

Abstract
Background:
The perioperative period is associated with a high risk for human ischaemic stroke. Although inflammatory mechanisms are known to have an important role in cerebral infarction in the nonoperative setting, their role in modulating perioperative risk remains unclear.
Methods:
In this prospective case-control study, we compared 10 patients (cases) who developed magnetic resonance imaging (MRI) evidence of cerebral infarction following transcatheter aortic valve implantation with 10 patients (controls) who underwent the same procedure without neurological complication. Blood sampling was performed preoperatively (baseline) and at 24 h, 48 h and 72 h postoperatively and analysed for specific cytokines, chemokines and complement factors.
Results:
Baseline serum assessments identified significant differences between the two cohorts for levels of complement C3, complement C4b, granulocyte-macrophage colony-stimulating factor, interleukin-15 and macrophage inflammatory protein-1β. Longitudinal regression analysis and best-fit polynomial curves of postoperative analyte profiles identified significantly higher levels of complement C3 and matrix metalloproteinase-9, and lower levels of interferon-γ and macrophage inflammatory protein-1β levels in cases versus controls.
Conclusions:
These results support a potentially important role for inflammatory mechanisms in MRI-defined perioperative stroke and reveal a potentially important role for complement components in this process.
Keywords: clinical trials observational study, embolism, immunology, infarction, inflammation, magnetic resonance imaging
Introduction
Over the past decades, there have been substantial advances in our understanding of the immunological mechanisms that result from, contribute to, and modify cerebrovascular disease. This has revealed a complex cascade of events, with inflammatory signalling an active and integral participant in all stages.1 Following ischaemic brain injury, deleterious events such as oxidative stress, neuronal excitotoxicity, blood–brain barrier dysfunction, microvascular injury and postischaemic inflammation transpire. These changes are orchestrated by a complex interplay between neurons, microglial cells, astrocytes, endothelial cells, and the innate and adaptive immune system, all mediated by synergy between pro-inflammatory prostaglandins, cytokines and chemokines (Figure 1). However, this understanding has been primarily derived from preclinical (predominantly murine) models, with data from human investigations comparatively scarce. Significant immunological differences between these two species are well described,2 which could explain why the successful translation of novel therapeutic targets to manage ischaemic stroke in humans has, to date, proven elusive.
Figure 1.

Inflammatory disequilibrium in stroke. The components shown in bold were analysed in this study. (Copyright © 2016 Studio Kayama.)
GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; IP-10, interferon gamma-induced protein 10; MCP-1, monocyte chemoattractant protein 1; MIP, macrophage inflammatory protein; MMP-9, matrix metallopeptidase-9; TNF-α; tumour necrosis factor alpha.
The limited data specifically from humans show that acute inflammation is a major aetiological factor for ischaemic stroke. For example, acute infection is associated with a subsequent eight-fold increase in repeat stroke risk that persists for at least 3 months.3 Furthermore, levels of several inflammatory markers are altered in the postischaemic period, and these levels seem to be independently associated with clinical outcome.4 No such studies have investigated the role of immunological and inflammatory associations with cerebral infarction in the perioperative period.
Unpredictability in the occurrence and timing of stroke in the clinical setting has hampered the study of the subsequent acute response in humans. These issues are largely overcome in the perioperative setting, where there is a high risk of stroke occurring within a relatively short and well-defined timeframe. For example, following the transcatheter aortic valve implantation (TAVI) procedure that is used to treat severe aortic stenosis, there is a greater than 60% incidence of new cerebral infarcts detected with magnetic resonance imaging (MRI) in postoperative relative to preoperative scans.5 This injury has been presumed to be primarily embolic in nature but the exact aetiology remains uncertain, as hypoperfusion and inflammation probably also precipitate or exacerbate embolic injury.
Given the important and potentially modifiable role of inflammation in cerebral infarction in nonoperative settings, we investigated whether inflammatory mechanisms also modulate the risk for, and occurrence of, peri-interventional cerebral infarction.
Methods
Experimental design
A total of 20 patients were prospectively enrolled at The Prince Charles Hospital between March 2014 and October 2016 and included in this an a priori prospective case-control sub-study of a larger study assessing perioperative neurological injury in patients undergoing TAVI.6 All 20 patients underwent TAVI with a SAPIEN-XT valve (Edwards Lifesciences, Irvine, CA, USA) via transfemoral access and under standardized general anaesthesia for the management of severe aortic stenosis. Subsequent to the TAVI procedure, consecutive participants with complete blood samples (see below) were grouped according to whether they had postoperative MRI evidence of cerebral infarction (cases), or did not (controls). Patients were excluded from enrolment if any of the following criteria were met: one or more previous cerebrovascular events; any autoimmune or chronic inflammatory disease (including vascular disease); primary or secondary immunodeficiency (from disorders including diabetes, renal failure, liver failure, malignancy and asplenia/hyposplenism); a significant history of atopy or thrombosis; any recent infection; the recent use of immune-modulating or antibiotic medications. This last criterion did not include the dual antiplatelet thromboprophylaxis regimen (aspirin and clopidogrel) that all participants commenced preoperatively, or the heparin/protamine and cephazolin regimen that was routinely administered intraoperatively. Prior to any data collection, protocol approval was granted by the institution’s Human Research and Ethics Committee (HREC/12/QPCH/291). All participants provided written informed consent prior to their study participation.
Radiological assessment
MRI examinations were performed at baseline (within the 24 h prior to the procedure) and at day 3 ± 1 postoperatively, using a 1.5 Tesla MAGNETOM Aera (Siemens Healthcare, Erlanger, Germany). Perioperative cerebral infarction was defined as any “new focus of restricted diffusion (high diffusion weighted imaging signal and low apparent diffusion coefficient) occurring in either white or grey matter, located in the cerebrum, cerebellum or brainstem” on the postoperative MRI scan that was not apparent at baseline.7
Clinical assessments
The National Institutes of Health Stroke Scale was used to detect the occurrence of clinically apparent stroke and the modified Rankin Scale was used to categorize stroke severity. These tests were administered within 24 h prior to the TAVI procedure, and daily for the first 3 days postoperative.
Blood sampling and processing
In the 24 h prior to surgery, and 24 h, 48 h and 72 h postoperatively, blood samples were collected by peripheral phlebotomy and transferred immediately to the in-house laboratory for processing. One 3 ml ethylenediamine tetraacetic acid and one serum separator tube (SST) 5 ml, were processed immediately for ‘standard’ laboratory blood tests, including a full blood count (haemoglobin, platelets, white cell count and differential), and assessments of renal (serum creatinine and urea) and hepatic function (aspartate and alanine aminotransferase, alkaline phosphatase, gamma-glutamyl transferase, lactate dehydrogenase and albumin). A second SST tube was centrifuged at 3000 × g for 15 min upon its arrival in the laboratory; the serum then was aliquoted and samples frozen at −80°C until the time of assay at the completion of subject recruitment.
Immunological profiling of serum
Serum levels of high-sensitivity C-reactive protein were measured using a commercial enzyme-linked immunosorbent assay kit (DE740011, Demeditec Diagnostics GmbH, Kiel, Germany). Measures for the remaining immunological analytes were determined on a Millipore MAGPIX System (Merck, Darmstadt, Germany) using the following dedicated panels, as per the manufacturer’s instructions:
human complement panels (HCMP1MA-G19K08 and HCMP2MAG19K02, Merck) were used to measure serum levels of mannose-binding lectin, factor D, factor I, factor B, factor H, properdin and complement components 1q (C1q), C2, C3, C3b/iC3b, C4, C4b, C5, C5a and C9;
a human cytokine/chemokine panel (HCYTOMAG60K23, Merck) was used to measure levels of the following analytes: interleukin-1beta (IL-1β), IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12p40, IL-12p70, IL-13, IL-15, IL-17, IL-1 receptor-alpha (IL-1Rα), tumour necrosis factor alpha (TNF-α), interferon alpha-2 (IFN-α2), interferon gamma (IFN-γ), interferon gamma-induced protein 10 (IP-10) also known as C-X-C motif chemokine 10 (CXCL10), monocyte chemoattractant protein 1 (MCP-1) also known as chemokine C-C motif ligand 2, macrophage inflammatory protein 1 alpha (MIP-1α), macrophage inflammatory protein 1 beta (MIP-1β), eotaxin and granulocyte-macrophage colony-stimulating factor (GM-CSF);
a human matrix metallopeptidase-9 (MMP-9) bead panel (HMMP2MA-G55K01, Merck) was used to measure serum levels of MMP-9.
Statistical analysis
Group means and 95% confidence intervals (CIs) were calculated for all baseline variables. Student’s t test was performed to determine the statistical significance of any difference between MRI-positive cases and MRI-negative controls. As the kinetic profile is different for each analyte and the exact sampling time varied for each participant, changes from baseline were plotted as changes in concentration versus time. A smoothing model was then used to select the best-fitting polynomial curve over time for cases versus controls. Six polynomial curves were tested for each group and the best combination chosen using the selecting model with the smallest deviance (best fit to the data). The best fit was plotted using the mean and 95% CI. For each variable, inferential testing to identify statistically significant mean differences in the two curves was performed at the 24 h postoperative time point. As a last step, longitudinal regression was used to test for overall differences between cases and controls over all time points. A random intercept was used to account for repeated results from the same patient. The dependent variable of interest was, once again, change from baseline (time 0), to either three or four follow-up time points (depending on the duration of follow up). Three regression parameters were determined: (a) the measured difference between those who had new cerebral infarction versus those who did not; (b) a linear time trend, scaled to 10 h (used to control for any general change over time in all patients); (c) the constant (or intercept), which is the estimated value at time zero for all patients (a positive value indicates that values generally increased from baseline).
Results
Baseline data
Patient and procedural characteristics
Of the 20 patients enrolled, 10 had postoperative MRI evidence of cerebral infarction (cases), whereas 10 did not (controls). Baseline patient and procedural characteristics for cases and controls are summarized in Table 1. No significant differences in either patient or procedure characteristics were identified that might have confounded subsequent comparisons between groups.
Table 1.
Baseline characteristics of study participants.
| Variable | Controls (n = 10) | Cases (n = 10) |
|---|---|---|
| Patient characteristics | ||
| Age, years | 83.3 ± 9.4 | 84.2 ± 3.2 |
| Female | 4 (40%) | 5 (50%) |
| Body mass index, kg/m2 | 31.4 ± 8.4 | 27 ± 5.9 |
| EuroSCORE II, % | 5.9 ± 2.6 | 5.1 ± 3.1 |
| STS, % | 6.6 ± 3.6 | 5.5 ± 2.7 |
| Arteriovenous anastomosis, cm2 | 0.8 ± 0.1 | 0.8 ± 0.1 |
| Peak jet velocity | 4.3 ± 0.4 | 4.4 ± 0.6 |
| Mean pressure gradient | 49.2 ± 11.4 | 47 ± 16.2 |
| Pre-operative left ventricular ejection fraction | 58.4 ± 9.2 | 51.3 ± 16.7 |
| Procedural characteristics | ||
| Device success | 10 (100%) | 10 (100%) |
| Average procedure time, min | 93 ± 68 | 61 ± 13 |
STS, Society of Thoracic Surgeons.
Radiological assessments
The participants with MRI-defined cerebral infarction typically had several lesions with a median ± interquartile range of 3 ± 3 (95% CI 1–8) lesions/participant and a volume of 197 ± 776 µl/participant (data not shown). Where perioperative cerebral infarcts were sustained, 83% of the volume of infarction occurred within the cortical grey matter, with just 2% in subcortical grey matter, 4% in subcortical white matter, and 9% within the cerebellar cortex.
Clinical assessments
No clinically apparent symptoms of stroke were evident from the serial neurological assessments.
Blood and serum assessments
Baseline blood and serum assessments are summarized in Table 2. Note that no statistically or clinically significant differences were apparent in any blood cell counts or any marker of renal or liver function. Significant differences between MRI-positive (cases) and MRI-negative (controls) were identified for serum levels of complement C3 (38 ± 22 versus 73 ± 40, respectively; p = 0.005), complement C4b (4011 ± 783 versus 2357 ± 804; p = 0.048), GM-CSF (0.98 ± 0.35 versus 0.56 ± 0.25; p = 0.009), IL-15 (5.42 ± 6.7 versus 2.575 ± 0.15; p = 0.047) and MIP-1β (39 ± 32 versus 14.9 ± 6.50; p = 0.038).
Table 2.
Summary statistics for baseline blood analyte measurements in cases versus controls.
| Analyte | Controls |
Cases |
||
|---|---|---|---|---|
| Mean | SD | Mean | SD | |
| Routine laboratory tests | ||||
| White blood cell count | 6.820 | 2.205 | 6.460 | 2.667 |
| Neutrophils | 4.371 | 2.227 | 3.892 | 2.245 |
| Lymphocytes | 1.646 | 0.413 | 1.717 | 0.467 |
| Monocytes | 0.582 | 0.124 | 0.565 | 0.185 |
| Creatinine | 101.300 | 33.330 | 107.400 | 28.640 |
| Haemoglobin | 126.700 | 10.478 | 121.600 | 12.076 |
| Platelets | 173.300 | 60.369 | 201.200 | 51.441 |
| C2 | 5212.300 | 1413.629 | 8723.800 | 5881.982 |
| C4b* | 3256.600 | 803.701 | 4010.560 | 783.449 |
| C5 | 90,399.900 | 33,301.216 | 1,11,882.100 | 59,453.757 |
| C5a | 4912.400 | 1177.679 | 5797.600 | 1877.560 |
| C9 | 4278.200 | 1147.436 | 3867.500 | 1123.123 |
| Factor D | 1120.800 | 11.361 | 1125.200 | 11.361 |
| MBL | 3527.278 | 4822.419 | 3443.722 | 3548.954 |
| C1q | 44.723 | 14.821 | 39.727 | 18.430 |
| C3** | 72.862 | 39.904 | 38.026 | 22.024 |
| C3b/iC3b | 350.452 | 125.924 | 428.461 | 242.295 |
| C4 | 544.511 | 199.553 | 459.482 | 136.783 |
| Factor B | 147.966 | 64.978 | 117.032 | 37.082 |
| Factor H | 313.943 | 86.472 | 266.233 | 93.867 |
| Factor I | 11,162.800 | 65.583 | 11,188.200 | 65.583 |
| Properdin | 18.307 | 6.733 | 17.135 | 8.379 |
| Cytokines/chemokines | ||||
| Eotaxin | 134.869 | 53.173 | 164.066 | 48.438 |
| GM-CSF** | 0.559 | 0.247 | 0.975 | 0.348 |
| IFN-α2 | 3.922 | 1.153 | 43.972 | 89.830 |
| IFN-γ | 3.170 | 1.613 | 22.689 | 29.608 |
| IL-10 | 6.121 | 5.843 | 6.338 | 6.361 |
| IL-12p40 | 4.790 | 1.110 | 23.614 | 37.595 |
| IL-12p70 | 2.914 | 0.201 | 13.216 | 32.658 |
| IL-13 | 6.597 | 10.657 | 97.719 | 275.431 |
| IL-15* | 2.575 | 0.145 | 5.420 | 6.703 |
| IL-17 | 2.466 | 0.134 | 5.281 | 6.155 |
| IL-1rα | 65.631 | 175.526 | 144.218 | 358.130 |
| IL-1b | 2.970 | 0.129 | 4.944 | 6.454 |
| IL-2 | 1.791 | 0.987 | 10.513 | 17.697 |
| IL-4 | 7.150 | 5.637 | 27.545 | 58.181 |
| IL-5 | 2.394 | 1.299 | 33.162 | 92.903 |
| IL-6 | 6.007 | 5.886 | 7.344 | 9.446 |
| IL-7 | 2.458 | 1.627 | 9.904 | 16.032 |
| IL-8 | 15.445 | 14.992 | 13.820 | 17.883 |
| IP-10 | 509.487 | 288.678 | 486.979 | 235.094 |
| MCP-1 | 627.069 | 139.342 | 562.416 | 269.159 |
| MIP-1α | 3.671 | 1.046 | 10.757 | 11.650 |
| MIP-1β* | 14.902 | 6.516 | 39.008 | 32.052 |
| TNF-α | 12.716 | 7.090 | 17.411 | 12.572 |
| Other inflammatory markers | ||||
| MMP-9 | 1,95,266.800 | 95,023.810 | 1,52,542.000 | 85,619.562 |
| hs-CRP | 5484.9 | 5035.4 | 2075.2 | 2362.8 |
p < 0.05; **p < 0.001. hs-CRP, high-sensitivity C-reactive protein; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; IP-10, interferon gamma-induced protein 10; MBL, mannose-binding lectin; MCP-1, monocyte chemoattractant protein 1; MIP, macrophage inflammatory protein; MMP-9, matrix metallopeptidase-9; SD, standard deviation; TNF-α; tumour necrosis factor alpha.
Postoperative changes
Longitudinal regression
The three regression parameters (difference, time and constant/intercept) are presented for each variable in Supplementary Table 1. Of note, C3 (mean difference 25.79; 95% CI 1.19–42.24) levels were significantly higher (p < 0.05) whereas IFN-γ (mean difference −14.17; 95% CI −27.8 to −0.57) and MIP-1β levels (mean difference −10.6; 95% CI −20.67 to −0.47) were significantly lower (p < 0.05) postoperatively over time in the MRI-positive group than in the control group.
Smoothing model analysis
The best-fit polynomial curves for the change from baseline data for select analytes of interest are presented in Figure 2, with the remainder available in Supplementary Figure 1. Results of inferential testing performed 24 h postoperatively are shown in Table 3. At this time point, significantly higher levels of MMP-9 (mean difference 72,435; 95% CI 6401–135,310; p = 0.0272) and complement C3 (mean difference 25.9; 95% CI 4.46–47.8; p = 0.0288) were identified in MRI-positive cases, whereas platelets (mean difference −26.7; 95% CI −52.3 to −3.74; p = 0.0272), and IFN-γ (mean difference −14.3; 95% CI −27.5 to −2.1; p = 0.0288) were lower.
Figure 2.

Cases versus controls polynomial best-fit curves for select analytes.
Table 3.
Results of 24 h significance testing for differences between groups extracted from the polynomial curve of best-fit smoothing models.
| Analyte | Difference between groups |
|
|---|---|---|
| Mean difference | 95% confidence interval | |
| Routine laboratory tests | ||
| White blood cell count | 0.711 | −1.67, 3.1 |
| Neutrophils | 0.318 | −2.27, 2.93 |
| Lymphocytes | 0.0238 | −0.477, 0.504 |
| Monocytes | 0.0504 | −0.325, 0.399 |
| Creatinine | −0.604 | −17, 15.3 |
| Haemoglobin | 1.98 | −7.55, 11 |
| Platelets* | −26.7 | −52.3, −3.74 |
| Complement cascade components | ||
| C2 | −1028 | −6632, 4740 |
| C4b | −766 | −2311, 766 |
| C5 | −26,221 | −62,308, 9688 |
| C5a | 1131 | −653, 2986 |
| C9 | 210 | −364, 793 |
| Factor D | 12.8 | −26.5, 52.9 |
| MBL | −427 | −3089, 2298 |
| C1q | 1.62 | −13.1, 16.3 |
| C3* | 25.9 | 4.46, 47.8 |
| C3b/iC3b | 352 | −232, 958 |
| C4 | 23.4 | −118, 159 |
| Factor B | 14.8 | −38.9, 68.9 |
| Factor H | 18 | −53.4, 90 |
| Factor I | 394 | −233, 1011 |
| Properdin | −1.32 | −7.17, 4.47 |
| Cytokines/chemokines | ||
| Eotaxin* | −36.8 | −74.2, 0.384 |
| GM-CSF | −6.8 | −27.7, 14.1 |
| IFN-α2 | −17.7 | −65, 30.8 |
| IFN-γ* | −14.3 | −27.5, −2.1 |
| IL-10 | 16.7 | −9.57, 44.2 |
| IL-12p40 | −8.06 | −24.6, 8.44 |
| IL-12p70 | −4.85 | −17.8, 8.01 |
| IL-13 | −20.6 | −47.9, 6.58 |
| IL-15 | −1.41 | −4.73, 1.98 |
| IL-17 | −0.767 | −1.84, 0.364 |
| IL-1rα | 19.2 | −51.4, 86.5 |
| IL-1b | −1.25 | −4.04, 1.68 |
| IL-2 | −6.96 | −15.3, 1.58 |
| IL-4 | −5.45 | −25.9, 16.1 |
| IL-5 | −7 | −16, 2.42 |
| IL-6 | 25.8 | −28.9, 84.1 |
| IL-7 | −6.51 | −13.7, 0.943 |
| IL-8 | 5.82 | −3.05, 14.3 |
| IP-10 | −4.05 | −213, 213 |
| MCP-1 | −13.6 | −153, 131 |
| MIP-1α | −2.67 | −5.76, 0.473 |
| MIP-1β | −8.25 | −18.8, 2.43 |
| TNFα | −1.45 | −5.76, 2.87 |
| Other inflammatory markers | ||
| hs-CRP | 3261 | −2924, 870 |
| MMP9* | 72,435 | 6401, 1,35,310 |
p < 0.05. hs-CRP, high-sensitivity C-reactive protein; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; IP-10, interferon gamma-induced protein 10; MBL, mannose-binding lectin; MCP-1, monocyte chemoattractant protein 1; MIP, macrophage inflammatory protein; MMP-9, matrix metallopeptidase-9; TNF-α, tumour necrosis factor alpha.
Discussion
In this study, we tested serum from a select group of prospectively enrolled TAVI patients during the perioperative period for a panel of immune markers, including cytokines, chemokines and complement components that have been implicated in the inflammatory response to ischaemic stroke.8 Serum levels from patients with MRI-documented cerebral infarction were compared with those from patients with no evidence of infarction. Over and above the inflammatory changes seen across the entire cohort, secondary to underlying aortic stenosis and the perioperative period, we detected significant differences in baseline levels of complement C3 and C4b, as well as cytokines GM-CSF, IL-15 and MIP-1β. Postoperatively, there were significant differences in serum MMP-9, complement C3, IFN-γ and MIP-1β between patients who sustained cerebral infarction and those who did not.
Over the past decade, matrix metalloproteinases, especially MMP-9, have been widely investigated for their role in the acute pathogenesis of blood–brain barrier disruption, vasogenic oedema and haemorrhage transformation following stroke.9–11 Immunohistochemical analysis of human stroke tissue has shown an overproduction of MMP-9 during the early and pro-inflammatory phases of stroke, as well as an increase in the number of neutrophils (the primary source of MMP-9), for up to 1 week post-stroke.12 MMP-9 has also been investigated as a biomarker for stroke, as it is measurably elevated in the serum of stroke patients, correlates positively with infarct volume, and is an effective marker of worse outcomes.13 Peak levels of MMP-9 generally occur between 12 h and 24 h after the stroke, but elevated levels persist for up to 5 days.9 These data are consistent with these present results for the perioperative period, which show significantly higher serum MMP-9 levels at 24 h postoperatively in those who sustain cerebral infarction.
Similarly, our data on changes in the perioperative complement profile agree with previously published findings in nonoperative settings. The complement system is a primordial and highly conserved component of the innate immune response to specific pathogen- and damage-associated molecular patterns, and accounts for approximately 10% of the globulin fraction within serum.14,15 It has multiple neurological functions in both health (e.g. elimination of synapses from maturing and axotomized neurons, neurogenesis and neuroprotection) and various disease states, and is implicated in the inflammatory disequilibrium and cerebral tissue damage that occurs during ischaemic stroke.1,16,17 In acute ischaemic stroke, complement activation results in the release of pro-inflammatory cytokines, leucosequestration, opsonization and membrane attack complex deposition on damaged and surrounding cells within the ischaemic penumbra (Figure 3).
Figure 3.
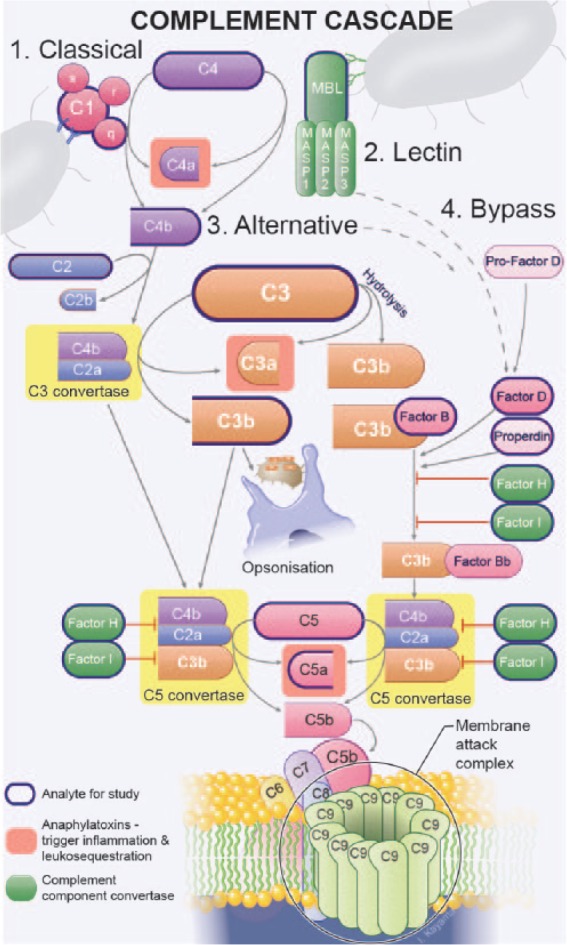
Overview of the complement cascade. (Copyright © 2016 Studio Kayama.)
As the central component for all activating pathways, changes in C3 levels can be considered a surrogate marker that reflects the global status of complement function. Our detection of higher C3 levels in the postoperative period among our cerebral infarction cohort is therefore noteworthy (Figure 2c). In fact, this positive difference reflects a less marked drop from baseline in the cerebral infarction group than in controls. Concurrently, on visual inspection of the polynomial curves, complement C3b/iC3b, the immediate by-product of C3 breakdown, is substantially elevated from baseline in cerebral infarction cases versus controls during the early postoperative phase (Figure 2d). The postoperative drop in C5 was more marked in the cases versus control group (Figure 2e), with corresponding separation between the two curves early postoperatively for the C5 by-product C5a (Figure 2f). Together, these observations support a distinct pattern of complement activation associated with perioperative cerebral infarction.
Despite the appearance of the polynomial curves, statistically significant differences were only evident for the C3 component of the complement system. However, this is not surprising given the short in vivo half-lives of complement activation products. For example, C5a has a half-life of approximately 1 min, due to rapid receptor binding, and although C3 activation product kinetics are slightly more favourable, they are still short-lived and their half-life of 2–3 h is much less than the 24 h sampling intervals used in this study.18,19 A more sensitive indicator of in vivo activation would therefore be a reduction in native component levels rather than the activation products alone.20,21 Consistent with this, we found that the aforementioned differences in complement components between cerebral infarct and control groups predicted by the best-fit polynomial curve smoothing model were most evident at the earliest time points.
Interestingly, the general pattern of expression for the cytokines/chemokines in this study was an initial transitory rise from baseline measured in the cerebral infarct versus control group, followed by a rapid reduction, often with levels dropping markedly lower than baseline levels. Three important exceptions to this were the pro-inflammatory cytokines IL-6 and IL-8, and the anti-inflammatory cytokine IL-10, for which cerebral infarct patients averaged more positive values than controls. Longitudinal regression analysis showed that the observed changes were statistically significant only for IFN-γ and MIP-1β, and only for IFN-γ at the 24 h time point in the smoothing model.
This pattern of expression and the notable drop in pro-inflammatory cytokines were unexpected, given that these markers are typically elevated following ischaemic stroke in preclinical models.1 However, such inconsistencies have been a common theme among human studies in this field, with the kinetic profiles of key pro-inflammatory cytokines often producing conflicting results. Although a small number of studies have demonstrated elevated levels of TNF-α and IL-1β in patients following ischaemic stroke,22,23 others have detected no change.24–27 IL-6 elevation is well documented but the timing of this elevation has varied widely, with peaks reported as early as 10 h and as late as 7 days post-stroke.26,28,29 In addition to their circulation half-life, factors that might influence the serum levels of these and other cytokine/chemokines assessed in this study include their location of expression, their presence in cerebrospinal fluid, disruption of the blood–brain barrier, extra-cranial release and a systemic acute phase response (including a hepatic chemokine response).
In addition to the changes we observed subsequent to cerebral infarction, our baseline results indicate key differences between those who ultimately would go on to sustain infarction and those who would not, suggesting potential risk factors or predictors of their occurrence. The most significant baseline differences, both statistically and biologically, were evident for C3 (case mean: 38 ± 22 versus control mean: 73 ± 40; p = 0.005) and for GM-CSF (case mean: 1 ± 0.3 versus control mean: 0.6 ± 0.2; p = 0.009).
As discussed above, complement factor C3 is both one of the most common blood proteins and the central component of the complement cascade (Figure 3). We observed baseline levels of C3 that were approximately 50% less in patients who go on to sustain cerebral infarction following TAVI compared with those who do not. Low C3 has been attributed to both depressed synthesis and increased consumption/hypercatabolism.21 Several autoimmune diseases have been implicated in acquired deficiencies of complement, secondary to accelerated consumption by immune complexes (e.g. systemic lupus erythematosus, antiphospholipid antibody syndrome, cryoglobulinaemia, various vasculitides). However, the rarity of these disorders means this is an unlikely explanation for the observations made in this study. C3 consumption may also result from coagulation and a prothrombotic state. Several interactions exist between the complement and coagulation cascades, including, most appreciably, platelet-mediated immune activation (Figure 1). This is most apparent clinically in the established anti-inflammatory effects of many of the antiplatelet medications, for example, clopidogrel.30 They are also both inextricably linked in the pathobiology of the response to cerebral ischaemic infarction.1 Indeed, during the postoperative period, platelet counts dropped significantly more in the cerebral infarction group, suggesting thromboses and increased consumption of C3 (the mean case-control difference on longitudinal regression analysis was −26.7; 95% CI −52.3 to −3.74; p = 0.027). Consumption of C3 might also be expected to result in comparatively low levels of C4 in those with cerebral infarction. Although this reduction was evident in the cerebral infarction group (mean: 459 ± 137 versus control mean: 545 ± 199), this difference was not statistically significant (p = 0.281). Usually, C3 levels are three- to six-fold higher than C4 levels.14,15 Therefore, the same percentage change in both (as would occur with consumption) could reduce C3 to a statistically significant extent, but not C4. Supporting this theory of baseline complement hyper-catabolism/consumption, the C4 activation product C4b was significantly higher in the cerebral infarction group (mean: 4011 ± 783 versus control mean: 3257 ± 804; p = 0.048).
Several factors with the potential to alter a patient’s inflammatory status were present in our study cohort. First, inflammation is a key propagator of calcific aortic stenosis.31 Complement activation probably plays a critical role, and has been demonstrated in stenotic aortic valves leading to increases in pro-inflammatory cytokines IL-6, IL-8 and MCP-1.32 Furthermore, the myocardium has been shown to be a major source of pro-inflammatory cytokines; as such, the increased left ventricular mass common in patients with severe aortic stenosis might also be a contributing factor.33 However, no differences existed between the aetiology (non-rheumatic, degenerative calcific) or severity of aortic stenosis between our two subject groups, or in their pretreatment health status. Second, all patients were on dual antiplatelet therapy (aspirin and clopidogrel) and statins, both of which have anti-inflammatory properties. However, this was consistent across the entire cohort of assessed patients and, as such, is unlikely to have confounded comparisons between the two groups.
Although we were able to detect some notable immunological differences between the MRI-positive and MRI-negative groups, our study was limited by the fact that we sampled blood every 24 h and for only 3 days of follow up, which could have resulted in peak serum level changes from baseline being missed. In addition, our study exclusively recruited a homogeneous group of highly selected patients limiting extrapolation of these findings to other settings (both operative and nonoperative). Furthermore, all cerebral infarcts considered were iatrogenic and subclinical limiting extrapolation of data to out-of-hospital clinically apparent stroke. Moreover, although the timing of cerebral infarction in this setting is assumed to be predominantly intraprocedural, the exact timing remains unknown. It is possible that infarcts occurred at any time between the patients’ baseline MRI scan and the day 3 postprocedure follow-up scan. Finally, the relatively small sample size of just 10 subjects per group is also likely to have limited our ability to detect significant intergroup differences.
Conclusion
This study provides evidence for notable differences in the complement activation and cytokine/chemokine responses associated with perioperative cerebral infarction. The significant baseline and postoperative differences in complement C3 between those who sustained cerebral infarction and those who did not is a particularly major finding, especially as this protein constitutes a major fraction of total blood protein. Our results should inspire further investigation focused on these identified complement and cytokine/chemokine signals and their potential to predict, detect and modulate neuroinflammation after perioperative cerebral infarction.
Supplemental Material
Supplemental material, Supplementary_material_V2 for Differential immunological profiles herald magnetic resonance imaging-defined perioperative cerebral infarction by Jonathon P. Fanning , Louise E. See Hoe, Margaret R. Passmore, Adrian G. Barnett, Barbara E. Rolfe, Jonathan E. Millar, Allan J. Wesley, Jacky Suen and John F. Fraser in Therapeutic Advances in Neurological Disorders
Acknowledgments
The authors would like to thank Mrs Cliona O’Sullivan, who assisted with the recruitment of patients and the collection of data; Professor Darren Walters for facilitating patient recruitment and assessment; Mrs Wendy Strugnell and the Department of Medical Imaging who performed the necessary neuroimaging; Queensland Pathology for assisting the collection and processing of blood samples; and, Professor Trent Woodruff for complement-specific intellectual input.
Footnotes
Funding: Funding for this study was provided by The Prince Charles Hospital Foundation and The University of Queensland. JPF and JFF are supported by Queensland Government–Health Innovation, Investment and Research Office Fellowships.
Conflict of interest statement: The authors declare no conflicts of interest in preparing this article.
ORCID iD: Jonathon P. Fanning  https://orcid.org/0000-0002-1675-0522
https://orcid.org/0000-0002-1675-0522
Contributor Information
Jonathon P. Fanning, Critical Care Research Group, Level 3 Clinical Sciences Building, The Prince Charles Hospital, Rode Road, Chermside, Brisbane, Queensland 4032, Australia.
Louise E. See Hoe, Critical Care Research Group, The Prince Charles Hospital, Brisbane, Queensland, Australia Faculty of Medicine, The University of Queensland, Brisbane, Queensland, Australia.
Margaret R. Passmore, Critical Care Research Group, The Prince Charles Hospital, Brisbane, Queensland, Australia Faculty of Medicine, The University of Queensland, Brisbane, Queensland, Australia.
Adrian G. Barnett, School of Public Health and Social Work, Queensland University of Technology, Brisbane, Queensland, Australia
Barbara E. Rolfe, Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, Queensland, Australia
Jonathan E. Millar, Critical Care Research Group, The Prince Charles Hospital, Brisbane, Queensland, Australia Faculty of Medicine, The University of Queensland, Brisbane, Queensland, Australia; Wellcome-Wolfson Centre For Experimental Medicine, Queen’s University, Belfast, UK.
Allan J. Wesley, Metro North Hospital and Health Service District, Queensland, Australia
Jacky Suen, Critical Care Research Group, The Prince Charles Hospital, Brisbane, Queensland, Australia; Faculty of Medicine, The University of Queensland, Brisbane, Queensland, Australia.
John F. Fraser, Critical Care Research Group, The Prince Charles Hospital, Brisbane, Queensland, Australia Faculty of Medicine, The University of Queensland, Brisbane, Queensland, Australia; Metro North Hospital and Health Service District, Queensland, Australia.
References
- 1. Petrovic-Djergovic D, Goonewardena SN, Pinsky DJ. Inflammatory disequilibrium in stroke. Circ Res 2016; 119: 142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mestas J, Hughes CCW. Of mice and not men: differences between mouse and human immunology. J Immunol 2004; 172: 2731–2738. [DOI] [PubMed] [Google Scholar]
- 3. Elkind MSV, Carty CL, O’Meara ES, et al. Hospitalization for infection and risk of acute ischemic stroke. Stroke 2011; 42: 1851–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Worthmann H, Tryc AB, Goldbecker A, et al. The temporal profile of inflammatory markers and mediators in blood after acute ischemic stroke differs depending on stroke outcome. Cerebrovasc Dis 2010; 30: 85–92. [DOI] [PubMed] [Google Scholar]
- 5. Fanning JP, Wesley AJ, Walters DL, et al. Neurological injury in intermediate-risk transcatheter aortic valve implantation. J Am Heart Assoc 2016; 5: e0042033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fanning JP, Wesley AJ, Platts DG, et al. The silent and apparent neurological injury in transcatheter aortic valve implantation study (SANITY): concept, design and rationale. BMC Cardiovasc Dis 2014; 14: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fanning JP, Wesley AJ, Wong AA, et al. Emerging spectra of silent brain infarction. Stroke 2014; 45: 3461–3471. [DOI] [PubMed] [Google Scholar]
- 8. Kim JY, Kawabori M, Yenari MA. Innate inflammatory responses in stroke: mechanisms and potential therapeutic targets. Curr Med Chem 2014; 21: 2076–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Romanic AM, White RF, Arleth AJ, et al. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 1998; 29: 1020–1030. [DOI] [PubMed] [Google Scholar]
- 10. Fujimura M, Gasche Y, Morita-Fujimura Y, et al. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res 1999; 842: 92–100. [DOI] [PubMed] [Google Scholar]
- 11. Gasche Y, Fujimura M, Morita-Fujimura Y, et al. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: a possible role in blood-brain barrier dysfunction. J Cereb Blood Flow Metab 1999; 19: 1020–1028. [DOI] [PubMed] [Google Scholar]
- 12. Rosell A, Cuadrado E, Ortega-Aznar A, et al. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 2008; 39: 1121–1126. [DOI] [PubMed] [Google Scholar]
- 13. Montaner J, Alvarez-Sabín J, Molina C, et al. Matrix metalloproteinase expression after human cardioembolic stroke: temporal profile and relation to neurological impairment. Stroke 2001; 32: 1759–1766. [DOI] [PubMed] [Google Scholar]
- 14. Walport MJ. Complement. First of two parts. N Engl J Med 2001; 344: 1058–1066. [DOI] [PubMed] [Google Scholar]
- 15. Walport MJ. Complement. Second of two parts. N Engl J Med 2001; 344: 1140–1144. [DOI] [PubMed] [Google Scholar]
- 16. Arumugam TV, Magnus T, Woodruff TM, et al. Complement mediators in ischemia–reperfusion injury. Clin Chim Acta 2006; 374: 33–45. [DOI] [PubMed] [Google Scholar]
- 17. Arumugam TV, Woodruff TM, Lathia JD, et al. Neuroprotection in stroke by complement inhibition and immunoglobulin therapy. Neuroscience 2009; 158: 1074–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oppermann M, Götze O. Plasma clearance of the human C5a anaphylatoxin by binding to leucocyte C5a receptors. Immunology 1994; 82: 516–521. [PMC free article] [PubMed] [Google Scholar]
- 19. Teisner B, Brandslund I, Grunnet N, et al. Acute complement activation during an anaphylactoid reaction to blood transfusion and the disappearance rate of C3c and C3d from the circulation. J Clin Lab Immunol 1983; 12: 63–67. [PubMed] [Google Scholar]
- 20. Nurnberger W, Bhakdi S. Plasma C3d/C3 quotient as a parameter for in vivo complement activation. J Immunol Methods 1984; 74: 87–91. [DOI] [PubMed] [Google Scholar]
- 21. Swaak AJ, Hannema A, Vogelaar C, et al. Determination of the half-life of C3 in patients and its relation to the presence of C3-breakdown products and/or circulating immune complexes. Rheumatol Int 1982; 2: 161–166. [DOI] [PubMed] [Google Scholar]
- 22. Intiso D, Stampatore P, Zarrelli MM, et al. Incidence of first-ever ischemic and hemorrhagic stroke in a well-defined community of southern Italy, 1993–1995. Eur J Neurol 2003; 10: 559–565. [DOI] [PubMed] [Google Scholar]
- 23. Zaremba J, Losy J. Early TNF-alpha levels correlate with ischaemic stroke severity. Acta Neurol Scand 2001; 104: 288–295. [DOI] [PubMed] [Google Scholar]
- 24. Montaner J, Rovira A, Molina CA, et al. Plasmatic level of neuroinflammatory markers predict the extent of diffusion-weighted image lesions in hyperacute stroke. J Cereb Blood Flow Metab 2003; 23: 1403–1407. [DOI] [PubMed] [Google Scholar]
- 25. Ormstad H, Aass HC, Lund-Sorensen N, et al. Serum levels of cytokines and C-reactive protein in acute ischemic stroke patients, and their relationship to stroke lateralization, type, and infarct volume. J Neurol 2011; 258: 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fassbender K, Schmidt R, Mossner R, et al. Pattern of activation of the hypothalamic-pituitary-adrenal axis in acute stroke. Relation to acute confusional state, extent of brain damage, and clinical outcome. Stroke 1994; 25: 1105–1108. [DOI] [PubMed] [Google Scholar]
- 27. Tarkowski E, Rosengren L, Blomstrand C, et al. Early intrathecal production of interleukin-6 predicts the size of brain lesion in stroke. Stroke 1995; 26: 1393–1398. [DOI] [PubMed] [Google Scholar]
- 28. Perini F, Morra M, Alecci M, et al. Temporal profile of serum anti-inflammatory and pro-inflammatory interleukins in acute ischemic stroke patients. Neurol Sci 2001; 22: 289–296. [DOI] [PubMed] [Google Scholar]
- 29. Waje-Andreassen U, Krakenes J, Ulvestad E, et al. IL-6: an early marker for outcome in acute ischemic stroke. Acta Neurol Scand 2005; 111: 360–365. [DOI] [PubMed] [Google Scholar]
- 30. Molero L, Lopez-Farre A, Mateos-Caceres PJ, et al. Effect of clopidogrel on the expression of inflammatory markers in rabbit ischemic coronary artery. Br J Pharmacol 2005; 146: 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abdelbaky A, Corsini E, Figueroa AL, et al. Early aortic valve inflammation precedes calcification: a longitudinal FDG-PET/CT study. Atherosclerosis 2015; 238: 165–172. [DOI] [PubMed] [Google Scholar]
- 32. Helske S, Oksjoki R, Lindstedt KA, et al. Complement system is activated in stenotic aortic valves. Atherosclerosis 2008; 196: 190–200. [DOI] [PubMed] [Google Scholar]
- 33. Wan S, DeSmet JM, Barvais L, et al. Myocardium is a major source of proinflammatory cytokines in patients undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg 1996; 112: 806–811. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Supplementary_material_V2 for Differential immunological profiles herald magnetic resonance imaging-defined perioperative cerebral infarction by Jonathon P. Fanning , Louise E. See Hoe, Margaret R. Passmore, Adrian G. Barnett, Barbara E. Rolfe, Jonathan E. Millar, Allan J. Wesley, Jacky Suen and John F. Fraser in Therapeutic Advances in Neurological Disorders