

Abstract
Hearing loss changes the auditory brain, sometimes maladaptively. When deprived of cochlear input, central auditory neurons become more active spontaneously and begin to respond more strongly and synchronously to better preserved sound frequencies. This spontaneous and sound-evoked central hyperactivity has been postulated to trigger tinnitus and hyperacusis, respectively. Localized hyperactivity has also been observed after long-term exposure to noise levels that do not damage the cochlea. Adult animals exposed to bands of nondamaging noise exhibited suppressed spontaneous and sound-evoked activity in the area of primary auditory cortex (A1) stimulated by the exposure band but had increased spontaneous and evoked activity in neighboring A1 areas. We hypothesized that the cortically suppressed frequencies should for some time after exposure be perceived as less loud than before (hypoacusis), whereas the hyperactivity outside of the exposure band might lead to frequency-specific hyperacusis or tinnitus. To investigate this, adult CBA/Ca mice were exposed for >2 months to 8 to 16 kHz noise at 70 or 75 dB sound pressure level and tested for hypo-/hyperacusis and tinnitus using tone and gap prepulse inhibition of the acoustic startle reflex. Auditory brainstem responses and distortion product otoacoustic emissions showed evidence of cochlear synaptopathy after exposure at 75 but not 70 dB, putting a lower bound on damaging noise levels for CBA/Ca mice. Contrary to hypothesis, neither exposure significantly shifted startle results from baseline. These negative findings nevertheless have implications for startle test methodology and for the putative role of central hyperactivity in hyperacusis and tinnitus.
Keywords: noise, cochlear synaptopathy, tinnitus, hyperacusis, acoustic startle reflex, prepulse inhibition
Introduction
Exposure to loud noise can permanently destroy cochlear outer hair cells (OHCs) and inner hair cells (IHCs). Although IHCs survive noise trauma better than OHCs, IHC synapses with auditory nerve fibers (ANFs) can be irreparably damaged by noise doses that spare the hair cells themselves. The end result is a reduction of ANF activity from traumatized cochlear frequency regions, mostly at lower sound pressure levels (SPLs) when OHCs are lost, and apparently mostly at higher SPLs when IHC synapses are lost (Henderson, Bielefeld, Harris, & Hu, 2006; Kujawa & Liberman, 2015; Ruggero, Rich, Robles, & Recio, 1990).
Chronically reduced ANF activity is a trigger for changes in the auditory brain. Deprived central auditory neurons typically become more active spontaneously and respond more strongly and synchronously to better preserved cochlear “lesion-edge frequencies,” increasing their “representational area” in the auditory brainstem and even more so in auditory cortex (ACx; Eggermont, 2017a). Lesion-edge frequencies consequently become somewhat easier to discriminate, in small part compensating for the hearing loss (Thai-Van et al., 2007). However, the increased spontaneous activity may give rise to tinnitus—phantom ringing or hissing perceived as originating in one or both ears or in the head (Eggermont, 2012), and the increased sound-evoked activity may lead to hyperacusis—a reduced tolerance of what are normally considered moderately loud sounds (Pienkowski et al., 2014; Tyler et al., 2014). The mechanisms of tinnitus and hyperacusis remain controversial (see, e.g., Krauss et al., 2016; Sedley, Friston, Gander, Kumar, & Griffiths, 2016), but hyperactivity in the auditory brain is a common endpoint of most available models.
Spontaneous hyperactivity in auditory areas of the brainstem (particularly the dorsal cochlear nucleus or DCN, and the central nucleus of the inferior colliculus or ICC) and cortex has been correlated with behavioral evidence of tinnitus in animals with permanent noise-induced hearing loss (NIHL; Coomber et al., 2014; Engineer et al., 2011; Kaltenbach, Zacharek, Zhang, & Frederick, 2004; Li, Choi, & Tzounopoulos, 2013; Longenecker & Galazyuk, 2016; Ropp, Tiedemann, Young, & May, 2014; Sturm, Zhang-Hooks, Roos, Nguyen, & Kandler, 2017), and even after apparently full recovery from temporary noise trauma (Basura, Koehler, & Shore, 2015; Wu, Martel, & Shore, 2016). Interestingly, salicylate, a reliable inducer of transient tinnitus, led to increased spontaneous firing rates (SFRs) in the secondary ACx (A2) of cats but to decreased SFRs in both cat primary ACx (A1) and anterior auditory field (AAF; Eggermont & Kenmochi, 1998), a finding later replicated by several studies in rats (Lu et al., 2011; Yang et al., 2007; Zhang, Yang, Cao, Qin, & Sato, 2011). Analogously, sound-evoked central hyperactivity has been correlated with animal models of hyperacusis after NIHL (Chen et al., 2013; Hickox & Liberman, 2014; Sun, Deng, Jayaram, & Gibson, 2012), salicylate injection (Sun et al., 2009; Turner & Parish, 2008), and in hereditary progressive hearing loss (Carlson & Willott, 1996; Ison, Allen, & O'Neill, 2007; Xiong et al., 2017). Nevertheless, aspects of these data remain puzzling (see Discussion section), and there is so far only sparse evidence from studies of the human brain linking spontaneous hyperactivity with tinnitus (Adjamian, Sereda, & Hall, 2009; Elgoyhen, Langguth, De Ridder, & Vanneste, 2015) and sound-evoked hyperactivity with hyperacusis (Gu, Halpin, Nam, Levine, & Melcher, 2010).
Spontaneous and sound-evoked hyperactivity are also observed in specific areas of A1 after prolonged exposure to nondamaging levels of noise (Eggermont, 2017b; Pienkowski & Eggermont, 2011). In a series of studies on adult cats exposed to various tone pip ensembles and noise bands at ∼70 dB SPL for weeks to months at a time, it was shown that A1 neural activity was strongly suppressed at frequencies within the exposure band (particularly near its edges) but was generally enhanced at frequencies above and below the exposure band (Pienkowski & Eggermont, 2009, 2010a, 2010b; Pienkowski, Munguia, & Eggermont, 2011, 2013; note that the seminal study by Noreña, Gourévitch, Aizawa, & Eggermont, 2006 used an exposure level of 80 dB SPL). We attributed this suppression to a compensatory reduction in central neural gains in response to the persistent exposure stimulus and the enhancement to decreased lateral inhibition from the suppressed region (Pienkowski & Eggermont, 2012). These cortical changes slowly reversed (also over weeks to months) after the end of the nondamaging exposure (Pienkowski & Eggermont, 2009, 2010a, 2010b). Interestingly, postexposure increases in spontaneous firing and neural synchrony were observed in the enhanced areas of A1 (Munguia, Pienkowski, & Eggermont, 2013; Noreña et al., 2006; Pienkowski & Eggermont, 2009, 2010b), not in the deprived area as is the case with permanent hearing loss (Eggermont, 2017a). Examples of these findings are reproduced in Figure 1. We wondered whether the cortical hyperactivity resulting from chronic, nondamaging noise exposure could lead to hyperacusis or tinnitus. Specifically, we hypothesized that frequencies within the exposure band should for some time be perceived as softer than before (hypoacusis), whereas frequencies outside of the exposure band might be perceived as louder than before (hyperacusis) and might become internalized as tinnitus.
Figure 1.

(a) Multiunit (MU) spike recordings from ACx of a representative unexposed control adult cat (left) and an adult cat exposed for 2 months to 4 to 20 kHz band-pass noise at ∼70 dB SPL (right). MU characteristic frequencies (CFs) are color-coded (scale top-center) and superposed on photographs of the cortical surface. Blank circles indicate electrode penetrations that yielded insignificant sound-evoked activity. A1 is outlined and lightly shaded to distinguish it from surrounding auditory fields (AAF, PAF, A2, DP; scale bars: 2 mm; D: dorsal; P: posterior). CF–distance plots for MUs recorded in A1 are shown centered between the cortical maps; black circles give the mean positions ±1 SD of A1 units in each of the seven color-coded, octave-wide bins. The bottom row shows histogram distributions of A1 CFs and scatterplots of A1 response thresholds versus the CF. Note that noise exposure led to a clear underrepresentation of A1 units with CFs in the exposure frequency range and to an overrepresentation of A1 units with CFs above and below that range. That is, units with high and low CFs covered larger than normal areas of A1, and the thresholds and suprathreshold firing rates of these units were on average better than normal. Adapted from Pienkowski et al. (2011). (b) A1 MU rate-level functions averaged at the best frequency for unexposed control (black traces) and 4 to 20 kHz (2 months at 70 dB SPL) noise-exposed cats (red traces). Data were pooled by MU location in A1: “Posterior A1” spans a distance of 0% to 30% along the tonotopic gradient toward AAF and represents neurons normally tuned to <4 kHz (i.e., below the exposure range); “Central A1” spans a distance of 30% to 70% and represents neurons normally tuned to 4 to 20 kHz (within the exposure range); “Anterior A1” spans a distance of 70% to 100% and represents neurons normally tuned to >20 kHz (above the exposure range). Error bars show ±1 SE, and asterisks indicate post hoc Bonferroni tests significant at p < .05 (two-way ANOVA). Noise exposure induced neural hyperactivity in posterior and anterior A1 and neural hypoactivity in central A1. Adapted from Pienkowski et al. (2014). (c) Long-term exposure of adult cats to three different tone pip ensembles and to a broadband “factory noise” at ∼70 dB SPL generally increased A1 SFRs in the frequency regions outside of the exposure band (i.e., in the regions of neural hyperacusis; Figure 1(b)). Error bars show +1 SE. Adapted from Munguia et al. (2013), based on data replotted from Pienkowski and Eggermont (2010b; 2–40 kHz, 4 and 16 kHz), Pienkowski and Eggermont (2009; 4–20 kHz), and Pienkowski et al. (2013; “factory noise”). (d) Long-term exposure of adult cats to a 4 to 20 kHz tone pip ensemble at ∼80 dB SPL led to an increase in pairwise neural synchrony (quantified by ρ; color-code at right), particularly in A1 frequency regions outside of the exposure band. Figure reproduced from Noreña et al. (2006).
ACx = auditory cortex; SPL = sound pressure level; CF = characteristic frequency; AAF = anterior auditory field; PAF = posterior auditory field; ANOVA = analysis of variance; SFR = spontaneous firing rate.
To investigate this possibility, adult CBA/Ca mice were exposed for >2 months to 8 to 16 kHz band-pass noise at either 70 or 75 dB SPL and tested for hypo-/hyperacusis and tinnitus using tone and gap prepulse inhibition (PPI) of the acoustic startle reflex (ASR). Auditory brainstem responses (ABRs) and distortion product otoacoustic emissions (DPOAEs) were used to show that the 70 dB exposure was nondamaging, whereas 75 dB appeared to cause a small but significant IHC synapse loss in the 8 to 16 kHz cochlear region. Contrary to hypothesis, prolonged exposure at either 70 or 75 dB SPL did not affect baseline tone or gap PPI results, implying an absence of any hyper/hypoacusis or tinnitus. These negative findings nevertheless have implications for PPI testing and for the putative role of central auditory hyperactivity in tinnitus and hyperacusis.
Methods
Animals and Noise Exposure
This work was approved by the Institutional Animal Care and Use Committee of Salus University (A-MP1502). Nine normal-hearing male CBA/Ca mice (Jackson Laboratories) served as subjects in the main experiment reported here and were exposed binaurally for 2 months, 24 hr/day, to sharply filtered 8 to 16 kHz noise at ∼70 dB SPL (measured with a Larson-Davis Model 824 sound level meter), beginning at about three months of age. Six mice served as unexposed controls, and another six were exposed to the same 8 to 16 kHz noise at ∼75 dB SPL.
The exposure stimulus was synthesized in Adobe Audition, converted to analog (Creative, Model SB1240), and played from a loudspeaker mounted just above the cages housing the mice (Tucker Davis Technologies—TDT, Model MF1; frequency response flat to 5 dB between 8 and 16 kHz). Mice were kept on a 12-hr light/dark schedule (light 8 a.m.–8 p.m.) and were given free access to food and water. They were rewarded for their participation in ASR testing with sunflower seeds. There were no apparent signs of long-term distress in any of the noise-exposed mice.
Assessment of Loudness Perception (Hypo-/Hyperacusis) and Tinnitus Using the ASR
The ASR is a protective reflex elicited by a sudden, loud sound (Koch, 1999). In mice, it involves a whole-body flinch and jump, the force of which was measured using a motion-sensitive platform in an anechoic foam-lined, sound-attenuating startle chamber (San Diego Instruments, SR-LAB).
ASR amplitudes can be reduced by preceding the loud startling sound with a nonstartling “prepulse,” known as PPI of the ASR. The degree of startle amplitude reduction, called the magnitude of the PPI, is related to the perceptual salience of the prepulse. For instance, the greater the perceived loudness of a tonal prepulse, the greater the magnitude of the PPI (e.g., Carlson & Willott, 1996). Thus, an animal with hyperacusis is expected to show greater PPI for a given prepulse SPL and vice versa for hypoacusis. In the gap variant of PPI, often abbreviated as GPIAS (Galazyuk & Hébert, 2015; Turner et al., 2006), the prepulse is a silent gap in a moderate-level, narrowband noise (NBN) background. It is assumed that a ringing tinnitus with a pitch similar to the NBN background can reduce the salience of the gap and decrease the magnitude of the PPI. Figure 2 illustrates these concepts and details the startle parameters used in the present study. For both tone and gap PPI testing, startle stimuli were 20-ms-long bursts of broadband noise (BBN) presented at 105 dB SPL. Tone prepulses were also 20 ms long (including 1 ms onset and offset cos2 ramps), were presented at 50 or 70 dB SPL, and preceded the startle BBN by 100 ms (measured between sound onsets). For GPIAS testing, silent gaps of 20 or 50 ms long were embedded in third-octave NBN presented at 65 dB SPL, and the gaps also preceded the startling BBN by 100 ms (onset-to-onset). These stimuli were synthesized in Adobe Audition, converted to analog (TASCAM, Model US–2x2), and played out by a HiVi Isodynamic Tweeter (Model RT2C-A; frequency response flat to 5 dB over the stimulus frequency range, from 4 to 32 kHz) that was mounted in the ceiling of the startle chamber. Stimuli were calibrated with a quarter-inch microphone (ACO Pacific, Model 7017) placed in the mouse restraining cylinder inside the startle box.
Figure 2.

Schematic diagrams of PPI ASR (top) and GPIAS (bottom), including the stimulus parameters used in the present study. Also shown are hypothetical PPI functions in animals with hypo- and hyperacusis (top-right), and hypothetical GPIAS results that are positive for tinnitus at 16 kHz (bottom-right).
PPI = prepulse inhibition; ASR = acoustic startle reflex; GPIAS = gap-prepulse inhibition of the acoustic startle; BBN = broadband noise; SPL = sound pressure level; NBN = narrowband noise.
Figure 3(a) shows the experiment timeline, and Figure 3(b) is a block diagram of a single startle session. Each session consisted of 362 startle trials with an average interstartle interval (ISI) of 5 s (range 3–7 s), for a total session time of ∼30 min. Gap PPI testing was conducted in NBN frequencies of 6, 8, 11, 16, 23, and 32 kHz, and in BBN. Tone PPI testing was conducted with prepulse frequencies of 4, 6, 8, 11, 16, 23, and 32 kHz. Each gap-in-noise or tone prepulse frequency was presented in a block of 21 trials in pseudorandom order, with seven startle-only trials, and seven trials each with 20 and 50 ms gap prepulses, or with 50 and 70 dB SPL tone prepulses. The ratio of the ASR amplitude in the presence of a gap or tone prepulse to that without a prepulse was calculated as an average for each block and constituted the “raw data” for that session. Any block that contained less than half (i.e., 3/7 or less) good startle responses (see the following) for either the startle-only or prepulse conditions was dropped and not included in the across-session average; this happened infrequently on 1.2% and 2.4% of preexposure tone and gap PPI blocks, respectively, and on 1.6% and 1.5% of postexposure blocks. In addition, three sets of “I/O functions” were run per session, in which startle-only amplitudes were measured in response to 20-ms-long tones (including 1 ms on/off ramps) at 4, 6, 8, 11, 16, and 23 kHz and to BBN, all presented at 85 and 105 dB SPL. Finally, two sets of startle-only trials to 105 dB SPL BBN were measured at the beginning and end of each session, to track within-session adaptation of the startle response. Each mouse completed many such sessions (see the following), with the order of the tone and gap PPI blocks interchanged and randomized between sessions to offset within-session habituation effects. All startle sessions were conducted during the day, but in darkness, with the lights off inside the startle box, as darkness potentiates the startle response.
Figure 3.

(a) Experiment timeline. (b) Block diagram of a single startle session. See text (Methods section) for detailed description.
ASR = acoustic startle reflex; SPL = sound pressure level; ABR = auditory brainstem response; DPOAE = distortion product otoacoustic emission; ISI = interstartle interval; GPIAS = gap-prepulse inhibition of the acoustic startle; PPI = prepulse inhibition; BBN = broadband noise.
Prior to the first session that was included in the averaged results, the mice were gradually acclimated to the startle box and test stimuli over a period of 2 weeks. During this time, startle amplitudes habituated to a more stable baseline, and both tone and gap PPI became more reliable in the sense that ASR amplitude ratios (with vs. without a prepulse) were more consistently <1. Of 24 mice subjected to this acclimation period, three were excluded from further testing on the basis of unreliable PPI effects; that is, their amplitude ratios continued to be highly variable and were almost as likely to be >1 as <1.
Each of the remaining 21 mice (6 control; 9 subsequently exposed to 70 dB SPL; 6 exposed to 75 dB SPL) was then tested during 12 startle sessions, as described earlier, and the results were averaged across sessions. Each mouse was limited to one 30-min test session per day and completed the 12 sessions over a 3 - to 4-week period. This baseline testing was followed by the 2-month noise exposure, and then another 12 sessions of ASR testing again over 3 to 4 weeks, after a shorter 1-week ASR reacclimation period. During the weeks of postexposure ASR testing, the 8 to 16 kHz noise was left on for 12 hr each night (8 p.m.–8 a.m.). Mice were tested in random order during the day, starting no earlier than 10 a.m., 2 hr after the noise had been switched off for the day. Maintaining the noise at night eliminated the potential confound on postexposure testing of the gradual reversal of noise-induced changes after the cessation of exposure (Pienkowski & Eggermont, 2009, 2010a, 2010b).
Startle response analysis was automated using custom software written in Mathematica (Wolfram Research, Champaign, IL, USA). Reliable responses were identified using a template-matching algorithm similar to that described by Grimsley et al. (2015). Thousands of startle trials were checked by eye, and the performance of the algorithm was found to be excellent, with misidentification of responses occurring on <1% of trials. Figure 4(a) shows a complete set of good startle responses from a single test session. The response amplitude was taken as the largest peak in each trace. Figure 4(b) shows the percentage of all startle trials that produced good responses, the percentage that were contaminated by nonstartle movements, and the percentage that produced no significant response; these data were averaged across sessions for each mouse, both pre- and postexposure. Pooled across mice, the fractions preexposure were 70.7%, 19.9%, and 9.4% for good, movement-contaminated, and nonstartling trials, respectively; corresponding numbers postexposure were 72.6%, 17.3%, and 10.1%. A trial reject rate due to nonstartle movements of less than 20% was acceptable given the short ISI of 3 to 7 s adopted here, as this permitted the recording of a large number of startle trials (n = 362) within each 30-min session. Short ISIs were also used with good effect in a recent study by Longenecker, Alghamdi, Rosen, and Galazyuk (2016).
Figure 4.

(a) Complete set of “good” trials from a single example startle session. (b) Percentage of all startle trials, pre- and postexposure that were good trials (as in Figure 4(a); black traces), that were contaminated by nonstartle movements (red traces), and that lacked clear responses (blue traces). Data are shown for the nine individual mice (error bars give ± 1 SE).
Trials contaminated by nonstartle movements were discarded from the record, but nonstartling trials were counted and were assigned an amplitude equal to the average noise floor for that recording session. Recall that at least four of seven clean startle responses were required, in each of three conditions (control and two prepulse), for a response block to count in the across-session average (else it was discarded). Keeping the few remaining nonstartling trials (∼5% of all trials) had the effect of slightly increasing PPI magnitudes. This is because trials with tone or gap prepulses were almost 3 times as likely to yield a nonstartle response than were control trials. Thus, on some trials, the prepulses didn’t just reduce the ASR, they appeared to suppress it completely; deleting nonstartling trials would have excluded this potentially important effect.
Individual mouse and group-averaged ASR results were compared pre- and postexposure using two-way analyses of variance (ANOVAs) with post hoc Bonferroni tests.
DPOAE and ABR Recording
DPOAEs and ABRs were measured from the left ears and mastoid areas 2 weeks following the completion of postexposure (or control group) startle sessions. Mice were anesthetized with a mixture of 50 mg/kg ketamine and 10 mg/kg xylazine, injected intraperitoneally, and were topped up with half doses of the mixture as needed to maintain a state of areflexia. They were placed on a homeothermic blanket (Harvard Apparatus) that kept their body temperature at 36.5°C, inside a single-walled sound-attenuating chamber (ETS-Lindgren).
ABRs were always measured first. Stimuli were tone bursts at 4, 6, 8, 11, 16, 23, and 32 kHz and were 3 ms in duration including 1 ms cos2 on and off ramps. Stimuli were synthesized using TDT software (SigGen), converted to analog (TDT, RZ6), and played out by a TDT MF1 speaker coupled to the animal’s left ear canal with a 10-cm tube and sealed probe (system flat to 20 dB from 4 to 32 kHz). Sound levels were calibrated with the probe coupled to a quarter-inch microphone (ACO Pacific, 7017) with an additional 7-mm-long plastic tube, intended to approximate the length of the mouse ear canal. Stimuli were presented at 10 to 90 dB SPL at each frequency, in 10 dB steps, with 512 repetitions per level and a presentation rate of 21.1/s. The ABR was recorded differentially between the left mastoid area and vertex (ground electrode in the nape of the neck) using subdermal needle electrodes (Rochester Electro-Medical, Lutz, FL, USA, Model S83018-R9). Potentials were amplified 5,000× (TDT, RA4LI headstage [20×] and RA4PA pre-amp [250×]), digitized, and filtered between 100 and 3,000 Hz (TDT, RZ6), under the control of TDT software (BioSig, Alachua, FL, USA). At low stimulus levels, measurements were repeated twice, and ABR threshold was defined as the lowest SPL that yielded a reproducible ABR, minus 5 dB (half the step size). Peak-to-trough amplitudes were then determined for mouse ABR Waves 1 to 4 at all suprathreshold SPLs, for distinct waves.
Following ABR recording, DPOAEs were measured from the left ear using an OAE probe coupled to a pair of TDT MF1 speakers and to an Etymotic Research microphone (ER-10B+). Stimuli were synthesized using TDT software (SigGen). The frequency of the higher primary tone (f2) was again set to 4, 6, 8, 11, 16, 23, or 32 kHz, and the frequency of the lower primary tone (f1) was given by f1 = f2/1.2. Levels of f1 (L1) ranged from 20 to 80 dB SPL in 10 dB steps, with L2 = L1 – 10 dB. DPOAEs at frequency 2f1 – f2 were amplified (ER-10B+ amp) and digitized (TDT, RZ6) using TDT software (BioSig). DPOAE amplitudes are reported in units of dB V, and DPOAE threshold was defined as the lowest level of L1 (again minus half the step size, or 5 dB) at which the DPOAE amplitude was above the 99% confidence interval for the microphone noise floor, averaged across the six frequency bins adjacent to 2f1 – f2.
Results
ABRs and DPOAEs
Nine normal-hearing adult male CBA/Ca mice were exposed 24 hr/day for 2 months and then 12 hr/day for 1 month to sharply filtered 8 to 16 kHz noise at 70 dB SPL. ABRs and DPOAEs were measured 2 weeks after the end of the 3-month exposure, to allow recovery from any potential temporary threshold shifts (TTS), although TTS was not confirmed. They were compared with measurements made at the same age in six unexposed control mice and in another six mice that were exposed to the same 8 to 16 kHz noise at 75 dB SPL. Group-averaged ABR results (± 1 standard error of the mean or SE) are shown in Figure 5(a). There were no significant differences in ABR thresholds between the three groups: p = .56 for the main effect of group across frequency in a two-way ANOVA. ABR Wave 1 input-output functions were not affected after exposure to 70 dB SPL noise but were significantly reduced after exposure to 75 dB SPL at frequencies of 8 kHz (p = .031), 11 kHz (p = .004), and 16 kHz (p = .007), that is, only at frequencies within the exposure band (all other frequencies were p > .05 as indicated). These p values reflect the main effect of group across ABR stimulus level in a two-way ANOVA and were not corrected for multiple comparisons at the various stimulus SPLs. Note that none of the differences at individual stimulus SPLs were significant at the p = .05 level after post hoc Bonferroni correction, only the main effects across SPL. Importantly, no significant differences were found between groups in the amplitudes of ABR Waves 2 to 4 (data not shown). Also, there were no significant differences (p > .05) between groups in DPOAE thresholds and DPOAE input-output functions at any primary tone frequency (Figure 5(b)). As will be discussed, these results are consistent with mild noise-induced cochlear synaptopathy following exposure at 75 dB SPL, but no cochlear damage after exposure at 70 dB SPL.
Figure 5.

(a) ABR audiograms and Wave 1 growth functions at different stimulus frequencies for unexposed control mice (black traces) and for mice exposed for 3 months to 8 to 16 kHz noise at 70 dB SPL (blue traces) and 75 dB SPL (red traces). Error bars show ±1 SE; p values are for the main effect of group across SPL (two-way ANOVA) and were not corrected for multiple comparisons at the different ABR frequencies. (b) As Figure 5(a) but showing DPOAE audiograms and growth functions at different stimulus frequencies for unexposed and exposed mice.
ABR = Auditory brainstem response; SPL = sound pressure level; ANOVA = analysis of variance; DPOAE = distortion product otoacoustic emission.
Tone PPI Testing for Hypo-/Hyperacusis
ASR results presented here are compared pre- and postexposure for the nine mice exposed to 8 to 16 kHz noise at the nondamaging level of 70 dB SPL. It was hypothesized that this exposure would cause sound frequencies within the noise band to be perceived as softer than before (hypoacusis), whereas frequencies above or below the noise band would be perceived as louder than before (hyperacusis).
The mean weight (± 1 SD) of the nine mice at the start of preexposure ASR testing was 37.1 ± 2.0 g, and for postexposure, it was 38.3 ± 2.2 g. In unexposed control mice, ASR results were unchanged over a 2-month waiting period between the test and retest sessions (data not shown), despite a similar small weight gain over that period.
Figure 6(a) shows group-averaged ASR amplitudes (±1 SE) in response to tonal and BBN startle stimuli presented at 85 dB SPL (black traces) and 105 dB SPL (gray traces). There were no significant differences pre- versus postexposure at any startle frequency or level (p > .05, as indicated, for the main effect of group across frequency; separate two-way ANOVAs were run at each SPL). This suggests an absence of hypo- or hyperacusis at 85 and 105 dB SPL. It is important to note that changes in startle amplitudes after noise trauma could potentially confound PPI testing (Lobarinas, Hayes, & Allman, 2013; Salloum et al., 2016), but this is not a concern here.
Figure 6.

(a) Group-averaged ASR amplitudes (± 1 SE) in response to BBN and tonal startle stimuli presented at 85 dB SPL (black traces) and 105 dB SPL (gray traces), pre- and postexposure. The average startle system noise floor is illustrated (avg. NF). The p values are for the main effect of exposure across startle stimulus (two-way ANOVA) and were not corrected for multiple comparisons at the two startle SPLs. (b) Group-averaged PPI results (± 1 SE) for prepulse levels of 50 dB SPL (black traces) and 70 dB SPL (gray traces). Again, p values compare PPI ratios before and after exposure across prepulse frequency.
ASR = acoustic startle reflex; BBN = broadband noise; SPL = sound pressure level; ANOVA = analysis of variance; PPI = prepulse inhibition.
Group-averaged PPI results (± 1 SE) are shown in Figure 6(b) for tone prepulse levels of 50 dB SPL (black traces) and 70 dB SPL (gray traces). Note that the PPI effect was highly significant (p ≪ .05) at the group level for all prepulse frequencies and SPLs, that is, ASR amplitude ratios with versus without prepulse are all ≪1. However, again there were no significant differences postexposure (p > .05, as indicated). Figure 7 shows the pre- and postexposure PPI results of the nine individual mice, averaged across the 12 startle sessions (± 1 SE), using the same legend as Figure 6. For prepulses at 70 dB SPL (gray traces), the PPI effect was highly robust in individual mice: In only 2% of results, counting across mice and frequencies, both pre- and postexposure, did the uncorrected 95% CI of the ASR amplitude ratio include the value of 1, indicating insignificant PPI. For prepulses at 50 dB SPL (black traces), the PPI effect in individual mice was somewhat less robust and was insignificant 29% of the time preexposure and 24% of the time postexposure. Despite the relatively robust PPI effect, especially at 70 dB SPL, the individual results do not support the hypothesis that at least some of the mice may have developed frequency-specific hypo- or hyperacusis postexposure: None of the differences at individual frequencies were significant at p < .05 after post hoc Bonferroni testing.
Figure 7.

Pre- and postexposure PPI results from the nine individual mice, averaged across the 12 startle sessions (± 1 SE), using the same legend as Figure 6.
PPI = prepulse inhibition; ASR = acoustic startle reflex.
Gap PPI (GPIAS) Testing for Tinnitus
GPIAS results are also reported here pre- and postexposure for the nine mice exposed to 8 to 16 kHz noise at the nondamaging level of 70 dB SPL. Figure 8 shows group-averaged ASR amplitude ratios (±1 SE) with and without 20 ms (black traces) and 50 ms (gray traces) silent gaps embedded in 1/3-octave NBN at a range of frequencies, and in BBN. As will be discussed further, GPIAS testing was performed using both 50 ms and 20 ms gaps because previous studies of auditory cortical ablation/inactivation have suggested that cortex is not essential for GPIAS with gaps of 50 ms or longer but is required at gap durations of <30 ms (Bowen, Lin, Taylor, & Ison, 2003; Ison, O’Connor, Bowen, & Bocirnea, 1991; Weible et al., 2014). However, there were no significant differences postexposure with either 50 or 20 ms gaps, despite the fact that the GPIAS effect itself was highly significant (p < <.05) at the group level for both gap durations in all NBN backgrounds (i.e., ASR amplitude ratios with vs. without gap are all ≪1). Figure 9 shows the pre- and postexposure GPIAS results of the nine individual mice, averaged across the 12 startle sessions (±1 SE), using the same legend as Figure 8. For 50 ms gaps (gray traces), the GPIAS effect was highly robust in individual mice: The uncorrected 95% CI of the ASR amplitude ratio included the value of 1 (indicating insignificant GPIAS) in only 3% of both pre- and postexposure results. For 20 ms gaps (black traces), the GPIAS effect in individual mice was less robust and was insignificant in 22% of measurements preexposure and 21% postexposure. Again, the individual results do not support the idea that at least some of the mice may have developed tinnitus postexposure, as none of the differences at individual frequencies remained significant after post hoc Bonferroni analysis.
Figure 8.
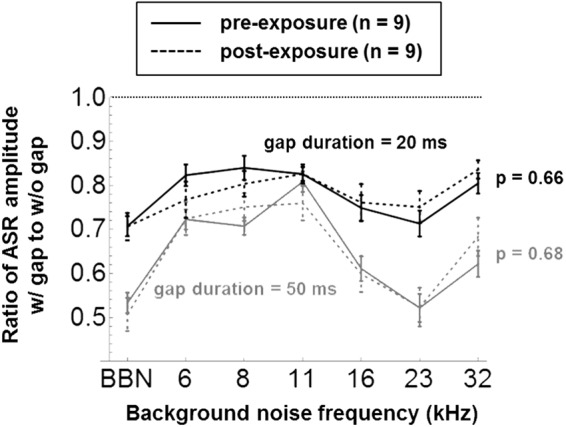
Group-averaged GPIAS results (± 1 SE) for gap durations of 20 ms (black traces) and 50 ms (gray traces). The p values compare GPIAS ratios before and after exposure across background noise frequency.
GPIAS = gap-prepulse inhibition of the acoustic startle; ASR = acoustic startle reflex; BBN = broadband noise.
Figure 9.

Pre- and postexposure GPIAS results from the nine individual mice, averaged across the 12 startle sessions (± 1 SE), using the same legend as Figure 8.
GPIAS = gap-prepulse inhibition of the acoustic startle; ASR = acoustic startle reflex; BBN = broadband noise.
Discussion
Effects of Prolonged Exposure to Moderately Loud Noise on the Auditory Periphery
There was evidence of cochlear synaptopathy in CBA/Ca mice following a 3-month exposure in adulthood to 8 to 16 kHz noise at 75 dB SPL, but not 70 dB SPL. Despite the absence of significant ABR threshold shifts 2 weeks after the end of either exposure, ABR Wave 1 amplitudes at suprathreshold stimulus levels were significantly reduced after the 75 dB SPL exposure, and this was specific to stimuli at 8, 11, and 16 kHz, that is, frequencies within the exposure band (Figure 5(a)). DPOAEs were not affected at any stimulus frequency (Figure 5(b)), nor were ABR Waves 2 to 4 amplitudes (data not shown). Note that histology was not performed.
This pattern of damage differs in a predictable way from that caused by shorter exposures to more intense noise. A study by Fernandez, Jeffers, Lall, Liberman, and Kujawa (2015) compared the effects on CBA/Ca mice of 2 hr exposures to 8 to 16 kHz noise at 100 and 91 dB SPL. By 2 weeks postexposure, DPOAEs and ABR thresholds had returned to preexposure baselines in both cases, while ABR Wave 1 amplitudes remained reduced only after the more intense, 100 dB SPL dose. However, at 1 day postexposure, temporary shifts were observed in both DPOAE and ABR thresholds: After the 100 dB exposure, maximum TTS was observed at the highest frequencies tested, >30 kHz, but after the 91 dB exposure, TTS was reduced and peaked at ∼20 kHz, only slightly above the 8 to 16 kHz exposure band. These differences are likely due to the “spread of excitation” (and damage) at higher exposure levels to more basal regions of the cochlea, as a result of the saturating nonlinearity in cochlear mechanics at the characteristic frequency (CF; Johnstone, Patuzzi, & Yates, 1986). At the more moderate exposure level of 75 dB SPL used here, this spread of excitation is limited, and the observed damage as assessed by Wave 1 reduction is smaller and restricted to the CF region of the exposure band. Maison, Usubuchi, and Liberman (2013) exposed CBA/Ca mice to 8 to 16 kHz noise at 84 dB SPL for 1 week and found reduced ABR Wave 1 amplitudes and IHC synapse counts 1 week postexposure. As in the present study, the damage appeared to be greatest at 8 to 16 kHz, although smaller changes were also seen at frequencies above and below the exposure band.
Another potential difference is that after exposure at 75 dB SPL, ABR Wave 1 amplitudes appeared reduced at all but the lowest stimulus levels (Figure 5(a)). Note again, however, that these reductions were significant (p < .05) only when averaged across levels (i.e., only as an ANOVA main effect), but not at individual levels after post hoc Bonferroni adjustment. Previous studies have reported greatest reductions of Wave 1 amplitudes at high stimulus SPLs (e.g., Kujawa & Liberman, 2009) and have attributed this to the greater vulnerability of the high-threshold ANFs compared with low-threshold ANFs (Furman, Kujawa, & Liberman, 2013). However, a study on guinea pigs using the neurotoxin ouabain showed that the contribution of high-threshold fibers to the compound action potential (an analog of ABR Wave 1) can be small, even at high stimulus levels, and that substantial reductions of the high-level compound action potential imply a loss of low-threshold fibers as well (Bourien et al., 2014). The mechanisms of noise-induced synaptopathy are not fully understood at present (Kujawa & Liberman, 2015), but it’s possible that the lower level, more chronic exposure used here was less selectively damaging to high-threshold ANFs.
The present study appears to be the first to report that cochlear synaptopathy can occur in CBA/Ca mice after prolonged exposure to noise levels as low as 75 dB SPL. A recent opinion piece has argued that because humans appear less susceptible to TTS than rodents, they should also be less vulnerable to synaptopathy (Dobie & Humes, 2017). Moreover, the octave-band exposures typically used in animal studies are not representative of real-world noise. While these are fair points, a recent study by Valero et al. (2017) showed that a single 4 hr exposure to 108 dB SPL NBN centered at 2 kHz produced synaptopathy in rhesus monkeys in the absence of OHC damage. It thus remains uncertain whether chronic exposure at the current occupational noise limit of ∼85 dB A for 8 hr/day (National Institute for Occupational Safety and Health, 1998; Occupational Safety and Health Administration, 2002) is indeed safe for the ear, avoiding both hair cell loss and synaptopathy.
Effects of Prolonged Exposure to Moderately Loud Noise on the Central Auditory System and on Auditory Perception
We have previously reported that adult cats chronically exposed to bandlimited tone pip ensembles and noise at ∼70 dB SPL exhibited a strong suppression of A1 responses to frequencies within the exposure band, and an enhancement of responses to frequencies above and below the exposure band, as illustrated in Figure 1 (Noreña et al., 2006; Pienkowski & Eggermont, 2009, 2010a, 2010b; Pienkowski et al., 2011, 2013). Preliminary data, not presented here, suggest similar noise-induced changes in mouse A1. These changes are also likely exhibited at the level of the thalamic medial geniculate body (MGB), as inferred from cortically recorded local field potentials (Pienkowski & Eggermont, 2011), which reflect synchronous postsynaptic potentials from thalamic inputs (e.g., Bruno & Sakmann, 2006; Mitzdorf, 1985). We have also previously reported that cat ABR Wave 4 (analogous to wave V in humans), which represents neural generators in the nuclei of the lateral lemniscus and the IC, appears unaffected by exposure to nondamaging noise (Pienkowski & Eggermont, 2009, 2010a, 2010b; Pienkowski et al., 2011, 2013). Lau, Zhang, McPherson, Pienkowski, and Wu (2015) performed whole brain functional magnetic resonance imaging following long-term exposure of adult rats to BBN at 65 dB SPL and found reduced noise-evoked activation of ACx and MGB, but no changes in the IC and lower brainstem. However, a follow-up functional magnetic resonance imaging study using tone stimulation did find evidence of an expansion of the IC area responsive to 40 kHz, above the cutoff frequency of the BBN exposure (Lau, Pienkowski, Zhang, McPherson, & Wu, 2015). Nevertheless, it could be that the central effects of moderate noise are limited mostly to the MGB and ACx, while those of traumatic noise are already prominent at the level of the IC and lower brainstem (Eggermont, 2017a).
Several previous studies have demonstrated clear effects of adulthood exposure to moderate noise on auditory perception. Zhou and Merzenich (2012) reported both cortical and perceptual temporal discrimination deficits in adult rats exposed to temporally random sequences of BBN bursts at 65 dB SPL. In another study of rats exposed to BBN at 65 dB SPL, Zheng (2012) observed frequency discrimination deficits in quiet but improved frequency discrimination in noise. Deficits in quiet but not in noise were also reported in a study of /pa/ – /ka/ syllable discrimination in a small sample of young shipyard workers and preschool teachers with an average 6 years of moderate-level occupational noise exposure but “clinically normal” audiograms (Kujala et al., 2004; see Pienkowski, 2017 for a wider discussion). These findings argue against synaptopathy as a contributing factor to the observed deficits, as synaptopathy has been linked with impaired hearing in noise but not in quiet (Bharadwaj, Verhulst, Shaheen, Liberman, & Shinn-Cunningham, 2014; Lobarinas, Spankovich, & Le Prell, 2017). Kamal, Holman, and de Villers-Sidani (2013) also exposed young adult rats for 2 months to BBN at 65 dB SPL and observed that A1 responses postexposure resembled those of naturally aged rats (de Villers-Sidani et al., 2010). Note again, however, that most of these changes slowly reverse following the end of exposure (Kamal et al., 2013; Pienkowski & Eggermont, 2009, 2010a, 2010b). It should also be mentioned that low to moderately loud noise can be an important component of therapies for improving hearing (Beste & Dinse, 2013; Green, Ohlemacher, & Rosen, 2016; Wright, Sabin, Zhang, Marrone, & Fitzgerald, 2010), expanding the auditory dynamic range after acquired hearing loss (Formby, Sherlock, Hawley, & Gold, 2017), slowing down progressive hereditary hearing loss (Willott & Bross, 2004; Willott & Turner, 1999), reducing the impact of a traumatic noise exposure (Canlon, Borg, & Flock, 1988; Noreña & Eggermont, 2005, 2006; Tanaka et al., 2009), and ameliorating tinnitus and hyperacusis (Noreña & Chery-Croze, 2007; Schaette, König, Hornig, Gross, & Kempter, 2010).
No Change in Tone and Gap PPI After Prolonged Exposure to Moderately Loud Noise
After chronic exposure to 8 to 16 kHz noise at 70 dB SPL, there were no significant changes in ASR amplitudes evoked by tones and BBN at 85 and 105 dB SPL (Figure 6(a)), and no changes in PPI amplitude ratios with tone prepulses at 50 and 70 dB SPL (Figures 6(b) and 7), nor with 20 and 50 ms silent gaps embedded in NBN and BBN backgrounds (Figures 8 and 9). Similar results were obtained in mice exposed at 75 dB SPL, using different startle protocols than reported here (Pienkowski, 2016), despite the evidence for mild synaptopathy caused by this exposure level (Figure 5). Thus, perhaps surprisingly, there was no ASR-based evidence of hypo-/hyperacusis or tinnitus in CBA/Ca mice exposed for 3 months to 8 to 16 kHz noise at up to 75 dB SPL.
It was hypothesized on the basis of our previous work (Figure 1) that sound frequencies within the exposure band should be perceived as softer than normal (hypoacusis), whereas frequencies outside of the exposure band might be perceived as louder than normal (hyperacusis) and might be internalized as tinnitus. However, in a study of normal human listeners exposed continuously for 2 weeks to noise with an effective bandwidth of 1 to 8 kHz, peaking at ∼50 dB SPL near 6 kHz, Formby, Sherlock, & Gold (2003) found reversible hypoacusis at both 0.5 and 2 kHz, that is, both below and within the exposure band, but no hyperacusis or tinnitus. Hypoacusis was also observed as an increase in loudness discomfort levels following low-level noise therapy in people with tinnitus and hyperacusis (Formby, Sherlock, Gold, & Hawley, 2007; Noreña & Chery-Croze, 2007), and following first-time hearing aid use (Munro & Merrett, 2013). Interestingly, the amelioration of hyperacusis observed in some of these studies was also not frequency-specific and occurred even at frequencies that were not stimulated by the noise generators or hearing aids. Furthermore, people with clinical hyperacusis often experience it quite uniformly across frequency, irrespective of the range and amount of their hearing loss (Sheldrake, Diehl, & Schaette, 2015). In contrast, auditory deprivation during a week or so of continuous earplug use did induce temporary hyperacusis (Formby et al., 2003) and tinnitus (Schaette, Turtle, & Munro, 2013) in the majority of normal-hearing subjects. Collectively, these human studies strongly suggest that the mice in the present study should at least have experienced hypoacusis after noise exposure (if not also hyperacusis, as hypothesized based on our previous work in cats).
A possible reason for why hypoacusis (and perhaps also hyperacusis) was not detected here is that, as mentioned earlier, nondamaging noise induces plasticity mainly at the thalamocortical level, whereas tone PPI of the ASR appears sensitive to changes mainly at the (preattentive) brainstem level, as evidenced by studies on decerebrate rats (Davis & Gendelman, 1977; Fox, 1979; Li & Frost, 2000). Thus, tone PPI may not be suitable for studying the perceptual correlates of neural changes that are observed mainly at the thalamocortical level, but not in the brainstem.
This important caveat may not apply entirely to gap PPI or GPIAS, which at present is by far the most popular way of testing for tinnitus in rodents (Galazyuk & Hébert, 2015; Turner et al., 2006). In the present study, GPIAS testing was performed using both 50 and 20 ms gaps, as previous work has suggested that ACx is not essential for GPIAS with the longer gap duration, but becomes important with the shorter gap duration (Bowen et al., 2003; Ison et al., 1991; Weible et al., 2014). Nevertheless, mice tested negative for tinnitus here using both gap durations. This is perhaps surprising given that several recent GPIAS studies have reported chronic tinnitus in guinea pigs after 2 hr unilateral exposures to NBN at 97 dB SPL, which produced only TTS with no apparent evidence of synaptopathy (Basura et al., 2015; Wu et al., 2016). To add to the mystery, Hickox and Liberman (2014) did not find GPIAS-based evidence for tinnitus in CBA/Ca mice after a proven synaptopathic noise dose (2 h, 100 dB SPL, 8–16 kHz). This last negative finding is especially surprising in light of a recent study showing that the 100 dB SPL noise dose increased SFRs in the mouse ICC by an even greater margin than exposure at 105 dB SPL, which caused more NIHL, including damage to hair cells (Hesse et al., 2016).
Does Central Auditory Hyperactivity Trigger Tinnitus or Hyperacusis?
An important article by Gao, Manzoor, and Kaltenbach (2016) reported that just 2 min of loud noise exposure can trigger transient increases in dorsal cochlear nucleus SFRs despite little or no TTS and proposed that this could underlie the transient tinnitus experienced by people after similarly brief noise doses (Atherley, Hempstock, & Noble, 1968). Nevertheless, the link between central hyperactivity and tinnitus remains somewhat tenuous. Recent studies on rats (Ropp et al., 2014), guinea pigs (Coomber et al., 2014), and mice (Longenecker & Galazyuk, 2016) found increased SFRs in the ICC of all noise-exposed animals, including those that tested negative for tinnitus using GPIAS. A potential explanation here is that the tinnitus can be silenced by feedback from other brain areas, without affecting auditory brainstem hyperactivity. In a study of rats exposed for 1 hr to 115 dB SPL, octave-band noise centered at 16 kHz, Engineer et al. (2011) found that 18 of 28 rats exhibited positive GPIAS results in the 8 to 10 kHz range. They then found that a treatment consisting of paired tone pip and vagus nerve stimulation reversed the positive tinnitus results, despite the fact that A1 SFRs increased further. Some of the findings related to hyperacusis also remain puzzling. Notably, direct salicylate application to the cochlea failed to produce the sound-evoked hyperactivity seen in A1 with systemic salicylate injection, although the temporary peripheral losses and the ASR-based evidence for hyperacusis were similar in both cases (Sun et al., 2009). Thus, while central hyperactivity appears to be a general consequence of NIHL, hyperactivity alone may not be sufficient for the tinnitus (or hyperacusis) percept to emerge.
Tinnitus as a Possible Consequence of Noise Exposure in the Absence of Hearing Loss, Including Synaptopathy
A number of studies have reported evidence of cochlear damage in people with tinnitus and clinically normal audiograms (Gu, Herrmann, Levine, & Melcher, 2012; Paul, Bruce, & Roberts, 2017; Schaette & McAlpine, 2011; Weisz, Hartmann, Dohrmann, Schlee, & Noreña, 2006), but tinnitus can also occur in the absence of any hearing loss as a symptom of head and neck injury (Folmer & Griest, 2003; Shore, 2011). In animals, acute noise exposure that produced only TTS but no apparent synaptopathy could also lead to tinnitus (Basura et al., 2015; Wu et al., 2016). A large study of Danish workers reported that the prevalence of tinnitus increased with occupational noise exposure level and duration in workers with hearing loss but was not associated with the noise dose in workers with clinically normal hearing (Rubak et al., 2008). On the other hand, Guest, Munro, Prendergast, Howe, and Plack (2017) found a link between tinnitus and noise exposure history in young adults with normal audiograms, but no evidence of synaptopathy or other cochlear damage. It thus remains to be confirmed whether or not chronic exposure to lower, borderline synaptopathic noise levels could be an additional path to tinnitus, in spite of the negative GPIAS results reported here.
Acknowledgments
My thanks to AuD students—Evan Draper, Amanda McVey, Trevor Simones, Jessica Strzepek, and Roksolana Voshchilo—for assisting with the data collection. Thanks also to Dr. Ryan Longenecker for several very helpful discussions on optimizing startle data collection and to Drs. Longenecker, Jos Eggermont, and Larry Roberts and two anonymous reviewers for their comments on a previous version of this article.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is financially supported by Pennsylvania Lions Hearing Research Foundation.
References
- Adjamian P., Sereda M., Hall D. A. (2009) The mechanisms of tinnitus: perspectives from human functional neuroimaging. Hearing Research 253: 15–31. DOI: 10.1016/j.heares.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Atherley C. C., Hempstock T. I., Noble W. G. (1968) Study of tinnitus induced temporarily by noise. Journal of the Acoustical Society of America 44: 1503–1506. [DOI] [PubMed] [Google Scholar]
- Basura G. J., Koehler S. D., Shore S. E. (2015) Bimodal stimulus timing-dependent plasticity in primary auditory cortex is altered after noise exposure with and without tinnitus. Journal of Neurophysiology 114: 3064–3075. DOI: 10.1152/jn.00319.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beste C., Dinse H. R. (2013) Learning without training. Current Biology 23: 489–499. DOI: 10.1016/j.cub.2013.04.044. [DOI] [PubMed] [Google Scholar]
- Bharadwaj H. M., Verhulst S., Shaheen L., Liberman M. C., Shinn-Cunningham B. G. (2014) Cochlear neuropathy and the coding of supra-threshold sound. Frontiers in System Neuroscience 8: 26 DOI: 10.3389/fnsys.2014.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourien J., Tang Y., Batrel C., Huet A., Lenoir M., Ladrech S., Desmadryl G., Nouvian R., Puel J. L., Wang J. (2014) Contribution of auditory nerve fibers to compound action potential of the auditory nerve. Journal of Neurophysiology 112: 1025–1039. DOI: 10.1152/jn.00738.2013. [DOI] [PubMed] [Google Scholar]
- Bowen G. P., Lin D., Taylor M. K., Ison J. R. (2003) Auditory cortex lesions in the rat impair both temporal acuity and noise increment thresholds, revealing a common neural substrate. Cerebral Cortex 13: 815–822. [DOI] [PubMed] [Google Scholar]
- Bruno R. M., Sakmann B. (2006) Cortex is driven by weak but synchronously active thalamocortical synapses. Science 312: 1622–1627. DOI: 10.1126/science.1124593. [DOI] [PubMed] [Google Scholar]
- Canlon B., Borg E., Flock A. (1988) Protection against noise trauma by pre-exposure to a low level acoustic stimulus. Hearing Research 34: 197–200. [DOI] [PubMed] [Google Scholar]
- Carlson S., Willott J. F. (1996) The behavioral salience of tones as indicated by prepulse inhibition of the startle response: relationship to hearing loss and central neural plasticity in C57BL/6J mice. Hearing Research 99: 168–175. [DOI] [PubMed] [Google Scholar]
- Chen G., Lee C., Sandridge S. A., Butler H. M., Manzoor N. F., Kaltenbach J. A. (2013) Behavioral evidence for possible simultaneous induction of hyperacusis and tinnitus following intense sound exposure. Journal of the Association for Research in Otolaryngology 14: 413–424. DOI: 10.1007/s10162-013-0375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coomber B., Berger J. I., Kowalkowski V. L., Shackleton T. M., Palmer A. R., Wallace M. N. (2014) Neural changes accompanying tinnitus following unilateral acoustic trauma in the guinea pig. European Journal of Neuroscience 40: 2427–2441. DOI: 10.1111/ejn.12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M., Gendelman P. M. (1977) Plasticity of the acoustic startle response in the acutely decerebrate rat. Journal of Comparative Physiology and Psychology 91: 549–563. [DOI] [PubMed] [Google Scholar]
- de Villers-Sidani E., Alzghoul L., Zhou X., Simpson K. L., Lin R. C., Merzenich M. M. (2010) Recovery of functional and structural age-related changes in the rat primary auditory cortex with operant training. Proceedings of the National Academy of Sciences USA 107: 13900–13905. DOI: 10.1073/pnas.1007885107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobie R. A., Humes L. E. (2017) Commentary on the regulatory implications of noise-induced cochlear neuropathy. International Journal of Audiology 56: 74–78. DOI: 10.1080/14992027.2016.1255359. [DOI] [PubMed] [Google Scholar]
- Eggermont J. J. (2017. a) Acquired hearing loss and brain plasticity. Hearing Research 343: 176–190. DOI: 10.1016/j.heares.2016.05.008. [DOI] [PubMed] [Google Scholar]
- Eggermont J. J. (2017. b) Effects of long-term non-traumatic noise exposure on the adult central auditory system. Hearing problems without hearing loss. Hearing Research 352: 12–22. DOI: 10.1016/j.heares.2016.10.015. [DOI] [PubMed] [Google Scholar]
- Eggermont J. J. (2012) The Neuroscience of Tinnitus, Oxford, England: Oxford University Press. [Google Scholar]
- Eggermont J. J., Kenmochi M. (1998) Salicylate and quinine selectively increase spontaneous firing rates in secondary auditory cortex. Hearing Research 117: 149–160. [DOI] [PubMed] [Google Scholar]
- Elgoyhen A. B., Langguth B., De Ridder D., Vanneste S. (2015) Tinnitus: perspectives from human neuroimaging. Nature Reviews Neuroscience 16: 632–642. DOI: 10.1038/nrn4003. [DOI] [PubMed] [Google Scholar]
- Engineer N. D., Riley J. R., Seale J. D., Vrana W. A., Shetake J. A., Sudanagunta S. P., Borland M. S., Kilgard M. P. (2011) Reversing pathological neural activity using targeted plasticity. Nature 470: 101–104. DOI: 10.1038/nature09656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez K. A., Jeffers P. W., Lall K., Liberman M. C., Kujawa S. G. (2015) Aging after noise exposure: Acceleration of cochlear synaptopathy in “recovered” ears. Journal of Neuroscience 35: 7509–7520. DOI: 10.1523/JNEUROSCI.5138-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmer R. L., Griest S. E. (2003) Chronic tinnitus resulting from head or neck injuries. Laryngoscope 113: 821–827. DOI: 10.1097/00005537-200305000-00010. [DOI] [PubMed] [Google Scholar]
- Formby C., Sherlock L. P., Hawley M. L., Gold S. L. (2017) A sound therapy-based intervention to expand the auditory dynamic range for loudness among persons with sensorineural hearing losses: case evidence showcasing treatment efficacy. Seminars in Hearing 38: 130–150. DOI: 10.1055/s-0037-1598069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formby C., Sherlock L. P., Gold S. L., Hawley M. L. (2007) Adaptive recalibration of chronic auditory gain. Seminars in Hearing 28: 295–302. [Google Scholar]
- Formby C., Sherlock L. P., Gold S. L. (2003) Adaptive plasticity of loudness induced by chronic attenuation and enhancement of the acoustic background. Journal of the Acoustical Society of America 114: 55–58. [DOI] [PubMed] [Google Scholar]
- Fox J. E. (1979) Habituation and prestimulus inhibition of the auditory startle reflex in decerebrate rats. Physiology & Behavior 23: 291–297. [DOI] [PubMed] [Google Scholar]
- Furman A. C., Kujawa S. G., Liberman M. C. (2013) Noise-induced cochlear neuropathy is selective for fibers with low spontaneous rates. Journal of Neurophysiology 110: 577–586. DOI: 10.1152/jn.00164.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galazyuk A., Hébert S. (2015) Gap-prepulse inhibition of the acoustic startle reflex (GPIAS) for tinnitus assessment: Current status and future directions. Frontiers in Neurology 6: 88 DOI: 10.3389/fneur.2015.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y., Manzoor N., Kaltenbach J. A. (2016) Evidence of activity-dependent plasticity in the dorsal cochlear nucleus, in vivo, induced by brief sound exposure. Hearing Research 341: 31–42. DOI: 10.1016/j.heares.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green D. B., Ohlemacher J., Rosen M. J. (2016) Benefits of stimulus exposure: developmental learning independent of task performance. Frontiers in Neuroscience 10: 263 DOI: 10.3389/fnins.2016.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsley C. A., Longenecker R. J., Rosen M. J., Young J. W., Grimsley J. M., Galazyuk A. V. (2015) An improved approach to separating startle data from noise. Journal of Neuroscience Methods 253: 206–217. DOI: 10.1016/j.jneumeth.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J. W., Herrmann B. S., Levine R. A., Melcher J. R. (2012) Brainstem auditory evoked potentials suggest a role for the ventral cochlear nucleus in tinnitus. Journal of the Association for Research in Otolaryngology 13: 819–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J. W., Halpin C. F., Nam E. C., Levine R. A., Melcher J. R. (2010) Tinnitus, diminished sound-level tolerance, and elevated auditory activity in humans with normal hearing sensitivity. Journal of Neurophysiology 104: 3361–3370. DOI: 10.1007/s10162-012-0344-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guest H., Munro K. J., Prendergast G., Howe S., Plack C. J. (2017) Tinnitus with a normal audiogram: Relation to noise exposure but no evidence for cochlear synaptopathy. Hearing Research 344: 265–274. DOI: 10.1016/j.heares.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson D., Bielefeld E. C., Harris K. C., Hu B. H. (2006) The role of oxidative stress in noise-induced hearing loss. Ear & Hearing 27: 1–19. DOI: 10.1097/01.aud.0000191942.36672.f3. [DOI] [PubMed] [Google Scholar]
- Hesse L. L., Bakay W., Ong H. C., Anderson L., Ashmore J., McAlpine D., Linden J., Schaette R. (2016) Non-monotonic relation between noise exposure severity and neuronal hyperactivity in the auditory midbrain. Frontiers in Neurology 7: 133 DOI: 10.3389/fneur.2016.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickox A. E., Liberman M. C. (2014) Is noise-induced cochlear neuropathy key to the generation of hyperacusis or tinnitus? Journal of Neurophysiology 111: 552–564. DOI: 10.1152/jn.00184.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ison J. R., Allen P. D., O'Neill W. E. (2007) Age-related hearing loss in C57BL/6J mice has both frequency-specific and non-frequency-specific components that produce a hyperacusis-like exaggeration of the acoustic startle reflex. Journal of the Association for Research in Otolaryngology 8: 539–550. DOI: 10.1007/s10162-007-0098-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ison J. R., O’Connor K., Bowen G. P., Bocirnea A. (1991) Temporal resolution of gaps in noise by the rat is lost with functional decortication. Behavioral Neuroscience 105: 33–40. [DOI] [PubMed] [Google Scholar]
- Johnstone B. M., Patuzzi R., Yates G. K. (1986) Basilar membrane measurements and the travelling wave. Hearing Research 22: 147–153. [DOI] [PubMed] [Google Scholar]
- Kaltenbach J. A., Zacharek M. A., Zhang J., Frederick S. (2004) Activity in the dorsal cochlear nucleus of hamsters previously tested for tinnitus following intense tone exposure. Neuroscience Letters 355: 121–125. [DOI] [PubMed] [Google Scholar]
- Kamal B., Holman C., de Villers-Sidani E. (2013) Shaping the aging brain: Role of auditory input patterns in the emergence of auditory cortical impairments. Frontiers in System Neuroscience 7: 52 DOI: 10.3389/fnsys.2013.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M. (1999) The neurobiology of startle. Progress in Neurobiology 59: 107–128. [DOI] [PubMed] [Google Scholar]
- Krauss P., Tziridis K., Metzner C., Schilling A., Hoppe U., Schulze H. (2016) Stochastic resonance controlled upregulation of internal noise after hearing loss as a putative cause of tinnitus-related neuronal hyperactivity. Frontiers in Neuroscience 10: 597 DOI: 10.3389/fnins.2016.00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujala T., Shtyrov Y., Winkler I., Saher M., Tervaniemi M., Sallinen M., Teder-Sälejärvi W., Alho K., Reinikainen K., Näätänen R. (2004) Long-term exposure to noise impairs cortical sound processing and attention control. Psychophysiology 41: 875–881. [DOI] [PubMed] [Google Scholar]
- Kujawa S. G., Liberman M. C. (2015) Synaptopathy in the noise-exposed and aging cochlea: Primary neural degeneration in acquired sensorineural hearing loss. Hearing Research 330: 191–199. DOI: 10.1016/j.heares.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujawa S. G., Liberman M. C. (2009) Adding insult to injury: cochlear nerve degeneration after "temporary" noise-induced hearing loss. Journal of Neuroscience 29: 14077–14085. DOI: 10.1523/JNEUROSCI.2845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C., Zhang J. W., McPherson B., Pienkowski M., Wu E. X. (2015. a) Functional magnetic resonance imaging of the adult rat central auditory system following long-term, passive exposure to non-traumatic acoustic noise. Neuroimage 107: 1–9. DOI: 10.1016/j.neuroimage.2014.11.048. [DOI] [PubMed] [Google Scholar]
- Lau C., Pienkowski M., Zhang J. W., McPherson B., Wu E. X. (2015. b) Chronic exposure to broadband noise at moderate sound pressure levels spatially shifts tone-evoked responses in the rat auditory midbrain. Neuroimage 122: 44–51. DOI: 10.1016/j.neuroimage.2015.07.065. [DOI] [PubMed] [Google Scholar]
- Li L., Frost B. J. (2000) Azimuthal directional sensitivity of prepulse inhibition of the pinna startle reflex in decerebrate rats. Brain Research Bulletin 51: 95–100. [DOI] [PubMed] [Google Scholar]
- Li S., Choi V., Tzounopoulos T. (2013) Pathogenic plasticity of Kv7.2/3 channel activity is essential for the induction of tinnitus. Proceedings of the National Academy of Sciences USA 110: 9980–9985. DOI: 10.1073/pnas.1302770110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobarinas E., Spankovich C., Le Prell C. G. (2017) Evidence of "hidden hearing loss" following noise exposures that produce robust TTS and ABR wave-I amplitude reductions. Hearing Research 349: 155–163. DOI: 10.1016/j.heares.2016.12.009. [DOI] [PubMed] [Google Scholar]
- Lobarinas E., Hayes S. H., Allman B. L. (2013) The gap-startle paradigm for tinnitus screening in animal models: limitations and optimization. Hearing Research 295: 150–160. DOI: 10.1016/j.heares.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longenecker R. J., Galazyuk A. V. (2016) Variable effects of acoustic trauma on behavioral and neural correlates of tinnitus in individual animals. Frontiers in Behavioral Neuroscience 10: 207 DOI: 10.3389/fnbeh.2016.00207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longenecker R. J., Alghamdi F., Rosen M. J., Galazyuk A. V. (2016) Prepulse inhibition of the acoustic startle reflex vs. auditory brainstem response for hearing assessment. Hearing Research 339: 80–93. DOI: 10.1016/j.heares.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., Lobarinas E., Deng A., Goodey R., Stolzberg D., Salvi R. J., Sun W. (2011) GABAergic neural activity involved in salicylate-induced auditory cortex gain enhancement. Neuroscience 189: 187–198. DOI: 10.1016/j.neuroscience.2011.04.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison S. F., Usubuchi H., Liberman M. C. (2013) Efferent feedback minimizes cochlear neuropathy from moderate noise exposure. Journal of Neuroscience 27: 5542–5552. DOI: 10.1523/JNEUROSCI.5027-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitzdorf U. (1985) Current source-density method and application in cat cerebral cortex: investigation of evoked potentials and EEG phenomena. Physiological Reviews 65: 37–100. [DOI] [PubMed] [Google Scholar]
- Munguia R., Pienkowski M., Eggermont J. J. (2013) Spontaneous firing rate changes in cat primary auditory cortex following long-term exposure to non-traumatic noise: Tinnitus without hearing loss? Neuroscience Letters 546: 46–50. DOI: 10.1016/j.neulet.2013.04.048. [DOI] [PubMed] [Google Scholar]
- Munro K. J., Merrett J. F. (2013) Brainstem plasticity and modified loudness following short-term use of hearing aids. Journal of the Acoustic Society of America 133: 343–349. DOI: 10.1121/1.4770234. [DOI] [PubMed] [Google Scholar]
- NIOSH (1998). Criteria for a recommended standard: Occupational noise exposure. National Institute for Occupational Safety and Health Publication No: 98–126. Retrieved from https://www.cdc.gov/niosh/docs/98-126/pdfs/98-126.pdf.
- Noreña A. J., Chery-Croze S. (2007) Enriched acoustic environment rescales auditory sensitivity. Neuroreport 18: 1251–1255. DOI: 10.1097/WNR.0b013e3282202c35. [DOI] [PubMed] [Google Scholar]
- Noreña A. J., Gourévitch B., Aizawa N., Eggermont J. J. (2006) Spectrally enhanced acoustic environment disrupts frequency representation in cat auditory cortex. Nature Neuroscience 9: 932–939. DOI: 10.1038/nn1720. [DOI] [PubMed] [Google Scholar]
- Noreña A. J., Eggermont J. J. (2006) Enriched acoustic environment after noise trauma abolishes neural signs of tinnitus. Neuroreport 17: 559–563. [DOI] [PubMed] [Google Scholar]
- Noreña A. J., Eggermont J. J. (2005) Enriched acoustic environment after noise trauma reduces hearing loss and prevents cortical map reorganization. Journal of Neuroscience 25: 699–705. DOI: 10.1523/JNEUROSCI.2226-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OSHA (2002). Hearing conservation. Occupational Safety and Health Administration, U.S. Department of Labor, Publication No: OSHA 3074. Retrieved from http://www.osha.gov/Publications/osha3074.pdf.
- Paul B. T., Bruce I. C., Roberts L. E. (2017) Evidence that hidden hearing loss underlies amplitude modulation encoding deficits in individuals with and without tinnitus. Hearing Research 344: 170–182. DOI: 10.1016/j.heares.2016.11.010. [DOI] [PubMed] [Google Scholar]
- Pienkowski M. (2017) On the etiology of listening difficulties in noise despite clinically normal audiograms. Ear & Hearing 38: 135–148. DOI: 10.1097/AUD.0000000000000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pienkowski, M. (2016). Acoustic startle reflex audiometry following non-traumatic noise exposure in CBA/CaJ mice (Abstract #186). Paper presented at the 39th Midwinter Meeting of the Association for Research in Otolaryngology, San Diego, CA.
- Pienkowski M., Tyler R. S., Roncancio E. R., Jun H. J., Brozoski T., Dauman N., Coelho C. B., Andersson G., Keiner A. J., Cacace A. T., Martin N., Moore B. C. (2014) A review of hyperacusis and future directions: Part II. Measurement, mechanisms, and treatment. American Journal of Audiology 23: 420–436. DOI: 10.1044/2014_AJA-13-0037. [DOI] [PubMed] [Google Scholar]
- Pienkowski M., Munguia R., Eggermont J. J. (2013) Effects of passive, moderate-level sound exposure on the mature auditory cortex: Spectral edges, spectrotemporal density, and real-world noise. Hearing Research 296: 121–130. DOI: 10.1016/j.heares.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Pienkowski M., Eggermont J. J. (2012) Reversible long-term changes in auditory processing in mature auditory cortex in the absence of hearing loss induced by passive, moderate-level sound exposure. Ear & Hearing 33: 305–314. DOI: 10.1097/AUD.0b013e318241e880. [DOI] [PubMed] [Google Scholar]
- Pienkowski M., Eggermont J. J. (2011) Cortical tonotopic map plasticity and behavior. Neuroscience & Biobehavioral Reviews 35: 2117–2128. DOI: 10.1016/j.neubiorev.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Pienkowski M., Munguia R., Eggermont J. J. (2011) Passive exposure of adult cats to bandlimited tone ensembles or noise leads to long-term response suppression in auditory cortex. Hearing Research 277: 117–126. DOI: 10.1016/j.heares.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Pienkowski M., Eggermont J. J. (2010. a) Intermittent exposure with moderate-level sound impairs central auditory function of mature animals without concomitant hearing loss. Hearing Research 261: 30–35. DOI: 10.1016/j.heares.2009.12.025. [DOI] [PubMed] [Google Scholar]
- Pienkowski M., Eggermont J. J. (2010. b) Passive exposure of adult cats to moderate-level tone pip ensembles differentially decreases AI and AII responsiveness in the exposure frequency range. Hearing Research 268: 151–162. DOI: 10.1016/j.heares.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Pienkowski M., Eggermont J. J. (2009) Long-term, partially-reversible reorganization of frequency tuning in mature cat primary auditory cortex can be induced by passive exposure to moderate-level sounds. Hearing Research 257: 24–40. DOI: 10.1016/j.heares.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Ropp T. J., Tiedemann K. L., Young E. D., May B. J. (2014) Effects of unilateral acoustic trauma on tinnitus-related spontaneous activity in the inferior colliculus. Journal of the Association for Research in Otolaryngology 15: 1007–1022. DOI: 10.1007/s10162-014-0488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubak T., Kock S., Koefoed-Nielsen B., Lund S. P., Bonde J. P., Kolstad H. A. (2008) The risk of tinnitus following occupational noise exposure in workers with hearing loss or normal hearing. International Journal of Audiology 47: 109–114. DOI: 10.1080/14992020701581430. [DOI] [PubMed] [Google Scholar]
- Ruggero, M. A., Rich, N. C., Robles, L., & Recio A. (1990). The effects of acoustic trauma, other cochlear injury, and death on basilar-membrane responses to sound. In: A. Axelsson, H. Borchgrevink, P. A. Hellström, D. Henderson, R. P. Hamernik, & R. Salvi (Eds.), Scientific Basis of Noise-Induced Hearing Loss (pp. 23–35). New NY, USA: Thieme Medical Publishers.
- Salloum R. H., Sandridge S., Patton D. J., Stillitano G., Dawson G., Niforatos J., Santiago L., Kaltenbach J. A. (2016) Untangling the effects of tinnitus and hypersensitivity to sound (hyperacusis) in the gap detection test. Hearing Research 331: 92–100. DOI: 10.1016/j.heares.2015.10.005. [DOI] [PubMed] [Google Scholar]
- Schaette R., Turtle C., Munro K. J. (2013) Reversible induction of phantom auditory sensations through simulated unilateral hearing loss. PLoS One 7: e35238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaette R., McAlpine D. (2011) Tinnitus with a normal audiogram: physiological evidence for hidden hearing loss and computational model. Journal of Neuroscience 31: 13452–13457. DOI: 10.1523/JNEUROSCI.2156-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaette R., König O., Hornig D., Gross M., Kempter R. (2010) Acoustic stimulation treatments against tinnitus could be most effective when tinnitus pitch is within the stimulated frequency range. Hearing Research 269: 95–101. DOI: 10.1016/j.heares.2010.06.022. [DOI] [PubMed] [Google Scholar]
- Sedley W., Friston K. J., Gander P. E., Kumar S., Griffiths T. D. (2016) An integrative tinnitus model based on sensory precision. Trends in Neurosciences 39: 799–812. DOI: 10.1016/j.tins.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrake J., Diehl P. U., Schaette R. (2015) Audiometric characteristics of hyperacusis patients. Frontiers in Neurology 6: 105 DOI: 10.3389/fneur.2015.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore S. E. (2011) Plasticity of somatosensory inputs to the cochlear nucleus – implications for tinnitus. Hearing Research 281: 38–46. DOI: 10.1016/j.heares.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm J. J., Zhang-Hooks Y. X., Roos H., Nguyen T., Kandler K. (2017) Noise trauma induced behavioral gap detection deficits correlate with reorganization of excitatory and inhibitory local circuits in the inferior colliculus and are prevented by acoustic enrichment. Journal of Neuroscience 37: 6314–6330. DOI: 10.1523/JNEUROSCI.0602-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W., Deng A., Jayaram A., Gibson B. (2012) Noise exposure enhances auditory cortex responses related to hyperacusis behavior. Brain Research 1485: 108–116. DOI: 10.1016/j.brainres.2012.02.008. [DOI] [PubMed] [Google Scholar]
- Sun W., Lu J., Stolzberg D., Gray L., Deng A., Lobarinas E., Salvi R. J. (2009) Salicylate increases the gain of the central auditory system. Neuroscience 159: 325–334. DOI: 10.1016/j.neuroscience.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka C., Chen G. D., Hu B. H., Chi L. H., Li M., Zheng G., Bielefeld E. C., Jamesdaniel S., Coling D., Henderson D. (2009) The effects of acoustic environment after traumatic noise exposure on hearing and outer hair cells. Hearing Research 250: 10–18. DOI: 10.1016/j.heares.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Thai-Van H., Micheyl C., Noreña A., Veuillet E., Gabriel D., Collet L. (2007) Enhanced frequency discrimination in hearing-impaired individuals: a review of perceptual correlates of central neural plasticity induced by cochlear damage. Hearing Research 233: 14–22. DOI: 10.1016/j.heares.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Turner J. G., Parish J. (2008) Gap detection methods for assessing salicylate-induced tinnitus and hyperacusis in rats. American Journal of Audiology 17: S185–S192. [DOI] [PubMed] [Google Scholar]
- Turner J. G., Brozoski T. J., Bauer C. A., Parrish J. L., Myers K., Hughes L. F., Caspary D. M. (2006) Gap detection deficits in rats with tinnitus: a potential novel screening tool. Behavioral Neuroscience 120: 188–195. DOI: 10.1037/0735-7044.120.1.188. [DOI] [PubMed] [Google Scholar]
- Tyler R. S., Pienkowski M., Roncancio E. R., Hyungjin-jun J., Brozoski T., Dauman N., Coelho C. B., Anderrson G., Keiner A. J., Cacace A., Martin N., Moore B. C. (2014) A review of hyperacusis and future directions. Part I. Definitions and manifestations. American Journal of Audiology 23: 402–419. DOI: 10.1044/2014_AJA-14-0010. [DOI] [PubMed] [Google Scholar]
- Valero M. D., Burton J. A., Hauser S. N., Hackett T. A., Ramachandran R., Liberman M. C. (2017) Noise-induced cochlear synaptopathy in rhesus monkeys (Macaca mulatta). Hearing Research 353: 213–223. DOI: 10.1016/j.heares.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weible A. P., Moore A. K., Liu C., DeBlander L., Wu H., Kentros C., Wehr M. (2014) Perceptual gap detection is mediated by gap termination responses in auditory cortex. Current Biology 24: 1447–1455. DOI: 10.1016/j.cub.2014.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz N., Hartmann T., Dohrmann K., Schlee W., Noreña A. (2006) High-frequency tinnitus without hearing loss does not mean absence of deafferentation. Hearing Research 222: 108–114. DOI: 10.1016/j.heares.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Willott J. F., Bross L. (2004) Effects of prolonged exposure to an augmented acoustic environment on the auditory system of middle-aged C57BL/6J mice: cochlear and central histology and sex differences. Journal of Comparative Neurology 472: 358 370. DOI: 10.1002/cne.20065. [DOI] [PubMed] [Google Scholar]
- Willott J. F., Turner J. G. (1999) Prolonged exposure to an augmented acoustic environment ameliorates age-related auditory changes in C57BL/6J and DBA/2J mice. Hearing Research 135: 78–88. [DOI] [PubMed] [Google Scholar]
- Wright B. A., Sabin A. T., Zhang Y., Marrone N., Fitzgerald M. B. (2010) Enhancing perceptual learning by combining practice with periods of additional sensory stimulation. Journal of Neuroscience 30: 12868–12877. DOI: 10.1523/JNEUROSCI.0487-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Martel D. T., Shore S. E. (2016) Increased synchrony and bursting of dorsal cochlear nucleus fusiform cells correlate with tinnitus. Journal of Neuroscience 36: 2068–2073. DOI: 10.1523/JNEUROSCI.3960-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong B., Alkharabsheh A., Manohar S., Chen G. D., Yu N., Zhao X., Salvi R., Sun W. (2017) Hyperexcitability of inferior colliculus and acoustic startle reflex with age-related hearing loss. Hearing Research 350: 32–42. DOI: 10.1016/j.heares.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G., Lobarinas E., Zhang L., Turner J., Stolzberg D., Salvi R., Sun W. (2007) Salicylate induced tinnitus: behavioral measures and neural activity in auditory cortex of awake rats. Hearing Research 226: 244–253. DOI: 10.1016/j.heares.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Zhang X., Yang P., Cao Y., Qin L., Sato Y. (2011) Salicylate induced neural changes in the primary auditory cortex of awake cats. Neuroscience 172: 232–245. DOI: 10.1016/j.neuroscience.2010.10.073. [DOI] [PubMed] [Google Scholar]
- Zheng W. (2012) Auditory map reorganization and pitch discrimination in adult rats chronically exposed to low-level ambient noise. Frontiers in System Neuroscience 6: 65 DOI: 10.3389/fnsys.2012.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Merzenich M. M. (2012) Environmental noise exposure degrades normal listening processes. Nature Communications 3: 843 DOI: 10.1038/ncomms1849. [DOI] [PubMed] [Google Scholar]