

Summary
Endothelial lipase (LIPG) plays a critical role in lipoprotein metabolism, cytokine expression, and the lipid composition of cells. Thus far, the extensive investigations of LIPG have focused on its mechanisms and involvement in metabolic syndromes such as atherosclerosis. However, recent developments have found that LIPG plays a role in cancer. This review summarizes the field of LIPG study. We focus on the role of LIPG in lipid metabolism and the inflammatory response, and highlight the recent insights in its involvement in tumor progression. Finally, we discuss the potential of targeting LIPG in therapeutic strategies.
Keywords: endothelial lipase, LIPG, cancer, inflammation, lipid metabolism
Introduction
Lipids are a category of macromolecules consisting of hydrophobic and amphipathic molecules that are divided into diverse groups based on structure, including: fatty acyls, glycerolipids, glycerophospholipids, sphingolipids, sterols, prenols, saccharolipids, and polyketides (Fahy et al., 2009). Lipids play critical roles in maintaining cell homeostasis and regulating proliferation, apoptosis, inflammation, and membrane synthesis (Baumann et al., 2013). Lipids are obtained through dietary intake or endogenous cell synthesis. After entering a cell, lipids go through intricate metabolic pathways, resulting in the synthesis or degradation of lipid signaling molecules and support of intercellular processes. These metabolic pathways are highly regulated and necessary to maintain cell homeostasis.
Complex lipids are made up of fatty acids, which are hydrophobic molecules characterized by their repeating methylene groups (Fahy et al., 2005). Fatty acids travel through the bloodstream both freely and complexed in lipoproteins, molecules composed of a hydrophilic membrane containing phospholipids, apolipoproteins, and free cholesterol centralized around a hydrophobic core. This hydrophilic outer layer emulsifies highly hydrophobic lipid molecules, and transports them through the blood stream. Lipoproteins are divided into five categories based on relative density, which is determined by their size and composition of lipids and proteins. These classes include: chlyomicrons, very low density lipoproteins (VLDL), intermediate density lipoproteins (IDL), low density lipoproteins (LDL), and high density lipoproteins (HDL). HDL has many anti-inflammatory and anti-atherogenic properties such as its promotion of cholesterol efflux from cells in the artery wall, inhibition of adhesion molecules in endothelial cells, and suppression of LDL oxidation (Barter et al., 2004). Although the functional properties of HDL have a clear value for the development of anti-atherogenic therapies, the clinical impact of therapies that raise HDL levels is still uncertain. Therefore, the mechanisms that regulate the anti-inflammatory effects of HDL require further study.
In order for fatty acids to enter the cell, circulating lipoproteins must be hydrolyzed. Extracellular enzymes called lipases bind to lipoproteins and catalyze their hydrolysis, releasing their contents. Free fatty acids are then taken up by cells, where they can directly enter metabolic pathways or be stored for later use in intracellular structures called lipid droplets. The triglyceride (TG) lipase gene family plays a critical role in plasma lipoprotein metabolism, and includes the lipases lipoprotein lipase (LPL), hepatic lipase (HL), and endothelial lipase (LIPG). Endothelial lipase is of particular interest, as it has been demonstrated to be upregulated in inflammatory conditions such as atherosclerosis, and is a primary determinant of plasma HDL levels.
While the roles of lipids in the pathology of disease are still widely undefined, there is increasing evidence that deregulation of lipid metabolism and changes in intercellular lipid composition play an essential role in the initiation and progression of diseases such as atherosclerosis and cancer. LIPG is widely accepted to have an important functional role in atherosclerosis and inflammation, however its contribution to cancer cell energetics is still unknown. In this review, we will summarize the key findings of LIPG in lipid metabolism, its role in metabolic diseases and inflammation, and explore its potential therapeutic value in targeting cancer cell metabolism.
Structure of LIPG
Endothelial lipase is encoded by the LIPG gene, and is a member of the triglyceride lipase family. LPL is mainly synthesized by adipocytes, skeletal muscle cells, and cardiac muscle cells, and HL is found in the liver. In contrast, LIPG is primarily synthesized by vascular endothelial cells (Jaye et al., 1999). It is abundantly expressed in tissues with high metabolic rates and vascularization, including the liver, lung, kidney, thyroid, ovary, testis, and placenta (Hirata et al., 1999; Jaye et al., 1999). LIPG is synthesized as a 55 kDa protein, and after post-translational glycosylation is secreted from the cell as a 68 kDa protein. After secretion, LIPG binds to proteoglycans on the cell surface where it exerts its function (Broedl et al., 2003). Outside the cell, it may undergo further cleavage by proprotein convertases, resulting in inactive N-terminal 40 kDa and C-terminal 28 kDa products (Jaye et al., 1999; Jin, Sun, et al., 2003; Miller et al., 2004; Gauster, Hrzenjak, et al., 2005).
The tertiary structure of LIPG has yet to be discovered. However, amino acid sequence alignments of LIPG with closely related TG lipase family members reveal a shared identity of 44% with LPL, 41% with HL, and 27% with pancreatic lipase (PL). All four enzymes share a similar catalytic region, with full conservation of the catalytic triad consisting of Serine 169, Aspartic Acid 193, and Histidine 274, which is necessary for lipid hydrolysis (Hirata et al., 1999; Jaye et al., 1999). Additionally, there is conservation of 10 cysteine residues involved in disulfide bond formation, as well as two hydrophobic stretches on both ends of the catalytic triad that may be necessary for substrate binding (Hirata et al., 1999). Much like other TG lipases, the LIPG protein sequence begins with a secretory signal peptide consisting of an 18-residue hydrophobic region. The conserved positively charged heparin binding sites in LIPG govern its binding to heparin sulphate proteoglycans on the cell surface, aided by five glycosylation sites indicated by the universal acceptor sequence: Asn-X-(Ser/Thr). These similarities suggest that there is a high level of structural and functional conservation within the TG family (Hirata et al., 1999; Jaye et al., 1999). The primary difference between LIPG and its TG lipase family members lies within the amino acid loop or “lid” region covering the catalytic site and governing substrate specificity (Dugi et al., 1992; Hirata et al., 1999; Jaye et al., 1999; Griffon et al., 2006). This structural difference indicates that contrary to the primary triglyceride lipase functions of LPL and HL, LIPG predominantly functions as a phospholipase, but maintains low levels of triglyceride lipase activity (McCoy et al., 2002).
LIPG possesses three unique splice isoforms, endothelial-derived lipases (EDL) 1a, 2a, and 2b (Ishida, Zheng, et al., 2004). EDL1a is the predominant 68 kDa full-length glycosylated form (500 amino acids), whereas 2a and 2b result from alternative splicing of the full-length gene. EDL2a is a truncated 480 amino acid version of the full-length isoform, which no longer contains the secretory signal peptide at the 5′ end. In comparison, the 346 amino acid long EDL2b isoform has a similar sequence to EDL2a, but has a 74 amino acid deleted region, which includes the amino-terminal portion of the lid region and the catalytic Asparagine residue. All three isoforms contain the GXSXG lipase motif and hydrophobic clusters. Studies by Ishida et al. revealed that 1a (68 kDa) was primarily found in the culture medium and to a lesser extent in the cell lysate, while the 2a (46 kDa) and 2b (38kDa) isoforms were only detected in the cell lysate, and localized in the cytosol. Importantly, assessment of the metabolic function of the isoforms found that only the full-length 1a isoform has high phospholipase and low triglyceride lipase activity, while 2a and 2b showed no significant enzymatic activity as compared to control cells. Therefore, due to the lack of the secretory peptide, EDL 2a and 2b splice variants are not secreted from the cell and do not possess lipase activity.
While the full-length protein is catalytically active by itself, LIPG commonly forms a head-to-tail homodimer prior to secretion (Griffon et al., 2009). Dimerization appears to both enhance LIPG activity and stabilize it from inactivation by proprotein convertases. Convertase cleavage results in N-terminal 40 kDa and C-terminal 28 kDa isoforms, separating the catalytic site from the substrate-binding region necessary for LIPG to bind to lipoproteins. Dimerized LIPG is more resistant to cleavage, and when cleaved can maintain partial activity (Gauster, Hrzenjak, et al., 2005; Griffon et al., 2009).
Enzymatic and Non-Enzymatic Functions of LIPG
The primary function of LIPG is its involvement in HDL metabolism. LIPG overexpression in transgenic mice dramatically reduced the serum concentration of HDL cholesterol (HDL-C) and apo A-I, while marginally reducing VLDL/LDL cholesterol levels (Jaye et al., 1999; Ishida et al., 2003). Conversely, antibody-directed inhibition of mouse LIPG activity in wild-type, apo A-transgenic, and HL-knockout mice resulted in a significant increase in HDL cholesterol and phospholipid levels, which was attributed to increased HDL particle size and reduced HDL phospholipid clearance (Jin, Millar, et al., 2003). Additionally, HDL cholesterol levels were significantly increased in LIPG knockout mice (Ishida et al., 2003; Ma et al., 2003). Therefore, it was observed that HDL is the preferred substrate of LIPG, but LIPG is still capable of hydrolyzing apo B-containing lipoproteins (VLDL/LDL) (McCoy et al., 2002; Broedl et al., 2004). In fact, Broedl et al. demonstrated that LIPG reduces the serum concentration of VLDL cholesterol, LDL cholesterol, phospholipids, and apo B in atherosclerosis-prone mouse models with elevated apo B-containing lipoproteins (Broedl et al., 2004). These data suggest that in addition to its role in HDL metabolism, LIPG may also contribute to VLDL and LDL metabolism.
Unlike its TG lipase family members, LIPG primarily functions as a phospholipase with minor triglyceride lipase activity. Hirata et al. observed the phospholipase activity of LIPG by incubating LIPG-overexpressing COS7 cells in the presence of phosphatidylcholine (PC) labeled at the sn-1 position, and measuring the level of free fatty acids released into the supernatant (Hirata et al., 1999). The LIPG-overexpressing cell supernatant had a two-fold increase in free fatty acid levels as compared to control supernatant, signifying its ability to hydrolyze apo A-I phospholipids like PC at the sn-1 position. Additionally, McCoy et al. confirmed the primary phospholipase activity and secondary triglyceride lipase activity by incubating conditioned medium from LIPG-overexpressing COS7 cells with either radiolabeled-dipalmitoylphosphatidylcholine (DPPC) or 2 triglyceride substrates: tributyrin and triolein (McCoy et al., 2002). Their studies found that although LIPG has triglyceride lipase activity, the ratio of triglyceride lipase to phospholipase activity was 0.65.
Through its catalytic activity, LIPG cleaves lipoproteins, liberating the free fatty acid lipid precursors. These can then be taken up by the cell and incorporated into endogenous lipids (Figure 1A). It was shown that in LPL-deficient mouse adipose tissue LIPG is upregulated, serving to increase the tissue supply of HDL-PC-derived fatty acids (Kratky et al., 2005). Strauss et al. showed that LIPG cleavage of HDL-PC supplies cells with non-esterified fatty acids (NEFA) for the biosynthesis of lipids (Strauss et al., 2003). Gauster et al. expanded these studies by reporting the ability of LIPG to release unsaturated and saturated fatty acids from HDL-PC through its sn-1 phospholipase A1 and lysophospholipase activity. These fatty acids are then used in the catabolism of endogenous lipids such as triglycerides and phospholipids (Gauster, Rechberger, et al., 2005). Additionally, released free fatty acids from HDL by LIPG hydrolysis can then mediate the detachment of LIPG from the cell surface (Strauss et al., 2002).
Figure 1. Enzymatic and Non-Enzymatic Functions of LIPG.
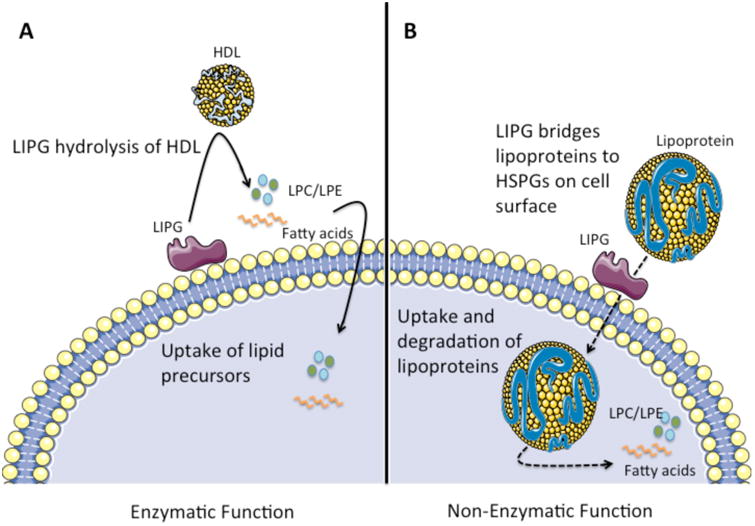
A) LIPG bound to the cell surface hydrolyzes extracellular HDL, releasing lipid precursors such as fatty acids and LPC/LPE. These lipid precursors are then taken up by the cell and used for the biosynthesis of lipids, contributing to multiple cellular processes. B) Independent of its enzymatic activity, LIPG serves as a bridging molecule between lipoproteins (HDL/LDL/VLDL) and heparin-sulfate proteoglycans (HSPGs) on the cell surface. This facilitates the internalization of HDL, LDL, and VLDL particles into the cell via endocytosis, leading to their catabolism, supplying the cell with the lipid precursors necessary for lipid biosynthesis. While LIPG promotes the binding and uptake of all lipoprotein species, LDL and VLDL are internalized more efficiently than HDL particles. Through this mechanism, LDL and VLDL provide lipid precursors to the cell, however the majority of HDL particles are bound and released without internalization. This figure was produced using Servier medical art, available from http://www.servier.com/Powerpoint-image-bank.
To examine the effect of LIPG on cell lipid composition, Riederer et al. overexpressed LIPG in human aortic endothelial cells (HAEC) and examined the extra- and intracellular lipid fractions. LIPG overexpression resulted in increased levels of extracellular lysophosphatidylcholine (LPC) and lysophosphatidylethanolamine (LPE) found in the culture medium, as well as increased intracellular levels of PC, LPC, and TG (Riederer et al., 2012). Riederer et al. concluded that LIPG supplies cells with HDL-derived LPC and LPE, which results in increased cellular TG and PC and decreased endogenous PC synthesis. Additionally, intracellular lipid composition was unaltered in the absence of HDL, further confirming the role of LIPG in HDL metabolism.
In addition to supporting cell metabolism, LIPG can promote the anti-inflammatory function of HDL in certain contexts. Ahmed et al. found that the hydrolysis of HDL by LIPG activates PPARα, which inhibits vascular cell adhesion molecule 1 (VCAM-1) expression in LIPG-overexpressing endothelial cells. This led to the suppression of leukocyte adhesion to the endothelium (Ahmed et al., 2006).
Interestingly, LIPG has also been shown to exert non-catalytic functions that are independent of its enzymatic activity (Figure 1B). Strauss et al. demonstrated the ability of LIPG to facilitate the binding and uptake of HDL holoparticles, and the selective uptake of HDL-cholesterol esters (CEs) in HepG2 cells (Strauss et al., 2002). In fact, inactivating LIPG enzyme activity through tetrahydrolipstatin (THL) treatment or overexpression of a mutant LIPG construct actually increased the binding and uptake of holoparticles, demonstrating that this is not dependent on the catalytic activity of LIPG. LIPG can also mediate the binding and uptake of apo A-I (i.e. HDL), and apo B (i.e. VLDL and LDL) containing plasma lipoproteins. This process is dependent on heparin sulfate proteoglycans (HSPGs) on the cell membrane, which bind to LIPG. In this way, LIPG serves as a bridging molecule between plasma lipoproteins and cells (Strauss et al., 2002; Fuki et al., 2003). LIPG is more efficient at bridging VLDL and LDL than HDL, and while binding of VLDL and LDL results in their uptake and degradation, binding of HDL does not, and the HDL is eventually released back into the medium (Fuki et al., 2003). This is due to endocytosis by HSPGs being dependent on their clustering by large ligands (i.e. lipoproteins). Therefore, the relatively larger surface area of the LDL particle may be more efficient at clustering HSPGs than smaller HDL particles (Fuki et al., 2003).
These findings demonstrate that LIPG has a multitude of crucial functions in the metabolism and uptake of extracellular lipids. LIPG facilitates the clearance of HDL from circulation, influences the intracellular and extracellular lipid profile, and provides lipid precursors for lipid synthesis. Accordingly, expression of LIPG is inversely correlated with plasma levels of HDL-Cholesterol (HDL-C). LIPG knockout mice have been shown to have increased fasting plasma HDL-C levels (Jaye et al., 1999; Ishida et al., 2003). Since high levels of HDL-C have been implicated as protective against cardiovascular disease, reduction of LIPG is an appealing therapeutic strategy.
Regulation of LIPG Biogenesis, Expression, and Activity
Functional Effects of Post-Translational Modifications on LIPG
Following translation, LIPG must undergo several post-translational modifications to become functional. First, the LIPG protein is glycosylated at 5 sites, increasing the molecular weight of the mature protein from 55 kDa to 68 kDa. Two of the glycosylation sites of LIPG (N60 and N373) are conserved in HL and LPL, while the remaining three (N116, N449, and N471) are unique. When cells were treated with glycosidases, LIPG was reduced from the full-length 68 kDa protein to 55 kDa, and LIPG was unable to leave the cell (Miller et al., 2004). To identify the role of each glycosylation site, Miller et al. performed site-directed mutagenesis of the glycosylation sites. This revealed that with the exception of N449, all glycosylation sites were necessary for LIPG secretion or catalytic activity. N60 was shown to be critical for LIPG secretion, but did not significantly alter LIPG activity. In contrast, mutated N116 significantly increased phospholipase activity, and mutated N373 reduced lipase activity, but neither affected the secretion of LIPG. Follow up studies revealed that glycosylation at N116 reduces the ability of LIPG to hydrolyze lipids in LDL and HDL2 (Brown et al., 2007).
LIPG homodimerization greatly increases the activity and stability of LIPG outside the cell. The endoplasmic reticulum (ER) membrane protein lipase maturation factor 1 (LMF1) was previously shown to be important for the post-translational maturation of lipases including HL and LPL. Therefore, Ben-Zeev et al. examined whether LMF1 was necessary for LIPG biogenesis (Ben-Zeev et al., 2011). When a loss-of-function LMF1 mutation was induced, cells were unable to generate active LIPG. Because LMF1 is specific for dimeric lipases, it is likely that it aids in the proper assembly or stabilization of the LIPG homodimer (Doolittle et al., 2010).
Mechanisms of Innate Regulation of LIPG Expression
Once mature LIPG is formed, its activity and substrate specificity is governed by sphingomyelin, a major phospholipid in lipoproteins. Yang et al. reported that sphingomyelin is a physiologic inhibitor of LIPG. They showed that spingomyelin inhibits LIPG hydrolysis of PC at the sn-1 position of lipoproteins (Yang et al., 2014). Furthermore, LIPG showed a higher hydrolysis for HDL-PC over LDL-PC, and inhibition of sphingomyelin abrogated this difference. Therefore, the higher concentration of sphingomyelin in VLDL and LDL may explain the specificity of LIPG for HDL.
Given that LIPG regulates the catabolism of HDL, which has anti-inflammatory and anti-oxidant functions, researchers have characterized the regulation of inflammatory signaling on LIPG expression. It was shown that tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β) can induce LIPG mRNA and protein expression in endothelial cells, and inhibition of the NFκB-pathway abrogated this induction (Hirata et al., 2000; Jin, Sun, et al., 2003). Studies by Kempe et al. further demonstrated that TNF-α induces recruitment of the NFκB transcription factors RelA/p65 to the LIPG promoter (Kempe et al., 2005). Additionally, angiotensin II (AngII) and phorbol 12-myristate 13-acetate (PMA) induce LIPG mRNA and protein expression in vascular smooth muscle cells of hypertensive rats, which may be dependent on NFκB and MAPK signaling pathways (Shimokawa et al., 2005; Zhang et al., 2014).
Multiple studies have examined how LIPG can regulate lipids, however very few have demonstrated how lipids can regulate LIPG expression and the inflammatory response. While investigating the roles of fatty acids and LIPG in inflammation, Jung et al. found that the saturated fatty acid palmitic acid (PA) can induce expression of LIPG in macrophages and mouse models (Jung et al., 2012). In contrast, the PUFA eicosapentaenoic acid (EPA) was able to decrease LIPG expression. The regulation of LIPG by fatty acids has important ramifications in inflammatory diseases, because saturated fatty acids have been implicated as pro-atherogenic and pro-inflammatory, while polyunsaturated fatty acids (PUFA) (i.e.: omega-3 fatty acids) are anti-inflammatory. It was further shown that in addition to LIPG expression changes, PA increased expression of the pro-atherogenic transcription factor peroxisome proliferator activated receptor-gamma (PPAR-γ), and decreased expression of the anti-inflammatory cytokine interleukin-10 (IL-10) in macrophages. EPA had the opposite effect, resulting in downregulation of PPAR-γ, increased expression of IL-10, and decreased expression of pro-inflammatory cytokines IL-6 and IL-12. Jung et al. confirmed these findings in vivo, using atherosclerosis prone low density lipoprotein receptor (LDLR) knockout mice fed a diet rich in saturated fats or omega-3 fat. When mice were fed a diet rich in saturated fat, they observed increased arterial expression of LIPG and pro-inflammatory markers, and decreased expression of anti-inflammatory markers compared to omega-3 fed mice. Therefore, it is likely that diets rich in saturated fats, in contrast to diets rich in omega-3 PUFA, can contribute to high LIPG expression and pro-inflammatory conditions that promote atherogenesis. However, the function of LIPG in these findings is not fully understood.
Functional Role of LIPG in Chronic Inflammatory Diseases
LIPG in Human Chronic Inflammatory Diseases
Several studies have attempted to define the role of LIPG in human metabolic syndromes such as obesity and coronary heart disease. Badellino et al. reported the association of high plasma LIPG levels with some symptoms of the metabolic syndrome and obesity, such as low HDL-C concentration, increased triglyceride serum-concentration, high fasting glucose, and hypertension (Badellino et al., 2006). Additionally, plasma levels of LIPG correlate with inflammatory markers. In a study examining healthy sedentary men, high LIPG expression was found to correlate with increased plasma expression of inflammatory markers C-reactive protein (CRP), interleukin 6 (IL-6), and plasma secretory phospholipase A(2) type IIA (sPLA2-IIa) (Paradis et al., 2006). Moreover, in a large cohort of healthy subjects with a history of family coronary artery disease, high LIPG expression positively correlated with pro-inflammatory markers: CRP, IL-6, tumor necrosis factor receptor II (TNFRII), soluble intercellular adhesion molecule, and leptin, while being conversely associated with adiponectin, an anti-inflammatory factor (Badellino et al., 2008). In support of these findings, the study induced endotoxemia in subjects by low intravenous doses of LPS, and observed an increase in LIPG concentration, and a decrease in HDL phospholipid levels.
High LIPG serum concentrations have also been seen in type 2 diabetic patients and patients with chronic subclinical inflammation (Shiu et al., 2008; Shiu et al., 2010). These studies found that low HDL levels and subclinical inflammation were associated with reduced serum capacity to induce cholesterol efflux in diabetic patients. High LIPG concentration had a weak but significant association with impaired cholesterol efflux. However since LIPG activity was not measured, one cannot exclude the possible causal role of LIPG.
The Role of LIPG in Macrophages
LIPG can contribute to inflammatory conditions by aiding in the adhesion of monocytes to the endothelium through interaction with heparan sulfate proteoglycans (Kojma et al., 2004). Furthermore, LIPG was overexpressed in the diseased aorta of apo E knockout mice, which is likely due to an increase in infiltrating macrophages (Ishida, Choi, et al., 2004).
Polyinosinic:polycytidylic acid (poly I:C) and lipopolysaccharide (LPS) have been shown to induce LIPG expression via activation of toll-like receptor 3 and 4 respectively (TLR3/4) in macrophages. Induction of LIPG led to downregulation of anti-inflammatory IL-10 and upregulation of pro-inflammatory interleukin-12 (IL-12) in macrophages (Wang et al., 2007; Yasuda et al., 2007). Knockdown of LIPG in THP-1 macrophages decreased secretion of pro-inflammatory cytokines: IL-1β, IL-6, monocyte chemoattractant protein-1 (MCP-1), and TNF-α. In addition to these anti-inflammatory effects, decreased LIPG expression affected intracellular lipid composition by reducing levels of cholesterol, triglycerides, and LPC, while increasing PC and other phospholipids (Qiu et al., 2007). Further studies found that LIPG promotes apo A-I-mediated cholesterol efflux in THP-1 macrophages, which is dependent on its hydrolytic and lipoprotein bridging functions (Qiu & Hill, 2009).
The Role of LIPG in Atherogenesis
LIPG expression has been demonstrated to have an important role in the pathogenesis of atherosclerosis through its inverse relationship with HDL. Studies in apo E knockout mice showed that LIPG knockdown modestly increases HDL-C levels, yet dramatically decreases the atherosclerotic plaque area compared to control apo E knockout mice (Ishida, Choi, et al., 2004). However, another group was unable to confirm the correlation between LIPG expression and atherosclerosis development (Ko et al., 2005). Ko et al. demonstrated that LIPG deficiency in apo E knockout mice and LDLR knockout mice fed a western diet, did not have a significant difference in atherosclerotic lesion areas compared to controls. However, it was confirmed in both mouse models that LIPG deficiency results in increased HDL-C levels. Therefore, although LIPG clearly affects HDL-C levels, its role in atherogenesis is still unclear.
It was further established that IL-6 dependent induction of LIPG stimulates the translocation of HDL through the endothelium, which is the first step in reverse cholesterol transport (Robert et al., 2013). According to these findings, upregulation of EL in inflammatory states, including atherosclerosis and the metabolic syndrome, may contribute to the low HDL-C levels seen in these conditions.
The Role of LIPG in Bronchial Asthma
Although LIPG is highly expressed in the lung, its function in this organ is poorly understood. Because the anti-inflammatory properties of HDL have potentially far-reaching effects in other chronic inflammatory diseases, Otera et al. evaluated the role of LIPG and HDL in allergic asthma. Their studies found that infiltration of inflammatory cells such as eosiniphils into the vessel wall, aided by cell adhesion proteins like VCAM-1 is one of the major mechanisms of bronchial asthma progression. During eosinophilic inflammation, LIPG expression is upregulated in epithelial cells, alveolar type II cells, and lung endothelial cells (Otera et al., 2009). Otera et al. demonstrated with an in vitro adhesion assay that LIPG expression on the cell surface of COS7 cells promoted the binding of eosinophils. Inactivation of LIPG reduced the allergic inflammatory response, decreased VCAM-1 expression, and inhibited the ligand-binding function of LIPG. Therefore, their studies support the contributing role of LIPG to the progression of bronchial asthma.
LIPG Inhibition Enhances Anti-Inflammatory Effects of HDL
Given the above studies, it is clear that a selective inhibitor against LIPG would be a useful therapeutic tool to regulate HDL-C metabolism. Hara et al. examined the effect of LIPG expression on HDL function. HDL particles isolated from LIPG deficient mice have enhanced anti-inflammatory properties with increased HDL phospholipid and fatty acid levels compared to wild-type mice (Hara et al., 2011). Additionally, HDL particles from LIPG deficient mice possessed a higher LPS-neutralizing capacity, and maintained HDL properties of inhibition of VCAM-1 expression, suppression of LDL oxidation, cholesterol efflux, and activity of HDL-associated anti-oxidative enzymes (i.e. PON-1 and PAF-AH) compared to wild-type mice. Therefore, targeted inhibition of LIPG would raise HDL levels with preserved anti-inflammatory functions.
LIPG Contributes to the Metabolic Reprograming of Cancer Cells
Cancer initiation and progression result from the accumulation of genetic mutations and epigenetic modifications, alterations of molecular signaling networks, and reprograming of metabolic pathways. These driving factors disrupt cell homeostasis and result in the development of oncogenic characteristics combined with the loss of protective mechanisms. One of the most important drivers of cancer is uncontrolled proliferation, resulting from constitutive growth signaling and silencing of growth suppressors and immune regulators (Hanahan & Weinberg, 2011). Cells undergoing rapid growth and proliferation require significant amounts of energy, drawn from the production of biosynthetic precursors such as ATP, nucleotides, amino acids, and lipids. While cancer cells have been demonstrated to exhibit metabolic dysregulation, the complete mechanisms that grant cancer cells the energy to sustain rapid growth and proliferation are still poorly understood. Research has largely focused on the balance of aerobic and anaerobic oxidation as detailed in the Warburg Effect and Hypothesis (Koppenol et al., 2011). However, due to the high level of energy contained within fatty acids, it is possible that they play a significant role in supporting the reprogramming of cancer cell energetics (Santos & Schulze, 2012; Carracedo et al., 2013). For example, adipocytes have been shown to provide ovarian cancer cells with fatty acids for rapid tumor growth (Nieman et al., 2011). Therefore, given the role of LIPG in supplying lipid precursors such as fatty acids to cells, it is likely that LIPG plays a fundamental role in cancer cell metabolism.
Very little research has explored the contribution of LIPG to the transformation of non-neoplastic cells and development of cancer. High LIPG mRNA and protein expression have been reported in testicular pre-invasive carcinoma in situ and testicular germ cell tumors, which may promote the supply of nutrients or provide cholesterol for the production of testosterone in the testes (Nielsen et al., 2010). Conversely, Dong et al. found an average 9.9-fold decrease of LIPG expression in the urine samples of gastric cancer patients compared to healthy control volunteers, establishing LIPG as a statistically significant biomarker for gastric cancer (Dong et al., 2013). However, they did not detect a correlation between LIPG expression and tumor grade or stage. With respect to breast cancer, Cadenas et al. studied alterations in gene expression following the induction of ErbB2 expression in the luminal breast cancer cell line MCF7. Through gene array analysis they identified the upregulation of many enzymes involved in lipid metabolism, including LIPG, in ErbB2-overexpressing MCF7 cells (Cadenas et al., 2012). Furthermore, cell free DNA derived from colon tumor epithelium induced LIPG mRNA expression in human colorectal adenocarcinoma cells HT29 (Fűri et al., 2015).
While these papers examined expression of LIPG in cancer samples, they did not investigate the function of LIPG in cancer. This was recently rectified by Slebe et al., when they investigated the mechanisms of LIPG on lipid metabolic adaptations in breast cancer (Slebe et al., 2016). Slebe et al. reported that the FoxA transcription factors, which are implicated in metabolic regulation, induce LIPG expression. They further demonstrated the dependence of breast cancer cells on extracellularly-derived lipid precursors supplied by LIPG for intracellular lipid production. LIPG downregulation suppressed breast cancer cell proliferation, and decreased the level of intracellular glycerolipid intermediates involved in TG synthesis such as PC, phosphatidylethanolamine, and phosphatidylglycerol, and their derivatives LPC and LPE. Their studies indicate the crucial role of LIPG in supporting the increased proliferation and high-energy demands of breast cancer cells.
In addition to its regulation of cell proliferation, LIPG has been found to be involved in HDL-induced angiogenesis. HDL lipoproteins contain sphingosine-1-phosphate (S1P), which is implicated in the vascular response and acts as a substrate for spingosine-1-phosphate receptors (S1PR) on endothelial cells (Rosen et al., 2013). LIPG hydrolysis of HDL releases and activates S1P, which binds to S1PR and promotes phosphorylation of protein kinase B (AKT) and endothelial nitric oxide synthase (eNOS). This leads to endothelial cell migration, tube formation, and angiogenesis (Tatematsu et al., 2013). This presents an additional mechanism of how LIPG could contribute to tumor angiogenesis.
It has been well established that the tumor microenvironment contains a wide variety of factors that contribute to cancer initiation and progression (Quail & Joyce, 2013). Adipocytes within the breast cancer microenvironment secrete adipokines such as leptin and IL-6, which can activate the epithelial-mesenchymal transition (EMT) and stem cell signaling through Janus kinase 2/signal transducer and activator of transcription 5 (JAK2/STAT5) (Wolfson et al., 2015). High LIPG expression correlated with increased levels of pro-inflammatory markers IL-6 and leptin, and IL-6 was shown to induce LIPG expression (Badellino et al., 2008; Robert et al., 2013). Therefore, it is possible that adipokine signaling by IL-6 and leptin could promote LIPG expression and activity, and targeting LIPG would present a method of preventing pro-tumor cancer cell-microenvironment crosstalk and signaling.
Finally, it has been demonstrated that the transcription factor NFκB binds to the LIPG promoter, activating LIPG transcription (Kempe et al., 2005). Constitutive activation of NFκB is a frequent oncogenic event, and NFκB is essential in tumor initiation and development due to its control of inflammation, survival, differentiation, and proliferation pathways (Hoesel & Schmid, 2013). LIPG was predicted to have two κB binding sites in the 5′ region upstream of the transcription start site at -467 (proximal) and -1250 (distal). While RelA/p65 binds both sites, only the distal site demonstrated strong binding by NFκB, with the proximal site exhibited weak binding. As LIPG expression is promoted by the inflammatory signals that also activate NFκB, NFκB could play a crucial role in the induction of LIPG-mediated tumorigenesis. This could be an important mechanism for the oncogenic impact of constitutive NFκB signaling, as LIPG may promote cancer inflammation, angiogenesis, and proliferation. These data demonstrate the need for further investigation on the role of LIPG in cancer, and its potential significance as a novel drug target (Figure 2).
Figure 2. The Potential Oncogenic Roles of LIPG.
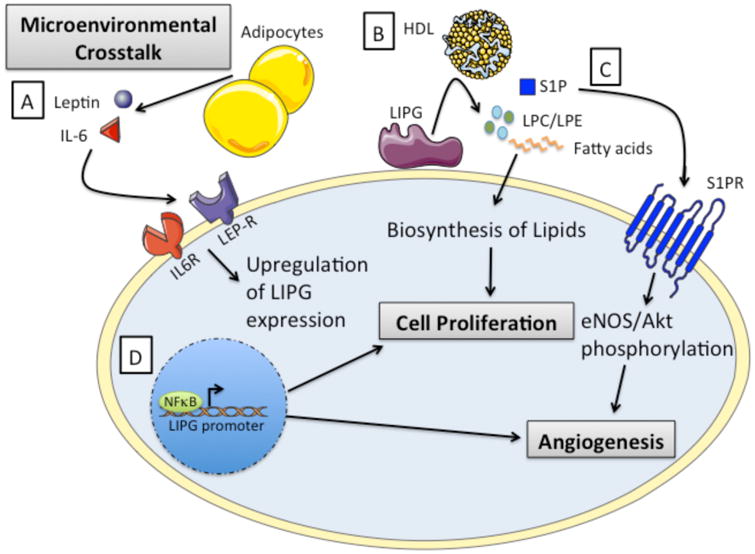
A) LIPG expression may be activated through microenvironmental crosstalk and signaling induced by adipokines such as IL-6 and leptin. B) Lipid precursors supplied to the cell through LIPG-mediated cleavage of HDL can promote proliferation and growth in cancer cells. C) LIPG catalysis of HDL releases Sphingosine-1-phosphate (S1P), which binds to sphingosine-1-phosphate receptors resulting in the phosphorylation and activation of Akt and eNOS. These pathways have been shown to promote endothelial cell migration, tube formation, and angiogenesis. D) Inflammatory signals can induce NFκB-mediated activation of LIPG transcription, which may have major implications in cancer development and progression. This figure was produced using Servier medical art, available from http://www.servier.com/Powerpoint-image-bank.
LIPG-Inhibiting Drugs
Due to the anti-inflammatory effects resulting from LIPG inactivation, several compounds have been developed to target LIPG. One such compound, Atvorastatin, was examined for its effect on LPL and LIPG expression in THP-1 macrophages. Atvorastatatin was shown to decrease LIPG expression through modulation of NFκB, and also decrease LPL through liver X receptor alpha (LXRα) (Qiu & Hill, 2007). Sulfonylfuran urea inhibitors designed by Goodman et al. and boronic acid inhibitors designed by O'Connell et al. have also demonstrated potency for LIPG, however both studies struggled to create compounds that specifically targeted LIPG (Goodman et al., 2009; O'Connell et al., 2012). Finally, the anthranilic acid XEN445 was reported to be highly selective for LIPG over HL and LPL, and exhibits high potency against LIPG (Sun et al., 2013). In addition, XEN445 administration was able to increase plasma HDL-C levels in wild-type mice. Further studies are necessary for optimization and to determine its efficacy in disease models before clinical evaluation.
Conclusion
Given the evidence presented in this review, it is clear that LIPG plays an essential role in regulating lipid metabolism and the inflammatory response. Hydrolysis of HDL by LIPG decreases HDL-C levels, which promotes inflammation through multiple mechanisms such as adhesion of monocytes to the endothelium through downregulation of adhesion molecule VCAM-1, and upregulation of pro-inflammatory factors IL-6 and IL-12. Furthermore, LIPG activity provides extracellular lipid precursors for intracellular lipid biosynthesis, which contributes to cell growth and proliferation. Therefore, in addition to its potential therapeutic role in metabolic diseases like atherogenesis and diabetes, LIPG poses an exciting and new opportunity for cancer therapy. Targeting LIPG could alter multiple pathways that contribute to cancer initiation and progression such as cell proliferation, angiogenesis, energetics, and inflammation. The far-reaching actions of LIPG could create a potent tumor suppressive response through its inactivation. Therefore, further elucidation on the role of LIPG in tumorigenesis could unlock its therapeutic potential.
References
- Ahmed W, Orasanu G, Nehra V, Asatryan L, Rader DJ, Ziouzenkova O, Plutzky J. High-density lipoprotein hydrolysis by endothelial lipase activates pparalpha: A candidate mechanism for high-density lipoprotein-mediated repression of leukocyte adhesion. Circ Res. 2006;98:490–498. doi: 10.1161/01.RES.0000205846.46812.be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badellino KO, Wolfe ML, Reilly MP, Rader DJ. Endothelial lipase concentrations are increased in metabolic syndrome and associated with coronary atherosclerosis. PLoS Med. 2006;3:e22. doi: 10.1371/journal.pmed.0030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badellino KO, Wolfe ML, Reilly MP, Rader DJ. Endothelial lipase is increased in vivo by inflammation in humans. Circulation. 2008;117:678–685. doi: 10.1161/CIRCULATIONAHA.107.707349. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Nicholls S, Rye KA, Anantharamaiah GM, Navab M, Fogelman AM. Antiinflammatory properties of hdl. Circ Res. 2004;95:764–772. doi: 10.1161/01.RES.0000146094.59640.13. [DOI] [PubMed] [Google Scholar]
- Baumann J, Sevinsky C, Conklin DS. Lipid biology of breast cancer. Biochim Biophys Acta. 2013;1831:1509–1517. doi: 10.1016/j.bbalip.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zeev O, Hosseini M, Lai CM, Ehrhardt N, Wong H, Cefalù AB, Noto D, Averna MR, Doolittle MH, Péterfy M. Lipase maturation factor 1 is required for endothelial lipase activity. J Lipid Res. 2011;52:1162–1169. doi: 10.1194/jlr.M011155. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Broedl UC, Maugeais C, Marchadier D, Glick JM, Rader DJ. Effects of nonlipolytic ligand function of endothelial lipase on high density lipoprotein metabolism in vivo. J Biol Chem. 2003;278:40688–40693. doi: 10.1074/jbc.M304367200. [DOI] [PubMed] [Google Scholar]
- Broedl UC, Maugeais C, Millar JS, Jin W, Moore RE, Fuki IV, Marchadier D, Glick JM, Rader DJ. Endothelial lipase promotes the catabolism of apob-containing lipoproteins. Circ Res. 2004;94:1554–1561. doi: 10.1161/01.RES.0000130657.00222.39. [DOI] [PubMed] [Google Scholar]
- Brown RJ, Miller GC, Griffon N, Long CJ, Rader DJ. Glycosylation of endothelial lipase at asparagine-116 reduces activity and the hydrolysis of native lipoproteins in vitro and in vivo. J Lipid Res. 2007;48:1132–1139. doi: 10.1194/jlr.M600535-JLR200. [DOI] [PubMed] [Google Scholar]
- Cadenas C, Vosbeck S, Hein EM, Hellwig B, Langer A, Hayen H, Franckenstein D, Büttner B, Hammad S, Marchan R, Hermes M, Selinski S, Rahnenführer J, Peksel B, Török Z, Vígh L, Hengstler JG. Glycerophospholipid profile in oncogene-induced senescence. Biochim Biophys Acta. 2012;1821:1256–1268. doi: 10.1016/j.bbalip.2011.11.008. [DOI] [PubMed] [Google Scholar]
- Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: Fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227–232. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Wang G, Zhang G, Ni Z, Suo J, Cui J, Cui A, Yang Q, Xu Y, Li F. The endothelial lipase protein is promising urinary biomarker for diagnosis of gastric cancer. Diagn Pathol. 2013;8:45. doi: 10.1186/1746-1596-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle MH, Ehrhardt N, Péterfy M. Lipase maturation factor 1: Structure and role in lipase folding and assembly. Curr Opin Lipidol. 2010;21:198–203. doi: 10.1097/MOL.0b013e32833854c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugi KA, Dichek HL, Talley GD, Brewer HB, Santamarina-Fojo S. Human lipoprotein lipase: The loop covering the catalytic site is essential for interaction with lipid substrates. J Biol Chem. 1992;267:25086–25091. [PubMed] [Google Scholar]
- Fahy E, Subramaniam S, Murphy RC, Nishijima M, Raetz CR, Shimizu T, Spener F, van Meer G, Wakelam MJ, Dennis EA. Update of the lipid maps comprehensive classification system for lipids. J Lipid Res. 2009;(50Suppl):S9–14. doi: 10.1194/jlr.R800095-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy E, Subramaniam S, Brown HA, Glass CK, Merrill AH, Murphy RC, Raetz CR, Russell DW, Seyama Y, Shaw W, Shimizu T, Spener F, van Meer G, VanNieuwenhze MS, White SH, Witztum JL, Dennis EA. A comprehensive classification system for lipids. J Lipid Res. 2005;46:839–861. doi: 10.1194/jlr.E400004-JLR200. [DOI] [PubMed] [Google Scholar]
- Fuki IV, Blanchard N, Jin W, Marchadier DH, Millar JS, Glick JM, Rader DJ. Endogenously produced endothelial lipase enhances binding and cellular processing of plasma lipoproteins via heparan sulfate proteoglycan-mediated pathway. J Biol Chem. 2003;278:34331–34338. doi: 10.1074/jbc.M302181200. [DOI] [PubMed] [Google Scholar]
- Fűri I, Kalmár A, Wichmann B, Spisák S, Schöller A, Barták B, Tulassay Z, Molnár B. Cell free dna of tumor origin induces a ‘metastatic’ expression profile in ht-29 cancer cell line. PLoS One. 2015;10:e0131699. doi: 10.1371/journal.pone.0131699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauster M, Hrzenjak A, Schick K, Frank S. Endothelial lipase is inactivated upon cleavage by the members of the proprotein convertase family. J Lipid Res. 2005;46:977–987. doi: 10.1194/jlr.M400500-JLR200. [DOI] [PubMed] [Google Scholar]
- Gauster M, Rechberger G, Sovic A, Hörl G, Steyrer E, Sattler W, Frank S. Endothelial lipase releases saturated and unsaturated fatty acids of high density lipoprotein phosphatidylcholine. J Lipid Res. 2005;46:1517–1525. doi: 10.1194/jlr.M500054-JLR200. [DOI] [PubMed] [Google Scholar]
- Goodman KB, Bury MJ, Cheung M, Cichy-Knight MA, Dowdell SE, Dunn AK, Lee D, Lieby JA, Moore ML, Scherzer DA, Sha D, Suarez DP, Murphy DJ, Harpel MR, Manas ES, McNulty DE, Annan RS, Matico RE, Schwartz BK, Trill JJ, Sweitzer TD, Wang DY, Keller PM, Krawiec JA, Jaye MC. Discovery of potent, selective sulfonylfuran urea endothelial lipase inhibitors. Bioorg Med Chem Lett. 2009;19:27–30. doi: 10.1016/j.bmcl.2008.11.033. [DOI] [PubMed] [Google Scholar]
- Griffon N, Budreck EC, Long CJ, Broedl UC, Marchadier DH, Glick JM, Rader DJ. Substrate specificity of lipoprotein lipase and endothelial lipase: Studies of lid chimeras. J Lipid Res. 2006;47:1803–1811. doi: 10.1194/jlr.M500552-JLR200. [DOI] [PubMed] [Google Scholar]
- Griffon N, Jin W, Petty TJ, Millar J, Badellino KO, Saven JG, Marchadier DH, Kempner ES, Billheimer J, Glick JM, Rader DJ. Identification of the active form of endothelial lipase, a homodimer in a head-to-tail conformation. J Biol Chem. 2009;284:23322–23330. doi: 10.1074/jbc.M109.037002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hara T, Ishida T, Kojima Y, Tanaka H, Yasuda T, Shinohara M, Toh R, Hirata K. Targeted deletion of endothelial lipase increases hdl particles with anti-inflammatory properties both in vitro and in vivo. J Lipid Res. 2011;52:57–67. doi: 10.1194/jlr.M008417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata K, Ishida T, Matsushita H, Tsao PS, Quertermous T. Regulated expression of endothelial cell-derived lipase. Biochem Biophys Res Commun. 2000;272:90–93. doi: 10.1006/bbrc.2000.2747. [DOI] [PubMed] [Google Scholar]
- Hirata K, Dichek HL, Cioffi JA, Choi SY, Leeper NJ, Quintana L, Kronmal GS, Cooper AD, Quertermous T. Cloning of a unique lipase from endothelial cells extends the lipase gene family. J Biol Chem. 1999;274:14170–14175. doi: 10.1074/jbc.274.20.14170. [DOI] [PubMed] [Google Scholar]
- Hoesel B, Schmid JA. The complexity of nf-κb signaling in inflammation and cancer. Mol Cancer. 2013;12:86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T, Choi S, Kundu RK, Hirata K, Rubin EM, Cooper AD, Quertermous T. Endothelial lipase is a major determinant of hdl level. J Clin Invest. 2003;111:347–355. doi: 10.1172/JCI16306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T, Choi SY, Kundu RK, Spin J, Yamashita T, Hirata K, Kojima Y, Yokoyama M, Cooper AD, Quertermous T. Endothelial lipase modulates susceptibility to atherosclerosis in apolipoprotein-e-deficient mice. J Biol Chem. 2004;279:45085–45092. doi: 10.1074/jbc.M406360200. [DOI] [PubMed] [Google Scholar]
- Ishida T, Zheng Z, Dichek HL, Wang H, Moreno I, Yang E, Kundu RK, Talbi S, Hirata K, Leung LL, Quertermous T. Molecular cloning of nonsecreted endothelial cell-derived lipase isoforms. Genomics. 2004;83:24–33. doi: 10.1016/s0888-7543(03)00181-2. [DOI] [PubMed] [Google Scholar]
- Jaye M, Lynch KJ, Krawiec J, Marchadier D, Maugeais C, Doan K, South V, Amin D, Perrone M, Rader DJ. A novel endothelial-derived lipase that modulates hdl metabolism. Nat Genet. 1999;21:424–428. doi: 10.1038/7766. [DOI] [PubMed] [Google Scholar]
- Jin W, Millar JS, Broedl U, Glick JM, Rader DJ. Inhibition of endothelial lipase causes increased hdl cholesterol levels in vivo. J Clin Invest. 2003;111:357–362. doi: 10.1172/JCI16146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Sun GS, Marchadier D, Octtaviani E, Glick JM, Rader DJ. Endothelial cells secrete triglyceride lipase and phospholipase activities in response to cytokines as a result of endothelial lipase. Circ Res. 2003;92:644–650. doi: 10.1161/01.RES.0000064502.47539.6D. [DOI] [PubMed] [Google Scholar]
- Jung UJ, Torrejon C, Chang CL, Hamai H, Worgall TS, Deckelbaum RJ. Fatty acids regulate endothelial lipase and inflammatory markers in macrophages and in mouse aorta: A role for pparγ. Arterioscler Thromb Vasc Biol. 2012;32:2929–2937. doi: 10.1161/ATVBAHA.112.300188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempe S, Kestler H, Lasar A, Wirth T. Nf-kappab controls the global pro-inflammatory response in endothelial cells: Evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005;33:5308–5319. doi: 10.1093/nar/gki836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko KW, Paul A, Ma K, Li L, Chan L. Endothelial lipase modulates hdl but has no effect on atherosclerosis development in apoe-/- and ldlr-/- mice. J Lipid Res. 2005;46:2586–2594. doi: 10.1194/jlr.M500366-JLR200. [DOI] [PubMed] [Google Scholar]
- Kojma Y, Hirata K, Ishida T, Shimokawa Y, Inoue N, Kawashima S, Quertermous T, Yokoyama M. Endothelial lipase modulates monocyte adhesion to the vessel wall. A potential role in inflammation. J Biol Chem. 2004;279:54032–54038. doi: 10.1074/jbc.M411112200. [DOI] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Kratky D, Zimmermann R, Wagner EM, Strauss JG, Jin W, Kostner GM, Haemmerle G, Rader DJ, Zechner R. Endothelial lipase provides an alternative pathway for ffa uptake in lipoprotein lipase-deficient mouse adipose tissue. J Clin Invest. 2005;115:161–167. doi: 10.1172/JCI15972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Cilingiroglu M, Otvos JD, Ballantyne CM, Marian AJ, Chan L. Endothelial lipase is a major genetic determinant for high-density lipoprotein concentration, structure, and metabolism. Proc Natl Acad Sci U S A. 2003;100:2748–2753. doi: 10.1073/pnas.0438039100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MG, Sun GS, Marchadier D, Maugeais C, Glick JM, Rader DJ. Characterization of the lipolytic activity of endothelial lipase. J Lipid Res. 2002;43:921–929. [PubMed] [Google Scholar]
- Miller GC, Long CJ, Bojilova ED, Marchadier D, Badellino KO, Blanchard N, Fuki IV, Glick JM, Rader DJ. Role of n-linked glycosylation in the secretion and activity of endothelial lipase. J Lipid Res. 2004;45:2080–2087. doi: 10.1194/jlr.M400162-JLR200. [DOI] [PubMed] [Google Scholar]
- Nielsen JE, Lindegaard ML, Friis-Hansen L, Almstrup K, Leffers H, Nielsen LB, Rajpert-De Meyts E. Lipoprotein lipase and endothelial lipase in human testis and in germ cell neoplasms. Int J Androl. 2010;33:e207–215. doi: 10.1111/j.1365-2605.2009.00988.x. [DOI] [PubMed] [Google Scholar]
- Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, Yamada SD, Peter ME, Gwin K, Lengyel E. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498–1503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell DP, LeBlanc DF, Cromley D, Billheimer J, Rader DJ, Bachovchin WW. Design and synthesis of boronic acid inhibitors of endothelial lipase. Bioorg Med Chem Lett. 2012;22:1397–1401. doi: 10.1016/j.bmcl.2011.12.043. [DOI] [PubMed] [Google Scholar]
- Otera H, Ishida T, Nishiuma T, Kobayashi K, Kotani Y, Yasuda T, Kundu RK, Quertermous T, Hirata K, Nishimura Y. Targeted inactivation of endothelial lipase attenuates lung allergic inflammation through raising plasma hdl level and inhibiting eosinophil infiltration. Am J Physiol Lung Cell Mol Physiol. 2009;296:L594–602. doi: 10.1152/ajplung.90530.2008. [DOI] [PubMed] [Google Scholar]
- Paradis ME, Badellino KO, Rader DJ, Deshaies Y, Couture P, Archer WR, Bergeron N, Lamarche B. Endothelial lipase is associated with inflammation in humans. J Lipid Res. 2006;47:2808–2813. doi: 10.1194/jlr.P600002-JLR200. [DOI] [PubMed] [Google Scholar]
- Qiu G, Hill JS. Atorvastatin decreases lipoprotein lipase and endothelial lipase expression in human thp-1 macrophages. J Lipid Res. 2007;48:2112–2122. doi: 10.1194/jlr.M600510-JLR200. [DOI] [PubMed] [Google Scholar]
- Qiu G, Hill JS. Endothelial lipase promotes apolipoprotein ai-mediated cholesterol efflux in thp-1 macrophages. Arterioscler Thromb Vasc Biol. 2009;29:84–91. doi: 10.1161/ATVBAHA.108.176487. [DOI] [PubMed] [Google Scholar]
- Qiu G, Ho AC, Yu W, Hill JS. Suppression of endothelial or lipoprotein lipase in thp-1 macrophages attenuates proinflammatory cytokine secretion. J Lipid Res. 2007;48:385–394. doi: 10.1194/jlr.M600304-JLR200. [DOI] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer M, Köfeler H, Lechleitner M, Tritscher M, Frank S. Impact of endothelial lipase on cellular lipid composition. Biochim Biophys Acta. 2012;1821:1003–1011. doi: 10.1016/j.bbalip.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert J, Lehner M, Frank S, Perisa D, von Eckardstein A, Rohrer L. Interleukin 6 stimulates endothelial binding and transport of high-density lipoprotein through induction of endothelial lipase. Arterioscler Thromb Vasc Biol. 2013;33:2699–2706. doi: 10.1161/ATVBAHA.113.301363. [DOI] [PubMed] [Google Scholar]
- Rosen H, Stevens RC, Hanson M, Roberts E, Oldstone MB. Sphingosine-1-phosphate and its receptors: Structure, signaling, and influence. Annu Rev Biochem. 2013;82:637–662. doi: 10.1146/annurev-biochem-062411-130916. [DOI] [PubMed] [Google Scholar]
- Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279:2610–2623. doi: 10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- Shimokawa Y, Hirata K, Ishida T, Kojima Y, Inoue N, Quertermous T, Yokoyama M. Increased expression of endothelial lipase in rat models of hypertension. Cardiovasc Res. 2005;66:594–600. doi: 10.1016/j.cardiores.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Shiu SW, Tan KC, Huang Y, Wong Y. Type 2 diabetes mellitus and endothelial lipase. Atherosclerosis. 2008;198:441–447. doi: 10.1016/j.atherosclerosis.2008.03.012. [DOI] [PubMed] [Google Scholar]
- Shiu SW, Zhou H, Wong Y, Tan KC. Endothelial lipase and reverse cholesterol transport in type 2 diabetes mellitus. J Diabetes Investig. 2010;1:111–116. doi: 10.1111/j.2040-1124.2010.00016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slebe F, Rojo F, Vinaixa M, García-Rocha M, Testoni G, Guiu M, Planet E, Samino S, Arenas EJ, Beltran A, Rovira A, Lluch A, Salvatella X, Yanes O, Albanell J, Guinovart JJ, Gomis RR. Foxa and lipg endothelial lipase control the uptake of extracellular lipids for breast cancer growth. Nat Commun. 2016;7:11199. doi: 10.1038/ncomms11199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JG, Hayn M, Zechner R, Levak-Frank S, Frank S. Fatty acids liberated from high-density lipoprotein phospholipids by endothelial-derived lipase are incorporated into lipids in hepg2 cells. Biochem J. 2003;371:981–988. doi: 10.1042/BJ20021437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JG, Zimmermann R, Hrzenjak A, Zhou Y, Kratky D, Levak-Frank S, Kostner GM, Zechner R, Frank S. Endothelial cell-derived lipase mediates uptake and binding of high-density lipoprotein (hdl) particles and the selective uptake of hdl-associated cholesterol esters independent of its enzymic activity. Biochem J. 2002;368:69–79. doi: 10.1042/BJ20020306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Dean R, Jia Q, Zenova A, Zhong J, Grayson C, Xie C, Lindgren A, Samra P, Sojo L, van Heek M, Lin L, Percival D, Fu JM, Winther MD, Zhang Z. Discovery of xen445: A potent and selective endothelial lipase inhibitor raises plasma hdl-cholesterol concentration in mice. Bioorg Med Chem. 2013;21:7724–7734. doi: 10.1016/j.bmc.2013.10.023. [DOI] [PubMed] [Google Scholar]
- Tatematsu S, Francis SA, Natarajan P, Rader DJ, Saghatelian A, Brown JD, Michel T, Plutzky J. Endothelial lipase is a critical determinant of high-density lipoprotein-stimulated sphingosine 1-phosphate-dependent signaling in vascular endothelium. Arterioscler Thromb Vasc Biol. 2013;33:1788–1794. doi: 10.1161/ATVBAHA.113.301300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Jin W, Rader DJ. Upregulation of macrophage endothelial lipase by tolllike receptors 4 and 3 modulates macrophage interleukin-10 and -12 production. Circ Res. 2007;100:1008–1015. doi: 10.1161/01.RES.0000263011.34709.c5. [DOI] [PubMed] [Google Scholar]
- Wolfson B, Eades G, Zhou Q. Adipocyte activation of cancer stem cell signaling in breast cancer. World J Biol Chem. 2015;6:39–47. doi: 10.4331/wjbc.v6.i2.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Belikova NA, Billheimer J, Rader DJ, Hill JS, Subbaiah PV. Inhibition of endothelial lipase activity by sphingomyelin in the lipoproteins. Lipids. 2014;49:987–996. doi: 10.1007/s11745-014-3944-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Hirata K, Ishida T, Kojima Y, Tanaka H, Okada T, Quertermous T, Yokoyama M. Endothelial lipase is increased by inflammation and promotes ldl uptake in macrophages. J Atheroscler Thromb. 2007;14:192–201. doi: 10.5551/jat.e502. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wu M, Jiang H, Hao J, Zhang Q, Zhu Q, Saren G, Zhang Y, Meng X, Yue X. Angiotensin ii upregulates endothelial lipase expression via the nf-kappa b and mapk signaling pathways. PLoS One. 2014;9:e107634. doi: 10.1371/journal.pone.0107634. [DOI] [PMC free article] [PubMed] [Google Scholar]