

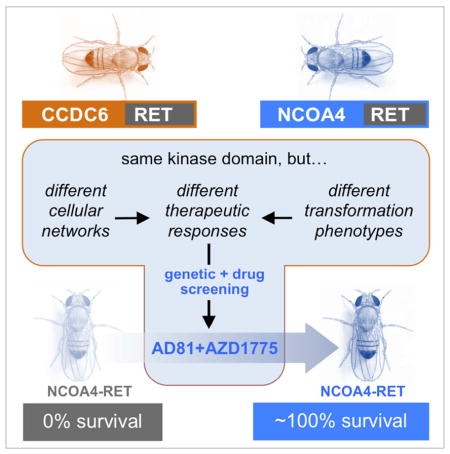
Summary
We generated and compared Drosophila models of RET fusions CCDC6-RET and NCOA4-RET. Both RET fusions directed cells to migrate, delaminate, and undergo EMT, and both resulted in lethality when broadly expressed. In all phenotypes examined NCOA4-RET was more severe than CCDC6-RET, mirroring their effects on patients. A functional screen against the Drosophila kinome and against a library of cancer drugs found that CCDC6-RET and NCOA4-RET acted through different signaling networks and displayed distinct drug sensitivities. Combining data from the kinome and drug screens identified the WEE1 inhibitor AZD1775 plus the multi-kinase inhibitor sorafenib as a synergistic drug combination that is specific for NCOA4-RET. Our work emphasizes the importance of identifying and tailoring a patient’s treatment to their specific RET fusion isoform and identifies a multi-targeted therapy that may prove effective against tumors containing the NCOA4-RET fusion.
eTOC Blurb
Levinson and Cagan examine two Drosophila RET-fusion models. They find that the N-terminus contributes to the overall function of fusion proteins, including their response to therapeutics. Genetic and chemical genetic screens identify a drug combination of sorafenib plus AZD1775 as effective against the NCOA4-RET fusion.
Introduction
Rates of thyroid cancer have tripled in the last few decades, making it the fastest growing cancer type (Brown et al., 2011). 80% of thyroid cancers are Papillary Thyroid Carcinoma (PTC), which arises from transformation of follicular cells in the thyroid. PTC is a relatively indolent disease with a five-year survival rate of 98%. However, radioiodine-refractory disease has no effective treatment, a five year survival rate of 50%, and results in the death of approximately 1500 patients in the US each year (Ricarte-Filho et al., 2009). 30% of PTC cases are driven by RET fusion proteins, generated as a result of a chromosomal translocation that results in fusion of the RET receptor tyrosine kinase to coiled-coiled domains of various cytoplasmic proteins. RET fusion proteins were first identified as oncogenes in Papillary Thyroid Carcinoma (PTC) (Grieco et al., 1990; Jhiang et al., 1996, 1992); more recently they been reported in other cancer types including lung adenocarcinomas (Kohno et al., 2012; Lipson et al., 2012; Takeuchi et al., 2012), chronic myelomonocytic leukemia, (Ballerini et al., 2012) and colorectal cancer (Le Rolle et al., 2015). Two RET fusion isoforms, CCDC6-RET and NCOA4-RET account for greater than 90% of PTC fusions (Santoro et al., 2006). Although both fusion proteins lead to activation of Ret, the two isoforms are associated with different PTC subtypes. CCDC6-RET is closely associated with the classic variant, a more benign subtype; NCOA4-RET is closely associated with the solid subtype, which is more aggressive and malignant (Basolo et al., 2002; Rabes, 2001; Thomas et al., 1999). The basis of this difference is poorly understood.
Full length RET phosphorylates downstream targets resulting in the activation of many signaling cascades including the Ras/ERK and PI3K/AKT pathways, which in turn regulate cellular differentiation, proliferation, and apoptosis (Wells and Santoro, 2009). Unlike their wild type counterpart, RET fusion proteins are not localized to the cell membrane. Nevertheless, RET fusions signal through at least some canonical RET signaling pathways (Castellone and Santoro, 2008), though the full downstream effects of RET fusions have not been fully explored (Knostman et al., 2007). MAPK inhibitors that show a strong effect against the Medullary Thyroid Carcinoma (MTC) human cancer cell line TT had a modest effect against the CCDC6-RET-harboring PTC human cancer cell line TPC1 (Gild et al., 2013). These differences in drug response—in addition to the differences in disease severity between fusion isoforms—suggest that different RET isoforms may act at least in part through distinct pathways, a testable hypothesis.
Previous work by our lab has validated Drosophila models of oncogenic RET isoforms that are associated with MTC; these models were used to explore function as well as identify lead therapeutic hits for RET isoforms activated by point mutation (Dar et al., 2012; Read et al., 2005; Vidal et al., 2005). Here we use Drosophila models to explore the role of RET fusions in transformation. Drosophila models provide a useful tool for examining oncogenes in situ: they provide powerful genetic tools, and flies are sensitive to cancer-relevant compounds (Edwards et al., 2011; Gladstone et al., 2012; Rudrapatna et al., 2012; Slack et al., 2015; Willoughby et al., 2013). Recently, multiple groups have studied the ability of these RET fusions to transform cells in culture (Gild et al., 2013; Knauf et al., 2003; Richardson et al., 2009; Wang et al., 2003), providing important insights on their activity. Whole animal studies can provide further important information on the interplay between tumor and non-tumor cells, aspects that are important both in the growth and metastasis of tumors and in the response of tumors to therapy (Wagstaff et al., 2013).
RET inhibitors have had moderate success in a small number of patients with PTC or lung adenocarcinoma, having shown considerable toxicity (Borrello et al., 2013; Horiike et al., 2016; Xing et al., 2013). Thus, therapeutics for PTC remain a significant unmet need. In this study we examine signaling downstream of the CCDC6-RET and NCOA4-RET fusions. Our data demonstrate that these two RET fusion genes are functionally different, both in the pathways that they utilize to promote transformation and in their sensitivity to clinically-relevant drugs.
Results
RET fusions promoted cell migration, delamination, and EMT in Drosophila
To generate Drosophila models of CCDC6-RET and NCOA4-RET we created transgenic fly lines that express the human fusion genes (Fig. 1A) under the control of the UAS promoter. Each construct was inserted into the same genomic site to ensure that the UAS-CCDC6-RET and UAS-NCOA4-RET transgenes would be expressed at similar levels. Crossing in the 765-Gal4 driver generated 765>CCDC6-RET and 765>NCOA4-RET, leading to expression of the RET fusion constructs throughout the developing wing disc. Staining with an antibody that recognizes intracellular Ret, we confirmed that both RET fusions were expressed at the same level (Fig. 2G, Fig. S1); both human RET fusions were activated as assessed with an antibody targeting the phosphorylated tyrosine epitope pTyr905, which is required for downstream RET signaling (Fig. 1D′–F′, Fig. 2D, Fig. S1). Interestingly, 765>NCOA4-RET displayed a higher level of activation than 765>CCDC6-RET (Fig. 2D, Fig. S1), mirroring the more severe disease presented by NCOA4-RET patients (Basolo et al., 2002; Rabes, 2001; Thomas et al., 1999). Known downstream effectors of RET signaling were also activated in animals expressing either RET fusion as shown by increased levels of phosphorylated Erk and Jnk and increased levels of total Rho1. However, one known effector, Rac1, was not upregulated (Fig. 2E, F, H, & I, Fig. S1).
Figure 1. CCDC6-RET and NCOA4-RET directed cell migration and whole animal lethality in Drosophila.

A. Schematic of the full length, wild-type RET gene and the two RET fusions CCDC6-RET and NCOA4-RET.
B. Histogram quantifying the percent larvae that matured to pupariation vs. balancer controls (Ctrl: tub>w-/Tubby, CC6-R: tub>CCDC6-RET/Tubby, NC4-R: tub>NCOA4-RET/Tubby). Animals expressing low levels of RET fusions under control of the tubulin-Gal4 driver died during larval stages. GAL4 activity—and therefore expression levels—were controlled by temperature. tub>CCDC6-RET was expressed at a higher level (27°C) yet led to only 42% larval lethality; tub>NCOA4-RET was expressed at lower levels (25°C) and led to 100% larval lethality. Data are represented as mean +/− SEM. Asterisks: ** p ≤ 0.01, **** p ≤ 0.0001.
C. Entire third instar larval wing disc expressing GFP in the ptc expression domain. Perimeter of wing is outlined to show shape of the disc. Region shown in D–F″ shown in yellow rectangle. Scale bar represents 315μm.
D–F″. Late, third instar larval wing discs expressing transgenes in the ptc expression domains as indicated; transformed cells were visualized with UAS-GFP. D′–F′. Localization with a phospho-RET antibody confirmed that human RET fusions are expressed and activated in Drosophila tissue. D″–F″. Merged panels highlight that only those cells expressing a RET fusion transgene activated RET. Scale bar in F represents 50 μm in D–F″.
G–I. Z-stack images of previously described wing discs showing migrating cells. Control animals (F) show no migration from endogenous ptc-GAL4 expression domain, while ptc>CCDC6-RET (G) and ptc>NCOA4-RET (H) have one or more cells that have left the original space and migrated into wild-type neighboring tissue; brackets show the distance cells have traveled from the posterior edge of the ptc domain and white arrows indicate cells that have migrated. Apical membrane is marked in blue with DE-Cadherin staining, emphasizing that migrating cells have lost their polarity to invade along the basal membrane. Scale bar in G represents 16.7μm in G–I.
J. Quantification of the severity of cell migration caused by each RET fusion expression under control of the ptc-GAL4 driver in larval wing discs. Wing discs were scored blind and binned into one of four categories (severe, moderate, weak, or no migration). Example images for each category is provided in Fig. S2. Again despite differences in temperature, more ptc>CCDC6-RET (27°C) was expressed in the wing disc and less ptc>NCOA4-RET (25°C), yet ptc>NCOA4-RET displayed a significantly more severe phenotype. Data are represented as mean +/− SEM. Asterisks: **** p ≤ 0.0001.
See also Figure S2
Figure 2. RET fusions directed EMT, upregulated canonical downstream targets.

A–C: Third instar larval wing discs expressing transgenes within the ptc-GAL4 expression domain. A′–C′: An antibody targeting MMP1 shows that migrating cells expressed matrix metalloproteinases, enzymes upregulated during mammalian EMT. A″–C″: An antibody against phosphorylated Src shows that expression of either RET fusion led to upregulation of activated Src, another marker of EMT. Note that ptc>NCOA4-RET displayed more cells expressing MMP1 and p-Src, correlating with its more severe cell migration phenotype. Scale bar in C represents 50 μm in A–C″.
D–I: Histograms quantifying western blots from third instar wing discs expressing either 765-Gal4 (control), 765>CCDC6-RET, or 765>NCOA4-RET. Lysates were from 20 wings discs from each genotype, and two biological replicates were averaged. All animals were raised at 25°C to ensure similar levels of Gal4 expression; Fig. S1 provides a representative blot confirming similar levels of RET fusion proteins in panel G. A phospho-RET antibody demonstrating that NCOA4-RET has a higher level of kinase activity as assessed by autophosphorylation (panel D). Downstream signaling factors showed elevated activation (phospho-Erk and phospho-Jnk) or expression (Rho1). Data are represented as mean +/− SEM.
See also Figures S1, S3, and S4
The RET fusions were next expressed in a central stripe of cells in the developing wing monolayer epithelium using the ptc-GAL4 driver; cells were visualized with a UAS-GFP reporter (Fig. 1C, D–I). Expression of either ptc>CCDC6-RET or ptc>NCOA4-RET resulted in cells migrating away from the posterior border of the ptc domain where ptc expression is highest (Fig. 1, 2). Migrating cells detached from the apical membrane and migrated through the basal layer of the epithelium (Fig. 1G–I). Flies expressing NCOA4-RET had significantly more migrating cells than CCDC6-RET (Fig. 1J, Fig. S2).
We have previously observed similar migration in other Drosophila models (Rudrapatna et al., 2013; Vidal et al., 2010); this migration was linked to an epithelial-to-mesenchymal transition (EMT). The migrating ptc>CCDC6-RET and ptc>NCOA4-RET cells stained positive for MMP1 and upregulated levels of phosphorylated Src, two markers of EMT (Fig. 2A–C″). Cells in ptc>CCDC6-RET and ptc>NCOA4-RET animals that were not yet migrating also expressed higher levels of pSrc (Fig. S3). Ptc>NCOA4-RET cells expressed higher levels of EMT markers (Fig. 2B–C′, Fig. S3), presumably reflecting the stronger activation of the RET fusion. Previous work in our lab has shown that caspase activity is a component of a JNK-Rho1-Actin signaling axis that drives cell migration (Rudrapatna et al., 2013). We found that migrating cells in ptc>CCDC6-RET and ptc>NCOA4-RET animals stained positive for cleaved caspase 3, indicating these cells are subject to a similar mechanism of migration (Fig. S4).
The stronger phenotype observed in NCOA4-RET flies was reflected in other phenotypes. Broad expression of either RET fusion via the tubulin-Gal4 promoter resulted in lethality: 42% of tub>CCDC6-RET animals died in larval stages, while 100% of tub>NCOA4-RET animals died as larvae (Fig. 1B). This difference in signaling, cell migration, and lethality correlates with the clinical observation that PTC patients with tumors harboring NCOA4-RET typically present with a worse prognosis than patients with CCDC6-RET tumors (Basolo et al., 2002; Rabes, 2001; Thomas et al., 1999). Overall, these data indicate that Drosophila can provide a useful model for exploring specific aspects of RET fusions in vivo.
CCDC6-RET and NCOA4-RET signal through shared and distinct kinases
Kinases are a common regulatory feature of most signaling networks. To explore the functional differences of CCDC6-RET vs. NCOA4-RET fusions in vivo, we conducted a genetic modifier screen against the full Drosophila kinome. Genetic modifier screens are a standard tool for identifying functional effectors of a signaling protein (e.g. (Müller et al., 2005; Simon, 1994; St Johnston, 2002; Ward et al., 2003)). One functional copy of each locus encoding a kinase was removed, one by one, in the context of tub>CCDC6-RET or tub>NCOA4-RET; we then assessed the ability of reduced kinase activity to rescue animal lethality (Fig. 3A). Despite sharing an identical RET tyrosine kinase domain, CCDC6-RET and NCOA4-RET displayed more functional differences than similarities in the signaling networks that promoted transformation (Fig. 3). Mutations in six genes were identified as significant genetic modifiers of tub>CCDC6-RET and eleven for tub>NCOA4-RET lethality; however, only two modifier loci were shared between the two RET fusions (Fig. 3B, Fig. S5). We identified regulators in a broad palate of signaling pathways including JNK, SRC, and STE20/Hippo (Fig. 3B–D). These mostly non-overlapping set of pathways indicate that a significant proportion of CCDC6-RET and NCOA4-RET signaling is distinct, and that the two diseases may have significant qualitative differences.
Figure 3. RET fusions directed similarities, differences in signaling networks.

A. Schematic of the kinome genetic modifier screen.
B. Venn Diagram showing genetic modifier hits from the kinome screen. In the background of overexpression of either tub>CCDC6-RET or tub>NCOA4-RET, kinases were assessed by (i) heterozygosity via a hypomorphic or amorphic allele, or (ii) RNA-interference (RNAi) mediated knockdown. Loci were considered hits that, when reduced, significantly (p<0.05) enhanced survival of the animal. Kinases listed in Venn Diagram significantly suppressed lethality induced by the specified RET fusion. Human orthologs of Drosophila kinases are listed in superscript; legend indicates primary signaling pathway for each hit. Note that most hits were specific to one RET fusion isoform. Figure S5 provides the exact level of rescue.
C. Histogram showing the genetic modifier hits for tub>CCDC6-RET. Data are represented as mean +/− SEM. Asterisks: * p ≤ 0.05, ** p ≤ 0.01.
D. Histogram showing the genetic modifier hits for tub>NCOA4-RET animals. Data are represented as mean +/− SEM. * p ≤ 0.05, ** p ≤ 0.01, **** p ≤ 0.0001.
See also Figure S5
CCDC6-RET and NCOA4-RET were sensitive to shared and distinct kinase inhibitors
Previous work has demonstrated that RET fusions can be sensitive to kinase inhibitors that target RET in cell culture (Carlomagno et al., 2002; Gild et al., 2013; Kim et al., 2006; Mologni et al., 2013; Verbeek et al., 2011). The differences we observed in signaling between CCDC6-RET and NCOA4-RET animals indicate that they may be differentially sensitive to kinase inhibitors when assayed in a whole animal context. We therefore screened a set of 55 FDA approved and experimental small molecule cancer therapies; larvae were fed by mixing compounds directly into the food. Compounds were fed at their maximum tolerated dose, which ranged from 1 μM to 200 μM. Maximum tolerated dose was determined as the maximum dose that had no detectable impact on wild-type fly viability. Five compounds rescued tub>CCDC6-RET and five rescued tub>NCOA4-RET lethality (Fig. 4A, C, D, Fig. S6, Table S1). Only three compounds rescued both, the multi-kinase FDA approved inhibitors ponatinib and regorafenib and the multi-kinase inhibitor AD81; relative sensitivities to these drugs differed between the fusion models (Fig. 4C, D, Fig. S6, Table S1). The major targets of all seven compounds that rescued either RET fusion is listed in Figure 4B.
Figure 4. RET fusions were sensitive to shared and distinct kinase inhibitors.

A. Venn Diagram of the seven hits from drug screen that significantly increased the percent of experimental animals that pupariated when compared to control balancer flies. Figure S6 provides level of rescue of each hit. Table S1 shows the results for all 55 drugs tested and shows exact p values of significant hits.
B. Table indicating major targets of each kinase inhibitor.
C. Histogram showing the drug screen hits for tub>CCDC6-RET. Data are represented as mean +/− SEM. Asterisks: * p ≤ 0.05, ** p ≤ 0.01.
D. Histogram showing the drug screen hits for tub>NCOA4-RET. Data are represented as mean +/− SEM. Asterisks: ** p ≤ 0.01, **** p ≤ 0.001.
E–E″. Third instar larval ptc>CCDC6-RET wing discs. The ptc domain is visualized with ptc>GFP. In contrast to DMSO controls, 100 μM vandetanib and 100 μM AD81 blocked low level cell invasion. Scale bar in E″ represents 50 μm in E–F″.
F–F″. Third instar larval ptc>NCOA4-RET wing discs. In contrast to DMSO controls, AD81 blocked the extensive cell invasion phenotype while vandetanib had no impact on migrating cells.
G. Quantification of the severity of cell migration in ptc>CCDC6-RET animals fed DMSO, vandetanib, or AD81. Figure S8 shows the number of wing discs that scored in each of the four categories and provides p values for both ptc>CCDC6-RET and ptc>NCOA4-RET.
H. Quantification of the severity of cell migration in ptc>NCOA4-RET animals fed DMSO, vandetanib, or AD81.
See also Figure S6 and S7, Table S1
Not all drug results matched our genetic screens; of note, all the screened drugs have significant ‘off target’ effects so a comparison is difficult. For example gefitinib, developed as an EGFR inhibitor, rescued tub>CCDC6-RET-mediated lethality yet heterozygosity of EGFR was not found to be a genetic modifier in the kinome screen. Gefitinib’s efficacy may reflect an off target effect or, alternatively, inhibiting EGFR activity more than a 50% gene reduction is required to significantly rescue lethality. Heterozygosity of pvr, encoding the Drosophila ortholog to VEGFR, significantly rescued tub>NCOA4-RET lethality and indeed several of the drugs that rescued this model are known inhibitors of VEGFR (Fig. 3B, 4A, and 4B). However, vandetanib has multiple kinase targets that include VEGFR but it failed to rescue tub>NCOA4-RET-mediated lethality. These discrepancies suggest that complexity in a drug’s kinase profile can affect its activity against specific oncogenes.
We investigated the effect that kinase inhibitors had on migrating wing cells by feeding drugs to ptc>CCDC6-RET and ptc>NCOA4-RET animals and visualizing transformed cells with UAS-GFP. We tested two compounds, vandetanib and AD81, because they displayed different abilities to rescue whole animal lethality induced by the RET fusions. Both compounds rescued tub>CCDC6-RET flies to adulthood; only AD81 was observed to rescue tub>NCOA4-RET flies. We observed results similar to our viability assays: both vandetinib and AD81 significantly reduced the number of cells leaving the ptc-GAL4 domain in ptc>CCDC6-RET larvae; however, only AD81 was observed to rescue wing cell migration in ptc>NCOA4-RET larvae (Fig. 4E–H, Fig. S7).
A rational approach to identifying synergistic drug combinations
Small molecule kinase inhibitors are a common tool for treating advanced tumors. Recently, interest has risen in the use of drug combinations to improve efficacy and to overcome resistance (Al-Lazikani et al., 2012; Iadevaia et al., 2010; Keith et al., 2005; Shi et al., 2011; Yan et al., 2010). However, identifying combinations in a rational manner presents a challenge. As a first step in identifying optimal drug combinations for RET fusion-based tumors in the context of the whole animal, we combined our genetic modifiers identified in the kinome screen with hits from our drug screen. More specifically we (i) removed a copy of each kinase that rescued lethality while simultaneously (ii) feeding tub>CCDC6-RET or tub>NCOA4-RET larvae each kinase inhibitor that rescued lethality (Fig. 5A). By testing each kinase functionally linked to a RET fusion, our goal was to rationally identify activities that pair synergistically with each active drug.
Figure 5. Rational identification of synergistic drug cocktails.

A. Schematic of the genetic modifier and drug screen.
B. Histogram showing percent of tub>NCOA4-RET larvae that matured to pupariation vs. balancer controls in the presence of drug plus heterozygosity for the indicated locus. Heterozygosity for most kinase loci synergized to rescue lethality when combined with AD81, sorafenib, or regorafenib. The most synergistic combinations included heterozygosity for btk29A (TEC ortholog), sqa (MYLK3 ortholog), or wee (WEE1 ortholog), which increased the efficacy of AD81 on average by 65.5%, sorafenib on average by 80%, and regorafenib on average by 57.3%. Other drugs had either no or minimal increased efficacy in the context of any of the kinase alleles. Table S2 provides the level of rescue of each genetic and drug combination.
C. Histogram showing percent of tub>CCDC6-RET animals that survived to adulthood in the presence of drug plus heterozygosity for the indicated locus. Baseline survival to adulthood (eclosure) for tub>CCDC6-RET animals is approximately 0%. The ability of most drugs to improve viability was synergistically enhanced with the removal of one functional copy of bsk (JNK ortholog). The best combination for tub>CCDC6-RET animals is a combination of bsk loss and AD81 treatment, which rescued survival to 64%. Table S3 provides the level of rescue of each genetic and drug combination.
D. Histogram showing drug combinations rescue of lethality of tub>NCOA4-RET flies; Ibrutinib (TEC/BTK inhibitor), AZD1775 (WEE1), and HA-100 (MYLK) were chosen from data in panel B. The optimal combination for tub>NCOA4-RET animals was HA-100 plus AD81. The optimal combination available to patients was AZD1775 plus sorafenib. Data are represented as mean +/− SEM. Table S4 shows level of rescue for each drug combination.
See also Table S2, S3, and S4
Our genetic screens proved successful in identifying candidate kinase targets effective against NCOA4-RET transformation. Most of our genetic modifiers of tub>NCOA4-RET synergized with AD81, sorafenib, and regorafenib to enhance survival to adulthood, since the combination of alleles and drugs increased viability more than the sum of either single treatment. For example, reducing one functional genomic copy of the cell cycle regulator wee (wee+/−) led to near 100% rescue when paired with AD81 and sorafenib and 68% rescue when paired with regorafenib, while reducing wee to heterozygosity when paired with DMSO control did not have any rescue of viability in this context. Heterozygosity for the TEC and MYLK3 orthologs btk29A and sqa also significantly synergized with these drugs (Fig. 5B). Ponatinib and cabozantinib displayed little to no synergy with any of the dominant modifier kinase hits we tested (Fig. 5B, Table S2).
In contrast, only reducing JNK activity (bsk+/−) led to synergistic rescue of tub>CCDC6-RET flies when combined with any drug tested. Of note, heterozygosity for bsk also synergized with many drugs that alone failed to detectably rescue tub>CCDC6-RET flies (Fig. 5C, Table S3), further emphasizing the sensitivity of CCDC6-RET fusion animals to reduction in JNK activity. This data is again consistent with the view that CCDC6-RET and NCOA4-RET fusions act through distinct networks, and further indicate that rationally selected drug combinations may prove useful as a therapeutic approach.
Synergistic kinome and drug combinations inform drug combinations
Of the genetic modifier hits that synergized with AD81, sorafenib, or regorafenib in the context of NCOA4-RET overexpression, a subset are targeted by validated kinase inhibitor compounds. Ibrutinib is an FDA approved inhibitor of TEC/BTK kinase used for B-cell tumors (Honigberg et al., 2010; Smith, 2015); AZD1775 is a WEE1 inhibitor currently in clinical trials for multiple solid tumors (e.g., Bridges et al., 2011; Do et al., 2015). HA-100 is a pan-MYLK inhibitor that is not in clinical use; we used this compound to determine whether chemical inhibition of MYLK3 can have clinical relevance.
Mixing two-drug cocktails into their food, tub>NCOA4-RET larvae were administered a cocktail of (i) AD81, sorafenib, or regorafenib plus (ii) Ibrutinib, AZD1775, or HA-100. All three of the latter drugs synergized with AD81, and AZD1775 displayed synergy with sorafenib (Fig. 5D, Table S4). The strongest rescues were observed with AD81 plus HA-100 or AD81 plus AZD1775. The strongest rescue of NCOA4-RET with patient available drugs was sorafenib plus AZD1775. The levels of improved rescue rose to the level of synergy, suggesting that drug combinations may provide improved outcome for patients with tumors harboring the NCOA4-RET fusion.
Discussion
As testing for the presence of chromosomal translocations has become more common, RET fusion genes have been identified in an increasing number of cancer types. Activating point mutations in RET represent a clinically actionable target with several FDA approved inhibitors (Borrello et al., 2013; Elisei et al., 2013; Kurzrock et al., 2011; Leboulleux et al., 2012). However, current RET inhibitors may not prove as effective in treating patients with RET fusions. Here we explore the properties of two common RET fusions in the context of a whole animal model.
Though both contain primarily coiled-coiled domains, our studies demonstrate that the N-terminus partner of the common RET fusions CCDC6-RET and NCOA4-RET differentially impact the function and drug sensitivity of the fusion protein. Multiple differences between the two fusions were observed in an in vivo context. First, expressing NCOA4-RET led to more severe phenotypes including whole body lethality and cell invasion, mirroring the more aggressive phenotypes observed in patients (Basolo et al., 2002; Rabes, 2001; Thomas et al., 1999). Second, using an unbiased genetic screen of the Drosophila kinome, we identified significant differences in the functional networks used by each fusion to direct transformation-like phenotypes (Figure 3).
The signaling differences between our two models led to a difference in their response to therapeutics: tub>NCOA4-RET and tub>CCDC6-RET animals displayed distinct responses to drugs both as single agents and as two-drug cocktails (Figures 4, 5). Regarding NCOA4-RET, the enhancement of sorafenib by reducing wee activity was mirrored by the strong efficacy of sorafenib plus AZD1775. This combination may prove useful in refractory PTC or lung adenocarcinoma patients harboring NCOA4-RET; additional mammalian in situ experiments will be required to further assess its potential. We were unable to identify a synergistic drug combination specific to CCDC6-RET, consistent with our genetic results indicating the importance of suppressing JNK activity in the presence of a broad spectrum of kinase inhibitors (Figure 5C). As clinically-relevant JNK inhibitors are developed (Bennett et al., 2001; Davies and Tournier, 2012; Kaoud et al., 2011), they may prove useful for patients with the less aggressive CCDC6-RET tumor isoforms. Also of note we focused on drugs and drug combinations that promoted viability, perhaps a functional equivalent of cytostatic activity. Future efforts to identify cytotoxic drugs would be initiated by screening for drugs that promote lethality including cell lethality.
A key observation of our work is that the specific RET fusion is important when considering tumor progression and appropriate therapeutics. This will become increasingly important as additional tumors are identified as harboring particular RET fusions. Whole animal models such as Drosophila provide a useful tool for understanding the mechanistic basis of how specific RET fusions direct transformation. They can also provide useful information on drugs and drug cocktails that attack the unique networks activated by each RET isoform.
Experimental Procedures
Generation of Drosophila models of CCDC6-RET and NCOA4-RET
CCDC6-RET and NCOA4-RET cDNA were isolated from vectors pUGH10-3-RET/PTC1 and pUGH10-3-RET/PTC3 (Knauf, et. al, 2003) which were provided by James Fagin, Memorial Sloan Kettering Cancer Center. Both cDNAs were amplified from the pUGH10-3 vectors with primers that introduced Not1 sites on the 5′ end and Xba1 sites on the 3′ end and a Drosophila Kozac sequence (CACC) to ensure efficient initiation of translation once inserted into the Drosophila genome.
F-CCDC6: 5′ GATCGCGGCCGCCACCATGGCGGACAGCGCCAGCG 3′
F-NCOA4: 5′ GATCGCGGCCGCACCATGAATACCTTCCAAGACCA 3′
R-RET: 5′ GATCTCTAGACTAGAATCTAGTAATGCATGGGAAATTCTACC 3′
pUAST-attB vector was cut with Not1 and Xba1 enzymes to clone in CCDC6-RET or NCOA4-RET DNA.
Sequenced pUAST-attB-CCDC6-RET and pUAST-attB-NCOA4-RET vectors were sent to BestGene to be injected in Drosophila embryos, targeting both the attP2 and attP40 sites. Stable fly lines with single vector inserts were kept as balanced stocks over (i) the CyO balancer for attP40 insertions or (ii) TM6B balancer for attP2 insertions.
Immunohistochemistry
Wing discs from third instar larvae were dissected on ice, fixed in 4% paraformaldehyde in PBS, washed in PBS containing 0.1% Triton X-100, and incubated with primary antibodies in PAXDG (PBS containing 1% BSA, 0.3% Triton X-100, 0.3% deoxycholate, and 5% goat serum), followed by washing and incubation with secondary antibodies in PAXDG. Tissues were mounted in Vectashield mounting media (Vector Laboratories). Antibodies used were directed against phosphorylated RET Tyr905 (Cell Signaling #3221), DE-Cadherin (Developmental Studies Hybridoma Bank #DCAD2), MMP1 (Developmental Studies Hybridoma Bank #3B8D12), and phosphorylated SRC (Invitrogen #44660G). Alexa Fluor secondary antibodies were used. Confocal imaging used a Leica DM5500 Q microscope, and image analysis was performed using Adobe Photoshop.
Western Blots
20 wing discs from third instar larvae expressing either CCDC6-RET or NCOA4-RET under control of the 765-Gal4 driver and raised at the same temperature of 25°C were dissected and dissolved in lysis buffer (50 mM Tris, 150 mM NaCl, 1% Triton X-100, and 1 mM EDTA) supplemented with protease-inhibitor cocktail and phosphatase-inhibitor cocktail (Sigma). Total protein was quantified using Bio-Rad protein assay. Samples were boiled, resolved on SDS-PAGE, and transferred by standard protocols. Antibodies used were directed against phospho-RET Tyr905 (Cell Signaling #3221), DE-Cadherin (Developmental Studies Hybridoma Bank #DCAD2), Total RET (Cell Signaling #3223), phospho-Erk (Sigma-Aldrich #M8159), phospho-JNK/SAPK (Cell Signaling #4668), Rho1 (Developmental Studies Hybridoma Bank #p1D9), Rac1 (BD Biosciences Pharmingen #610651) and Syntaxin (Developmental Studies Hybridoma Bank #8B-3). ImageJ software was used for quantification.
Cell Migration
Wing discs from third instar larvae were dissected and fixed in 4% formaldehyde. Approximately 20 discs per genotype were mounted to a slide and the severity of migration was scored in a blinded format.
Kinome Screen
Flies overexpressing either CCDC6-RET (UAS-CCDC6-RET;tubulin-Gal4/Gal80ts) or NCOA4-RET (UAS-NCOA4-RET;tubulin-Gal4/Gal80ts) were crossed to loss-of-function alleles for each Drosophila kinase. Positive hits were scored for ratio of wild-type pupae to balancer (Tubby) pupae.
Drug Studies
Drugs were dissolved in DMSO to their maximum tolerated dose as described in Table S1. Drug was added to molten (50°C)-enriched fly food and then aliquoted into 12 × 75 mm, 5 ml test tubes (Sarstedt catalog no. B00471). After allowing to solidify at room temperature each tube contained 1 ml fly food and drug at a final DMSO concentration of 0.1%. Five female flies tubulinGal4/TM6B and five male flies UAS-CCDC6-RET or UAS-NCOA4-RET (both inserted at the attP40 chromosomal site) were pre-mated for 3 days and then allowed to lay 30–60 embryos. Drug vials were kept at 25°C (NCOA4-RET) or 27°C (CCDC6-RET) and scored for ratio of wild-type pupae to balanced (Tubby) pupae. Drug studies were conducted in smaller vials as described above due to cost of the compounds. These smaller vials induce more stress on the animal, which raises the level of lethality slightly. Therefore, several of the hits from the kinome screens did not show a rescue on their own when combined with DMSO in these small vials, as they were originally identified with animals being raised in their larger vials.
Supplementary Material
Figure S1. RET fusions activated canonical signaling pathways in Drosophila. Related to Figure 2.
A representative Western blot showing that 765>CCDC6-RET and 765>NCOA4-RET animals induced activation of Erk and Jnk and upregulated levels of Rho1 but not Rac1. This blot also shows that similar levels of either RET fusion were expressed (Total Ret) and that at these levels of expression the NCOA4-RET isoform had a higher level of activation (pRet).
Figure S2. Migration phenotype significantly worse in ptc>NCOA4-RET animals than ptc>CCDC6-RET animals. Related to Figure 1.
Representative images of the four categories (none, moderate, weak, and severe) used to quantify cell migration caused by each RET fusion expression under control of the ptc-GAL4 driver in larval wing discs. Upper panels are an apical slice of the wing disc, lower panels are a basal slice through the same wing disc. Migration was scored by setting the boundary based on the apical region, and then counting cells that migrated away from this boundary by looking in the basal region. Wing discs were scored in a blinded format. Scale bar represents 50 μm in all panels.
Figure S3. RET fusions upregulate levels of phosphorylated Src in migrating and non-migrating cells. Related to Figure 2.
Third instar larval wing discs expressing transgenes within the ptc-GAL4 expression domain. An antibody against phosphorylated Src shows that expression of either RET fusion led to upregulation of activated Src, a marker of EMT, in cells that had not yet migrated away from their endogenous space. 2A′–C′ shows that pSrc is upregulated in migrating cells, as well. Note that ptc>NCOA4-RET displayed higher levels of pSrc, correlating with its more severe cell migration phenotype. Scale bar represents 50 μm in all panels.
Figure S4. Migrating cells stain positive for cleaved caspase 3. Related to Figure 2.
Third instar larval wing discs expressing transgenes within the ptc-GAL4 expression domain. An antibody against cleaved caspase 3 shows that expression of either RET fusion led to migrating cells that are also activating caspases. Cleaved caspase 3 is a marker of apoptosis, indicating that some of the migrating cells are dying. Previous work in our lab has shown that caspase activity is a component of a JNK-Rho1-Actin signaling axis that drives cell migration (Rudrapatna et al., 2013). These migrating cells may be activating a similar signaling cascade as a part of their acquired migratory phenotype. Scale bar represents 50 μm in all panels.
Figure S5. RET fusions directed similarities, differences in signaling networks. Related to Figure 3.
Tables indicating the level of rescue caused by heterozygosity of different kinase alleles in the background of either tub>CCDC6-RET or tub>NCOA4-RET. Note that while two kinase alleles were hits for both RET fusion isoforms their sensitivities differed.
Figure S6. RET fusions were sensitive to shared and distinct kinase inhibitors. Related to Figure 4.
Tables indicating the level of rescue caused by different kinase inhibitors when fed to either tub>CCDC6-RET or tub>NCOA4-RET animals.
Figure S7. Kinase inhibitors rescued cell migration in wing epithelium. Related to Figure 4.
Tables showing how kinase inhibitors vandetanib and AD81 were able to reduce the severity of the cell migration phenotype observed in ptc>CCDC6-RET and ptc>NCOA4-RET animals. Third instar larval wing discs were dissected, fixed, and mounted and cells were visualized with GFP. Slides were scored in a blinded format and binned into one of four qualitative categories (none, weak, moderate, and severe). ptc>CCDC6-RET animals had significantly fewer wing discs with weak or moderate cell migration when fed either vandetanib or AD81. ptc>NCOA4-RET had significantly fewer wing discs with moderate or severe cell migration when fed AD81, however no significant change in severity of phenotype when fed vandetanib. Note that both vandetanib and AD81 were able to rescue lethality in tub>CCDC6-RET animals, yet only AD81 was able to rescue tub>NCOA4-RET animals (Figure 4).
Table S1. RET fusions were sensitive to shared and distinct kinase inhibitors. Related to Figure 4.
Table showing results from the drug screen. All drugs were tested at their predetermined Drosophila maximum tolerated dose. Significant p values are listed (p>0.05).
Table S2. Rational identification of synergistic genetic drug combinations for NCOA4-RET. Related to Figure 5.
Table showing the level of rescue conferred by the combination of heterozygosity of each kinase allele identified from the kinome screen and feeding of each drug identified from the drug screen in tub>NCOA4-RET animals. Percent pupariation was calculated by dividing the number of tub>NCOA4-RET animals to the internal control (Tubby balancer). Values greater than 100% indicate that the combination genetic heterozygosity and drug treatment allowed more tub>NCOA4-RET animals to survive to pupariation than Tubby balancer controls, suggesting some reduction of viability from the latter.
Table S3. Rational identification of synergistic genetic drug combinations for CCDC6-RET. Related to Figure 5.
Table showing the level of rescue conferred by the combination of heterozygosity of each kinase allele identified from the kinome screen and feeding of each drug identified from the drug screen in tub>CCDC6-RET animals. The baseline for adult survival is usually 0% for this model, however when a copy of JNK ortholog bsk is removed adult survival is rescued up to 15%, which is increased to 64% in the context of AD81.
Table S4. Identification of synergistic drug cocktails for NCOA4-RET animals. Related to Figure 5.
Table showing level of rescue combinations of drugs had on tub>NCOA4-RET animals. Flies were fed the same concentration of primary drug (AD81, sorafenib, or regorafenib) that was able to rescue in the previous drug screen and a concentration of secondary drug (ibrutinib, AZD1775, HA-100) that had no effect on lethality. This way an increased efficacy in the drug combination would be identified as a synergistic effect. Percent pupariation was calculated by dividing the number of tub>NCOA4-RET animals to the internal control (Tubby balancer). Values greater than 100% indicate that the combinatorial drug treatment allowed more tub>NCOA4-RET animals to survive to pupariation than Tubby balancer controls.
Highlights.
Drosophila RET fusion cancer models have multiple transformation phenotypes
Similar to patients, NCOA4-RET present with more severe phenotypes than CCDC6-RET
CCDC6-RET and NCOA4-RET activate distinct pathways and have different drug sensitivity
A drug treatment, sorafenib plus AZD1775 (MK-1775), was effective for NCOA4-RET animals
Acknowledgments
We thank members of the Cagan laboratory for technical assistance and for helpful discussions. We thank Vienna Drosophila Resource Center and the Bloomington Drosophila Stock Center for Drosophila reagents. Microscopy was performed in part at the Microscopy Shared Resource Facility at the Icahn School of Medicine at Mount Sinai. This research was supported by National Institutes of Health grants NCI F31CA189794 to S.L and R01CA170495, R01CA170495, R01CA109730, and U54OD020353 to R.C.
Footnotes
Author Contributions
Conceptualization, Methodology, and Writing, S.L. and R.L.C; Investigation, S.L.; Funding Acquisition, S.L. and R.L.C; Supervision, R.L.C.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30:679–692. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- Ballerini P, Struski S, Cresson C, Prade N, Toujani S, Deswarte C, Dobbelstein S, Petit a, Lapillonne H, Gautier EF, Demur C, Lippert E, Pages P, Mansat-De Mas V, Donadieu J, Huguet F, Dastugue N, Broccardo C, Perot C, Delabesse E. RET fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia. 2012;26:2384–2389. doi: 10.1038/leu.2012.109. [DOI] [PubMed] [Google Scholar]
- Basolo F, Giannini R, Monaco C, Melillo RM, Carlomagno F, Pancrazi M, Salvatore G, Chiappetta G, Pacini F, Elisei R, Miccoli P, Pinchera A, Fusco A, Santoro M. Potent Mitogenicity of the RET/PTC3 Oncogene Correlates with Its Prevalence in Tall-Cell Variant of Papillary Thyroid Carcinoma. Am J Pathol. 2002;160:247–254. doi: 10.1016/S0002-9440(10)64368-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrello MG, Ardini E, Locati LD, Greco A, Licitra L, Pierotti Ma. Review RET inhibition: implications in cancer therapy. 2013:1–17. doi: 10.1517/14728222.2013.758715. [DOI] [PubMed] [Google Scholar]
- Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, Buchholz TA, Molkentine JM, Mason KA, Meyn RE. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res. 2011;17:5638–48. doi: 10.1158/1078-0432.CCR-11-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RL, de Souza JA, Cohen EE. Thyroid cancer: burden of illness and management of disease. J Cancer. 2011;2:193–199. doi: 10.7150/jca.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlomagno F, Vitagliano D, Guida T, Ciardiello F, Tortora G, Vecchio G, Ryan AJ, Fontanini G, Fusco A, Santoro M. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62:7284–90. [PubMed] [Google Scholar]
- Castellone MD, Santoro M. Dysregulated RET signaling in thyroid cancer. Endocrinol Metab Clin North Am. 2008;37:363–74. viii. doi: 10.1016/j.ecl.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Dar AC, Das TK, Shokat KM, Cagan RL. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature. 2012;486:80–84. doi: 10.1038/nature11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies C, Tournier C. Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem Soc Trans. 2012;40:85–9. doi: 10.1042/BST20110641. [DOI] [PubMed] [Google Scholar]
- Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, Collins J, Chen AP, Doroshow JH, Kummar S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J Clin Oncol. 2015;33:3409–15. doi: 10.1200/JCO.2014.60.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards A, Gladstone M, Yoon P, Raben D, Frederick B, Su TT. Combinatorial effect of maytansinol and radiation in Drosophila and human cancer cells. Dis Model Mech. 2011;4:496–503. doi: 10.1242/dmm.006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elisei R, Schlumberger MJ, Müller SP, Schöffski P, Brose MS, Shah MH, Licitra L, Jarzab B, Medvedev V, Kreissl MC, Niederle B, Cohen EEW, Wirth LJ, Ali H, Hessel C, Yaron Y, Ball D, Nelkin B, Sherman SI. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31:3639–46. doi: 10.1200/JCO.2012.48.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gild ML, Landa I, Ryder M, Ghossein Ra, Knauf Ja, Fagin Ja. Targeting mTOR in RET mutant medullary and differentiated thyroid cancer cells. Endocr Relat Cancer. 2013;20:659–667. doi: 10.1530/ERC-13-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladstone M, Frederick B, Zheng D, Edwards A, Yoon P, Stickel S, DeLaney T, Chan DC, Raben D, Su TT. A translation inhibitor identified in a Drosophila screen enhances the effect of ionizing radiation and taxol in mammalian models of cancer. Dis Model Mech. 2012;5:342–50. doi: 10.1242/dmm.008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, Pierotti Ma, Della Porta G, Fusco a, Vecchio G. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell. 1990;60:557–563. doi: 10.1016/0092-8674(90)90659-3. [DOI] [PubMed] [Google Scholar]
- Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107:13075–80. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiike A, Takeuchi K, Uenami T, Kawano Y, Tanimoto A, Kaburaki K, Tambo Y, Kudo K, Yanagitani N, Ohyanagi F, Motoi N, Ishikawa Y, Horai T, Nishio M. Sorafenib treatment for patients with RET fusion-positive non-small cell lung cancer. Lung Cancer. 2016;93:43–6. doi: 10.1016/j.lungcan.2015.12.011. [DOI] [PubMed] [Google Scholar]
- Iadevaia S, Lu Y, Morales FC, Mills GB, Ram PT. Identification of optimal drug combinations targeting cellular networks: integrating phospho-proteomics and computational network analysis. Cancer Res. 2010;70:6704–14. doi: 10.1158/0008-5472.CAN-10-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhiang SM, Caruso DR, Gilmore E, Ishizaka Y, Tahira T, Nagao M, Chiu IM, Mazzaferri EL. Detection of the PTC/retTPC oncogene in human thyroid cancers. Oncogene. 1992;7:1331–7. [PubMed] [Google Scholar]
- Jhiang SM, Sagartz JE, Tong Q, Parker-Thornburg J, Capen CC, Cho JY, Xing S, Ledent C. Targeted expression of the ret/PTC1 oncogene induces papillary thyroid carcinomas. Endocrinology. 1996;137:375–8. doi: 10.1210/endo.137.1.8536638. [DOI] [PubMed] [Google Scholar]
- Kaoud TS, Mitra S, Lee S, Taliaferro J, Cantrell M, Linse KD, Van Den Berg CL, Dalby KN. Development of JNK2-selective peptide inhibitors that inhibit breast cancer cell migration. ACS Chem Biol. 2011;6:658–66. doi: 10.1021/cb200017n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith CT, Borisy AA, Stockwell BR. Multicomponent therapeutics for networked systems. Nat Rev Drug Discov. 2005;4:71–8. doi: 10.1038/nrd1609. [DOI] [PubMed] [Google Scholar]
- Kim DW, Jo YS, Jung HS, Chung HK, Song JH, Park KC, Park SH, Hwang JH, Rha SY, Kweon GR, Lee SJ, Jo KW, Shong M. An orally administered multitarget tyrosine kinase inhibitor, SU11248, is a novel potent inhibitor of thyroid oncogenic RET/papillary thyroid cancer kinases. J Clin Endocrinol Metab. 2006;91:4070–6. doi: 10.1210/jc.2005-2845. [DOI] [PubMed] [Google Scholar]
- Knauf JA, Kuroda H, Basu S, Fagin JA. RET/PTC-induced dedifferentiation of thyroid cells is mediated through Y1062 signaling through SHC-RAS-MAP kinase. Oncogene. 2003;22:4406–12. doi: 10.1038/sj.onc.1206602. [DOI] [PubMed] [Google Scholar]
- Knostman KaB, Jhiang SM, Capen CC. Genetic Alterations in Thyroid Cancer: The Role of Mouse Models. Vet Pathol. 2007;44:1–14. doi: 10.1354/vp.44-1-1. [DOI] [PubMed] [Google Scholar]
- Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, Sakamoto H, Tsuta K, Furuta K, Shimada Y, Iwakawa R, Ogiwara H, Oike T, Enari M, Schetter AJ, Okayama H, Haugen A, Skaug V, Chiku S, Yamanaka I, Arai Y, Watanabe S, Sekine I, Ogawa S, Harris CC, Tsuda H, Yoshida T, Yokota J, Shibata T. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18:375–377. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzrock R, Sherman SI, Ball DW, Forastiere AA, Cohen RB, Mehra R, Pfister DG, Cohen EEW, Janisch L, Nauling F, Hong DS, Ng CS, Ye L, Gagel RF, Frye J, Müller T, Ratain MJ, Salgia R. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011;29:2660–6. doi: 10.1200/JCO.2010.32.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Rolle A-F, Klempner SJ, Garrett CR, Seery T, Sanford EM, Balasubramanian S, Ross JS, Stephens PJ, Miller VA, Ali SM, Chiu VK. Identification and characterization of RET fusions in advanced colorectal cancer. Oncotarget. 2015 doi: 10.18632/oncotarget.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leboulleux S, Bastholt L, Krause T, de la Fouchardiere C, Tennvall J, Awada A, Gómez JM, Bonichon F, Leenhardt L, Soufflet C, Licour M, Schlumberger MJ. Vandetanib in locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 2 trial. Lancet Oncol. 2012;13:897–905. doi: 10.1016/S1470-2045(12)70335-2. [DOI] [PubMed] [Google Scholar]
- Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, Curran Ja, Balasubramanian S, Bloom T, Brennan KW, Donahue A, Downing SR, Frampton GM, Garcia L, Juhn F, Mitchell KC, White E, White J, Zwirko Z, Peretz T, Nechushtan H, Soussan-Gutman L, Kim J, Sasaki H, Kim HR, Park S, Ercan D, Sheehan CE, Ross JS, Cronin MT, Jänne Pa, Stephens PJ. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–384. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mologni L, Redaelli S, Morandi A, Plaza-Menacho I, Gambacorti-Passerini C. Ponatinib is a potent inhibitor of wild-type and drug-resistant gatekeeper mutant RET kinase. Mol Cell Endocrinol. 2013;377:1–6. doi: 10.1016/j.mce.2013.06.025. [DOI] [PubMed] [Google Scholar]
- Müller D, Kugler SJ, Preiss A, Maier D, Nagel AC. Genetic modifier screens on Hairless gain-of-function phenotypes reveal genes involved in cell differentiation, cell growth and apoptosis in Drosophila melanogaster. Genetics. 2005;171:1137–52. doi: 10.1534/genetics.105.044453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabes HM. Gene rearrangements in radiation-induced thyroid carcinogenesis. Med Pediatr Oncol. 2001;36:574–82. doi: 10.1002/mpo.1133. [DOI] [PubMed] [Google Scholar]
- Read RD, Goodfellow PJ, Mardis ER, Novak N, Armstrong JR, Cagan RL. A Drosophila model of multiple endocrine neoplasia type 2. Genetics. 2005;171:1057–81. doi: 10.1534/genetics.104.038018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricarte-Filho JC, Ryder M, Chitale Da, Rivera M, Heguy a, Ladanyi M, Janakiraman M, Solit D, Knauf Ja, Tuttle RM, Ghossein Ra, Fagin Ja. Mutational Profile of Advanced Primary and Metastatic Radioactive Iodine-Refractory Thyroid Cancers Reveals Distinct Pathogenetic Roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009;69:4885–4893. doi: 10.1158/0008-5472.CAN-09-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson DS, Gujral TS, Peng S, Asa SL, Mulligan LM. Transcript Level Modulates the Inherent Oncogenicity of RET/PTC Oncoproteins. Cancer Res. 2009;69:4861–4869. doi: 10.1158/0008-5472.CAN-08-4425. [DOI] [PubMed] [Google Scholar]
- Rudrapatna Va, Bangi E, Cagan RL. Caspase signalling in the absence of apoptosis drives Jnk-dependent invasion. EMBO Rep. 2013;14:172–177. doi: 10.1038/embor.2012.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudrapatna Va, Cagan RL, Das TK. Drosophila cancer models. Dev Dyn. 2012;241:107–118. doi: 10.1002/dvdy.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro M, Melillo RM, Fusco a. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology Prize Lecture. Eur J Endocrinol. 2006;155:645–653. doi: 10.1530/eje.1.02289. [DOI] [PubMed] [Google Scholar]
- Shi H, Kong X, Ribas A, Lo RS. Combinatorial treatments that overcome PDGFRβ-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res. 2011;71:5067–74. doi: 10.1158/0008-5472.CAN-11-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MA. Signal transduction during the development of the Drosophila R7 photoreceptor. Dev Biol. 1994;166:431–42. doi: 10.1006/dbio.1994.1327. [DOI] [PubMed] [Google Scholar]
- Slack C, Alic N, Foley A, Cabecinha M, Hoddinott MP, Partridge L. The Ras-Erk-ETS-Signaling Pathway Is a Drug Target for Longevity. Cell. 2015;162:72–83. doi: 10.1016/j.cell.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR. Ibrutinib in B lymphoid malignancies. Expert Opin Pharmacother. 2015;16:1879–87. doi: 10.1517/14656566.2015.1067302. [DOI] [PubMed] [Google Scholar]
- St Johnston D. The art and design of genetic screens: Drosophila melanogaster. Nat Rev Genet. 2002;3:176–88. doi: 10.1038/nrg751. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, Lim Choi Y, Satoh Y, Okumura S, Nakagawa K, Mano H, Ishikawa Y. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378–381. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- Thomas GA, Bunnell H, Cook HA, Williams ED, Nerovnya A, Cherstvoy ED, Tronko ND, Bogdanova TI, Chiappetta G, Viglietto G, Pentimalli F, Salvatore G, Fusco A, Santoro M, Vecchio G. High prevalence of RET/PTC rearrangements in Ukrainian and Belarussian post-Chernobyl thyroid papillary carcinomas: a strong correlation between RET/PTC3 and the solid-follicular variant. J Clin Endocrinol Metab. 1999;84:4232–8. doi: 10.1210/jcem.84.11.6129. [DOI] [PubMed] [Google Scholar]
- Verbeek HHG, Alves MM, de Groot JWB, Osinga J, Plukker JTM, Links TP, Hofstra RMW. The effects of four different tyrosine kinase inhibitors on medullary and papillary thyroid cancer cells. J Clin Endocrinol Metab. 2011;96:E991–5. doi: 10.1210/jc.2010-2381. [DOI] [PubMed] [Google Scholar]
- Vidal M, Salavaggione L, Ylagan L, Wilkins M, Watson M, Weilbaecher K, Cagan R. A role for the epithelial microenvironment at tumor boundaries: evidence from Drosophila and human squamous cell carcinomas. Am J Pathol. 2010;176:3007–14. doi: 10.2353/ajpath.2010.090253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Wells S, Ryan A, Cagan R. ZD6474 suppresses oncogenic RET isoforms in a Drosophila model for type 2 multiple endocrine neoplasia syndromes and papillary thyroid carcinoma. Cancer Res. 2005;65:3538–41. doi: 10.1158/0008-5472.CAN-04-4561. [DOI] [PubMed] [Google Scholar]
- Wagstaff L, Kolahgar G, Piddini E. Competitive cell interactions in cancer: a cellular tug of war. Trends Cell Biol. 2013;23:160–167. doi: 10.1016/j.tcb.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Wang J, Knauf JA, Basu S, Puxeddu E, Kuroda H, Santoro M, Fusco A, Fagin JA. Conditional expression of RET/PTC induces a weak oncogenic drive in thyroid PCCL3 cells and inhibits thyrotropin action at multiple levels. Mol Endocrinol. 2003;17:1425–36. doi: 10.1210/me.2003-0041. [DOI] [PubMed] [Google Scholar]
- Ward RE, Evans J, Thummel CS. Genetic modifier screens in Drosophila demonstrate a role for Rho1 signaling in ecdysone-triggered imaginal disc morphogenesis. Genetics. 2003;165:1397–415. doi: 10.1093/genetics/165.3.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells Sa, Santoro M. Targeting the RET Pathway in Thyroid Cancer. Clin Cancer Res. 2009;15:7119–7123. doi: 10.1158/1078-0432.CCR-08-2742. [DOI] [PubMed] [Google Scholar]
- Willoughby LF, Schlosser T, Manning SA, Parisot JP, Street IP, Richardson HE, Humbert PO, Brumby AM. An in vivo large-scale chemical screening platform using Drosophila for anti-cancer drug discovery. Dis Model Mech. 2013;6:521–9. doi: 10.1242/dmm.009985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing M, Haugen BR, Schlumberger M. Progress in molecular-based management of differentiated thyroid cancer. Lancet. 2013;381:1058–1069. doi: 10.1016/S0140-6736(13)60109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Zhang B, Li S, Zhao Q. A formal model for analyzing drug combination effects and its application in TNF-alpha-induced NFkappaB pathway. BMC Syst Biol. 2010;4:50. doi: 10.1186/1752-0509-4-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. RET fusions activated canonical signaling pathways in Drosophila. Related to Figure 2.
A representative Western blot showing that 765>CCDC6-RET and 765>NCOA4-RET animals induced activation of Erk and Jnk and upregulated levels of Rho1 but not Rac1. This blot also shows that similar levels of either RET fusion were expressed (Total Ret) and that at these levels of expression the NCOA4-RET isoform had a higher level of activation (pRet).
Figure S2. Migration phenotype significantly worse in ptc>NCOA4-RET animals than ptc>CCDC6-RET animals. Related to Figure 1.
Representative images of the four categories (none, moderate, weak, and severe) used to quantify cell migration caused by each RET fusion expression under control of the ptc-GAL4 driver in larval wing discs. Upper panels are an apical slice of the wing disc, lower panels are a basal slice through the same wing disc. Migration was scored by setting the boundary based on the apical region, and then counting cells that migrated away from this boundary by looking in the basal region. Wing discs were scored in a blinded format. Scale bar represents 50 μm in all panels.
Figure S3. RET fusions upregulate levels of phosphorylated Src in migrating and non-migrating cells. Related to Figure 2.
Third instar larval wing discs expressing transgenes within the ptc-GAL4 expression domain. An antibody against phosphorylated Src shows that expression of either RET fusion led to upregulation of activated Src, a marker of EMT, in cells that had not yet migrated away from their endogenous space. 2A′–C′ shows that pSrc is upregulated in migrating cells, as well. Note that ptc>NCOA4-RET displayed higher levels of pSrc, correlating with its more severe cell migration phenotype. Scale bar represents 50 μm in all panels.
Figure S4. Migrating cells stain positive for cleaved caspase 3. Related to Figure 2.
Third instar larval wing discs expressing transgenes within the ptc-GAL4 expression domain. An antibody against cleaved caspase 3 shows that expression of either RET fusion led to migrating cells that are also activating caspases. Cleaved caspase 3 is a marker of apoptosis, indicating that some of the migrating cells are dying. Previous work in our lab has shown that caspase activity is a component of a JNK-Rho1-Actin signaling axis that drives cell migration (Rudrapatna et al., 2013). These migrating cells may be activating a similar signaling cascade as a part of their acquired migratory phenotype. Scale bar represents 50 μm in all panels.
Figure S5. RET fusions directed similarities, differences in signaling networks. Related to Figure 3.
Tables indicating the level of rescue caused by heterozygosity of different kinase alleles in the background of either tub>CCDC6-RET or tub>NCOA4-RET. Note that while two kinase alleles were hits for both RET fusion isoforms their sensitivities differed.
Figure S6. RET fusions were sensitive to shared and distinct kinase inhibitors. Related to Figure 4.
Tables indicating the level of rescue caused by different kinase inhibitors when fed to either tub>CCDC6-RET or tub>NCOA4-RET animals.
Figure S7. Kinase inhibitors rescued cell migration in wing epithelium. Related to Figure 4.
Tables showing how kinase inhibitors vandetanib and AD81 were able to reduce the severity of the cell migration phenotype observed in ptc>CCDC6-RET and ptc>NCOA4-RET animals. Third instar larval wing discs were dissected, fixed, and mounted and cells were visualized with GFP. Slides were scored in a blinded format and binned into one of four qualitative categories (none, weak, moderate, and severe). ptc>CCDC6-RET animals had significantly fewer wing discs with weak or moderate cell migration when fed either vandetanib or AD81. ptc>NCOA4-RET had significantly fewer wing discs with moderate or severe cell migration when fed AD81, however no significant change in severity of phenotype when fed vandetanib. Note that both vandetanib and AD81 were able to rescue lethality in tub>CCDC6-RET animals, yet only AD81 was able to rescue tub>NCOA4-RET animals (Figure 4).
Table S1. RET fusions were sensitive to shared and distinct kinase inhibitors. Related to Figure 4.
Table showing results from the drug screen. All drugs were tested at their predetermined Drosophila maximum tolerated dose. Significant p values are listed (p>0.05).
Table S2. Rational identification of synergistic genetic drug combinations for NCOA4-RET. Related to Figure 5.
Table showing the level of rescue conferred by the combination of heterozygosity of each kinase allele identified from the kinome screen and feeding of each drug identified from the drug screen in tub>NCOA4-RET animals. Percent pupariation was calculated by dividing the number of tub>NCOA4-RET animals to the internal control (Tubby balancer). Values greater than 100% indicate that the combination genetic heterozygosity and drug treatment allowed more tub>NCOA4-RET animals to survive to pupariation than Tubby balancer controls, suggesting some reduction of viability from the latter.
Table S3. Rational identification of synergistic genetic drug combinations for CCDC6-RET. Related to Figure 5.
Table showing the level of rescue conferred by the combination of heterozygosity of each kinase allele identified from the kinome screen and feeding of each drug identified from the drug screen in tub>CCDC6-RET animals. The baseline for adult survival is usually 0% for this model, however when a copy of JNK ortholog bsk is removed adult survival is rescued up to 15%, which is increased to 64% in the context of AD81.
Table S4. Identification of synergistic drug cocktails for NCOA4-RET animals. Related to Figure 5.
Table showing level of rescue combinations of drugs had on tub>NCOA4-RET animals. Flies were fed the same concentration of primary drug (AD81, sorafenib, or regorafenib) that was able to rescue in the previous drug screen and a concentration of secondary drug (ibrutinib, AZD1775, HA-100) that had no effect on lethality. This way an increased efficacy in the drug combination would be identified as a synergistic effect. Percent pupariation was calculated by dividing the number of tub>NCOA4-RET animals to the internal control (Tubby balancer). Values greater than 100% indicate that the combinatorial drug treatment allowed more tub>NCOA4-RET animals to survive to pupariation than Tubby balancer controls.