S, Treanor BL, et al. . CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nat Immunol. 2008;9(1):63-72. [DOI] [PubMed] [Google Scholar]
- 11.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376(6538):352-355. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Carter RH. CD19 regulates B cell maturation, proliferation, and positive selection in the FDC zone of murine splenic germinal centers. Immunity. 2005;22(6):749-761. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Brooks SR, Li X, Anzelon AN, Rickert RC, Carter RH. The physiologic role of CD19 cytoplasmic tyrosines. Immunity. 2002;17(4):501-514. [DOI] [PubMed] [Google Scholar]
- 14.Martin F, Kearney JF. Positive selection from newly formed to marginal zone B cells depends on the rate of clonal production, CD19, and btk. Immunity. 2000;12(1):39-49. [DOI] [PubMed] [Google Scholar]
- 15.Mulloy JC, Cancelas JA, Filippi MD, Kalfa TA, Guo F, Zheng Y. Rho GTPases in hematopoiesis and hemopathies. Blood. 2010;115(5):936-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo F, Cancelas JA, Hildeman D, Williams DA, Zheng Y. Rac GTPase isoforms Rac1 and Rac2 play a redundant and crucial role in T-cell development. Blood. 2008;112(5):1767-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo F, Velu CS, Grimes HL, Zheng Y. Rho GTPase Cdc42 is essential for B-lymphocyte development and activation. Blood. 2009;114(14):2909-2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fried S, Matalon O, Noy E, Barda-Saad M. WIP: more than a WASp-interacting protein. J Leukoc Biol. 2014;96(5):713-727. [DOI] [PubMed] [Google Scholar]
- 19.de la Fuente MA, Sasahara Y, Calamito M, et al. . WIP is a chaperone for Wiskott-Aldrich syndrome protein (WASP). Proc Natl Acad Sci USA. 2007;104(3):926-931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konno A, Kirby M, Anderson SA, Schwartzberg PL, Candotti F. The expression of Wiskott-Aldrich syndrome protein (WASP) is dependent on WASP-interacting protein (WIP). Int Immunol. 2007;19(2):185-192. [DOI] [PubMed] [Google Scholar]
- 21.Bai X, Zhang Y, Huang L, et al. . The early activation of memory B cells from Wiskott-Aldrich syndrome patients is suppressed by CD19 downregulation. Blood. 2016;128(13):1723-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janssen E, Tohme M, Hedayat M, et al. . A DOCK8-WIP-WASp complex links T cell receptors to the actin cytoskeleton. J Clin Invest. 2016;126(10):3837-3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang SJ, Cui HY, Liu YM, et al. . CD147 promotes Src-dependent activation of Rac1 signaling through STAT3/DOCK8 during the motility of hepatocellular carcinoma cells. Oncotarget. 2015;6(1):243-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harada Y, Tanaka Y, Terasawa M, et al. . DOCK8 is a Cdc42 activator critical for interstitial dendritic cell migration during immune responses. Blood. 2012;119(19):4451-4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Symons M, Derry JM, Karlak B, et al. . Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell. 1996;84(5):723-734. [DOI] [PubMed] [Google Scholar]
- 26.Aspenström P, Lindberg U, Hall A. Two GTPases, Cdc42 and Rac, bind directly to a protein implicated in the immunodeficiency disorder Wiskott-Aldrich syndrome. Curr Biol. 1996;6(1):70-75. [DOI] [PubMed] [Google Scholar]
- 27.Westerberg L, Greicius G, Snapper SB, Aspenström P, Severinson E. Cdc42, Rac1, and the Wiskott-Aldrich syndrome protein are involved in the cytoskeletal regulation of B lymphocytes. Blood. 2001;98(4):1086-1094. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.