

Abstract
The fruit fly, Drosophila melanogaster, has been a favorite experimental system of developmental biologists for more than a century. One of the most attractive features of this model system is the clarity by which one can analyze mutant phenotypes. Most genes are found in single copies and loss-of-function mutants have obvious phenotypes that can be analyzed during development and in adulthood. A significant fraction of Drosophila genes are used during both embryonic and post-embryonic development. Null mutants of these genes often die during embryogenesis precluding the analysis of post-embryonic tissues. For several decades researchers worked around this problem by either studying gynandromorphs or irradiating chromosomes carrying mutations in the hope of inducing mitotic recombination which would then allow for the analysis of mutant phenotypes in smaller populations of cells. However, clonal induction with the latter method occurs at relatively low frequencies making this method laborious. The introduction of the yeast FLP/FRT site directed recombination system to Drosophila made generating loss-of-function mosaic clones simple and easy. Over the years several variants of this method have allowed for developmental biologists to remove genes, over-express genes, and even express one gene in patches of cells that are mutant for a second gene. In this review we will briefly discuss some of various FLP/FRT based approaches that are being used to manipulate gene expression in Drosophila. The individual FLP/FRT based methods are described in the papers that are cited herein. We will outline the procedure that our lab uses to prepare and analyze mosaic clones in Drosophila eye-antennal imaginal discs.
Keywords: site specific recombinase, FLP/FRT, Drosophila, imaginal discs, gene expression, mosaic clones
Introduction
Genetic mosaics of have been used to study development in Drosophila for just about a century. The first mosaics to seen and described in flies were gynandromorphs, organisms that contain both male and female tissue (Morgan, 1914; Morgan and Bridges, 1919). It was immediately recognized that such mosaic animals could be used to study normal fly development. In particular, there was interest in tracing the developmental history of adult structures back through the imaginal discs and onto the embryo. Alfred Sturtevant took the first deliberate steps to generate gynandromorphs by using the loss of unstable X chromosomes in somatic tissue to generate organisms consisting of both male and female genotypes (Sturtevant, 1929). Using externally visible markers such as bristle shape and body pigments, he and others generated developmental fate maps in which the origin of individual adult structures such as the eyes, antennae, legs, and wings could be traced with a fair degree of precision back to regions of the embryonic blastoderm (Baker, 1978; Garcia-Bellido and Merriam, 1969; Parks, 1936; Patterson and Stone, 1938; Ripoll, 1972; Sturtevant, 1929). The use of this genetic system replaced other efforts to trace development such as cell ablation and histological analysis (Geigy, 1931; Howland and Child, 1935; Lohs-Schardin et al., 1979a; Lohs-Schardin et al., 1979b; Poulson, 1950). Other uses for gynandromorphs included the determination of where genes were acting. For example, gynandromorphs were used to show that the visual system defects associated with several mutants were due to the loss of the gene of interest within the compound eyes (Hotta and Benzer, 1970).
Although gynandromorphs were a popular tool for developmental studies, the discovery that mitotic recombination takes place between homologous chromosomes provided fly developmental biologists with any even more powerful method of generating mosaics (Stern, 1936). Flies heterozygous for an individual mutation could be rendered mosaic if a mitotic recombination event occurred within a cell at some point in development. While most cells within the animal will remain heterozygous, there will be patches of cells that are homozygous for the wild type chromosome and other patches that are homozygous for the mutant chromosome. These latter two cell populations are the products of mitotic recombination events. If the mutation in question had no other affect than to alter bristle morphology or body pigment then resulting mosaics could be (and were) used to study basic developmental questions such as how many cells are set aside during embryogenesis to give rise to each tissue type; when does cell proliferation within each tissue begin; what is the rate of cell proliferation within a given tissue; and when is each tissue committed to its final fate (Cohen, 1993).
If a mutation affects tissue growth or cell fate decisions then mosaic clones could be used to study the effect that an individual gene has on development. Extremely large sections of the gynandromorph body are mosaic because the unstable chromosomes are lost during the earliest embryonic divisions (Sturtevant, 1929). Therefore genes essential for embryonic development often could not be studied within later developmental contexts. In contrast, mitotic recombination events are not restricted to the early embryonic divisions thus genes essential for early development could be studied in post-embryonic tissues (if the recombination event took place after embryogenesis is complete). One serious drawback to this method was the rarity with which recombination events occurred spontaneously. Even with ionizing radiation, which increased the frequency of mitotic recombination events to approximately 1% (Friesen, 1936; Patterson, 1929), this was still a fairly laborious way to study development and gene function. Furthermore reductions in viability and fertility, increased levels of apoptosis, and decreases in cell proliferation were some of the side effects of treating flies with radiation. These posed additional challenges to studying the role that genes played in tissue growth and development.
The introduction of the yeast FLP (flippase recombinase) - FRT (FLP recombination target) system to flies eliminated these problems and revolutionized the use of mosaics to study development. The first demonstration that directed recombination could be achieved at high frequency came when a transgene carrying the white (w) pigment gene flanked with FRT elements was excised from the genome by the FLP enzyme. The resulting compound eyes contained patches of red (no excision) and white (excision) ommatidia (Golic and Lindquist, 1989). Subsequent efforts proved that the FLP/FRT system could be used to induce recombination between homologous chromosomes and that it could take place within both somatic and germline tissues (Chou and Perrimon, 1992; Golic, 1991). These papers showed that the FLP/FRT system could be used to generate mosaics at high frequencies and without the unwanted side effects of radiation. FRT sequences were then placed near the base of five out of the six chromosome arms and this allowed for the analysis of virtually the entire fly genome (Xu and Rubin, 1993, 2012). All that was required was for the mutation of interest to be brought onto a FRT bearing chromosome by meiotic recombination.
The original system had expression of FLP under the control of a heat inducible promoter (hsp70). As a result mutant clones were generated throughout the entire animal and that often would lead to early embryonic lethality or other unwanted developmental defects in tissues that were not relevant to the the particular. Unfortunately, these were some of the same issues that plagued the use of X-ray induced mitotic recombination. To limit mosaic clone induction to desired temporal windows and spatial locations, transcription of FLP was placed under the control of genomic enhancers that drive expression in very specific patterns (Duffy et al., 1998). FLP can be placed under direct control of a regulatory element (Newsome et al., 2000) or it can be indirectly coupled to the genomic enhancer using the UAS/GAL4 system (Brand and Perrimon, 1993; Duffy et al., 1998). Mosaic clones could now be induced specifically in any tissue or population of cells for which there is an appropriate genomic enhancer.
Over the last quarter century the original FLP/FRT system has been modified numerous times. Each modification has dramatically increased the versatility of the system - some examples are listed within Table 1. In addition to being able to analyze loss-of-function clones we can now use the FLP/FRT recombination system to conduct F1 screens for growth mutants (Kango-Singh et al., 2002; Tapon et al., 2001; Wu et al., 2003); to generate entire tissues/organs that are homozygous for an individual loss-of-function mutation (Newsome et al., 2000); to over-express transgenes within patches of cells (Ito et al., 1997; Pignoni and Zipursky, 1997; Struhl and Basler, 1993); to examine the morphology of an individual cell and its descendants (Lee and Luo, 1999); to simultaneously lineage trace multiple cell populations within a single tissue (Bosch et al., 2015; Hadjieconomou et al., 2011; Worley et al., 2013; Yu et al., 2009); to determine epistatic relationships between genes (Lee and Luo, 1999); to compare two different mutant cell populations to each other within a single tissue (Bosch et al., 2015); as well as to trace and concurrently analyze the historical and real-time expression patterns of genomic enhancers (Evans et al., 2009).
Table 1.
A list of FLP/FRT systems and their applications for studying fly development.
| Genetic Purpose | Reference | System Name |
|---|---|---|
| loss-of-function clones | Golic and Lindquist, 1989 | |
| Golic, 1991 | ||
| Chou and Perrimon, 1992 | ||
| Xu and Rubin, 1993 | ||
|
| ||
| directed mosaics | Duffy et al., 1998 | |
|
| ||
| induction of whole mutant tissues | Newsome et al., 2000 | |
|
| ||
| FLP/FRT F1 screens | Tapon et al., 2001 | |
| Kango-Singh et al., 2002 | ||
| Wu et al., 2003 | ||
|
| ||
| FLP-out over-expression clones | Stuhl and Basler, 1993 | |
| Ito et al., 1997 | ||
| Pignoni and Zipursky, 1997 | ||
|
| ||
| lineage tracing | Lee and Luo, 1999 | MARCM |
| Yu et al., 2009 | twin spot MARCM | |
| Evans et al., 2009 | G-trace | |
| Hadjieconomou et al., 2011 | Flybow | |
| Worley et al., 2013 | TIE-DYE | |
|
| ||
| FLP/FRT RNAi screens & two mutant clone comparison | Bosch et al., 2015 | COIN-FLP |
In many instances the modified FLP/FRT based recombination systems that are described in the above papers have been used to re-examine questions that were once addressed by earlier generations of developmental biologists with either gynandromorphs or with irradiation induced mosaic clones. For example, the TIE-DYE system has been applied to an old question: how many cells are set aside during embryogenesis for the formation of the wing disc and approximately how many cell doublings are necessary for the formation of the adult wing (Worley et al., 2013). The estimates from TIE-DYE suggest that the wing disc is derived from fewer than expected founder cells and as a consequence the generation of a full-sized adult wing requires an additional cell doubling.
Other FLP/FRT systems have allowed fly developmental biologists to address questions that could never have been answered with either gynandromorphs or standard mosaic clone analysis. For example, the COIN-FLP system allows for two mutant populations of cells to be generated and compared within a single tissue (Bosch et al., 2015). The G-trace method is used to simultaneously compare the real-time expression pattern of a gene with a composite of its expression history (Evans et al., 2009). Several methods such as MARCM, twin-spot MARCM, TIE-DYE, and Flybow can be used to trace the lineage of cells thereby essentially replacing earlier efforts that used actual physical dyes (Hadjieconomou et al., 2011; Lee and Luo, 1999; Worley et al., 2013; Yu et al., 2009). Lastly, rather sophisticated epistatic analysis can be conducted using MARCM (and its derivatives) – a gene can be expressed within a population of cells that are mutant for a second gene. The development of the mutant clone itself can be analyzed as can the expression of additional downstream genes (Lee and Luo, 1999, 2001).
Each of the above FLP/FRT based recombination systems has been used to generate mosaics in numerous distinct tissues such as imaginal discs, larval and adult brains, as well as germline cells. Our lab uses the following protocol to examine mosaic clones within Drosophila eye-antennal imaginal discs. It can be easily adapted for the analysis of other imaginal discs as well as for other larval tissues such as the brain, salivary glands, fat bodies and gut.
Materials
List of Required Materials
Sylgard 184 Silicone Elastomer Kit and Pyrex Glass Petri Dish 150 x 20mm
#5 Dissecting Forceps
9-well watch glass
50ml Erlenmeyer Flasks
50ml Conical Tubes
1.5ml Microfuge Tubes
Paraformaldehyde
Sodium Phosphate Monobasic and Sodium Phosphate Dibasic
Lysine
Sodium Periodate
Triton X-100
Sodium Hydroxide
100% Normal Goat Serum
Primary and Secondary Antibodies
Vectashield
Microscope Slides and Glass Cover Slips
Kimwipe Tissue
Paint Brush 000
List of Required Solutions
8% Paraformaldehyde
0.2M Sodium Phosphate Monobasic and 0.2M Sodium Phosphate Disbasic
1N Sodium Hydroxide
10% Triton
Distilled Water
0.1M Sodium Phosphate Buffer (Dissection Buffer)
0.1M Sodium Phosphate Buffer + 0.1% Triton (Wash Buffer)
Lysine Buffer
2% Paraformaldehyde-Lysine-Periodate Fixative (PLP)
10% Normal Goat Serum
Preparation of 8% Paraformaldehyde
To a 50ml Erlenmeyer flask add the following: 2.0 g paraformaldehyde, 23.0ml of distilled water and 4.0 drops of 1N sodium hydroxide (from a glass pasteur pipet). Mix and heat on a stir plate until solution reaches a gentle boil. Allow to gently boil until paraformaldehyde is completely dissolved. Place on ice until cold (make fresh prior to each dissection)
Preparation of 0.1M Phosphate Buffer (Dissection (P) Buffer)
To a 50ml conical tube add the following: 18.0ml 0.2M Sodium Phosphate dibasic, 7.0ml 0.2M Sodium Phosphate monobasic, and 25.0ml distilled water. Store at 4°C.
Preparation of 0.1M Phosphate + Detergent Buffer (Wash (W) Buffer)
To a 50ml conical tube add the following: 18.0ml 0.2M Sodium Phosphate dibasic, 7.0ml 0.2M Sodium Phosphate monobasic, 25.0ml distilled water, and 0.5ml 10% Triton. Store at room temperature.
Preparation of Lysine (L) Buffer
To a 50ml conical tube add the following: 0.36g Lysine, 1.2ml 0.2M Sodium Phosphate dibasic, 8.0ml Dissection (P) Buffer, and 10.0ml distilled water. Vortex solution until lysine is completely dissolved. Place on ice until cold (make fresh prior to each dissection).
Preparation of 2% Paraformaldehyde/Lysine/Periodate (PLP) fixative
To a 50ml conical tube add the following: 50mg Sodium Periodate, 15.0ml of Lysine (L) buffer, and 5.0ml of 8% Paraformaldehyde. Vortex solution well until sodium periodate is completely dissolved. Place on ice until cold (make fresh prior to each dissection).
Methods
Preparation and coarse dissection of eye-antennal discs
Fill a 35mm Petri dish with dissection buffer. Place 20–30 larvae in the Petri dish and allow them to swim around for a few minutes (self-cleaning step). Transfer the larvae to a pool of dissection buffer on a silicone-based dissection plate. This pool should be at one edge of the plate. The dissection plate consists of a silicone solution that has been poured and hardened within a glass Petri dish. Using a pasteur-pipet, place a larger pool of dissection buffer in the middle of the dissection plate. Make sure that you have pre-chilled the P buffer by placing it on ice for 30min prior to beginning dissections. The cooler temperatures prevent the larvae from squirming during the dissection process.
Using #5 forceps, transfer a single cleaned larva from the small pool to the larger pool of dissection buffer. While the larva is still within the large pool of dissection buffer, clasp the larva with the forceps. One pair of forceps should be used to grab the mouth hooks while the other pair of forceps is used to hold the animal still (grab the larva gently at 1/3 body length). Hold steady the pair of forceps containing the cuticle near the mouth hooks while quickly pulling the rest of the body away with the second pair of forceps. When the larva begins to tear apart you will feel a slight release in tension. Release the larva from the forceps and allow for the “guts” of the larva to spill out. This allows for the imaginal discs to remain in their normal conformation and prevents them from being deformed.
With one pair of forceps grasp the mouth hooks again to hold the front end of the larva in place. Using the other pair of forceps remove the lower 2/3 of the larvae including the inner guts. A complex containing the mouth hooks, the eye-antennal discs, brain hemispheres, ventral ganglion, the salivary glands, some leg discs, and the overlying cuticle will remain. With a pair of forceps gently remove overlying cuticle, salivary glands, leg discs and other tissue. The only tissues that should remain are the mouth hooks, eye-antennal discs, brain hemispheres, and ventral ganglion.
Repeat the above steps with additional larvae for 15–20min. Imaginal discs that remain in dissection buffer for longer periods of time tend to degrade and ultimately will appear as less than ideal specimens to be photographed. Thus, after a maximum of 20min all dissected tissues should be transferred to the PLP fixative. If you are interested in other imaginal discs and/or other larval tissues you can simply retrieve them at this stage and transfer them to the PLP fixative.
Fixation and staining of tissue with antibodies
Using a P-200 pipetman transfer the dissected tissues to a watch glass containing cold PLP fixative. Limit the volume of transferred dissection buffer to under 50μl to minimize dilution of the PLP. Be sure to cut off the end of the yellow tip with a razor blade so that the tip opening is large enough to accommodate the dissected tissues. Using a pair of forceps or a tungsten needle ensure that the dissected tissues are completely submerged in order for proper fixation to occur. Incubate dissected tissues in cold PLP fixative for 45min. This incubation can take place at room temperature without agitation.
Using a P-200 pipetman transfer the dissected tissues to wash buffer using another cut yellow tip for 45min. Limit the volume of transferred PLP to under 50μl to minimize dilution of the wash buffer. All dissected tissues should be completely submerged. Transfer 20–30 sets of dissected tissue to a 1.5ml microfuge tube. For optimal results no more than 20–30 eye-antennal disc complexes should be present within a single tube. If larger numbers of eye-antennal disc complexes are combined within the tube, there is a possibility that the tissue at the bottom of the tube will not be exposed properly to the antibodies. The dissected tissue will settle to the bottom of the microfuge tube. In subsequent steps use a pipetman to remove and replace wash buffer, blocking solution, and antibodies.
If your mosaic clones are marked with fluorescent markers such as GFP or RFP you can directly visualize these molecules with a fluorescent microscope and therefore you do not need to use antibodies. If this is the case then you can proceed directly to the fine dissection steps (below). However, if your microscope is not fitted with the appropriate filters for direct detection of these molecules or if you are using non-fluorescent markers such as lacZ, then you will need to follow the steps listed below for using primary and secondary antibodies.
Remove the wash buffer and replace with 100μl of blocking solution: 10% normal goat serum in wash buffer. Incubate at room temperature with gentle rotation on a table top rotator for 10min. Remove the blocking solution and replace with 100μl of primary antibodies that have been appropriately diluted in 10% normal goat serum. Incubate at room temperature with gentle rotation for 12–16hr. Remove the primary antibodies and replace with 750μl of wash buffer. Place tubes on a nutator and allow to nutate at room temperature for 10min. The primary antibody can be saved (store at 4°C) and reused later. Reusing antibodies multiple times can help in reducing non-specific binding. Please note that all antibodies cannot be reused – many will break down over time so monitor the quality of the images that you recover over time to determine if the antibody in question can be reused.
Allow heads to settle to the bottom of the tube, then remove wash buffer and add 100μl of secondary antibodies that have been appropriately diluted in 10% normal goat serum. Incubate at room temperature with gentle rotation for 2–4hrs. Remove the secondary antibody solutions from the tissue and replace with 500μl of wash buffer. Allow tissue to settle to the bottom of the tube.
Using a P-200 and a cut yellow tip, transfer all dissected tissues to a pool of wash buffer that has been placed on the dissecting dish. While proceeding with the next step of fine dissection, the tissue will incubate in the wash buffer. This helps to remove excess secondary antibodies.
Fine dissection of eye-antennal discs and mounting onto slides
Use one pair of forceps to clasp the cuticle by the mouth hooks. With a second pair of forceps remove the two brain lobes by closing the second forcep in the space between the brain and eye discs and swiftly pulling the brain away from the mouth hooks. While continuing to hold onto the mouth hooks, pinch off the tissue as close as possible to the connection between the antennal section of the eye-antennal disc and the mouth hooks. The eye-antennal disc complexes should be free of all tissue. Continue until all desired tissue that can be placed onto slides has been dissected. Similar procedures can be used to isolate and clean wing, haltere, leg and genital imaginal discs.
Add two small pieces of tissue paper (size of coverslips) approximately 3 inches apart on the dissecting dish. Place a glass slide onto the dish - the two pieces of tissue paper should be under the ends of the slide. This will prevent the slide from sticking to the silicone base of the dissecting dish. Using a P-20 with an uncut tip, add 9μl of an anti-bleaching agent to the middle of the glass microscope slide. This prevents bleaching of the tissue when viewed with fluorescent light. Using the same uncut tip, gather all eye-antennal discs and add them to the drop of anti-bleaching reagent. Minimize the amount of wash buffer that is carried over. Try to limit the amount of wash buffer to 10μl or less. Use a pair of forceps to separate and spread out the eye-antennal discs within the drop of anti-bleaching reagent.
Allow discs to incubate in anti-bleaching reagent for several minutes. The imaginal discs will turn clear as the tissue absorbs the anti-bleaching reagent. Using a fine paintbrush or your fingers gently lower a coverslip onto the specimen. Slowly lowering the coverslip onto the bubble of anti-bleaching reagent will prevent the formation of air bubbles. Store slides at −20°C until ready to view the eye-antennal discs using fluorescent microscopy.
Photography and image processing
Adjust the microscope for the appropriate magnification. A 10X lens is sufficient for viewing the entire eye-antennal disc - it is also appropriate for leg, wing, haltere, and genital discs. It is easiest if the eye-antennal disc is oriented in the 9 to 3 positions on a clock where the optic stalk is positioned to the left (9 o’clock) and the maxillary palp is positioned to the right (3 o’clock). You can simple rotate the stage until the eye-antennal disc is in the correct orientation. Bring the disc into focus by ensuring that the edge of the eye-antennal disc, the morphogenetic furrow, and the edges of the tissue folds within the antennal field are sharp.
Adjust the brightness/contrast and exposure times so that the non-disc parts of the screen are jet-black and the brightest portion of the stains are just below saturating levels. Photograph each visualized molecule separately and then combine your images into a merged file using your favorite image software such as Adobe Photoshop. When presenting your images be sure to orient the disc so that the anterior is to the right and dorsal is up. If you follow the above instructions for orienting the disc on the microscope you will have already place the disc in the correct anterior-posterior orientation. To determine the dorsal/ventral axis you should focus on the antennal disc, which is made up of several concentric folds of tissue. The central ring is not located with the exact middle of the disc. Instead it is positioned close to the ventral edge of the tissue. Orient your disc so that the ventral side is down and the dorsal side is up. You can adjust the A/P and D/V axes either on the microscope or on your computer.
An example of a loss-of-function clone is provided in Figure 1. The mutant clones are marked by the absence of GFP. In addition, an example of a FLP-out over-expression clones is provided in Figure 2. The over-expression clones are marked by the presence of GFP. Additional examples of loss-of-function clones, FLP-out clones, and MARCM clones that were generated with this protocol can be found within the following publications: (Anderson et al., 2012; Spratford and Kumar, 2013, 2015).
Figure 1.

An example of FLP/FRT mediated loss-of-function clones. (A–D) Light microscope images of a third larval instar eye-antennal disc containing dac4 loss-of-function clones. The clones are marked by the absence of GFP. Visualized molecules are listed within the figure. Anterior is to the right.
Figure 2.
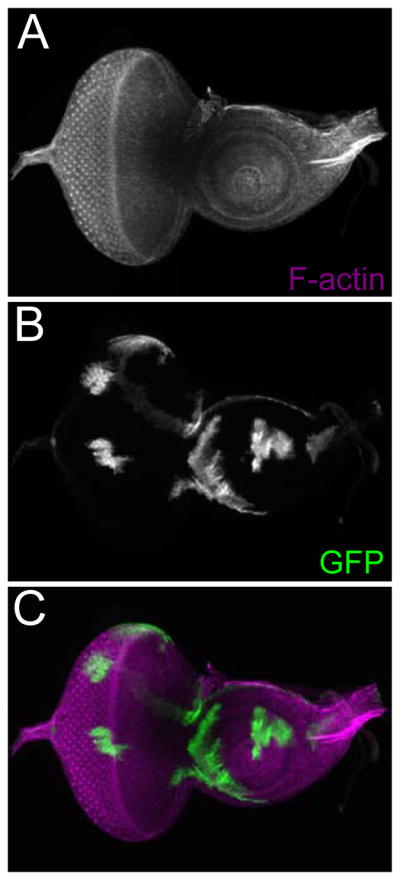
An example of FLP/FRT mediated over-expression clones. (A–C) Light microscope images of a third larval instar eye-antennal disc containing neutral over-expression clones. The clones are marked by the presence of GFP. Visualized molecules are listed within the figure. Anterior is to the right.
Notes
The papers that are cited within this article provide outstanding descriptions of each FLP/FRT recombination system. The required stocks are available from the Bloomington Drosophila Stock Collection or from the laboratory that generated them. Each of these methods has been used and vetted by numerous laboratories. Thus, the key to visually optimizing your results is to address potential issues in the dissection, fixation, and staining aspects of this protocol.
This procedure provides the best results when used on tissues of limited thickness such as imaginal discs that are monolayer epithelia. However, this method can be used with thicker tissues such as the brain and gonads. In such cases either increasing the concentration of paraformaldehyde or incubating the tissue in the PLP fixative for longer time periods may be necessary. The exact fixative concentration and/or time spent in PLP will depend on the particular tissue. Another way of preserving the tissue is to dissect for shorter periods of time and transfer the thicker tissue to the PLP fixative as soon as possible. You can assess the quality of tissue fixation by treating your tissue with fluorescently labeled phalloidin, a molecule that is found in mushrooms and binds to strongly to cortical F-actin. A well-preserved tissue is one in which the phalloidin stain (outlines of individual cells and the overall tissue) are extremely sharp.
Another potential pitfall is the buffers and fixatives themselves. In our hands, a wide-range of primary antibodies work well with the buffers and fixatives described in this protocol. However, there are examples of antibodies that do not – one such example is a monoclonal antibody that recognizes the Drosophila Rough (Ro) protein. This antibody works best with a PIPES-EGTA-MgSO4 (PEM) buffer (Dokucu et al., 1996). If you encounter an antibody that does not work with the buffers and fixatives listed here, then you should try other fixatives and buffers starting with, but limited to, PEM.
The working concentration of the antibodies being used may also have to be adjusted. Antibodies can work at a wide range of concentrations. We routinely use antibodies that must be used at high working concentrations (1:5) but have also encountered antibodies that can be used at quite dilute concentrations (1:3000). It should be noted that the same antibody may need to be used at different concentrations in different tissues. We have encountered situations in which a single antibody is used at one concentration in embryos but at an entirely different concentration within the imaginal discs. It is also been our experience that the recommended working concentration can vary from lab to lab even if you are working with the same tissue. This may be because the buffers and fixatives that are used in each lab may vary. We suggest that you start with the recommend working dilution and then increase or decrease the concentration as necessary.
Some antibodies display non-specific binding and this can manifest itself as a high level of background staining within the tissue. You can reduce this by several methods. One possible avenue is to either reduce the working concentration of the primary or secondary antibodies. You can also pre-adsorb your antibody against larval or embryonic carcasses. Lastly, you can try to affinity purify your antibody with the appropriate affinity columns.
Acknowledgments
We would like to thank Donald Ready and Kevin Moses for the original protocol for the dissection of eye-antennal imaginal discs. This work is supported by a grant from the National Eye Institute (R01 EY014863) to Justin P. Kumar.
Literature Cited
- Anderson AM, Weasner BM, Weasner BP, Kumar JP. Dual transcriptional activities of SIX proteins define their roles in normal and ectopic eye development. Development. 2012;139:991–1000. doi: 10.1242/dev.077255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker WK. A fine-structure gynandromorph fate map of the Drosophila head. Genetics. 1978;88:743–754. doi: 10.1093/genetics/88.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch JA, Tran NH, Hariharan IK. CoinFLP: a system for efficient mosaic screening and for visualizing clonal boundaries in Drosophila. Development. 2015;142:597–606. doi: 10.1242/dev.114603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Chou TB, Perrimon N. Use of a yeast site-specific recombinase to produce female germline chimeras in Drosophila. Genetics. 1992;131:643–653. doi: 10.1093/genetics/131.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SM. In: Imaginal disc development. Bate M, Martinez Arias A, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1993. pp. 747–841. [Google Scholar]
- Dokucu ME, Zipursky SL, Cagan RL. Atonal, rough and the resolution of proneural clusters in the developing Drosophila retina. Development. 1996;122:4139–4147. doi: 10.1242/dev.122.12.4139. [DOI] [PubMed] [Google Scholar]
- Duffy JB, Harrison DA, Perrimon N. Identifying loci required for follicular patterning using directed mosaics. Development. 1998;125:2263–2271. doi: 10.1242/dev.125.12.2263. [DOI] [PubMed] [Google Scholar]
- Evans CJ, Olson JM, Ngo KT, Kim E, Lee NE, Kuoy E, Patananan AN, Sitz D, Tran P, Do MT, et al. G-TRACE: rapid Gal4-based cell lineage analysis in Drosophila. Nature methods. 2009;6:603–605. doi: 10.1038/nmeth.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen H. Spermatogoniales Crossing-over bei Drosophila. Z Indukt Abstammungs Vererbungsl. 1936;71:501–526. [Google Scholar]
- Garcia-Bellido A, Merriam JR. Cell lineage of the imaginal discs in Drosophila gynandromorphs. J Exp Zool. 1969;170:61–75. doi: 10.1002/jez.1401700106. [DOI] [PubMed] [Google Scholar]
- Geigy R. Erzeugung rein imaginaler Defekte durch Ultraviolete Eibestrahlung bei Drosophila melanogaster. Roux’ Arch Entw Mech Organ. 1931;125:406–447. doi: 10.1007/BF00576360. [DOI] [PubMed] [Google Scholar]
- Golic KG. Site-specific recombination between homologous chromosomes in Drosophila. Science. 1991;252:958–961. doi: 10.1126/science.2035025. [DOI] [PubMed] [Google Scholar]
- Golic KG, Lindquist S. The FLP recombinase of yeast catalyzes site-specific recombination in the Drosophila genome. Cell. 1989;59:499–509. doi: 10.1016/0092-8674(89)90033-0. [DOI] [PubMed] [Google Scholar]
- Hadjieconomou D, Rotkopf S, Alexandre C, Bell DM, Dickson BJ, Salecker I. Flybow: genetic multicolor cell labeling for neural circuit analysis in Drosophila melanogaster. Nature methods. 2011;8:260–266. doi: 10.1038/nmeth.1567. [DOI] [PubMed] [Google Scholar]
- Hotta Y, Benzer S. Genetic dissection of the Drosophila nervous system by means of mosaics. Proc Natl Acad Sci U S A. 1970;67:1156–1163. doi: 10.1073/pnas.67.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland RB, Child GP. Experimental studies on development in Drosophila melanogaster. I. Removal of protoplasmic materials during late cleavage and early embryonic stages. J Exp Zool. 1935;3:33–56. [Google Scholar]
- Ito K, Awano W, Suzuki K, Hiromi Y, Yamamoto D. The Drosophila mushroom body is a quadruple structure of clonal units each of which contains a virtually identical set of neurones and glial cells. Development. 1997;124:761–771. doi: 10.1242/dev.124.4.761. [DOI] [PubMed] [Google Scholar]
- Kango-Singh M, Nolo R, Tao C, Verstreken P, Hiesinger PR, Bellen HJ, Halder G. Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila. Development. 2002;129:5719–5730. doi: 10.1242/dev.00168. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- Lohs-Schardin M, Cremer C, Nusslein-Volhard C. A fate map for the larval epidermis of Drosophila melanogaster: localized cuticle defects following irradiation of the blastoderm with an ultraviolet laser microbeam. Dev Biol. 1979a;73:239–255. doi: 10.1016/0012-1606(79)90065-4. [DOI] [PubMed] [Google Scholar]
- Lohs-Schardin M, Sander K, Cremer C, Cremer T, Zorn C. Localized ultraviolet laser microbeam irradiation of early Drosophila embryos: fate maps based on location and frequency of adult defects. Dev Biol. 1979b;68:533–545. doi: 10.1016/0012-1606(79)90224-0. [DOI] [PubMed] [Google Scholar]
- Morgan TH. Mosaics and gynandromorphs in Drosophila. Proc Soc Exp Biol Med. 1914;11:171–172. [Google Scholar]
- Morgan TH, Bridges CB. The origin of gynandromorphs. Carn Inst Wash Publ. 1919;278:1–22. [Google Scholar]
- Newsome TP, Asling B, Dickson BJ. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 2000;127:851–860. doi: 10.1242/dev.127.4.851. [DOI] [PubMed] [Google Scholar]
- Parks HB. Cleavage patterns in Drosophila and mosaic formation. Am Entom Soc. 1936;29:350–392. [Google Scholar]
- Patterson JT. The production of mutations in somatic cells of Drosophila melanogaster by means of X-rays. J Exp Zool. 1929;53:327–372. [Google Scholar]
- Patterson JT, Stone W. Gynandromorphs in Drosophila melanogaster. Univ Texas Publ. 1938;3825:1–67. [Google Scholar]
- Pignoni F, Zipursky SL. Induction of Drosophila eye development by decapentaplegic. Development. 1997;124:271–278. doi: 10.1242/dev.124.2.271. [DOI] [PubMed] [Google Scholar]
- Poulson DF. Histogenesis, organogenesis and differentiation in the embryo of Drosophila melanogaster. In: Demerec M, editor. Biology of Drosophila. New York: Wiley; 1950. pp. 168–274. [Google Scholar]
- Ripoll P. The embryonic organization of the imaginal wing disk of Drosophila melanogaster. Wilhelm Roux Arch Entwicklungsmech Organismen. 1972;1969:200–215. doi: 10.1007/BF00582553. [DOI] [PubMed] [Google Scholar]
- Spratford CM, Kumar JP. Extramacrochaetae imposes order on the Drosophila eye by refining the activity of the Hedgehog signaling gradient. Development. 2013;140:1994–2004. doi: 10.1242/dev.088963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratford CM, Kumar JP. Inhibition of Daughterless by Extramacrochaetae mediates Notch-induced cell proliferation. Development. 2015;142:2058–2068. doi: 10.1242/dev.121855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern C. Somatic crossing over and segregation in Drosophila melanogaster. Genetics. 1936;21:625–730. doi: 10.1093/genetics/21.6.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G, Basler K. Organizing activity of wingless protein in Drosophila. Cell. 1993;72:527–540. doi: 10.1016/0092-8674(93)90072-x. [DOI] [PubMed] [Google Scholar]
- Sturtevant AH. The claret mutant type of Drosophila simulans: a study of chromosome elimination and cell lineage. Z Wiss Zool Abt A. 1929;135:323–356. [Google Scholar]
- Tapon N, Ito N, Dickson BJ, Treisman JE, Hariharan IK. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell. 2001;105:345–355. doi: 10.1016/s0092-8674(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Worley MI, Setiawan L, Hariharan IK. TIE-DYE: a combinatorial marking system to visualize and genetically manipulate clones during development in Drosophila melanogaster. Development. 2013;140:3275–3284. doi: 10.1242/dev.096057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114:445–456. doi: 10.1016/s0092-8674(03)00549-x. [DOI] [PubMed] [Google Scholar]
- Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- Xu T, Rubin GM. The effort to make mosaic analysis a household tool. Development. 2012;139:4501–4503. doi: 10.1242/dev.085183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HH, Chen CH, Shi L, Huang Y, Lee T. Twin-spot MARCM to reveal the developmental origin and identity of neurons. Nature neuroscience. 2009;12:947–953. doi: 10.1038/nn.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]