

Key Points
HLA-lacking iPSC-derived HSCs from aplastic anemia patients show a hematopoietic ability similar to wild-type iPSC-HSCs.
iPSC-HSCs that lack HLA-B4002 escape specific T-cell attack.
Abstract
Hematopoietic stem cells (HSCs) that lack HLA-class I alleles as a result of copy-number neutral loss of heterozygosity of the short arm of chromosome 6 (6pLOH) or HLA allelic mutations often constitute hematopoiesis in patients with acquired aplastic anemia (AA), but the precise mechanisms underlying clonal hematopoiesis induced by these HLA-lacking (HLA−) HSCs remain unknown. To address this issue, we generated induced pluripotent stem cells (iPSCs) from an AA patient who possessed HLA-B4002–lacking (B4002−) leukocytes. Three different iPSC clones (wild-type [WT], 6pLOH+, and B*40:02-mutant) were established from the patient’s monocytes. Three-week cultures of the iPSCs in the presence of various growth factors produced hematopoietic cells that make up 50% to 70% of the CD34+ cells of each phenotype. When 106 iPSC-derived CD34+ (iCD34+) cells with the 3 different genotypes were injected into the femoral bone of C57BL/6.Rag2 mice, 2.1% to 7.3% human multilineage CD45+ cells of each HLA phenotype were detected in the bone marrow, spleen, and peripheral blood of the mice at 9 to 12 weeks after the injection, with no significant difference in the human:mouse chimerism ratio among the 3 groups. Stimulation of the patient’s CD8+ T cells with the WT iCD34+ cells generated a cytotoxic T lymphocyte (CTL) line capable of killing WT iCD34+ cells but not B4002− iCD34+ cells. These data suggest that B4002− iCD34+ cells show a repopulating ability similar to that of WT iCD34+ cells when autologous T cells are absent and CTL precursors capable of selectively killing WT HSCs are present in the patient’s peripheral blood.
Visual Abstract
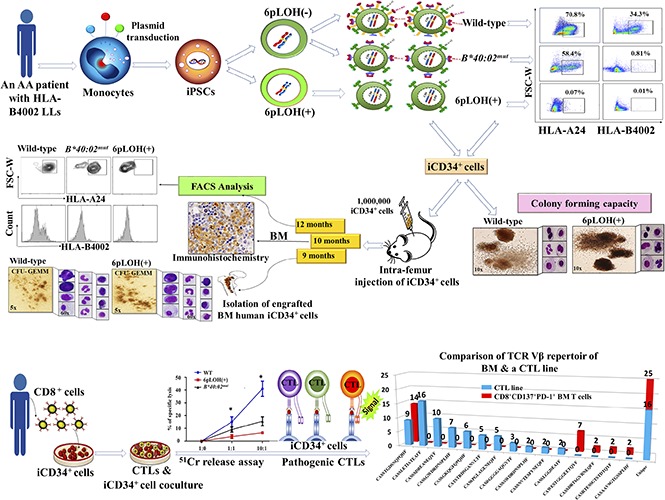
Introduction
Acquired aplastic anemia (AA) is a bone marrow (BM) failure syndrome characterized by peripheral pancytopenia and BM hypoplasia that has been etiologically linked to an idiopathic autoimmune response, mainly elicited by pathogenic cytotoxic T lymphocytes (CTLs), which trigger the destruction of hematopoietic stem cells (HSCs) in the BM.1 The fact that most patients with AA treated with immunosuppressive therapy achieve a good clinical response, along with the presence of various immune alterations in patients with AA, including the accumulation of T cells with specific clonotypes2-4 within the BM and the presence of dysregulated CD8+ cytotoxic T cells,5 constitutes important biological evidence that supports an autoimmune etiology in this disease. In this model, CTLs that recognize autoantigens presented on HSCs through their class I HLA molecules are considered to play a central role in the autoimmune reactions that ultimately lead to the destruction of HSCs. However, the precise molecular mechanisms that trigger this autoimmune response remain unclear.
Clonal hematopoiesis is frequently observed in patients with AA, and this phenomenon seems to be directly associated with the pathogenesis of this disease.6 The copy-number neutral loss of heterozygosity in the short arm of chromosome 6 (6pLOH) caused by acquired uniparental disomy is a common abnormality documented in AA patients with clonal hematopoiesis.7-12 Although 6pLOH+ HSCs are thought to escape the CTL attack and induce clonal hematopoiesis by HLA class I–lacking (HLA−) leukocytes, the precise mechanisms underlying such clonal hematopoiesis remain unknown.
One important historic limitation for systematically studying AA has been the lack of target tissues suitable for studies that aim to identify potential autoantigens or search for intrinsic molecular abnormalities in HSCs because a low number of HSCs is inherent in this disease. Somatic cell reprogramming into induced pluripotent stem cells (iPSCs) represents a valuable tool for research as well as diagnostic and therapeutic applications, and generating HSCs from patient-specific iPSCs is of great importance in studying tissue-depleted diseases such as AA.
In previous studies, we showed that the HLA alleles that are most frequently lost in patients with AA are HLA-A*02:01, -A*02:06, -A*31:01, and -B*40:02.6,9,10 Taking advantage of a new monoclonal antibody (mAb) specific to HLA-B4002, we recently reported that granulocytes lacking B4002 (B4002− granulocytes) are produced not only by 6pLOH+ HSCs but also by HSCs that undergo various mutations in HLA-B*40:02.10 The presence of such B*40:02mut leukocytes in addition to 6pLOH+ leukocytes suggests that B4002 is a critical HLA molecule that presents autoantigens of HSCs. B4002− HSCs derived from AA patients and their B4002+ counterparts may therefore be useful for investigating the pathogenesis of AA.
To better understand clonal hematopoiesis induced by HLA− HSCs in AA, we generated iPSCs from the monocytes of an AA patient who possessed B4002− leukocytes as a result of 6pLOH and/or allelic mutation. Our study revealed that the B4002− CD34+ cells induced from iPSCs (iCD34+ cells) possess hematopoietic potential in vitro and in vivo similar to that of their B4002+ counterparts. Precursors of CTLs capable of killing wild-type (WT) but not B4002− iCD34+ cells were detected among the patient’s CD8+ T cells.
Materials and methods
Patients
The characteristics and the details of our single patient’s HLA− leukocytes are described in the supplemental Data and in supplemental Tables 1 and 2.
Generation of iPSCs from an AA patient
Heparinized blood (21 mL) was drawn after signed informed consent was obtained from an AA patient (KANA1) who was dependent on cyclosporine but was in convalescence 14 years after antithymocyte globulin therapy. This study was approved by the ethics committee of Kanazawa University Institute of Medical, Pharmaceutical, and Health Sciences. iPSCs were generated from the patient’s peripheral blood mononuclear cells (PBMCs) at the Center for iPS Cell Research and Application (Kyoto University, Kyoto, Japan) using a nonintegrating method as described previously with some modifications.13 PBMCs were transfected with episomal vector mixtures and cultured in non–T-cell medium that mainly stimulates the proliferation of monocytes (details are provided in supplemental Methods).
Maintenance and expansion of iPSCs
iPSCs were maintained in an undifferentiated state by coculturing them with mitomycin C–treated SNL feeder cells in Dulbecco’s modified Eagle medium/F12 supplemented with 20% knockout serum replacement medium, fibroblast growth factor (10 ng/mL), 2 mM l-glutamine, nonessential amino acids, and 1% penicillin and streptomycin.
Induction of HSCs from iPSCs
CD34+ cells were generated from iPSCs by 4 different methods: (1) coculture with mitomycin-C–treated OP9 cells as described previously, (2) a classical feeder-free system (StemPro-34 serum-free medium supplemented with cytokines and growth factors), (3) culture in the conditioned medium (CM) derived from OP9 cells, and (4) culture in the CM derived from WEHI cells. Preparation of the CM is described in supplemental Methods (supplemental Figure 1). Detailed protocols for flow cytometry (FCM) analysis, HLA genotyping, reverse transcription polymerase chain reaction (RT-PCR) analysis, and detection of HLA allele mutations are provided in the supplemental Data, and the mAbs and primer sets used for this study are provided in supplemental Tables 3-6.
Colony-forming unit assay
iCD34+ cells obtained from feeder and feeder-free culture systems were isolated by using human CD34 Microbeads (Miltenyi Biotec, Tokyo, Japan) or an Aria Fusion fluorescence-activated cell sorter (BD Biosciences, Franklin Lakes, NJ). Enriched cells were re-suspended at a density of 40 000 cells per mL. A total of 400 μL of the cell re-suspension was mixed with 3 mL of MethoCult GF H4034 (STEMCELL Technologies, Vancouver, BC, Canada), and 1 mL was plated into 35-mm dishes in duplicate and incubated at 37°C with 5% CO2 for 14 to 16 days. an An inverted microscope was used to count and characterize colony-forming unit (CFU)–derived colonies. Lineage assignment was determined by a morphologic analysis of Giemsa-stained individual colonies.
Transplantation of human iCD34+ cells into immunodeficient mice
A total of 1 × 106 iCD34+ cells was injected into the femurs of sublethally (150 cGy) irradiated C57BL/6.Rag2nullIl2rgnull NOD-Sirpa (BRGS) mice lacking T, B, and natural killer lymphocytes14,15 as described previously. In some experiments, adult mice (4-5 months of age) were used instead of young mice (5-6 weeks of age). The animals were maintained as described previously16 and were euthanized at 9, 10, or 12 weeks after transplantation. The peripheral blood (PB), BM, spleens, and thymus were obtained and prepared for FCM and immunohistochemical analyses. Additional mice injected with phosphate-buffered saline were used as negative controls. The detailed protocols are provided in supplemental Methods. The mAbs used for this study are listed in supplemental Table 7. The engraftment of human cells was assessed by PCR for the α-satellite region of human chromosome 17 as described previously16 or by detecting human CD45 and HLA genes using genomic DNA from the BM and spleens of transplanted mice as templates. PCR was carried out with HotstarTaq Plus Master Mix (QIAGEN, Venlo, The Netherlands) using the primer sets listed in supplemental Table 6. These animal studies were approved by the Institutional Animal Care and Use Committee at Kanazawa University.
Establishment of CTL lines
CTLs were induced basically as described before17 with the following minor modifications: PBMCs from KANA1 were isolated using Lymphoprep, and the CD8+ cell fraction was purified using a CD8+ T-cell isolation kit (BioLegend, San Diego, CA). A total of 1 × 106 purified CD8+ cells was cultured in RPMI 1640 plus 10% pooled human serum and recombinant interleukin-7 (IL-7) (PeproTech, Rocky Hill, NJ) for 48 hours in the presence of 0.5 × 106 mitomycin-treated iCD34+ cells. The cells were then cultured in RPMI 1640 medium containing 10% pooled human serum and 50 U/mL of recombinant IL-2 (PeproTech) and re-stimulated with 0.2 × 106 iCD34+ cells once every 3 days for 2 weeks. Cytotoxicity was evaluated by using a classical 51Cr release assay,18 by using a CD107 degranulation assay as described previously,19 by coculturing the 0.5 × 106 cultured CD8+ T cells with either autologous 6pLOH+ iCD34+ or WT iCD34+ cells at different effector:target ratios, or by measuring the release of interferon-γ (IFN-γ) from the CTLs cocultured with target cells (6pLOH+ or WT iCD34+ cells) overnight at an effector:target ratio of 10:1. The levels of IFN-γ in the cultured supernatants were determined by using an enzyme-linked immunosorbent assay (Mabtech, Nacka Strand, Sweden) according to the manufacturer’s recommendations. In some experiments, the cytotoxicity of the CD8+ T cells against iCD34+ cells was assessed by directly observing the mixture of T cells and iCD34+ (6pLOH+ or WT) cells cultured in 96-well round-bottom plates for 5 days in RPMI 1640 medium supplemented with 10% human serum and recombinant IL-2 (PeproTech) along with IL-3, Flt3 ligand, and granulocyte-macrophage colony-stimulating factor (all from ProsPec, Rehobot, Israel) under an inverted microscope.
T-cell receptor repertoire analysis
CD8+ cells from a CTL line and CD8+CD137+PD-1+ cells from the patient’s BM that were obtained before immunosuppressive therapy were subjected to single-cell sorting using a FACSAria cell sorter (Becton Dickinson) followed by T-cell receptor (TCR) complementary DNA analysis (supplemental Figure 2) as described previously.20
Data analyses
Statistical analyses were performed using the GraphPad Prism software package, version 5.02 (San Diego, CA). The results were analyzed using Student t test. For all experiments, values were shown as the mean of the individual sample ± standard error of the mean. Findings were judged to be statistically significant if P < .05.
Results
Generation of HLA-lacking iPSCs from an AA patient possessing HLA-B4002− leukocytes
To investigate the biological relevance of HLA− leukocytes in patients with AA, we performed somatic cell reprograming of a patient’s monocytes to generate iPSC clones with normal and abnormal HLA genotypes. Our patient had 11.3% to 40.9% HLA-A24–lacking (6pLOH+) cells in all lineages of leukocytes (Figure 1A), and his monocytes consisted of 3 different populations, including WT, A24+B4002−, and A24−B4002− (6pLOH+) cells at rates indicated in supplemental Figures 1B and 3. Among the 14 iPSC clones generated from the patient’s monocytes, 10 clones were 6pLOH+, and 4 clones were 6pLOH−, as demonstrated by quantitative PCR (Figure 1C) and qualitative PCR (supplemental Figure 4A-B). Deep sequencing revealed that 1 of the 4 6pLOH− iPSC clones had a mutation at the start codon of B*40:02 (Figure 1D), thereby showing an A24+B4002− phenotype, and confirmed that the 10 clones had 6pLOH (supplemental Figure 4C). Figure 1E and supplemental Table 8 summarize the genotypes of the 14 iPSC clones. Morphologically, the WT and HLA− iPSC clones were indistinguishable (supplemental Figure 5A-G). Consistent with previous studies, HLA molecules were not detectable on the surface of iPSCs in either WT or B4002− iPSC clones (supplemental Figure 5H).
Figure 1.

Establishment of iPSCs with different HLA genotypes from the monocytes of a patient with acquired AA. (A) HLA-A*24:02–lacking cells in the different lineages of cells. Histograms of HLA-A*24:02-expressing cells in different lineages of the patient (green lines) and a healthy individual (blue lines) are shown. (B) HLA-B4002 expression in monocyte fractions of the patient. Three different populations including A24+B4002+ (WT), A24+B4002− (B*40:02mut), and A24−B4002− (6pLOH+) monocytes are shown. (C) Results of TaqMan PCR on 14 iPSC clones showing 10 6pLOH+ iPSC clones (red dots) and 4 6pLOH− iPSC clones (green dots) that include 1 B4002− clone; the blue dots represent control DNA. The x-axis and y-axis represent the amplified product amounts of B*40:02 and B*35:01, respectively. (D) Next-generation sequencing findings for B4002− iPSC clone G2 showing a start loss mutation in B*40:02. The results of the HLA-B allelic sequencing of WT iPSC clone E3 is shown as a control. The green bar represents the start loss mutation locus of the B*40:02mut clone. (E) The percentages of WT, B*40:02mut, and 6pLOH+ iPSC clones are shown.
Because the hematopoietic potential of iPSCs derived from AA patients is not known, we first evaluated the capacity of a WT iPSC clone derived from KANA1 (clone G1) to generate hematopoietic cells after coculture with OP9 feeder cells. An FCM analysis performed at 3 weeks of culture showed that these cells expressed various hematopoietic markers on their surface, including CD34, CD45, CD38, glycophorin-A, and CD90 (supplemental Figure 6A-B), and a substantial proportion of these iCD34+ cells were positive for CD45, CD108, and CXCR-4 and vigorously expressed HLA-ABC on their surface (Figure 2A). A time kinetic study showed the progressive increase in the expression of the hematopoietic markers and the concomitant loss of the immature iPSC markers (SSEA-4 and TRA2-49) as the iPSCs differentiated into hematopoietic cells in vitro (Figure 2 B-C; supplemental Figure 6C). Of note, the expression of hematopoietic cell markers varied among different iPSCs (supplemental Figure 6D). A gene expression analysis showed the upregulation of hematopoietic lineage genes HoxA9, CD45, and CD41 and the concomitant downregulation of pluripotency-specific markers (OCT3) in cultured iPSCs (Figure 2D).
Figure 2.

Induction of HSC differentiation from iPSCs using different culture systems. (A) Representative FCM results showing the expression of various surface markers on iPSCs obtained by coculturing WT iPSC clone G1 with OP9 cells for 21 days. (B) The percentages of SSEA-4–, CD34+, CD108+, CD184+, and CD45+ expressed by iPSCs during the differentiation process of iPSCs cultured with OP9 cells. The percentages represent the mean ± standard deviation (SD) of the data from 5 experiments using 3 different WT iPSC clones analyzed at 7, 14, and 21 days of culture. (C) Changes in the SSEA-4 expression of WT clone G1 with time after culture. (D) HSC- and iPSC-related gene expression patterns of iCD34+ cells. Total RNA extracted from original iPSC clones (WT and 6pLOH clones), iCD34+ cells induced from the same iPSCs, CD34+ cord blood cells, and a stem cell-like acute monoblastic leukemia (AML) cell line (AML-MT) was subjected to an RT-PCR analysis for 4 genes: HoxA9, CD45, CD41, and OCT3. (E) Representative images of hematopoietic cells induced from iPSCs in various hematopoietic differentiation media (StemPro-34 serum-free medium [SFM] and OP9, WEHI, and OP9 and WEHI conditioned media). Images were captured by using a light microscope at day 14. (F) The expression of cell surface markers on iPSCs cultured under the 4 different methods. Cells were harvested and analyzed by FCM at the indicated time points (0, 7, 14, 21, and 28 days). The data represent results from 2 independent experiments and are expressed as the mean ± standard error of the mean (SEM) of the percentage of cells expressing the given marker. FSC-W, forward scatter width; SFM, serum-free medium; SSC-H, side scatter height; UCB, umbilical cord blood.
Generation of iCD34+ cells using various hematopoietic differentiation methods
The potential of iPSCs to differentiate into HSCs was examined by using several different culture and differentiation systems. Comparative differentiation capacity was observed when the iPSCs were co-cultured with OP9 feeder cells in Alpha minimum essential medium or when they were cultured in StemPro-34 serum-free medium supplemented with hematopoietic cytokines and growth factors (data not shown). Of note, cultured iPSCs generated in the feeder-free system tended to have a higher HLA expression than those generated with OP9 cells (supplemental Figure 7). Importantly, we successfully generated iCD34+ cells by culturing iPSCs in CM derived from either OP9 or WEHI cells or a combination of both (Figure 2E). Cultured iPSCs generated with those CMs showed similar morphologic characteristics and essentially equal phenotypes, although a higher proportion of CD45+ cells among iCD34+ cells was generated with WEHI and OP9 and WEHI CM, and a higher proportion of CD34+ and HLA-ABC+ cells was generated with StemPro-34 serum-free medium (Figure 2F; supplemental Figure 8A).
Generation of iCD34+ cells from WT and HLA-B4002− iPSC clones
Next, we characterized the iCD34+ cells generated from WT or B4002− iPSC clones. FCM confirmed the normal HLA expression by WT iCD34+ and the absence of HLA-A24 in hematopoietic stem/progenitor cells (HSPCs) derived from a 6pLOH+ iPSC clone (Figure 3A). Notably, cultured iPSCs derived from WT or 6pLOH+ monocytes showed comparable expression of various hematopoietic markers, such as CD34, CD45, CD38, and CD90, and showed comparable expression of HLA molecules (HLA-ABC, -A2, -DR, -E, and -G) (Figure 3B). As expected, iCD34+ cells with the 6pLOH+ genotype showed a higher expression of the retained allele (HLA-A2), which was compatible with duplication of HLA-A*02:01 due to 6pLOH. FCM revealed the absence of HLA-A24 and/or B4002 in iCD34+ cells corresponding to the genotype of the original iPSCs (Figure 3C-D). Notably, increasing passage number (up to17 passages) of iPSC clones or culture methods did not alter the differentiation potential or the HLA expression in the generated iCD34+ cells, although it did result in varying expression of hematopoietic markers (supplemental Figure 8B-C).
Figure 3.

Characterization of HSPs derived from iPSCs with different HLA genotypes. (A) Expression of A*02:01 and A*24:02 by WT and 6pLOH+ iCD34+ cells. WT and 6pLOH+ iPSCs cultured with OP9 cells for 21 days were examined for HLA-A24 and A2 expression on the gated CD34+ cells. (B) HSC induction from WT and 6pLOH+ iPSCs and HLA expression by the iCD34+ cells. Cultured WT (blue column) and 6pLOH+ (red column) with OP9 cells were analyzed for the percentage of CD34+ cells and HLA expression by the gated CD34+ cells. The data show the mean ± SEM from 3 independent experiments. (C-D) B4002 expression by CD34+ cells derived from 3 different iPSCs with different HLA genotypes. iPSCs cultured with a feeder system (OP9 cells) for 21 days were examined for the expression of A2402 and B4002. HSCs derived from a B4002− iPSC clone lacked B4002 but retained A2402 as expected. A representative set of (C) scattergrams and (D) the percentages of CD34+ cells and HLA-A allele+ cells are shown. The columns represent the mean ± SEM of the values determined by FMC. (E) Hematopoietic colony-forming capacities of iCD34+ cells. The pie chart shows CFU, granulocyte, erythrocyte, megakaryocyte, macrophage (CFU-GEMM), CFU-granulocyte (CFU-G), CFU, granulocyte-macrophage (CFU-GM), CFU, macrophage (CFU-M), and burst-forming unit erythroid (BFU-E)–derived colonies generated from the WT-iCD34+ cells. (F) The plating efficiencies of iCD34+ cells derived from (left panel) 3 WT iPSC clones are compared among (middle panel) 3 different feeder-free systems with StemPro or CM from OP9 or WEHI cells. (Right panel) Summary of the plating efficiencies of iCD34+ cells with 3 different HLA genotypes. The data indicate the mean ± SEM of the CFU percentages obtained from 3 independent experiments. The plating efficiency was defined as the frequency of colonies generated from 5000 iCD34+ seeded cells (total number of colonies per 5 × 103 cells seeded). *P < .05; **P < .01; ***P < .001. NS, not significant; SSC-W, side scatter width.
Clonogenic potential of iCD34+ cells with different HLA genotypes
iCD34+ cells generated from 3 WT iPSC clones (clones E2, E3, and G1) gave rise to CFUs, including CFU, granulocyte-macrophage (CFU-GM); burst-forming unit erythroid (BFU-E); CFU, macrophage (CFU-M); and CFU, granulocyte, erythrocyte, megakaryocyte, macrophage (CFU-GEMM), at comparable plating efficiencies (Figure 3E-F, left; supplemental Figure 9A). Staining of individual cells from selected colonies confirmed the presence of various myeloid and erythroid cells (supplemental Figure 9B). The CFU-inducing capacity was comparable between iCD34+ cells generated in feeder-plus and feeder-free systems (supplemental Figure 10A). iCD34+ cells obtained by differentiation in the CM efficiently generated CFUs and, except for a lower percentage of erythroid colonies in the WEHI CM–derived cells, the iCD34+ cells generated from CM showed clonogenicity equal to that derived from StemPro-34 serum-free medium (Figure 3F, middle; supplemental Figure 10B-C). The genetic background of the iPSC clones did not affect the clonogenic potential of their iCD34+ cells, because cells derived from WT, B*40:02mut, and 6pLOH+ clones formed colonies at comparable efficiency (Figure 3F, right; supplemental Figure 11).
Engraftment of WT and B4002− iCD34+ cells in BRGS mice
To assess the in vivo clonogenicity of iPSC-derived HSCs from a patient with AA, we isolated iCD34+ cells derived from WT, B*40:02mut, or 6pLOH+ iPSC clones and injected the cells into the femoral BM of BRGS mice to assess the human cell engraftment at weeks 9, 10, and 12 after transplantation. FCM analyses of single-cell suspensions obtained from various mouse organs, including the BM, spleen, and blood, revealed the successful engraftment of human CD45+ (hCD45+) leukocytes, with similar engraftment capacity among WT, B*40:02mut, and 6pLOH+ iCD34+ cells (Figure 4A, left and right). Immunohistochemistry studies that used human myeloperoxidase staining confirmed the presence of human myeloid cells in the mouse BM (Figure 4B). Young and adult mice injected with iCD34+ cells showed a greater increase in body weight than control mice (supplemental Figure 12A-B). FCM (supplemental Figure 12C-D) and a PCR analysis of the human chromosome and the hCD45 gene (supplemental Figure 12E-F) further confirmed the presence of human cells in young or adult transplanted mice. Deep analyses of the BM, spleen, and PB demonstrated the presence of hCD45+ cells (2.1%, 4.5%, and 7.3%, respectively) with persistent chimerism of hCD45+ cells in the BM of the mice injected with B4002− iCD34+ cells for up to 12 weeks (supplemental Figure 13A). Notably, after unilateral intrafemoral injection of iCD34+cells, we observed comparable human hematopoietic cell engraftment in both femurs, indicating repopulation of the contralateral femur through migration and homing of HSCs (supplemental Figure 13B). All mice engrafted from WT and 6pLOH+ iCD34+ cells showed multilineage reconstitution of myeloid cells (CD33+), B cells (CD19+), and T cells (CD3+) in the BM (supplemental Figure 13C-E), spleen (supplemental Figure 13F-G), PB (supplemental Figure 14A-C), and lymphoid cells in the thymus (supplemental Figure 15A-B). Similar percentages of CD33+, CD19+, or CD3+ cells in the BM, spleen, and PB were seen in the mice reconstituted with human WT or B4002− iCD34+ cells (Figure 4C). Giemsa staining of cytospin preparations of PB (supplemental Figure 16) of young mice reconstituted with human WT and B4002− iCD34+ cells showed human glycophorin-A+ erythroid cells and CD41a+ platelets in the spleen and PB of the BRGS mice (supplemental Figure 17A-D). In addition, no marked difference in the number of erythroid cells and platelets was observed among 3 mice injected with different iCD34+ cells, including B*40:02mut iCD34+ cells (supplemental Figure 18A-C). hCD45+ cells collected from the spleen in the mice transplanted with WT, B*40:02mut, or 6pLOH+ iCD34+ cells showed corresponding phenotypes, indicating that each mouse carried human cells with HLA profiles similar to those of the parental iCD34+ cells (Figure 4D; supplemental Figure 19A-B).
Figure 4.

Engraftment of AA iPSC WT, B*40:02mut, and 6pLOH+HSCs (CD34+iPSC HSPCs) in immunodeficient mice. (A) Human CD34+ and CD45+ cells in different organs of young BRGS mice at 10 to 12 weeks after transplantation of WT, B*40:02mut and 6pLOH+ CD34+ iPSCs. (Left panel) The percentages of human hematopoietic cells represent the mean ± SEM of 3 WT, 2 B*40:02mut, and 3 6pLOH+ cell recipients. (Right panel) A representative set of FCM results is shown. The numbers below the gated area indicate the mean ± SEM of the positive cell percentages. (B) Human myeloid cells in the BM of a young mouse transplanted with 6pLOH+ iCD34+ cells. Representative results of (upper panel) hematoxylin and eosin staining and (lower panel) human myeloperoxidase staining are shown. (C) Engraftment of myeloid and lymphoid lineage cells in the different organs. The percentages of each cell lineage in (top left panel) the BM, (top right panel) spleen, and (bottom panel) PB are shown. The data represent the mean ± SEM of the percentages of each marker-positive cell obtained from 3 WT, 2 B*40:02mut, and 3 6pLOH+ cell recipient mice. (D) HLA expression profiles of human B cells isolated from the spleen of the transplanted mice with the indicated iCD34+, (upper panels) HLA-A2402, and (lower panels) HLA-B4002 expression by CD19+ lymphoid cells in the spleens. (E) Morphology of CFU-GEMM–derived colonies from the BM iCD34+ cells. (Left) CFU-GEMM–derived colonies and (right) the constituent cells prepared by the cytospin are shown. FSC-A, forward scatter area; FSC-H, forward scatter height.
Clonogenic capacity of hCD34+ cells isolated from chimeric mice
When CD34+ cells isolated from the BM of the transplanted mice were cultured in semisolid medium, WT and B4002− iCD34+ cells formed colonies with similar clonogenic capacities (Figure 4E; supplemental Figure 20A,C). Giemsa staining of cytospin preparations revealed the presence of various cell types within the CFUs, such as megakaryocytes (Figure 4E; supplemental Figure 20C). An FCM analysis revealed that the cells collected from the culture plates were bona fide CD45+ human cells (supplemental Figure 20D). In keeping with the higher percentages of CD33+ cells in the BM and PB of the mice engrafted with 6pLOH+ iCD34+ cells than in mice with WT iCD34+ cells, the percentage of CD33+ cells in the cells collected from the culture of 6pLOH+ CD34+ cells was higher than the percentage of WT CD34+ cells (supplemental Figure 21).
Escape of B4002− iCD34+ cells from attack by autologous CTLs specific to B4002+ iCD34+ cells
As a proof of concept, we next examined whether or not patient-derived CTLs are capable of killing autologous HSCs and, more importantly, if they are able to discriminate between B4002+ and B4002− iCD34+ cells in coculture experiments. Cultures of CTLs with iCD34+ cells for up to 5 days under culture conditions favorable for the growth of iCD34+cells showed vigorous proliferation of B4002− iCD34+ cells that outnumbered the CTLs. In contrast, the cultures containing WT iCD34+ cells failed to proliferate, and the centers of the culture wells were occupied by small round cells that looked like activated lymphocytes (Figure 5A). In a CD107 degranulation assay, a higher proportion of CD107+ cells was detected in CTLs that were exposed to WT iCD34+ cells than in those exposed to B4002− iCD34+ cells (Figure 5B). In addition, CTLs secreted threefold more IFN-γ in response to co-culture with WT iCD34+ than in response to coculture with the B4002− counterparts (Figure 5C), and they more efficiently lysed WT iCD34+ than B4002− iCD34+ target cells in a cytotoxic assay (Figure 5D). We then analyzed the TCR β repertoire of the CTL line capable of preferentially killing WT iCD34+ cells as well as T cells in the BM of the patient obtained before immunosuppressive therapy and found expanded T cells that expressed the same TCR β chain in each population (Figure 5E). Surprisingly, the third-most recurrent TCRVβ CDR3 sequence of the CTL line was identical to that of the most predominant T-cell in the patient’s BM. When we analyzed the TCR α-chain complementary DNA of the 2 T cells from the CTL line and BM T cells, they expressed the same TCR α-chain (Figure 5F). This finding strongly indicates that the T-cell clone with this particular TCR was involved in the killing of the patient’s HSPCs as well as the escape of HLA class I–lacking HSPCs.
Figure 5.

Escape of B4002−iCD34+cells from the attack by autologous CTLs specific to B4002+iCD34+cells. (A) CTLs were cocultured with iCD34+ cells possessing either the 6pLOH+ or the WT genotypes in 96-well round-bottom plates for 5 days and visualized using a light microscope. Representative images of experiments are shown; black arrows indicate iCD34+ cells; white arrows indicate CTLs. Top panels, original magnification ×60; bottom panels, original magnification ×20. (B) CTLs were cocultured with iCD34+ cells possessing either the 6pLOH+ or the WT genotypes in 96-well round-bottom plates at a 10:1 effector:target (E:T) ratio for 4 hours and then analyzed by FCM. The analysis was performed by gating cells as 7-aminoactinomycin D–negative (live cells), CD3+, CD8+, or CD107a+ cells. (C) CTLs were cocultured in 96-well round-bottom plates with iCD34+ cells with 6pLOH+, WT, or B*40:02mut genotypes at the indicated E:T ratios. The levels of IFN-γ in the supernatants from overnight cocultures were determined by enzyme-linked immunosorbent assay. Summarized data (mean ± SEM) from 3 independent experiments are shown. (D) iCD34+ cells possessing the 6pLOH+, B*40:02mut, or WT genotypes were labeled with 51Cr and then cocultured with autologous CTLs in 96-well round-bottom plates for 4 hours. Summarized data (mean ± SEM) from 3 independent experiments are shown. (E) Clonotype analysis (Vβ cDNA sequencing) of the CTL line and BM T cells of the patient before receiving immunosuppressive therapy using single T-cell RT-PCR indicate that the third-most recurrent TCRVβ CDR3 sequence of the CTL line was identical to that of the most predominant T-cell clone in the BM. (F) CDR3α sequence of T cells with the same CDR3β sequence derived from the CTL line and BM T cells. The T cell expressed 2 TCRα chain mRNAs; 1 was functional and the other was unproductive. *P < .05.
Discussion
In this study, we used somatic cell reprogramming to generate iPSCs from a patient with acquired AA and used them to successfully produce a large number of iCD34+ cells. These patient-derived iPSC HSCs displayed robust hematopoietic potential in vitro and in vivo, generating multiple blood cell lineages. In addition, we were able to generate iPSC clones from HLA-B4002− monocytes that were derived from 6pLOH+ or B40:04mut HSCs and use them to assess the cytotoxic activity of patient-derived CTLs against autologous iCD34+ cells. Failure of the CTLs to eliminate B4002− iCD34+ cells constitutes experimental proof that an immune attack mediated by pathogenic CTL cells against HSCs is the root of the pathogenesis of acquired AA. This study is the first to demonstrate that HLA-B4002–lacking iCD34+ cells derived from an AA patient that evade CTL attack can reconstitute hematopoiesis as well as WT iCD34+ cells in immunodeficient mice.
In this study we developed a very efficient method for in vitro differentiation of iPSCs into HSCs. By using this method, we successfully generated a large number of iCD34+ cells with no requirement for special cell culture medium or expensive growth factors. The method is therefore easy, rapid, and cost-effective, which are obvious advantages over its predecessors. Because this method requires only the use of supernatants from OP9 or WEHI cells, it can be implemented in any laboratory equipped with the minimal facilities required for human cell culture.
Previous studies have reported generating iPSCs from patients with myelodysplastic syndrome.21 By combining somatic reprogramming with gene editing, a conditional PIGA knockout model in human iPSCs was also successfully generated that allowed for the production of glycosylphosphatidylinositol-anchored protein–deficient blood cells.22 In contrast to the reported ineffective hematopoiesis, reduced or absent clonogenicity, and increased cell death of iPSCs derived from myelodysplastic syndrome with del(7q) or the PIGA knockout model,21,22 the iPSC lines generated from monocytes of AA patients showed excellent hematopoietic potential both in vitro and in vivo with virtually no differences between the WT and B4002− iCD34+ cells.
Previous reports have also shown that human iPSCs display low expression of HLA class I molecules23 and that iCD34+ cells express nonclassical major histocompatibility complex molecules, such as HLA-G and HLA-E.24 In this study, we also observed that iPSCs derived from monocytes of AA patients poorly expressed HLA molecules, but the HLA expression gradually increased as the cells differentiated into iCD34+ cells, which allowed for the investigation of their sensitivity to CTLs.
The use of immunodeficient mice capable of supporting the engraftment of human hematopoietic cells represents a valuable tool for studying the engraftment of iCD34+ cells that lack HLA expression in vivo. A major advantage of this model is that we can measure and characterize the behavior of WT and B4002− iCD34+ cells without the influence of autologous mature T cells in vivo. Because equal numbers of 2 different iCD34+ cell lines were transplanted into the mice, we were able to determine the in vivo behavior of the individual clones in the mice, resulting in an unanticipated consistency of clonal behavior in the recipients. A recent study reported that HSC behavior is highly cell autonomous and suggested that cell autonomy dominates the in vivo behavior of HSCs.25 Our data indicated that all donor cells detected in mouse organs retained their original phenotypes and confirmed the in vivo clone-specific features. These results are consistent with recent data suggesting that single isolated cells can be serially transplanted without losing their cell behavior.26
Our study confirmed that B4002− HSCs can escape immune attack by autologous CTLs through effectively deleting the particular HLA allele recognized by the immune cells.10 Although the hematopoiesis derived from iCD34+ cells may be primitive rather than definitive hematopoiesis, the CTL line was able to discriminate WT iCD34+ cells from B4002-lacking iCD34+ cells in vitro, suggesting that our xenograft model can serve as an AA model in which antigen-specific T cells may selectively eliminate WT HSPCs in vivo. This study thus added a new piece of evidence to support the immune-mediated destruction of HSCs as a central pathogenic mechanism in this disease and opens new venues for the identification of autoantigens that trigger the autoimmune attack. Because attempts to generate T-cell clones from the pathogenic CTL line were unsuccessful, we were unable to identify putative antigens of the HSCs that are recognized by the CTLs. However, our clonotype analyses support the assumption that the T-cell clone with this particular TCR that was enriched in vitro after coculture with WT iCD34+ cells was involved in killing the patient’s HSPCs as well as the escape of HLA class I–lacking HSPCs. Whether or not the TCR ligand is associated with B4002 needs to be determined by future investigation.
In conclusion, we successfully generated iPSCs with different HLA genotypes from an AA patient possessing B4002− leukocytes and demonstrated that B4002− CD34+ cells induced from these iPSC clones have normal hematopoietic potential in vivo and in vitro. Our study may contribute to modeling rare human blood diseases with potential research applications, including the identification of autoantigens and testing the efficacy of pharmacologic agents aimed at restoring the hematopoietic function in patients with these disorders.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors are indebted to Misato Nishikawa at CiRA for her excellent technical support with the generation of induced pluripotent stem cells.
This study was supported by Grant-in-Aid No. 2604419-00 for Scientific Research from the Japan Society for the Promotion of Science (JSPS), by an FY2014 JSPS Postdoctoral Fellowship for Foreign Researchers (Kanazawa University Foreign Researcher) (J.L.E. and S.N.), by the Ministry of Education, Culture, Sports, Science and Technology Grant-in-Aid for Scientific Research (B, 24390243) (S.N.), by the Cultural Affairs and Missions Sectors of Egyptian Ministry of Higher Education (M.I.E.), and by the 2014-2015 missions plan for joint supervision missions (Sohag University).
Authorship
Contribution: J.L.E., H.A.H., A.K.A.N. and S.N. designed the study; J.L.E. and M.I.E. performed most of the in vitro and in vivo experiments; K.C. and Y.Y. generated induced pluripotent stem cells; T.K. set up in vivo experiments; K.H. performed immunohistochemical analyses; N.N., Y.Z., and T.I. performed HLA genotyping; K.T. and K.A. established BRGS mice; Y.A. performed cytotoxic T lymphocyte assays; H.H. and H.K. performed T-cell receptor repertoire analyses; J.L.E., M.I.E., and S.N. wrote the manuscript; and all authors critically reviewed the manuscript and checked the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Shinji Nakao, Hematology/Respiratory Medicine, Faculty of Medicine, Institute of Medical, Pharmaceutical and Health Sciences, Kanazawa University, 13-1 Takara-machi, Kanazawa 920-8641, Japan; email: snakao8205@staff.kanazawa-u.ac.jp.
References
- 1.Young NS. Current concepts in the pathophysiology and treatment of aplastic anemia. Hematology Am Soc Hematol Educ Program. 2013;2013:76-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakao S, Takami A, Takamatsu H, et al. Isolation of a T-cell clone showing HLA-DRB1*0405-restricted cytotoxicity for hematopoietic cells in a patient with aplastic anemia. Blood. 1997;89(10):3691-3699. [PubMed] [Google Scholar]
- 3.Risitano AM, Maciejewski JP, Green S, Plasilova M, Zeng W, Young NS. In-vivo dominant immune responses in aplastic anaemia: molecular tracking of putatively pathogenetic T-cell clones by TCR beta-CDR3 sequencing. Lancet. 2004;364(9431):355-364. [DOI] [PubMed] [Google Scholar]
- 4.Zeng W, Nakao S, Takamatsu H, et al. Characterization of T-cell repertoire of the bone marrow in immune-mediated aplastic anemia: evidence for the involvement of antigen-driven T-cell response in cyclosporine-dependent aplastic anemia. Blood. 1999;93(9):3008-3016. [PubMed] [Google Scholar]
- 5.Sun YX, Li H, Feng Q, et al. Dysregulated miR34a/diacylglycerol kinase ζ interaction enhances T-cell activation in acquired aplastic anemia. Oncotarget. 2017;8(4):6142-6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katagiri T, Sato-Otsubo A, Kashiwase K, et al. ; Japan Marrow Donor Program. Frequent loss of HLA alleles associated with copy number-neutral 6pLOH in acquired aplastic anemia. Blood. 2011;118(25):6601-6609. [DOI] [PubMed] [Google Scholar]
- 7.Betensky M, Babushok D, Roth JJ, et al. Clonal evolution and clinical significance of copy number neutral loss of heterozygosity of chromosome arm 6p in acquired aplastic anemia. Cancer Genet. 2016;209(1-2):1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Afable MG II, Wlodarski M, Makishima H, et al. SNP array-based karyotyping: differences and similarities between aplastic anemia and hypocellular myelodysplastic syndromes. Blood. 2011;117(25):6876-6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maruyama H, Katagiri T, Kashiwase K, et al. Clinical significance and origin of leukocytes that lack HLA-A allele expression in patients with acquired aplastic anemia. Exp Hematol. 2016;44(10):931-939.e3. [DOI] [PubMed] [Google Scholar]
- 10.Zaimoku Y, Takamatsu H, Hosomichi K, et al. Identification of an HLA class I allele closely involved in the autoantigen presentation in acquired aplastic anemia. Blood. 2017;129(21):2908-2916. [DOI] [PubMed] [Google Scholar]
- 11.Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373(1):35-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young NS, Ogawa S. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373(17):1675-1676. [DOI] [PubMed] [Google Scholar]
- 13.Okita K, Yamakawa T, Matsumura Y, et al. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells. 2013;31(3):458-466. [DOI] [PubMed] [Google Scholar]
- 14.Yamauchi T, Takenaka K, Urata S, et al. Polymorphic Sirpa is the genetic determinant for NOD-based mouse lines to achieve efficient human cell engraftment. Blood. 2013;121(8):1316-1325. [DOI] [PubMed] [Google Scholar]
- 15.Takenaka K, Prasolava TK, Wang JC, et al. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat Immunol. 2007;8(12):1313-1323. [DOI] [PubMed] [Google Scholar]
- 16.Espinoza JL, Elbadry MI, Taniwaki M, et al. The simultaneous inhibition of the mTOR and MAPK pathways with Gnetin-C induces apoptosis in acute myeloid leukemia. Cancer Lett. 2017;400:127-136. [DOI] [PubMed] [Google Scholar]
- 17.Inaguma Y, Akatsuka Y, Hosokawa K, et al. Induction of HLA-B*40:02-restricted T cells possessing cytotoxic and suppressive functions against haematopoietic progenitor cells from a patient with severe aplastic anaemia. Br J Haematol. 2016;172(1):131-134. [DOI] [PubMed] [Google Scholar]
- 18.Lu X, Ohata K, Kondo Y, Espinoza JL, Qi Z, Nakao S. Hydroxyurea upregulates NKG2D ligand expression in myeloid leukemia cells synergistically with valproic acid and potentially enhances susceptibility of leukemic cells to natural killer cell-mediated cytolysis. Cancer Sci. 2010;101(3):609-615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Espinoza JL, Nguyen VH, Ichimura H, et al. A functional polymorphism in the NKG2D gene modulates NK-cell cytotoxicity and is associated with susceptibility to human papilloma virus-related cancers. Sci Rep. 2016;6(1):39231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi E, Mizukoshi E, Kishi H, et al. A new cloning and expression system yields and validates TCRs from blood lymphocytes of patients with cancer within 10 days. Nat Med. 2013;19(11):1542-1546. [DOI] [PubMed] [Google Scholar]
- 21.Kotini AG, Chang CJ, Boussaad I, et al. Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat Biotechnol. 2015;33(6):646-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan X, Braunstein EM, Ye Z, et al. Generation of glycosylphosphatidylinositol anchor protein-deficient blood cells from human induced pluripotent stem cells. Stem Cells Transl Med. 2013;2(11):819-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suárez-Alvarez B, Rodriguez RM, Calvanese V, et al. Epigenetic mechanisms regulate MHC and antigen processing molecules in human embryonic and induced pluripotent stem cells. PLoS One. 2010;5(4):e10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim EM, Manzar G, Zavazava N. Human iPS cell-derived hematopoietic progenitor cells induce T-cell anergy in in vitro-generated alloreactive CD8(+) T cells. Blood. 2013;121(26):5167-5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu VWC, Yusuf RZ, Oki T, et al. Epigenetic memory underlies cell-autonomous heterogeneous behavior of hematopoietic stem cells. Cell. 2017;168(5):944-945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Picelli S, Björklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10(11):1096-1098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.