

Abstract
Macrophages are a major component of the tumor microenvironment and orchestrate various aspects of immunity. Within tumors, macrophages can reversibly alter their endotype in response to environmental cues, including hypoxia and stimuli derived from other immune cells, as well as the extracellular matrix. Depending on their activation status, macrophages can exert dual influences on tumorigenesis by either antagonizing the cytotoxic activity immune cells or by enhancing antitumor responses. In most solid cancers, increased infiltration with tumor-associated macrophages (TAMs) has long been associated with poor patient prognosis, highlighting their value as potential diagnostic and prognostic biomarkers in cancer. A number of macrophage-centered approaches to anticancer therapy have been investigated, and include strategies to block their tumor-promoting activities or exploit their antitumor effector functions. Integrating therapeutic strategies to target TAMs to complement conventional therapies has yielded promising results in preclinical trials and warrants further investigation to determine its translational benefit in human cancer patients. In this review, we discuss the molecular mechanisms underlying the pro-tumorigenic programming of macrophages and provide a comprehensive update of macrophage-targeted therapies for the treatment of solid cancers.
Keywords: macrophages, immunotherapy, macrophage polarization, inflammation, cancer
Introduction
Tumors are complex tissues where cancer cells maintain intricate interactions with their surrounding stroma. Important components of the tumor stroma include macrophages, which are intimately involved in tumor rejection, promotion, and metastasis. In some cases, macrophages can comprise up to 50% of the tumor mass, and their abundance is associated with a poor clinical outcome in most cancers. Tumor-associated macrophages (TAMs) promote tumor growth by facilitating angiogenesis, immunosuppression, and inflammation, and can also influence tumor relapse after conventional anticancer therapies. Strategies aimed at targeting TAMs have shown great promise in mouse models, and a number of these agents are currently under clinical investigation. Here, we review current understanding of how TAMs regulate tumor progression and provide a comprehensive update of therapies targeting macrophages for the treatment of solid cancers. We also evaluate the contribution of TAMs in moderating the effectiveness of different anticancer treatment modalities and reflect on the challenges that need to be addressed to successfully incorporate the targeting of TAMs into current anticancer regimens.
The Ontogeny of TAMs
Macrophages are required to maintain homeostasis in the organs they occupy. Given the specific needs of each tissue microenvironment, there are many different types of macrophages with morphologically and functionally distinct characteristics. Prototypical examples include liver Kupffer cells, brain microglia, and lung alveolar macrophages, which together reflect the versatility of the mononuclear phagocytic system.
Tissue-resident macrophages were long considered to be recruited from bone-marrow progenitors that differentiated into mature cells upon seeding into tissues (1). However, new evidence indicates that these macrophages are derived from yolk sac precursors, which arise during early development and persist locally via self-renewal (2). In a similar vein, TAMs were once hypothesized to originate from circulating monocytes that were recruited in response to chemotactic signals released from tumor cells. While monocyte-derived TAMs are continuously replenished by peripheral recruitment, a small proportion of TAMs can also arise from tissue-resident macrophages that are partially maintained through in situ proliferation (3, 4).
Circulating cells that are recruited into tissues and subsequently differentiate into TAMs include inflammatory monocytes and monocyte-related, myeloid-derived suppressor cells (MDSCs). The differentiation of inflammatory Ly6CHigh monocytes into TAMs depends on RBPJ, the transcriptional regulator of Notch signaling (3). Genetic ablation of the Rbpj gene reduced tumor burden in a spontaneous mouse model of breast cancer, indicating the absolute requirement of these monocyte-derived TAMs in supporting tumor growth (3). A smaller subset of TAMs may also arise from Ly6CLow monocytes, which include cells that express the angiopoietin-2 (ANG2) receptor TIE2 (5). These TIE2-expressing cells are recruited in response to the secretion of ANG2 by tumor endothelial cells and play non-redundant roles during tumor neovascularization (6). By contrast, inhibition of STAT3 caused by upregulation of CD45 phosphatase activity is a key process that mediates the differentiation of MDSCs into mature TAMs (7). MDSCs may exhibit a Ly6CHighLy6GNeg monocytic or a Ly6CIntLy6GHigh granulocytic endotype (8). Since the monocytic MDSCs strongly resemble Ly6CHigh monocytes, it is hypothesized that these cells represent a precursor functional state of these inflammatory cells (8).
Tissue-resident macrophages coexist with recruited macrophages in tumors with potentially distinct roles. In glioblastoma, TAMs are comprised of a mixed population of cells including resident microglia and bone marrow-derived monocytes and macrophages (9). The relative contribution of these populations in glioma progression was investigated in a genetically engineered mouse model, in which the chemokine CX3CR1/CX3CL1 signaling was ablated in both microglia and inflammatory monocytes (9). CX3CR1 is expressed by circulating monocytes and exclusively by microglia in the central nervous system, while its ligand CX3CL1 is expressed by neurons and serves as a chemotactic signal. Loss of Cx3cr1 in the host microenvironment facilitated the recruitment of Ly6CHigh “inflammatory monocytes” into tumor tissues, which were responsible for increased tumor incidence and shorter survival times in glioma-bearing mice. By contrast, selective ablation of Cx3cr1 in microglia had no impact on glioma growth (9). These results suggest that the tumor-promoting effect observed upon Cx3cr1 ablation is conferred by infiltrating inflammatory monocytes and highlights the contrasting roles of bone marrow-derived and tissue resident-derived TAMs. However, since this may also depend on tumor type, the contribution of tissue-resident versus recruited TAMs in tumorigenesis warrants further investigation.
TAM Function and Diversity
Tumor-associated macrophage heterogeneity is not only dependent on the nature of their monocytic precursor, but also on their functional diversity. To coordinate complex processes to promote immunity, while also minimizing damage to tissues where these responses occur, macrophages can reversibly alter their endotype in response to environmental cues. These environmental cues include stimuli derived from pathogens, parenchymal, and immune cells, as well as the extracellular matrix (10, 11).
Similar to the Th1/Th2 T-cell dichotomy, macrophages may be broadly classified into two groups, referred to as “classically activated M1” (CAM) or “alternatively activated M2” (AAM) endotypes. Much our understanding of macrophage polarization has relied on in vitro techniques, whereby macrophages are stimulated with M1- or M2-polarizing signals (12). For M1 this typically involves stimulation with IFNγ or lipopolysaccharide (LPS), while M2 polarization usually involves stimulation with IL4 or IL13 (12). Changes in gene expression, cell-surface markers and signaling pathways have subsequently been used to distinguish the various activation states (Table 1), and the contribution of some of these factors in mediating CAM/AAM characteristics has been validated in genetically engineered mouse models (Table 2). However, given the heterogeneity of tissues, macrophage polarization should be regarded as a complex process that occurs over a continuum (10, 13).
Table 1.
Characteristics of classically activated M1 (CAM) and alternatively activated M2 (AAM) endotypes.
| CAM |
AAM |
||||
|---|---|---|---|---|---|
| M1 | M2a | M2b | M2c | M2d | |
| Stimuli | IFNγ Lipopolysaccharide GM-CSF |
IL4 IL13 Fungal/helminth infection |
IL1R | IL10 TGFβ GCs |
IL6 LIF Adenosine |
| Markers | CD40 CD86 CD80 CD68 MHC II IL1R TLR2 TLR4 SOCS3 |
CD163 MHC II SR CD206 YM1a FIZZ1a ARG1a |
CD86 MHC II MerTK |
CD163 TLR1 TLR8 |
VEGF |
| Cytokine secretion | TNFα IL1 IL6 IL12 IL23 |
IL10 TGFβ |
IL1 IL6 IL10 TNFα |
IL10 TGFB |
IL10 IL12 TNFα TGFβ |
| Chemokine secretion | CCL10 CCL11 CCL5 CCL8 CCL4 CXCL9 CXCL10 |
CCL17 CCL22 CCL24 |
CCL1 | CCR2 | CCL5 CXCL10 CXCL16 |
| Function | Inflammation, tissue damage, and pathogen clearance | Allergic inflammation, tissue repair, tissue remodeling, and fibrosis | Anti-inflammation, tissue remodeling, and fibrosis | Anti-inflammation | Tissue repair, angiogenesis |
aDenotes markers that are only found in mouse macrophages.
Table 2.
Genetic mouse models of macrophage polarization.
| Protein/gene | Genetic manipulation | Effect on macrophage polarization | Reference |
|---|---|---|---|
| IRF5/Irf5 | KO and conditional LysM-Cre KO | ↓↓ M1 | (14, 15) |
| JUNB/JunB | Conditional LysM-Cre KO | (16) | |
| KLF4/Klf4 | Conditional LysM-Cre KO | ↑ M1/↓ M2 | (17) |
| TSC1/Tsc1 | Conditional LysM-Cre KO | (18) | |
| DAB2/Dab2 | Conditional LysM-Cre KO | (19) | |
| let-7c (mIR) | Knockdown and overexpression | (20) | |
| mIR-223/mir223 | KO | (21) | |
| Rictor/Rictor | Conditional LysM-Cre KO | ↑↑ M1 | (22) |
| AKT1/Akt1 | KO | (23) | |
| IL4RA/Il4ra | KO and conditional LysM-Cre KO | ↓↓ M2 | (24, 25) |
| HCK/Hck | KO and knockdown | (26, 27) | |
| STAT6/Stat6 | KO | (28) | |
| IRF4/Irf4 | KO | (29) | |
| PPARy/Pparg | KO | (30) | |
| JMJD3/Jmjd3 | KO | (29) | |
| P50/P105/NfKb | KO | (31) | |
| PI3Kγ/Pi3kγ | KO | (32) | |
| KLF6/Klf6 | Conditional LysM-Cre KO | ↑ M2/↓M1 | (33) |
| mIR-33/Mir33 | KO | (34) | |
| MyD88/myD88 | KO | (35) | |
| AKT2/Akt2 | KO | ↑↑ M2 | (23) |
| SHIP/Inpp5d | KO | (36) | |
| SHP-2/Ptpm6 | KO | (37) | |
| p16 INK4a/Cdkn2a | KO | (38) | |
| TNFR1/Tnfrsf1a | KO | (35) | |
| TNF/Tnf | KO | (35, 39) |
The current classification of CAM or M1 macrophages is in part based on their response to stimulation with bacterial LPS, TNFα, and/or IFNγ (Table 1). TNFα is produced by antigen presenting cells upon recognition of pathogenic signals, while IFNγ is produced by innate and adaptive immune cells such as natural killer (NK) and Th1 cells (10, 40). Once activated, CAMs secrete pro-inflammatory cytokines (IL1, IL6, and TNFα) and effector molecules (including reactive nitrogen intermediates) and express chemokines such as CXCL9 and CXCL0 (10). These molecules exert and amplify antimicrobial and tumoricidal activities alongside increased Th1 adaptive immune responses through enhanced antigen presentation. Because these cytokines play an important role in immune defense, their inappropriate release can result in chronic inflammation and extensive tissue damage (41).
Alternatively activated M2 macrophages are broadly characterized by their anti-inflammatory and wound-healing endotype (42). While these functional outputs are important for the maintenance of tissue homeostasis, aberrant AAM activation can trigger allergic reactions, promote tumor growth, and delay immune responses toward pathogens (43–45). Among the most important activators of AAMs are IL4, IL10, and IL13; however, several other stimuli and signaling pathways can also induce AAM polarization (Table 1). Thus, AAMs can be further divided into M2a, M2b, M2c, and M2d (12, 46). The M2a subtype is stimulated in response to IL4, IL13, as well as fungal and helminth infections. M2a macrophages express high levels of mannose receptor (CD206) and secrete large amounts of pro-fibrotic factors including fibronectin, insulin-like growth factor and TGFβ, which are all involved in wound healing and tissue repair. M2b macrophages are stimulated by immune complexes and bacterial LPS and exhibit upregulated expression of CD206 and the MER receptor tyrosine kinase. They primarily produce IL10, IL1β, IL6, and TNFα, which exert anti-inflammatory effects. M2c macrophages are activated by IL10, TGFβ, and glucocorticoids and are also generally thought to be anti-inflammatory in nature. Finally, differentiation of M2d macrophages occurs in response to co-stimulation with TLR ligands and adenosine (47). M2d macrophages express low levels of CD206 but are high producers of IL10 and VEGF. In light of these findings, it is now appreciated that the “AAM” terminology encompasses a functionally diverse group of macrophages that share the functional outputs of tumor progression by stimulating immunosuppression and angiogenesis.
Macrophages in Cancer Initiation and Promotion
Although macrophages are crucial for promoting host defenses, inappropriate or prolonged activation can result in damage to the host, immune dysregulation, and disease (48). In cancers, the role of macrophages in tumor progression remains to be fully elucidated, in part due to the contrasting roles they play depending on their polarization. On the one hand, studies have shown that macrophages are capable of exerting tumoricidal activity in vitro (49). Indeed, in colorectal cancer, TAMs are predominantly polarized toward a classically activated endotype and express pro-inflammatory cytokines such as IFNγ, which activate cytotoxic CD8+ T-cell responses to promote tumor destruction (50). Another route by which TAMs can cause the death of tumor cells involves the production of macrophage migration inhibitor factor (MIF). In addition to inhibiting the recruitment of macrophages (51), MIF stimulates key tumoricidal functions such as phagocytosis (52), cellular toxicity and the release of TNFα and IL1β (53). The secretion of IL18 and IL22 by TAMs has also been associated with tumor cell killing as they can amplify cytokine production (particularly IFNγ and IL2) and by augmenting the cytotoxic activity of NK cells (54, 55).
Many macrophage depletion studies have highlighted the importance of TAMs in tumor development and progression (56, 57). Genetic ablation of the Csf1 gene (encoding, macrophage colony-stimulating factor, and required for macrophage maturation) in mice susceptible to mammary carcinoma delayed metastasis and decreased tumor growth, while the transgenic expression of the corresponding CSF1 protein accelerated cancer progression and promoted pulmonary metastasis (56). Similar findings were also observed in a genetic model of thyroid cancer and in mice transplanted with human osteosarcoma cancer cells (58, 59). These findings suggest that there is a delicate balance between the tumoricidal and tumor-promoting functions of TAMs. To date, the tumor-promoting mechanisms of TAMs that have been well characterized include chronic inflammation, immune suppression, angiogenesis, and invasion/metastasis (Figure 1).
Figure 1.
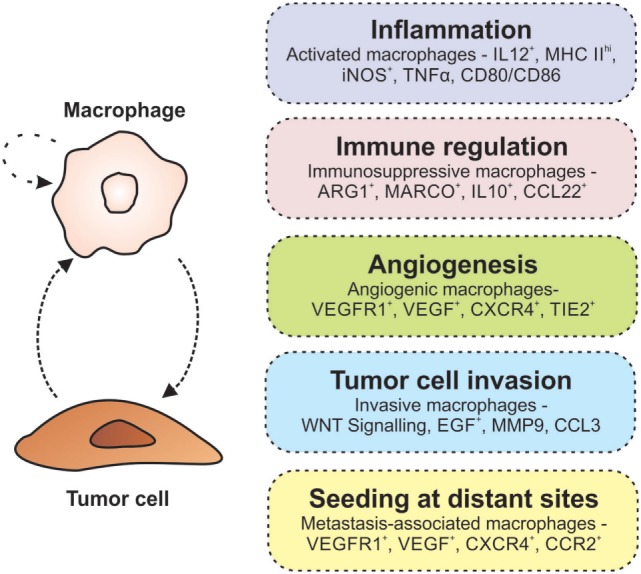
Macrophages promote tumorigenesis. The interaction between macrophages and tumor cells results in an autocrine/paracrine loop that enhances their pro-tumorigenic properties. Within the tumor microenvironment, macrophages are involved in many activities associated with tumor growth and progression including inflammation, immune regulation, angiogenesis, invasion, and metastasis (indicated in each of the boxes on the right).
Chronic Inflammation
Chronic inflammation is associated with some solid cancers (60). Patients with inflammatory bowel disease including ulcerative colitis and Crohn’s disease have an increased risk of developing neoplasia (61) owing to the production of TNFα (62), IL6 (63), and IL1β (64) by TAMs. This link between chronic inflammation and tumorigenesis is similarly observed in hepatocellular carcinoma (65), gastric cancer (66), and lung cancer (67). In these scenarios, the secretion of pro-inflammatory cytokines by macrophages in response to pathogens (e.g., HBV and Helicobacter pylori) and irritants (cigarette smoke) creates a mutagenic environment in the sub-epithelial stroma. These transformed neoplastic cells consequently produce inflammatory mediators including TNFα (68) and IL1β (69) that form a closed paracrine loop to perpetuate this tumor-reactive microenvironment.
Immune Suppression
Macrophages comprise a key component of the host immune responses, and they can facilitate tumor death by promoting cytotoxicity. For instance, stimulation of macrophages with granulocyte macrophage colony-stimulating factor GM-CSF or bacterial-derived CpG has been shown to activate toll-like receptor and enhance the secretion of immune-stimulatory cytokines that impair tumor growth and metastasis (70, 71). However, in the vast majority of cancers, macrophages exhibit an immunosuppressive endotype characterized by low levels of inflammatory molecules (IL18, IL12, and TNFα), and an increased expression of transcripts expressed by AAMs (Il10, Stat3, and ll13) (72, 73). This immunosuppressive effect has been proposed to occur due to STAT3 activation in AAMs opposing STAT1-driven Th1 antitumor responses (74). Likewise, expression of MHC class II molecules on TAMs is actively downregulated by tumor cell-derived TGFβ1, IL10, and PGE2 and results in decreased Th1 differentiation (48).
The direct suppression of immune responses by TAMs has also been described. IL10, for example, upregulates the expression of programmed-death ligand (PD)-L1 on the surface of monocytes and TAMs (75). Although naïve T-cells can be stimulated by PDL1, its most prominent role is the inhibition of activated effector T-cells by ligation with the PD1 receptor. Indeed, high tumor expression of PDL1 is associated with increased tumor aggressiveness and mortality in renal cell carcinoma and ovarian cancer, with an inverse correlation between PDL1 expression and intraepithelial CD8+ T-cell infiltration (76, 77). The expression of PDL1 and PDL2 by TAMs also triggers the expression of the regulatory molecules B7-H4 and VISTA in T-cells to elicit similar immunosuppressive functions (78). More recently, it has been shown that PI3Kγ signaling in TAMs inhibits NFκB activation while stimulating C/EBPβ, thereby triggering a transcriptional program that promotes immune suppression during inflammation and tumor growth (79). Another indirect mechanism by which TAMs may promote immune suppression is the recruitment of other immune cells into the tumor milieu. Specifically, the production of chemokines including CCL17 and CCL22 attract Th2 and regulatory T-cells (Tregs) that steer monocyte differentiation toward an anti-inflammatory AAM endotype (80). Macrophage-derived CXCL13, CCL16, and CCL18 can also bind to their CXCR5, CCR1, and CCR8 receptors to promote the recruitment of eosinophils and naïve T-cells that suppress immune responses and promote tissue remodeling (80, 81).
Angiogenesis
The benign-to-malignant transition of most solid cancers is marked by a significant increase in blood vessel formation, known as the “angiogenic switch” (82). Hypoxia is a major driver of angiogenesis, and TAMs preferentially accumulate in poorly vascularized regions during early tumor formation (83). The transcription factor HIF1α is constitutively expressed in macrophages and acts as a major regulator of hypoxic stress by inducing a switch from aerobic to anaerobic metabolism (83). These changes correlate with an increased expression of the HIF1 target genes Cxcr4, Ccl2, and endothelins that enhance macrophage recruitment into tumors (83, 84).
Macrophages are central to the angiogenic switch, and their increased tumor infiltration directly correlates with blood vessel density in human tumors (85). Likewise, Csf1 knockout mice that have reduced macrophage numbers are less susceptible to tumorigenesis, while Csf1 overexpression results in macrophage accumulation, enhanced angiogenesis, and accelerated malignant transformation (86). Proangiogenic macrophages are associated with an AAM endotype and secrete a diverse range of factors including TGFβ, VEGF, PDGF, and fibrin (74, 87–89). They express an enrichment of transcripts that encode for angiogenic molecules, and the ablation of these genes inhibits the angiogenic switch (72, 90–92). A subset of AAMs characterized by cell-surface expression of TIE2, a marker of mature endothelial cells, has been shown to play an indispensable role in blood vessel formation (93). Co-injection of TIE2-expressing macrophages with tumor cells significantly enhanced angiogenesis (93), while therapeutic targeting of TIE2 resulted in tumor vasculature regression and inhibited the progression of late-stage, metastatic mammary tumors, and pancreatic carcinomas (94). Because these data strongly support a role for macrophages in promoting angiogenesis, inhibiting pathways involved in these processes provide a promising therapeutic approach for the treatment of cancer.
Tumor Cell Invasion and Metastasis
Metastasis represents the most important cause of cancer mortality and occurs when cancer cells dissociate from the primary tumor and spread to distal organs (95). While it is well established that macrophages constitute a major population at metastatic niches, their role in metastasis has only recently been appreciated (41). Metastatic progression is dependent on the cross talk between macrophages and cancer cells. For example, secretion of the extracellular matrix proteoglycan versican by the primary tumor stimulates metastasis in the Lewis Lung Carcinoma model through TLR2 and TNFα signaling in macrophages (96). Likewise, tumor cells also induce the expression of matrix metalloproteinase (MMP)-9 in macrophages to promote the release of matrix-bound VEGF, which enhances angiogenesis and metastasis (97).
Macrophages are the predominant cells at sites of basement membrane degradation during early tumorigenesis and at the invasive front of tumors during malignant transformation (95). They are a rich source of proteases including cathepsins, MMPs, and serine proteases that promote extracellular matrix degradation and allow the escape of tumors from the basement membrane through the dense stroma (98, 99). Furthermore, upregulation of CSF1 by tumor cells stimulates macrophage recruitment and the production of epidermal growth factor (EGF), which in turn promotes tumor cell migration. This paracrine loop involving EGF and CSF1 is crucial for tumor invasion, and the inhibition of either signaling pathway inhibits the migration of both cell types (95, 100). Consistent with this, CSF1 expression in human cancers is highest at the invasive edge where macrophages are most abundant (56). Other factors that drive macrophage-mediated tumor invasion include Wnt5a, which acts through the non-canonical WNT pathway to stimulate cancer cell motility (101), and SPARC/Osteonectin, which regulates the deposition of collagen fibers and expression of MMPs (102).
A distinct population of macrophages known as metastasis-associated macrophages (MAMs), which are recruited by CCL2, have been identified (103). Activation of the CCL2/CCR2 axis increased secretion of CCL3 by MAMs, which in turn facilitated metastatic seeding of breast cancer cells in the lung (103). Interestingly, MAM-derived CCL3 was also shown to act as an autocrine mediator for MAMs by prolonging their retention in metastatic foci and resulting in the enhanced extravasation of cancer cells to other organs (103). The CCL2/CCR2 axis between cancer cells and MAMs may also promote bone metastasis of prostate cancer by supporting the activation of osteoclasts (104). The destruction of bone by osteoclasts triggers the release of growth factors that support tumor growth (105), while the inhibition of these cells with neutralizing antibodies or shRNAs for CCL2 significantly impairs prostate cancer-induced formation of osteoclasts and bone resorption (106, 107). In another example, expression of vascular cell adhesion protein 1 on cancer cells enhanced tumor growth and lung metastasis through interaction with α4-integrin expressed by MAMs (108). Collectively, these studies provide unequivocal evidence for the multidimensional role of macrophages in the establishment of metastatic niches as well as the extravasation of tumor cells to secondary organs.
Macrophages as Diagnostic and Prognostic Biomarkers
The extent of macrophage infiltration serves as an important diagnostic and prognostic biomarker in many human cancers. The identification and quantification of TAMs can be performed through various methods, ranging from morphological discrimination to gene expression analysis and cell-surface marker profiling. Human TAMs are typically identified by CD68 expression; however, CD163, CD206, and CD204 are also commonly used to distinguish those of the AAM endotype (109, 110). By contrast, macrophages with a CAM endotype in humans can be identified by CD40 (111) and HLA-DR expression (112).
Increased macrophage infiltration is associated with advanced stage disease and worse overall survival in breast (113), pancreatic (110) and bladder cancer (114). On the other hand, high macrophage density correlates with a favorable outcome in colorectal cancer (115). TAM density may also be used as a prognostic marker to predict chemotherapy response. In Hodgkin lymphoma, overexpression of a macrophage gene signature in diagnostic lymph-node samples is associated with primary treatment failure (116). The increased presence of CD68+ macrophages was also negatively correlated with survival and secondary treatment outcome (116). In pancreatic cancer, TAMs are reported to be critical determinants of prognostic responsiveness to postsurgical adjuvant chemotherapy due to the re-education of TAMs to restrain tumor progression (110). Thus, the quantification of TAMs may also be used to stratify patients who are more likely to respond to postsurgical chemotherapy.
Macrophages as a Therapeutic Target
Tumor initiation and progression is driven by interactions between stromal and immune cells within the tumor microenvironment. Thus, multitargeted approaches in which several of these cell types are simultaneously inhibited may represent a more efficient method to treat cancer, especially when used in conjunction with other strategies such as chemotherapy. One major advantage of targeting the tumor microenvironment is the genetic stability of non-tumor cells, which is in contrast to tumor cells that are often highly unstable and can rapidly accumulate adaptive mutations that confer drug resistance. Given the indispensable role of macrophages in tumorigenesis and their correlation with a poor overall survival, these findings provide a strong basis for targeting these cells within the tumor microenvironment. Indeed, the pharmacological inhibition of macrophages has shown great promise in mouse models (Table 3), and a number of these agents are currently under clinical investigation (Table 4). Major strategies targeting macrophages within the tumor microenvironment include macrophage depletion, modifying macrophage recruitment and macrophage reprogramming.
Table 3.
Selected targets of macrophage inhibition in mouse models.
| Pathway targeted | Drug or target | Effect | Reference |
|---|---|---|---|
| Macrophage depletion | Trabectedin | Selective cytotoxicity in mononuclear phagocytes and inhibition of tumor-promoting cytokines | (117) |
| Clodronate ± anti-VEGF mAB | Tumor regression and reduced angiogenesis | (118) | |
| Macrophage recruitment | CCL2 | Reduced tumor growth and metastasis in prostate and breast cancer | (119, 120) |
| CXCL12/CXCR4 | Reduced tumor growth and metastasis in breast and prostate cancer | (121, 122) | |
| CSF1 receptor (CSF1R) | Antiangiogenic and antimetastatic effects in melanoma and mammary xenograft tumors and improved chemotherapeutic responses | (123–125) | |
| CD11b | Enhanced tumor responses to radiation | (126) | |
| Macrophage reprogramming [suppressing alternatively activated M2 (AAM)] | Jumonji | Impaired AAM differentiation and recruitment | (29) |
| STAT6 | Enhanced tumor immunity | (127) | |
| STAT3 | Inhibited immunosuppressive cytokine profile of AAMs | (128, 129) | |
| Superoxide [O(2−)] | Impaired AAM differentiation | (130) | |
| IL4Rα | Less aggressive skin tumors | (131) | |
| COX2 | Suppression of breast cancer metastasis | (132) | |
| PI3Kγ | Stimulation of T-cell-mediated tumor suppression and inhibition of tumor cell invasion and metastasis | (32) | |
| CSF1R | Increased survival and regressed established GBM tumors by reducing AAM polarization, but without affecting tumor-associated macrophage numbers in treated tumors | (125) | |
| HCK | Suppression of AAM polarization, enhanced tumor immunity in colon cancer | (27) | |
| Macrophage reprogramming (classically activated M1 stimulating) | IFNα | Reduced tumor growth and promoted near complete abrogation of breast cancer metastasis | (133) |
| CD40 | Tumor regression and increased survival | (134) | |
| Histidine-rich glycoprotein | Reduced pancreatic and breast cancer metastasis and increased survival | (135) | |
| NFκB | Tumor regression | (136) | |
Table 4.
Summary of selected NIH clinical trials of macrophage inhibition.
| Target | Phase | Trial number | Tumor type | Drug name/pharmacompany | Effect | Reference |
|---|---|---|---|---|---|---|
| CSF1/CSF1R | I/II | NCT01346358 | Advanced solid tumors | IMC-CS4/Eli Lilly Inc. | CSF1 R-blocking antibody | (137) |
| NCT01444404 | Advanced solid tumors | AMG 820/Merck | CSF1 R-blocking antibody | (138) | ||
| NCT01804530 | Pancreatic cancer | PLX7486/Plexxikon Inc. | Kinase inhibitor of CSF1R and Trk | (139) | ||
| NCT01004861 | Advanced solid tumors | PLX3397/Plexxikon Inc. | Kinase inhibitor of CSF1R and cKit | (140) | ||
| CCL2/CCR2 | II | NCT01015560 | Bone metastasis | MLN1202//Millennium Pharmaceuticals Inc. | Anti-CCR2 antibody | (141) |
| NCT01413022 | Locally advanced pancreatic cancer | PF-04136309//Pfizer Inc. | CCR2 antagonist | (142) | ||
| IL6R | I/II | NCT01637532 | Ovarian cancer | Tocilizumab and Peg-Intron/Genentech | IL6R monoclonal antibody | (143) |
| DNA repair mechanisms | III | NCT01692678 | Liposarcoma and leimyosarcoma | YONDELIS (Trabectedin)/PharmaMar | DNA backbone cleavage and cell apoptosis | (144) |
| II | NCT01772979 | Ovarian cancer | YONDELIS | DNA backbone cleavage and cell apoptosis | (145) | |
| I | NCT01426633 | Liposarcoma and leimyosarcoma | YONDELIS | DNA backbone cleavage and cell apoptosis | (146) | |
| CD40 | I/II | NCT01433172 | Lung cancer | (GM.CD40L) vaccine in combination with CCL21 | Boosts the immune system | (147) |
| I/II | NCT01103635 | Metastatic melanoma | Tremelimumab and CP-870, CP-893/AstraZeneca | CD40 agonist mAb | (148) | |
| STAT3 | I | NCT01839604 | Metastatic hepatocellular carcinoma | AZD9150/AstraZeneca | Antisense oligonucleotide inhibitor of STAT3 | (149) |
Macrophage Depletion
High TAM density is associated with a poor patient outcome and therapy resistance in most cancers. Macrophage depletion studies have shown great success in limiting tumor growth and metastatic spread, as well as restoring chemotherapeutic responsiveness (117, 118, 150). Trabectedin is a DNA-binding agent that exerts selective cytotoxicity to circulating monocytes and TAM populations by activating the extrinsic TRAIL apoptotic pathway. Monocytes in particular are sensitive to TRAIL as they express very low levels of TRAIL decoy receptors (151). In four different mouse tumor models, trabectedin significantly inhibited the production of cytokines including CCL2 and IL6, which are important in promoting tumor growth (117). Bisphosphonates comprise another class of drugs that exert myeloid cell cytotoxicity. These drugs are typically used in the clinic for the treatment of osteoporosis and complications arising from bone metastases; however, macrophages in mammary tumors also display sensitivity to bisphosphonate-mediated apoptosis (152). In the clinic, bisphosphonates have been used to treat breast cancer and other solid malignancies in combination with chemotherapy and hormone therapy. This approach has substantially reduced disease recurrence and improved survival in treated patients compared with chemotherapy/hormone therapy alone (153).
In mice, clodronate-liposome-mediated depletion of TAMs significantly reduced tumorigenesis. When combined with anti-angiogenic therapy, administration of clodronate and anti-VEGF antibodies further enhanced TAM depletion and augmented tumor inhibition (118). Thus, macrophage depletion may represent a novel strategy for an indirect cancer therapy specifically aimed at tumor-promoting cells within the microenvironment. However, the challenge with this approach is to find ways for local administration of such drugs to the tumor. Indeed, a major disadvantage of most macrophage depletion studies is the systemic clearance of macrophages, which is unfavorable in clinical applications when host immune responses are already compromised by chemotherapy.
Limiting Macrophage Recruitment and Localization
Another option for targeting TAMs is by inhibiting their recruitment to the primary tumor. CCL2 is a chemokine that regulates the migration of monocytes and macrophages. In mice, interference with the CCL2/CCR2 axis significantly reduced the growth of hepatocellular and renal cell carcinomas (154, 155), and abrogated breast cancer metastasis (119). Interestingly, cessation of CCL2 inhibition accelerated breast cancer metastasis by promoting the infiltration of bone-marrow monocytes into tumors (156), indicating the importance of CCL2 signaling in regulating metastatic growth. In the clinic, antibodies that selectively target CCL2 have completed Phase I and II clinical trials (Table 4). In a Phase I trial, administration of the anti-CCL2 antibody carlumab (CNTO 888) was well tolerated and showed promising antitumor activity in patients with advanced disease. However, this response was not observed in the Phase II study involving patients with castration-resistant prostate cancer. Furthermore, preclinical studies combining anti-CCL2 with the antimitotic chemotherapy agent Docetaxel enhanced antitumor responses (157); however, combining anti-CCL2 with conventional chemotherapy has produced mixed results in Phase IB clinical trials. In one trial, administration of the anti-CCL2 agent carlumab in combination with four chemotherapy regimens was well tolerated although no significant tumor response was observed (158). By contrast, combining the oral CCR2 small-molecule antagonist PF-04136309 with conventional chemotherapy resulted in partial tumor responses (49%) with local tumor control in 97% of patients with advanced pancreatic ductal adenocarcinoma (PDAC). None of the patients in the chemotherapy-alone group achieved an objective response (159).
CXCL12 is a chemokine that facilitates the migration of macrophages through endothelial barriers and into the tumor milieu. The secretion of CXCL12 by stromal cells also attracts the movement of cancer cells by upregulating their expression of CXCR4 (121). For this reason, inhibition of CXCL12/CXCR4 signaling represents a promising strategy to modulate macrophage infiltration and prevent metastasis. Indeed, targeting CXCR4 in mouse models of breast and prostate cancer significantly reduced total tumor burden and metastases (121, 122). However, the therapeutic efficacy of inhibiting CXCL12 in human patients has yet to be tested in clinical trials.
In addition to targeting chemokines, antibodies against macrophage surface receptors such as CD11b and CSF1 receptor (CSF1R) may be used to impair macrophage recruitment (126, 160). In the case of CD11b, administration of a neutralizing CD11b monoclonal antibody reduced tumor growth in a mouse model of spontaneous intestinal adenoma, and enhanced antitumor responses to radiation by reducing myeloid cell infiltration (126, 161). However, since CD11b is also expressed on other immune cells including neutrophils, this approach is limited in its specificity against TAMs.
Targeting the CSF1–CSF1R axis represents a more specific strategy, since CSF1R is exclusively expressed on cells of the monocytic lineage and therefore provides a viable target to specifically inhibit TAMs (162). As a single agent, treatment of mice with the humanized anti-CSF1R antibody emactuzumab (RG7155) selectively reduced TAM infiltration and promoted CD8+ T-cell expansion. Administration of emactuzumab to patients similarly led to a striking reduction of macrophages in tumor tissue, which translated to a marked clinical benefit for patients with diffuse-type giant cell tumors (163).
CSF1 receptor blockade in combination with conventional cancer treatments has also shown to improve the efficacy of radiotherapy, immunotherapy and chemotherapy. Locally recurrent disease and/or metastatic spread following radiotherapy has been attributed to an influx of bone marrow-derived monocytes that drive vasculature regrowth (164, 165). In mice harboring glioblastoma multiforme (GBM) xenografts, treatment with pexidartinib (PLX3397) augmented tumor responsiveness to radiotherapy by reducing the recruitment of bone marrow-derived TAMs (165). Pexidartinib also improved the antitumor efficacy of adoptive cell therapy in a syngeneic mouse model of BRAF (V600E)-driven melanoma (166). In agreement with previous studies of breast cancer models (167), inhibition of macrophage recruitment by CSF1R blockade enhanced the therapeutic efficacy of gemcitabine in a chemoresistant transgenic model of pancreatic cancer (168). Collectively, these results provide evidence for targeting the infiltration of TAMs as a complementary strategy to enhance the efficacy of conventional cancer therapies.
Macrophage Reprogramming
One key feature of macrophages is the plasticity of their endotype. Thus, the reprogramming of macrophages toward a tumoricidal CAM endotype has gained widespread interest as an attractive therapeutic strategy against cancer. This can either be achieved by preventing TAMs from adopting an AAM endotype or by promoting the repolarization of macrophages with an AAM endotype toward a tumoricidal CAM endotype.
Large-scale transcriptome studies performed on AAMs have identified key genes and signaling pathways that play a critical role in macrophage polarization. Jumonji domain containing-3 (JMJD3) protein, for example, is a histone 3 Lys27 demethylase that has been implicated in AAM activation (29). Loss of JMJD3 results in defective expression of Irf4 and other AAM-associated macrophage markers, and the impaired differentiation and recruitment of AAMs in response to helminth infection (29). The role of the myeloid-specific Src family kinase member HCK as a key regulator of gene expression in AAM human monocytes has also been described (26). Increased HCK activity in mice promotes colon tumorigenesis by enhancing angiogenesis and facilitating alternative macrophage polarization, while the genetic ablation or pharmacologic inhibition of HCK suppressed AAM polarization and impaired the growth of endogenous mouse and human colorectal cancer xenografts (27).
STAT3 and STAT6 play an important role in tumor-promoting macrophage polarization. A small-molecule inhibitor of STAT3 significantly reduced AAM polarization in patients with malignant glioma (169), while use of multitargeted tyrosine kinase inhibitors such as sunitinib and sorafenib promoted cancer cell apoptosis and reversed the immunosuppressive cytokine profile of AAMs by indirectly inhibiting signaling of downstream STAT3 (128, 129). Likewise, TAMs from STAT6 deficient mice display a CAM endotype and enhanced antitumor immunity (127). Together, these data suggest that the suppression of AAM endotypes can promote antitumor activities by reversing the immunosuppressive microenvironment.
Enhancing the CAM endotype of TAMs is another promising approach. IFNα has long been shown to exert tumoricidal effects and acts as a strong inducer of CAM polarization. When targeted to orthotopic human gliomas and spontaneous mouse mammary carcinomas, IFNα reduced tumor growth and abrogated metastasis (133). Similarly, systemic activation of CAMs with an agonist CD40 monoclonal antibody in combination with gemcitabine chemotherapy effectively circumvented tumor-mediated immune suppression and increased survival in patients with surgically incurable PDAC (134). In this study, it was shown that CD40-activated macrophages rapidly infiltrated tumors and exerted antitumor cytotoxicity (134). Subsequent Phase I clinical trials with a fully humanized CD40 agonist antibody (CP-870,893) in combination with gemcitabine showed well-tolerated responses and the activation of antitumor immune responses (170). Repolarization of TAMs from AAM toward a CAM endotype has also been achieved by inhibiting PI3Kγ in mice bearing PDACs, resulting in reduced tumor growth and metastasis (32). Genes associated with an AAM profile were strongly expressed in myeloid cells isolated from PDAC tumors; however, treatment with a PI3Kγ inhibitor significantly reduced the expression of these markers in PDAC tumors and in the corresponding TAMs. Conversely, the expression of immune-stimulatory factors such as IFNγ was significantly upregulated in animals treated with PI3Kγ inhibitors, consistent with enhanced CD8+ T-cell-mediated antitumor immune responses (32). Collectively, these molecular targets, alongside histidine-rich glycoprotein HRG (135) and the NFκB signaling cascade (136), provide promising mechanisms to promote the reprogramming of macrophages away from a tumor-promoting endotype.
Influence of Macrophages on Treatment Responses
Increasing evidence has supported a dual role for TAMs to affect the effectiveness of anticancer therapies by either antagonizing the activity of these treatments or enhancing the overall cytotoxic effect. Thus, targeting TAMs might amplify the susceptibility of cancer cells to such interventions and improve the clinical outcome.
Chemotherapy
A major challenge for successful cancer treatment is tumor resistance to chemotherapy. Preclinical models and clinical studies have revealed an important role of macrophages in modulating the adaptive immune response to improve chemotherapeutic responses. In an aggressive transgenic mouse model of mammary adenocarcinoma, administration of chemotherapy in combination with TAM blockade promoted antitumor immunity and cytotoxic T-cell infiltration, resulting in a significant decrease of pulmonary metastases and improved overall survival compared with chemotherapy alone (167). Likewise, the anti-proliferative agent trabectedin induces cell-cycle arrest in cancer cells by selectively depleting monocytes in soft tissue sarcoma (117).
Antiangiogenic Therapy
The development and use of antiangiogenic therapies has become an integral component of anti-cancer regimens. However, such therapies have shown limited durability due to acquired resistance. One mechanism of drug resistance suggested by preclinical studies is the recruitment of TAMs, since increased macrophage recruitment is frequently observed in resistant tumors (171, 172). In GBM patients, resistance to the antiangiogenic agent bevacizumab is driven by reduced expression of MIF at the tumor edge, causing the expansion of AAMs, which promote tumor growth (171). Similarly, secretion of MMP9 by intratumoral macrophages is associated with resistance to aflibercept, an anti-VEGF and anti-placental growth factor drug (173).
Treatment-induced hypoxia caused by vessel regression can similarly mediate resistance to antiangiogenic therapy by triggering the compensatory recruitment of myeloid cells to repair the vascular bed. In a mouse model of GBM, the hypoxia induced transcription factor HIF1α attracted bone marrow-derived TIE2- and VEGFR-expressing myeloid cells to promote neovascularization (174). These cell populations were diminished in HIF1α knockout tumors, which displayed normal and functional vasculature (174). Indeed, the angiogenic and hypoxic profiles of tumors is also used to predict radiographic response and survival benefit of GBM patients undergoing chemotherapy (175).
Targeting of macrophages in combination with anti-angiogenic therapies to restore or augment anti-tumor responses has yielded promising preclinical results. ANG2 is a member of the angiopoietin family that primarily signals through the TIE2 receptor. In addition to providing an escape mechanism to anti-VEGF therapy, ANG2 signaling modulates the activity of TIE2-expressing proangiogenic TAMs. In mice carrying orthotopic mammary tumors, ANG2 blockade inhibited tumor angiogenesis, growth, and metastasis, and impaired the activity of proangiogenic TIE2+ macrophages (94). Of note, ANG2 blockade also inhibited angiogenesis and tumor growth in mouse models that are prone to develop resistance to anti-VEGF/VEGFR therapy (94). Likewise, dual inhibition of ANG2 and VEGF normalized the tumor vasculature and prolonged survival in murine GBM models in part by altering TAM polarization (176, 177). When combined with anti-PD1 checkpoint inhibition, combined ANG2 and VEGF blockade with a bispecific antibody further enhanced the antitumor response (178). Thus, integration of TAM-targeting strategies to complement antiangiogenic therapies may improve treatment efficacy and patient survival.
Immunotherapy
Immune checkpoint therapies aim to reverse the immunosuppressive nature of the tumor microenvironment and restore cytotoxic immune cell functions against cancer cells. Clinically validated checkpoint targets include PD1, PDL1, and CTLA4, and their inhibition has been shown to exert significant antitumor responses in cancers as diverse as melanoma and Hodgkin’s lymphoma (179, 180). However, there are still many cancers that remain refractory to immunotherapy.
Macrophages are a key component of the immunosuppressive pathway targeted by immune checkpoint inhibitors. In response to various stimuli including cytokines (181) and hypoxia (182), TAMs can express the PD1 ligands PDL1 and PDL2, as well as ligands for CTLA4 (B7-1 and B7-2). Ligation of PDL1 to PD1 on the surface of cytotoxic T-cells leads to the inactivation of these immune effectors and facilitates immune escape. Mouse and human TAMs also express PD1, and the expression of this protein increases over time with disease severity (180). Interestingly, the majority of PD1+ TAMs exhibit an AAM endotype, which can be reversed to a CAM-like endotype by PD1–PDL1 blockade to restore phagocytic activity and antitumor immunity. These results suggest that activation of the PD1–PDL1 pathway in TAMs impairs their cytotoxic ability (180).
Inhibition of CTLA4, an inhibitory receptor expressed on the surface of T-cells, has emerged as an effective therapy for metastatic melanoma. Analysis of the mechanism by which anti-CTLA4 therapy exerts its antitumor effects has revealed an important role of macrophages in driving these responses (183). In melanoma patients, anti-CTLA-dependent cell-mediated cytotoxicity is mediated by CD16-expressing macrophages (179). Of note, ipilimumab responders displayed significantly higher baseline peripheral frequencies of CD16+ cells and a selective enrichment in tumor-infiltrating CD68+CD16+ (CAM-like) macrophages compared with non-responder patients. These results were consistent with a decrease in Treg cell numbers following immune checkpoint inhibition (179).
Challenges and Therapeutic Perspectives
Of Mice and Not Men: Differences in Mouse and Human Immunology
Mice provide a mainstay of in vivo experiments and have contributed significantly to our understanding of human immunology. Comparative analysis of the mouse and human genome has revealed a striking level of conservation. Despite this, there are major discrepancies between our innate and adaptive immune systems in terms of development, activation and function. Such differences are unsurprising since the divergence of mice and humans occurred more than 60 million years ago, resulting in the evolution of both species under different ecological niches and environmental pressures. Thus, while there are many parallels between mouse and human biology, it is also important to recognize the fundamental differences, especially when translating preclinical findings from bench to bedside. For example, expression of the cell-surface marker F4/80 is exclusively found in mouse macrophages and is undetectable on human cells (184). An alternative marker commonly used to distinguish human macrophages is CD68, however, since CD68 can also be expressed by some stromal and cancer cells, particular care should be taken when using this marker to identify TAMs (185).
Differences also exist when comparing the transcriptional profile of mouse and human macrophages following exposure to stimulating cytokines in vitro. For example, polarization of mouse macrophages toward an immunosuppressive AAM endotype is usually modeled by stimulation with IL4 and/or IL13. This results in the upregulation of M2-associated markers including FIZZ1, ARG1, and YM1; however, this response is not observed in human AAMs (46). Likewise, competitive metabolism of the amino acid arginine by NOS2 and ARG1 is used to delineate between pro-inflammatory CAM and immunosuppressive AAM endotypes in mouse macrophages, but this discriminative criteria does not apply to human cells (46). Thus, mouse and human macrophages exhibit distinct differences that should be taken into consideration to best translate our findings obtained from mouse models to human situations.
Monotherapy or Complement Therapies
Whether macrophage-targeting therapies will be most efficacious as monotherapies or as a combinatorial approach with chemotherapy and immunotherapy is still unclear. Considering that antigens are released by dying tumor cells following chemotherapy (186), the cross-representation of tumor antigens by TAMs could be exploited to enhance antitumor CD8+ T-cell responses and stimulate immunological memory. Likewise, TAM-targeting strategies may also complement the efficacy of immune checkpoint inhibitors by removing additional inhibitory factors that may further restrict T-cell function. In preclinical models of PDAC, anti-PD1 and anti-CTLA4 antagonists showed limited efficacy as monotherapies to restrain tumor growth, but the use of these agents in combination with CSF1R blockade resulted in tumor regression (187).
Predicting Clinical Response
Since macrophage-targeted approaches elicit distinct effects based on tumor type, another aspect that should be considered is the identification of cancers and stratification of patient cohorts that are most likely to respond to treatment. In one study, high TAM density in metastatic lymph nodes predicted better disease-free survival in stage III colorectal cancer patients undergoing 5-fluorouracil adjuvant therapy (188). On the other hand, increased TAM infiltration is significantly associated with an unfavorable outcome for esophageal cancer patients undergoing neoadjuvant chemotherapy (189). Thus, a clearer understanding of how macrophages contribute to tumor progression across different cancers is crucial to maximize clinical benefit. The timing and duration of macrophage-targeted therapies could similarly have profound effects on patient response and overall treatment efficacy, and warrants further investigation.
Minimizing the Side Effects of Targeting TAMs
The development of localized treatment options for the primary tumor represents a significant hurdle, since the systemic depletion of macrophages in an immunocompromised patient undergoing chemotherapy may increase their vulnerability to infections. Furthermore, long-term depletion may also perturb the behavior of other immune cells that rely on macrophages to guide their functions. For instance, systemic inflammation has been observed as a result of excessive neutrophil infiltration in the absence of macrophages (190). Likewise, resident macrophages play a critical role in maintaining homeostasis in tissues in which they reside (191, 192), and the prolonged-depletion of these cells may severely impair organ function. Kupffer cells, for example, are involved in the breakdown of red blood cells in the liver (191), and their depletion results in aggravated liver lesions (193). By contrast, the loss of alveolar macrophages increases morbidity and respiratory failure in mice following influenza infection (194). While macrophage reprogramming represents a more viable option, the delicate balance between the tumoricidal and tumor-promoting functions of these cells also needs to be carefully considered. Excessive reprogramming of TAMs toward a CAM endotype could result in an excess of cytotoxic cytokines, inflammation, and tissue damage. While AAMs are essential for wound healing, the loss of AAMs might result in impaired tissue repair responses.
Macrophage-targeting strategies currently encompass a range of antibodies and small-molecule inhibitors; however, these two classes of drugs exhibit major differences in their pharmacological properties. Owing to their larger molecule weight, monoclonal antibodies often have a reduced efficiency for tissue penetration, but extended tumor retention and clearance from the blood compared with small-molecule inhibitors (195). However, small-molecule inhibitors tend to be less specific than monoclonal antibodies with an increased risk of toxicity, although these adverse effects are generally mild (195). These factors should be carefully considered when developing new drugs to maximize the therapeutic efficacy of these compounds.
Concluding Remarks
Macrophages are a major component of solid cancers and can promote tumorigenesis by facilitating angiogenesis, immunosuppression, invasion, and metastasis. Given the association between high macrophage infiltration and poor survival in most cancers, these cells represent promising targets for anticancer therapy. Strategies aimed at targeting TAMs have shown success in clinical trials and include macrophage depletion, modifying macrophage recruitment, and the reprogramming of macrophages away from an AAM endotype. These macrophage-directed therapies have also shown complementary effects when combined with chemo- and immunotherapies, suggesting the additive benefit of targeting TAMs alongside other cell populations to augment antitumor immunity. For this reason, a greater understanding of the complex interactions between TAMs and their surrounding microenvironment is vital to identify additional pathways that can be targeted in parallel.
One major hurdle of targeting TAMs is to minimize the occurrence of negative side effects in the patient. Given their multifaceted roles of maintaining homeostasis, the systemic depletion of macrophages may lead to increased infections or impaired ability of tissue-resident cells to carry out their normal function. Thus, the identification of TAM-specific markers or molecules that are predominantly produced by AAMs and/or MAMs will enable the development of more sophisticated therapies that can be targeted specifically to tumors without affecting the function of other tissue-resident immune cells. In the same way, strategies aimed at reprogramming macrophages should also aim to conserve the ability of these cells to carry out phagocytosis and wound healing in non-tumor tissues.
Author Contributions
AP and ME wrote the manuscript and designed the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work in the laboratory of ME is supported by Ludwig Cancer Research, the Victorian State Government Operational Infrastructure Support, the IRIISS scheme of the National Health and Medical Research Council of Australia (NHMRC), and NHMRC grants #1069024, #1067244, and #1081373. AP is supported by a Cancer Council of Victoria Post-doctoral Research Fellowship. ME is an NHMRC Principal Research Fellow. The authors apologize that due to space limitations only selected original research articles could be cited.
References
- 1.van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ (1972) 46:845–52. [PMC free article] [PubMed] [Google Scholar]
- 2.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science (2012) 336:86–90. 10.1126/science.1219179 [DOI] [PubMed] [Google Scholar]
- 3.Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science (2014) 344:921–5. 10.1126/science.1252510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bottazzi B, Erba E, Nobili N, Fazioli F, Rambaldi A, Mantovani A. A paracrine circuit in the regulation of the proliferation of macrophages infiltrating murine sarcomas. J Immunol (1990) 144:2409–12. [PubMed] [Google Scholar]
- 5.Pucci F, Venneri MA, Biziato D, Nonis A, Moi D, Sica A, et al. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood (2009) 114:901–14. 10.1182/blood-2009-01-200931 [DOI] [PubMed] [Google Scholar]
- 6.Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res (2010) 70:5270–80. 10.1158/0008-5472.CAN-10-0012 [DOI] [PubMed] [Google Scholar]
- 7.Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity (2016) 44:303–15. 10.1016/j.immuni.2016.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111:4233–44. 10.1182/blood-2007-07-099226 [DOI] [PubMed] [Google Scholar]
- 9.Feng X, Szulzewsky F, Yerevanian A, Chen Z, Heinzmann D, Rasmussen RD, et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget (2015) 6:15077–94. 10.18632/oncotarget.3730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol (2010) 11:889–96. 10.1038/ni.1937 [DOI] [PubMed] [Google Scholar]
- 11.Mantovani A. Macrophages, neutrophils, and cancer: a double edged sword. N J Sci (2014) 214:1–14. 10.1155/2014/271940 [DOI] [Google Scholar]
- 12.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. 10.1016/j.immuni.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mosser D, Edwards J. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8:958–69. 10.1038/nri2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol (2011) 12:231–8. 10.1038/ni.1990 [DOI] [PubMed] [Google Scholar]
- 15.Dalmas E, Toubal A, Alzaid F, Blazek K, Eames HL, Lebozec K, et al. Irf5 deficiency in macrophages promotes beneficial adipose tissue expansion and insulin sensitivity during obesity. Nat Med (2015) 21:610–8. 10.1038/nm.3829 [DOI] [PubMed] [Google Scholar]
- 16.Fontana MF, Baccarella A, Pancholi N, Pufall MA, Herbert DR, Kim CC. JUNB is a key transcriptional modulator of macrophage activation. J Immunol (2015) 194:177–86. 10.4049/jimmunol.1401595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, et al. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest (2011) 121:2736–49. 10.1172/JCI45444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol (2015) 27:286–96. 10.1016/j.smim.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adamson SE, Griffiths R, Moravec R, Senthivinayagam S, Montgomery G, Chen W, et al. Disabled homolog 2 controls macrophage phenotypic polarization and adipose tissue inflammation. J Clin Invest (2016) 126:1311–22. 10.1172/JCI79590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banerjee S, Xie N, Cui H, Tan Z, Yang S, Icyuz M, et al. MicroRNA let-7c regulates macrophage polarization. J Immunol (2013) 190:6542–9. 10.4049/jimmunol.1202496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ying W, Tseng A, Chang RC, Morin A, Brehm T, Triff K, et al. MicroRNA-223 is a crucial mediator of PPARgamma-regulated alternative macrophage activation. J Clin Invest (2015) 125:4149–59. 10.1172/JCI81656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Festuccia WT, Pouliot P, Bakan I, Sabatini DM, Laplante M. Myeloid-specific Rictor deletion induces M1 macrophage polarization and potentiates in vivo pro-inflammatory response to lipopolysaccharide. PLoS One (2014) 9:e95432. 10.1371/journal.pone.0095432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci U S A (2012) 109:9517–22. 10.1073/pnas.1119038109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vannella KM, Barron L, Borthwick LA, Kindrachuk KN, Narasimhan PB, Hart KM, et al. Incomplete deletion of IL-4Ralpha by LysM(Cre) reveals distinct subsets of M2 macrophages controlling inflammation and fibrosis in chronic schistosomiasis. PLoS Pathog (2014) 10:e1004372. 10.1371/journal.ppat.1004372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herbert DR, Holscher C, Mohrs M, Arendse B, Schwegmann A, Radwanska M, et al. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity (2004) 20:623–35. 10.1016/S1074-7613(04)00107-4 [DOI] [PubMed] [Google Scholar]
- 26.Bhattacharjee A, Pal S, Feldman GM, Cathcart MK. Hck is a key regulator of gene expression in alternatively activated human monocytes. J Biol Chem (2011) 286:36708–23. 10.1074/jbc.M111.291492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poh AR, Love CG, Masson F, Preaudet A, Tsui C, Whitehead L, et al. Inhibition of hematopoietic cell kinase activity suppresses myeloid cell-mediated colon cancer progression. Cancer Cell (2017) 31:563.e–75.e. 10.1016/j.ccell.2017.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rutschman R, Lang R, Hesse M, Ihle JN, Wynn TA, Murray PJ. Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J Immunol (2001) 166:2173–7. 10.4049/jimmunol.166.4.2173 [DOI] [PubMed] [Google Scholar]
- 29.Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol (2010) 1:936–44. 10.1038/ni.1920 [DOI] [PubMed] [Google Scholar]
- 30.Chawla A. Control of macrophage activation and function by PPARs. Circ Res (2010) 106:1559–69. 10.1161/CIRCRESAHA.110.216523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A (2009) 106:14978–83. 10.1073/pnas.0809784106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, et al. Macrophage PI3Kgamma drives pancreatic ductal adenocarcinoma progression. Cancer Discov (2016) 6:870–85. 10.1158/2159-8290.CD-15-1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Date D, Das R, Narla G, Simon DI, Jain MK, Mahabeleshwar GH. Kruppel-like transcription factor 6 regulates inflammatory macrophage polarization. J Biol Chem (2014) 289:10318–29. 10.1074/jbc.M113.526749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB, et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest (2015) 125:4334–48. 10.1172/JCI81676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kratochvill F, Neale G, Haverkamp JM, Van de Velde LA, Smith AM, Kawauchi D, et al. TNF counterbalances the emergence of M2 tumor macrophages. Cell Rep (2015) 12:1902–14. 10.1016/j.celrep.2015.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rauh MJ, Ho V, Pereira C, Sham A, Sly LM, Lam V, et al. SHIP represses the generation of alternatively activated macrophages. Immunity (2005) 23:361–74. 10.1016/j.immuni.2005.09.003 [DOI] [PubMed] [Google Scholar]
- 37.Zheng C, Yang Q, Xu C, Shou P, Cao J, Jiang M, et al. CD11b regulates obesity-induced insulin resistance via limiting alternative activation and proliferation of adipose tissue macrophages. Proc Natl Acad Sci U S A (2015) 112:E7239–48. 10.1073/pnas.1500396113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cudejko C, Wouters K, Fuentes L, Hannou SA, Paquet C, Bantubungi K, et al. p16INK4a deficiency promotes IL-4-induced polarization and inhibits proinflammatory signaling in macrophages. Blood (2011) 118:2556–66. 10.1182/blood-2010-10-313106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schleicher U, Paduch K, Debus A, Obermeyer S, Konig T, Kling JC, et al. TNF-mediated restriction of arginase 1 expression in myeloid cells triggers type 2 NO synthase activity at the site of infection. Cell Rep (2016) 15:1062–75. 10.1016/j.celrep.2016.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro M, et al. Macrophage polarisation in tumour progression. Semin Cancer Biol (2008) 18:349–55. 10.1016/j.semcancer.2008.03.004 [DOI] [PubMed] [Google Scholar]
- 41.Qian B-Z, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141:39–51. 10.1016/j.cell.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis (2014) 17:109–18. 10.1007/s10456-013-9381-6 [DOI] [PubMed] [Google Scholar]
- 43.Venosa A, Malaviya R, Choi H, Gow AJ, Laskin JD, Laskin DL. Characterization of distinct macrophage subpopulations during nitrogen mustard-induced lung injury and fibrosis. Am J Respir Cell Mol Biol (2016) 54:436–46. 10.1165/rcmb.2015-0120OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Y, Zhang S, Wang Q, Zhang X. Tumor-recruited M2 macrophages promote gastric and breast cancer metastasis via M2 macrophage-secreted CHI3L1 protein. J Hematol Oncol (2017) 10:36. 10.1186/s13045-017-0408-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holscher C, Arendse B, Schwegmann A, Myburgh E, Brombacher F. Impairment of alternative macrophage activation delays cutaneous leishmaniasis in nonhealing BALB/c mice. J Immunol (2006) 176:1115–21. 10.4049/jimmunol.176.2.1115 [DOI] [PubMed] [Google Scholar]
- 46.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol (2006) 177:7303–11. 10.4049/jimmunol.177.10.7303 [DOI] [PubMed] [Google Scholar]
- 47.Duluc D, Delneste Y, Tan F, Moles MP, Grimaud L, Lenoir J, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood (2007) 110:4319–30. 10.1182/blood-2007-02-072587 [DOI] [PubMed] [Google Scholar]
- 48.Lewis C, Pollard J. Distinct role of macrophages in different tumor microenvironments. Cancer Res (2006) 15:605–12. 10.1158/0008-5472.CAN-05-4005 [DOI] [PubMed] [Google Scholar]
- 49.Klimp A, Vries ED, Scherphof G, Daemen T. A potential role of macrophage activation in the treatment of cancer. Crit Rev Oncol Hematol (2002) 44:143–61. 10.1016/S1040-8428(01)00203-7 [DOI] [PubMed] [Google Scholar]
- 50.Ong S-M, Tan Y-C, Beretta O, Jiang D, Yeap W-H, Tai JJY, et al. Macrophages in human colorectal cancer are pro-inflammatory and prime T cells towards an anti-tumour type-1 inflammatory response. Eur J Immunol (2012) 42:89–100. 10.1002/eji.201141825 [DOI] [PubMed] [Google Scholar]
- 51.Shimizu T, Abe R, Nakamura H, Ohkawara A, Suzuki M, Nishihira J. High expression of macrophage migration inhibitory factor in human melanoma cells and its role in tumour cell growth and angiogenesis. Biochem Biophys Res Commun (1999) 264:751–8. 10.1006/bbrc.1999.1584 [DOI] [PubMed] [Google Scholar]
- 52.Onodera S, Suzuki K, Matsuno T, Kaneda K, Takagi M, Nishihira J. Macrophage migration inhibitory factor induces phagocytosis of foreign particles by macrophages in autocrine and paracrine fashion. Immunology (1997) 92:131–7. 10.1046/j.1365-2567.1997.00311.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pozzi L, Weiser W. Human recombinant migration inhibitory factor activates human macrophages to kill tumour cells. Cell Immunol (1992) 145:372–9. 10.1016/0008-8749(92)90339-Q [DOI] [PubMed] [Google Scholar]
- 54.Osaki T, Hashimoto W, Gambotto A, Okamura H, Robbins P, Kurimoto M, et al. Potent anti-tumour effects mediated by local expression of the mature form of the interferone-gamma inducing factor, interleukin-18 (IL-18). Gene Ther (1999) 6:808–15. 10.1038/sj.gt.3300908 [DOI] [PubMed] [Google Scholar]
- 55.Netea M, Kullberg B, Verschueren I, Meer JVD. Interleukin-19 induces production of proinflammatory cytokines in mice: no intermediate role for the cytokines of the tumour necrosis factor family and interleukin-1beta. Eur J Immunol (2000) 30:3057–60. [DOI] [PubMed] [Google Scholar]
- 56.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med (2001) 193:727–40. 10.1084/jem.193.6.727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pyonteck S, Gadea B, Wang H, Gocheva V, Hunter K, Tang L, et al. Deficiency of the macrophage growth factor CSF-1 disrupts pancreatic neuroendocrine tumor development. Oncogene (2012) 31:1459–67. 10.1038/onc.2011.337 [DOI] [PubMed] [Google Scholar]
- 58.Kubota Y, Takubo K, Shimizu T, Ohno H, Kishi K, Shibuya M, et al. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J Exp Med (2009) 206:1089–102. 10.1084/jem.20081605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ryder M, Gild M, Hohl TM, Pamer E, Knauf J, Ghossein R, et al. Genetic and Pharmacological Targeting of CSF-1/CSF-1R inhibits tumor-associated macrophages and impairs BRAF-induced thyroid cancer progression. PLoS One (2013) 8:e54302. 10.1371/journal.pone.0054302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation and cancer. Cell (2010) 140:883–99. 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gupta RB, Harpaz N, Itzkowitz S, Hossain S, Matula S, Kornbluth A, et al. Histologic inflammation is a risk factor for progression to colorectal neoplasia in ulcerative colitis: a cohort study. Gastroenterology (2007) 133:1099–105. 10.1053/j.gastro.2007.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Popivanova B, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest (2008) 118:560–70. 10.1172/JCI32453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Y, Wang K, Han G, Wang R, Xiao H, Hou C, et al. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunol (2014) 7:1106–15. 10.1038/mi.2013.126 [DOI] [PubMed] [Google Scholar]
- 64.Ning C, Li Y, Wang Y, Han G, Wang R, Xiao H, et al. Complement activation promotes colitis-associated carcinogenesis through activating intestinal IL-1β/IL-17A axis. Mucosal Immunol (2015) 8(6):1275–84. 10.1038/mi.2015.18 [DOI] [PubMed] [Google Scholar]
- 65.Matsuzaki K, Murata M, Yoshida K, Sekimoto G, Uemura Y, Sakaida N, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology (2007) 46:48–57. 10.1002/hep.21672 [DOI] [PubMed] [Google Scholar]
- 66.Kaparakis M, Walduck AK, Price JD, Pedersen JS, van Rooijen N, Pearse MJ, et al. Macrophages are mediators of gastritis in acute helicobacter pylori infection in C57BL/6 mice. Infect Immun (2008) 76:2235–9. 10.1128/IAI.01481-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Doll R, Hill B. Smoking and carcinoma of the lung. Br Med J (1950) 2:739–48. 10.1136/bmj.2.4682.739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zins K, Abraham D, Sioud M, Aharinejad S. Colon cancer cell-derived tumor necrosis factor-alpha mediates the tumor growth-promoting response in macrophages by up-regulating the colony-stimulating factor-1 pathway. Cancer Res (2007) 67:1038–45. 10.1158/0008-5472.CAN-06-2295 [DOI] [PubMed] [Google Scholar]
- 69.Hou Z, Falcone D, Subbaramaiah K, Danneberg A. Macrophages induce COX-2 expression in breast cancer cells: role of IL-1b autoamplification. Carcinogenesis (2011) 32:695–702. 10.1093/carcin/bgr027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eubank T, Roberts R, Khan M, Curry J, Nuovo G, Kuppusamy P, et al. Granulocyte macrophage colony-stimulating factor inhibits breast cancer growth and metastasis by invoking an anti-angiogenic program in tumor educated macrophages. Cancer Res (2009) 49:2133–40. 10.1158/0008-5472.CAN-08-1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guiducci C, Vicari A, Sangaletti S, Trinchieri G, Colombo M. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res (2005) 65(8):3437–46. 10.1158/0008-5472.CAN-04-4262 [DOI] [PubMed] [Google Scholar]
- 72.Ojalvo L, King W, Cox D, Pollard J. High density gene expression analysis of tumor-associated macrophages from mouse mammary tumors. Am J Pathol (2009) 174:1048–64. 10.2353/ajpath.2009.080676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood (2006) 107:2112–22. 10.1182/blood-2005-01-0428 [DOI] [PubMed] [Google Scholar]
- 74.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophage as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23:549–55. 10.1016/S1471-4906(02)02302-5 [DOI] [PubMed] [Google Scholar]
- 75.Kuang D, Zhao Q, Peng C, Xu J, Zhang J, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med (2009) 206:1327–37. 10.1084/jem.20082173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, Webster WS, et al. Costimulatory B7-H1 in renal cell carcinoma patients: indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci U S A (2004) 101:17174–9. 10.1073/pnas.0406351101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A (2007) 104:3360–5. 10.1073/pnas.0611533104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med (2011) 208:577–92. 10.1084/jem.20100619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature (2016) 539:437–42. 10.1038/nature19834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer (2004) 4:540–50. 10.1038/nrc1388 [DOI] [PubMed] [Google Scholar]
- 81.Mantovania A, Sica A, Sozzani S, Allavena P, Vecchi A, Locatia M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25:677–86. 10.1016/j.it.2004.09.015 [DOI] [PubMed] [Google Scholar]
- 82.Hanahan D, Christofori G, Naik P, Arbeit J. Transgenic mouse models of tumour angiogenesis: the angiogenic switch, its molecular controls and prospects for preclinical therapeutic models. Eur J Cancer (1996) 32A:2386–93. 10.1016/S0959-8049(96)00401-7 [DOI] [PubMed] [Google Scholar]
- 83.Murdoch C, Muthana M, Coffelt S, Lewis C. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer (2008) 8:618–31. 10.1038/nrc2444 [DOI] [PubMed] [Google Scholar]
- 84.Murdoch C, Giannoudis A, Lewis C. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischaemic tissues. Blood (2004) 104:2224–34. 10.1182/blood-2004-03-1109 [DOI] [PubMed] [Google Scholar]
- 85.Leek R, Lewis C, Whitehouse R, Greenall M, Clarke J, Harris A. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res (1996) 56:4625–9. [PubMed] [Google Scholar]
- 86.Lin E, Li J, Gnatovskiy L, Deng Y, Zhu L, Grzesik F, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res (2006) 66:11238–46. 10.1158/0008-5472.CAN-06-1278 [DOI] [PubMed] [Google Scholar]
- 87.Schoppmann S, Birner P, Stockl J, Kalt R, Ullrich R, Caucig C, et al. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am J Pathol (2002) 161:947–56. 10.1016/S0002-9440(10)64255-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hotchkiss K, Ashton A, Klein R, Lenzi M, Zhu G, Schwartz E. Mechanisms by which tumor cells and monocytes expressing the angiogenic factor thymidine phosphorylase mediate human endothelial cell migration. Cancer Res (2003) 63:527–33. [PubMed] [Google Scholar]
- 89.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer (2006) 42:717–27. 10.1016/j.ejca.2006.01.003 [DOI] [PubMed] [Google Scholar]
- 90.Ojalvo L, Whittaker C, Condeelis J, Pollard J. Gene expression analysis of macrophages that facilitate tumor invasion supports a role for Wnt-signaling in mediating their activity in primary mammary tumors. J Immunol (2010) 184:702–12. 10.4049/jimmunol.0902360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zabuawala T, Taffany D, Sharma S, Merchant A, Adair B, Srinivasan R, et al. An ets2-driven transcriptional program in tumor-associated macrophages promotes tumor metastasis. Cancer Res (2010) 70:1323–33. 10.1158/0008-5472.CAN-09-1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stockman C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg J, et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature (2008) 456:814–8. 10.1038/nature07445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Palma MD, Venneri M, Galli R, Sergi LS, Politi L, Sampaolesi M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell (2005) 8:211–26. 10.1016/j.ccr.2005.08.002 [DOI] [PubMed] [Google Scholar]
- 94.Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell (2011) 19:512–26. 10.1016/j.ccr.2011.02.005 [DOI] [PubMed] [Google Scholar]
- 95.Wyckoff J, Wang Y, Lin E, Li L, Goswami S, Stanley E, et al. Direct visualisation of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res (2007) 67:2649–56. 10.1158/0008-5472.CAN-06-1823 [DOI] [PubMed] [Google Scholar]
- 96.Kim S, Takahashi H, Lin W-W, Descargues P, Grivennikov S, Kim Y, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature (2009) 457:102–6. 10.1038/nature07623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, et al. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell (2002) 2:289–300. 10.1016/S1535-6108(02)00153-8 [DOI] [PubMed] [Google Scholar]
- 98.Chalmers S, Wang Y, Gertz R, Kacinski B. Macrophage colony-stimulating factor mediates invasion of ovarian cancer cells through urokinase. Cancer Res (1995) 55:1578–85. [PubMed] [Google Scholar]
- 99.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell (2010) 141:52–67. 10.1016/j.cell.2010.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wyckoff J, Wang Y, Lin E, Pixley F, Stanley E, Graf T, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res (2004) 64:7022–9. 10.1158/0008-5472.CAN-04-1449 [DOI] [PubMed] [Google Scholar]
- 101.Pukrop T, Klemm F, Hagemann T, Gradl D, Schulz M, Siemes S, et al. Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc Natl Acad Sci U S A (2006) 103:5454–9. 10.1073/pnas.0509703103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gilles C, Bassuk JA, Pulyaeva H, Sage H, Foidart J-M, Thompson EW. SPARC/osteonectin induces matrix metalloproteinase 2 activation in human breast cancer cell lines. Cancer Res (1998) 58:5529–36. [PubMed] [Google Scholar]
- 103.Kitamura T, Qian B-Z, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med (2015) 212:1043–59. 10.1084/jem.20141836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mizutani K, Sud S, McGregor NA, Martinovski G, Rice BT, Craig MJ, et al. The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia (2009) 11:1235–42. 10.1593/neo.09988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Roodman G. Mechanisms of bone metastasis. N Engl J Med (2004) 350:1655–64. 10.1056/NEJMra030831 [DOI] [PubMed] [Google Scholar]
- 106.Lu Y, Cai Z, Xiao G, Keller E, Mizokami A, Yao Z, et al. Monocyte chemotactic protein-1 mediates prostate cancer-induced bone resorption. Cancer Res (2007) 67:3646–53. 10.1158/0008-5472.CAN-06-1210 [DOI] [PubMed] [Google Scholar]
- 107.Lu Y, Chen Q, Corey E, Xie W, Fan J, Mizokami A, et al. Activation of MCP-1/CCR2 axis promotes prostate cancer growth in bone. Clin Exp Metastasis (2008) 26:161–9. 10.1007/s10585-008-9226-7 [DOI] [PubMed] [Google Scholar]
- 108.Chen Q, Zhang X, Massague J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell (2011) 20:538–49. 10.1016/j.ccr.2011.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer (2013) 108:914–23. 10.1038/bjc.2013.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Di Caro G, Cortese N, Castino GF, Grizzi F, Gavazzi F, Ridolfi C, et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut (2016) 65:1710–20. 10.1136/gutjnl-2015-309193 [DOI] [PubMed] [Google Scholar]
- 111.Vogel DY, Glim JE, Stavenuiter AW, Breur M, Heijnen P, Amor S, et al. Human macrophage polarization in vitro: maturation and activation methods compared. Immunobiology (2014) 219:695–703. 10.1016/j.imbio.2014.05.002 [DOI] [PubMed] [Google Scholar]
- 112.Buchacher T, Ohradanova-Repic A, Stockinger H, Fischer MB, Weber V. M2 polarization of human macrophages favors survival of the intracellular pathogen Chlamydia pneumoniae. PLoS One (2015) 10:e0143593. 10.1371/journal.pone.0143593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Campbell MJ, Tonlaar NY, Garwood ER, Huo D, Moore DH, Khramtsov AI, et al. Proliferating macrophages associated with high grade, hormone receptor negative breast cancer and poor clinical outcome. Breast Cancer Res Treat (2011) 128:703–11. 10.1007/s10549-010-1154-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hanada T, Nakagawa M, Emoto A, Nomura T, Nasu N, Nomura Y. Prognostic value of tumor-associated macrophage count in human bladder cancer. Int J Urol (2000) 7:263–9. 10.1046/j.1442-2042.2000.00190.x [DOI] [PubMed] [Google Scholar]
- 115.Forssell J, Oberg A, Henriksson ML, Stenling R, Jung A, Palmqvist R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res (2007) 13:1472–9. 10.1158/1078-0432.CCR-06-2073 [DOI] [PubMed] [Google Scholar]
- 116.Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med (2010) 362:875–85. 10.1056/NEJMoa0905680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell (2013) 23:249–62. 10.1016/j.ccr.2013.01.008 [DOI] [PubMed] [Google Scholar]
- 118.Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjällman AHM, Ballmer-Hofer K, Schwendener RA. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br J Cancer (2006) 95:272–81. 10.1038/sj.bjc.6603240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011) 475:222–5. 10.1038/nature10138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang J, Patel L, Pienta K. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumourigenesis and metastasis. Cytokine Growth Factor Rev (2010) 21:41–8. 10.1016/j.cytogfr.2009.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wong D, Kandagatla P, Korz W, Chinni SR. Targeting CXCR4 with CTCE-9908 inhibits prostate tumor metastasis. BMC Urol (2014) 14:12–6. 10.1186/1471-2490-14-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Huang E, Singh B, Cristofanili M, Gelovani J, Wei C, Vincent L, et al. A CXCR4 antagonist CTCE-9908 inhibits primary tumor growth and metastasis of breast cancer. J Surg Res (2009) 155:231–6. 10.1016/j.jss.2008.06.044 [DOI] [PubMed] [Google Scholar]
- 123.Aharinejad S, Paulus P, Sioud M, Hofmann M, Zins K, Schäfer R, et al. Colony-stimulating factor-1 blockade by antisense oligonucleotides and small interfering RNAs suppresses growth of human mammary tumor xenografts in mice. Cancer Res (2004) 64:5378–84. 10.1158/0008-5472.CAN-04-0961 [DOI] [PubMed] [Google Scholar]
- 124.Paulus P, Stanley R, Schäfer R, Abraham D, Aharinejad S. Colony-stimulating factor-1 antibody reverses chemoresistance in human MCF-7 breast cancer xenografts. Cancer Res (2006) 66:4349–56. 10.1158/0008-5472.CAN-05-3523 [DOI] [PubMed] [Google Scholar]
- 125.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med (2013) 19:1264–72. 10.1038/nm.3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ahna G-O, Tsenga D, Liaoa C-H, Doriea MJ, Czechowiczb A, Browna JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci U S A (2010) 107:8363–8. 10.1073/pnas.0911378107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sinha P, Clements V, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol (2005) 174:636–45. 10.4049/jimmunol.174.2.636 [DOI] [PubMed] [Google Scholar]
- 128.Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res (2009) 69:2506–13. 10.1158/0008-5472.CAN-08-4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Edwards J, Emens L. The multikinase inhibitor sorafenib reverses the suppression of IL-12 and enhancement of IL-10 by PGE(2) in murine macrophages. Int Immunopharmacol (2010) 10:1220–8. 10.1016/j.intimp.2010.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, Liu Z-G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res (2013) 23:898–914. 10.1038/cr.2013.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Linde N, Lederle W, Depner S, Rooijen NV, Gutschalk CM, Mueller MM. Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J Pathol (2012) 227:17–28. 10.1002/path.3989 [DOI] [PubMed] [Google Scholar]
- 132.Na Y-R, Yoon Y-N, Son D-I, Seok S-H. Cyclooxygenase-2 inhibition blocks M2 macrophage differentiation and suppresses metastasis in murine breast cancer model. PLoS One (2013) 8:e63451. 10.1371/journal.pone.0063451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Palma MD, Mazzieri R, Politi LS, Pucci F, Zonari E, Sitia G, et al. Tumor-targeted Interferon-α delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell (2008) 14:299–311. 10.1016/j.ccr.2008.09.004 [DOI] [PubMed] [Google Scholar]
- 134.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (2011) 331:1614–6. 10.1126/science.1198443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell (2011) 19:31–44. 10.1016/j.ccr.2010.11.009 [DOI] [PubMed] [Google Scholar]
- 136.Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. “Re-educating” tumor-associated macrophages by targeting NF-κB. J Exp Med (2008) 205:1261–8. 10.1084/jem.20080108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.A Study of IMC-CS4 in Subjects with Advanced Solid Tumors. Available from: www.clinicaltrials.gov/show/NCT01346358
- 138.A Study of AMG 820 in Subjects with Advanced Solid Tumors. Available from: www.clinicaltrials.gov/show/NCT01444404
- 139.Phase 1 Study of PLX7486 as Single Agent and with Gemcitabine Plus Nab-Paclitaxel in Patients With Advanced Solid Tumors. Available from: https://clinicaltrials.gov/ct2/show/NCT01804530
- 140.Safety Study of PLX108-01 in Patients with Solid Tumors. Available from: https://clinicaltrials.gov/ct2/show/NCT01004861
- 141.S0916, MLN1202 in Treating Patients with Bone Metastases. Available from: https://clinicaltrials.gov/ct2/show/NCT01015560
- 142.FOLFIRINOX Plus PF-04136309 in Patients With Borderline Resectable and Locally Advanced Pancreatic Adenocarcinoma. Available from: https://clinicaltrials.gov/ct2/show/NCT01413022
- 143.Feasibility of the Combination of Chemotherapy (Carbo/Caelyx or Carbo/Doxorubicin) with Tocilizumab (mAb IL-6R) and Peg-Intron in Patients with Recurrent Ovarian Cancer (PITCH). Available from: https://clinicaltrials.gov/ct2/show/NCT01637532
- 144.A Study of Trabectedin (YONDELIS) in Patients with Locally Advanced or Metastatic Liposarcoma or Leiomyosarcoma. Available from: https://clinicaltrials.gov/ct2/show/NCT01692678
- 145.Study with Trabectedin in BRCA1 and BRCA2 Mutation Carrier and BRCAness Phenotype Ovarian Cancer (MITO15). Available from: https://clinicaltrials.gov/ct2/show/NCT01772979
- 146.Combination Therapy of Gemcitabine and Trabectedin in L-Sarcomas. Available from: https://clinicaltrials.gov/ct2/show/NCT01426633
- 147.Combination Immunotherapy of GM.CD40L Vaccine with CCL21 in Lung Cancer. Available from: https://clinicaltrials.gov/ct2/show/NCT01433172
- 148.Tremelimumab and CP-870,893 in Patients with Metastatic Melanoma. Available from: https://clinicaltrials.gov/ct2/show/NCT01103635
- 149.A Phase I/Ib Study of AZD9150 (ISIS-STAT3Rx) in Patients with Advanced/Metastatic Hepatocellular Carcinoma. Available from: https://clinicaltrials.gov/ct2/show/NCT01839604
- 150.Fritz JM, Tennis MA, Orlicky DJ, Yin H, Ju C, Redente EF, et al. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front Immunol (2014) 5:587. 10.3389/fimmu.2014.00587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Liguori M, Buracchi C, Pasqualini F, Bergomas F, Pesce S, Sironi M, et al. Functional TRAIL receptors in monocytes and tumor-associated macrophages: a possible targeting pathway in the tumor microenvironment. Oncotarget (2016) 7:41662–76. 10.18632/oncotarget.9340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Junankar S, Shay G, Jurczyluk J, Ali N, Down J, Pocock N, et al. Real-time intravital imaging establishes tumor-associated macrophages as the extraskeletal target of bisphosphonate action in cancer. Cancer Discov (2015) 5:35–42. 10.1158/2159-8290.CD-14-0621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Van Acker HH, Anguille S, Willemen Y, Smits EL, Van Tendeloo VF. Bisphosphonates for cancer treatment: mechanisms of action and lessons from clinical trials. Pharmacol Ther (2016) 158:24–40. 10.1016/j.pharmthera.2015.11.008 [DOI] [PubMed] [Google Scholar]
- 154.Teng K-Y, Han J, Zhang X, Hsu S-H, He S, Wani N, et al. Blocking the CCL2-CCR2 axis using CCL2 neutralizing antibody is an effective therapy for hepatocellular cancer in a mouse model. Mol Cancer Ther (2017) 16:312–22. 10.1158/1535-7163.MCT-16-0124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Arakaki R, Yamasaki T, Kanno T, Shibasaki N, Sakamoto H, Utsunomiya N, et al. CCL2 as a potential therapeutic target for clear cell renal cell carcinoma. Cancer Med (2016) 5:2920–33. 10.1002/cam4.886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature (2014) 515:130–3. 10.1038/nature13862 [DOI] [PubMed] [Google Scholar]
- 157.Loberg R, Ying C, Crag M, Day L, Sargent S, Neeley C, et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumour regression in vivo. Cancer Res (2007) 67:9417–24. 10.1158/0008-5472.CAN-07-1286 [DOI] [PubMed] [Google Scholar]
- 158.Brana I, Calles A, LoRusso PM, Yee LK, Puchalski TA, Seetharam S, et al. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol (2015) 10:111–23. 10.1007/s11523-014-0320-2 [DOI] [PubMed] [Google Scholar]
- 159.Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol (2016) 17:651–62. 10.1016/S1470-2045(16)00078-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Manthey CL, Johnson DL, Illig CR, Tuman RW, Zhou Z, Baker JF, et al. JNJ-28312141, a novel orally active colony-stimulating factor-1 receptor/FMS-related receptor tyrosine kinase-3 receptor tyrosine kinase inhibitor with potential utility in solid tumors, bone metastases, and acute myeloid leukemia. Mol Cancer Ther (2009) 8:3151–61. 10.1158/1535-7163.MCT-09-0255 [DOI] [PubMed] [Google Scholar]
- 161.Zhang Q-Q, Hu X-W, Liu Y-L, Ye Z-J, Gui Y-H, Zhou D-L, et al. CD11b deficiency suppresses intestinal tumor growth by reducing myeloid cell recruitment. Sci Rep (2015) 5:15948. 10.1038/srep15948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Richardsen E, Uglehus RD, Johnsen SH, Busund LT. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. Anticancer Res (2015) 35:865–74. [PubMed] [Google Scholar]
- 163.Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell (2014) 25:846–59. 10.1016/j.ccr.2014.05.016 [DOI] [PubMed] [Google Scholar]
- 164.Formenti SC, Demaria S. Systemic effects of local radiotherapy. Lancet Oncol (2009) 10:718–26. 10.1016/S1470-2045(09)70082-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Stafford JH, Hirai T, Deng L, Chernikova SB, Urata K, West BL, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol (2016) 18:797–806. 10.1093/neuonc/nov272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res (2014) 74:153–61. 10.1158/0008-5472.CAN-13-1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov (2011) 1(1):54–67. 10.1158/2159-8274.CD-10-0028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Weizman N, Krelin Y, Shabtay-Orbach A, Amit M, Binenbaum Y, Wong RJ, et al. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene (2014) 33:3812–9. 10.1038/onc.2013.357 [DOI] [PubMed] [Google Scholar]
- 169.Hussain SF, Kong L-Y, Jordan J, Conrad C, Madden T, Fokt I, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res (2007) 67:9630–6. 10.1158/0008-5472.CAN-07-1243 [DOI] [PubMed] [Google Scholar]
- 170.Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res (2013) 19:6286–95. 10.1158/1078-0432.CCR-13-1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Castro BA, Flanigan P, Jahangiri A, Hoffman D, Chen W, Kuang R, et al. Macrophage migration inhibitory factor downregulation: a novel mechanism of resistance to anti-angiogenic therapy. Oncogene (2017) 36:3749–59. 10.1038/onc.2017.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol (2013) 15:1079–87. 10.1093/neuonc/not082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.de Groot JF, Piao Y, Tran H, Gilbert M, Wu H-K, Liu J, et al. Myeloid biomarkers associated with glioblastoma response to anti-vascular endothelial growth factor therapy with aflibercept. Clin Cancer Res (2011) 17:4872–81. 10.1158/1078-0432.CCR-11-0271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell (2008) 13:206–20. 10.1016/j.ccr.2008.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sathornsumetee S, Cao Y, Marcello JE, Herndon JE, II, McLendon RE, Desjardins A, et al. Tumor angiogenic and hypoxic profiles predict radiographic response and survival in malignant astrocytoma patients treated with bevacizumab and irinotecan. J Clin Oncol (2008) 26:271–8. 10.1200/JCO.2007.13.3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kloepper J, Riedemann L, Amoozgar Z, Seano G, Susek K, Yu V, et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc Natl Acad Sci U S A (2016) 113:4476–81. 10.1073/pnas.1525360113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Peterson TE, Kirkpatrick ND, Huang Y, Farrar CT, Marijt KA, Kloepper J, et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc Natl Acad Sci U S A (2016) 113:4470–5. 10.1073/pnas.1525349113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med (2017) 9:eaak9670. 10.1126/scitranslmed.aak9670 [DOI] [PubMed] [Google Scholar]
- 179.Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A (2015) 112:6140–5. 10.1073/pnas.1417320112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545:495–9. 10.1038/nature22396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Loke PN, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A (2003) 100:5336–41. 10.1073/pnas.0931259100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211:781–90. 10.1084/jem.20131916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med (2013) 210:1695–710. 10.1084/jem.20130579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol (1981) 11:805–15. 10.1002/eji.1830111013 [DOI] [PubMed] [Google Scholar]
- 185.Ruffell B, Coussens Lisa M. Macrophages and therapeutic resistance in cancer. Cancer Cell (2015) 27:462–72. 10.1016/j.ccell.2015.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. 10.1146/annurev-immunol-032712-100008 [DOI] [PubMed] [Google Scholar]
- 187.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res (2014) 74:5057–69. 10.1158/0008-5472.CAN-13-3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Malesci A, Bianchi P, Celesti G, Basso G, Marchesi F, Grizzi F, et al. Tumor-associated macrophages and response to 5-fluorouracil adjuvant therapy in stage III colorectal cancer. Oncoimmunology (2017) 6:e1342918. 10.1080/2162402X.2017.1342918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sugimura K, Miyata H, Tanaka K, Takahashi T, Kurokawa Y, Yamasaki M, et al. High infiltration of tumor-associated macrophages is associated with a poor response to chemotherapy and poor prognosis of patients undergoing neoadjuvant chemotherapy for esophageal cancer. J Surg Oncol (2015) 111:752–9. 10.1002/jso.23881 [DOI] [PubMed] [Google Scholar]
- 190.Lee B, Qiao L, Kinney B, Feng GS, Shao J. Macrophage depletion disrupts immune balance and energy homeostasis. PLoS One (2014) 9:e99575. 10.1371/journal.pone.0099575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int (2006) 26:1175–86. 10.1111/j.1478-3231.2006.01342.x [DOI] [PubMed] [Google Scholar]
- 192.Wright JR. Clearance and recycling of pulmonary surfactant. Am J Physiol (1990) 259:L1–12. [DOI] [PubMed] [Google Scholar]
- 193.Golbar HM, Izawa T, Wijesundera KK, Bondoc A, Tennakoon AH, Kuwamura M, et al. Depletion of hepatic macrophages aggravates liver lesions induced in rats by thioacetamide (TAA). Toxicol Pathol (2016) 44:246–58. 10.1177/0192623315621191 [DOI] [PubMed] [Google Scholar]
- 194.Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, et al. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog (2014) 10:e1004053. 10.1371/journal.ppat.1004053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer (2006) 6:714. 10.1038/nrc1913 [DOI] [PubMed] [Google Scholar]