

Abstract
Human drug-metabolizing cytochrome P450 3A4 (CYP3A4) is a dynamic enzyme with a large and highly malleable active site that can fit structurally diverse compounds. Despite extensive investigations, structure–function relationships and conformational dynamics in CYP3A4 are not fully understood. This study was undertaken to engineer a well-expressed and functionally active cysteine-depleted CYP3A4 that can be used in biochemical and biophysical studies. cDNA codon optimization and screening mutagenesis were utilized to boost the level of bacterial expression of CYP3A4 and identify the least harmful substitutions for all six non-heme-ligating cysteines. The C58A/C64M/C98A/C239T/C377A/C468S (Cysless) mutant was found to be expressed as highly as the optimized wild-type (opt-WT) CYP3A4. The high-resolution X-ray structures of opt-WT and Cys-less CYP3A4 revealed that gene optimization leads to a different folding in the Phe108 and Phe189 regions and promotes binding of the active site glycerol that interlocks Ser119 and Arg212, critical for ligand association, and the hydrophobic cluster adjacent to Phe108. Crowding and decreased flexibility of the active site, as well as structural alterations observed at the C64M, C239T, and C468S mutational sites, might be responsible for the distinct ligand binding behavior of opt-WT and Cys-less CYP3A4. Nonetheless, the Cys-less mutant could be used for structure–function investigations because it orients bromoergocryptine and ritonavir (a high-affinity substrate and a high-potency inhibitor, respectively) like the WT and has a higher activity toward 7-benzyloxy(4-trifluoromethyl)coumarin.
Graphical abstract
Human cytochrome P450 3A4 (CYP3A4) plays a central role in drug metabolism.1,2 Despite its structural plasticity and low substrate specificity, CYP3A4 can oxidize chemically diverse compounds in a regio- and stereoselective manner. Specific orientation and limitation of the motional freedom of small substrate molecules can be achieved through conformational changes that decrease the active site volume, as observed in the midazolam (MDZ)-bound CYP3A4.3 Association of large or several small compounds, on the other hand, could cause an expansion of the active site. For instance, the bulky ritonavir, ketoconazole, and erythromycin molecules increase the active site volume by displacing the central part of the I helix and/or the 369–371 and F′–G′ fragments.4,5
One limitation of X-ray crystallography is that it can capture only selective protein conformers but not the full range of dynamic motion. A site-specific attachment of environment-sensitive fluorescent/phosphorescent probes and spin-labels is one of the alternative approaches that allowed investigation of the conformational variety, oligomerization state, and dynamic structural changes in CYP3A4.6–13 Measurement of the ligand-dependent interprobe distances helped to map binding sites of the effector α-naphthoflavone8,9 and fluorescent ligand fluorol-7GA6 and to investigate transition between the open and closed states in CYP3A413 and its interaction with other CYP enzymes.12 In all these studies, the probes were chemically attached to the native or newly introduced cysteine residues after partial or complete depletion of the native ones in Δ3–12 CYP3A4 or its S18F variant.
CYP3A4 has six non-heme-ligating cysteines, all of which are fully or partially solvent exposed (Figure S1): Cys58 and Cys64 are part of the A helix, Cys377 is at the end of the β-strand comprising the β-domain, and the most reactive cysteines 98, 239, and 468 are located in the flexible B–B′, G′–G, and C-terminal loops, respectively. Cys239 is solely alkylated by the raloxifene metabolite and the SH group reactive iodoacetamine and N-(1-pyrene)iodoacetamide,14,15 whereas cysteines 98 and 468 are selectively modified by lapachenole in photolabeled CYP3A4.16 Importantly, maleimides can conjugate with all non-heme-ligating cysteines but no more than two or three at the same time,17 because formation of multiple Cys adducts leads to protein denaturation.9 Menard and co-workers reported that the C98S/C239S/C468G mutant is 1.5-fold more active than the WT and undergoes maleimide modification primarily at the Cys64 site without a significant loss of the enzymatic activity.17 Based on the latter mutant, a poorly expressed C58T/C64A/C98S/C239S/C468G variant was built for mechanistic studies of the allostery and binding cooperativity in CYP3A4.18 Another group investigated C64S/C98S, C98S/C377S/C468G, C98S/C239S/C468G, C98S/C239S/C377S/C468A, and C58T/C64A/C98S/C239S/C377S/C468A variants6,7,9,10,12 and noticed that elimination of more than four native cysteines markedly lowers the level of CYP3A4 expression.7 However, the underlying cause was not investigated.
Our long-term objective is elucidation of structure–function relations, conformational dynamics, and the ligand binding mechanism in CYP3A4. The goal of this study was to engineer a well-expressed and functionally active Cys-depleted CYP3A4 for biochemical and biophysical investigations. As described here, cDNA codon optimization and mutagenesis screening allowed the preparation of large amounts of the highly purified wild type (opt-WT) and C58A/C64M/C98A/C239T/C377A/C468S (Cys-less) variant of Δ3–22 CYP3A4 for structural and functional characterization and comparison with the WT protein expressed using the original human cDNA.
EXPERIMENTAL PROCEDURES
CYP3A4 Gene Synthesis, Mutagenesis, and Protein Purification
Codon optimization for Escherichia coli expression and synthesis of the full-length CYP3A4 gene (Figure S2) were conducted by Genewiz. The Δ3–22 fragment with the C-terminal four-histidine tag (opt-WT) was used as a template for introduction of single mutations and various combinations of multiple cysteine mutations, which was performed with the QuikChange kit (Agilent). The non-optimized wild type (WT), opt-WT, and all CYP3A4 mutants were co-expressed with GroESL in E. coli C41(DE3) cells. After the cells had been broken with a microfluidizer, CYP3A4 was extracted with 0.5% IGEPAL in 0.1 M phosphate (pH 7.4), 0.1 M NaCl, 20% glycerol, 1 μM leupeptin, and 5 mM β-mercaptoethanol and purified using Ni-affinity and cation-exchange chromatography. Protein fractions with A417/A280 ratios of ≥1.6 were concentrated and stored at −80 °C in 0.1 M phosphate (pH 7.4), 0.1 M NaCl, 20% glycerol, and 2 mM dithiothreitol.
Equilibrium Titrations
Spectral changes in 1 μM opt-WT and Cys-less CYP3A4 induced by ritonavir (RIT), bromoergocryptine (BEC), or MDZ were monitored on a Cary 300 spectrophotometer in 0.1 or 0.6 M phosphate buffer (pH 7.4). Spectral dissociation constants (Ks) were derived from the hyperbolic or quadratic fits to the plots of the maximal absorbance change versus ligand concentration using IgorPro version 6.05 (WaveMetrics, Portland, OR).
Ligand Binding Kinetics
Ligand binding kinetics were measured in an SX.18MV stopped flow apparatus (Applied Photophysics) by monitoring the spin transition in the heme iron. Solutions of 2 μM CYP3A4 in 0.1 or 0.6 M phosphate (pH 7.4) were mixed with 1–40 μM RIT, BEC, or MDZ, and an absorbance decrease at 420 nm was monitored over time. Kinetic data were analyzed using the PROKIN software (Applied Photophysics, Leatherhead, U.K.).
CYP3A4 Activity Assays
The BFC O-debenzylase activity of CYP3A4 was assessed fluorimetrically in a lipid-free reconstituted system with rat cytochrome P450 reductase (CPR) in 0.1–0.6 M phosphate (pH 7.4), as described in detail previously.19
Kinetics of CYP3A4 Reduction with CPR
Kinetics of CYP3A4 reduction with CPR were measured under anaerobic conditions at ambient temperature on a Cary 300 spectrophotometer. The reaction solution contained 0.1 M phosphate (pH 7.4), 1 μM CYP3A4 and CPR, 30 μM MDZ, and an oxygen-scavenging system consisting of 10 mM glucose, glucose oxidase, and catalase (5 units/mL each). The reaction mixture was degassed in a sealed cuvette and saturated with CO. Fe2+−CO adduct formation was followed at 450 nm after injection of 100 μM NADPH (final concentration). Kinetic data were analyzed with SigmaPlot version 11.0 (Systal Software, San Jose, CA).
Crystallization of CYP3A4
Opt-WT, ligand-free and RIT-bound Cys-less CYP3A4, and the WT–RIT complex were crystallized by a microbatch method under oil after 0.4 μL of the protein solution [50–100 mg/mL in 20–100 mM phosphate (pH 7.4), 20% glycerol, and 100 mM NaCl] was mixed with 0.4 μL of solution 2–7, 2–43, and 1–7 from the Morpheus screening kit (Molecular Dimensions) or solution 15 from the PegIon2 kit (Hampton Research), respectively. Drops were covered with paraffin oil and produced crystals overnight. The BEC-bound Cys-less CYP3A4 was crystallized by a vapor diffusion method against Morpheus solution 1–7. All crystals were cryoprotected with Paratone-N oil and flash-frozen in liquid nitrogen. X-ray diffraction data were collected at Stanford Synchrotron Radiation Lightsource beamline 12-2 and Advanced Light Source beamlines 8.2.1 and 8.2.2. The water-bound structure of opt-WT CYP3A4 was the first to be determined by molecular replacement with PHASER20 and the WT CYP3A4 structure [Protein Data Bank (PDB) entry 1TQN] as a search model. All structures of Cys-less CYP3A4 were determined using opt-WT as a search model. The RIT-bound model of WT CYP3A4 was determined using the structure of PDB entry 4K9W for molecular replacement. The initial models were refined with COOT,21 REFMAC,22 and PHENIX23 (versions 0.8, 1.8.4-1496, and 5.8.0049, respectively). Simulated annealing omit maps and occupancies were calculated with PHENIX. Data collection and refinement statistics are summarized in Table S1. The atomic coordinates and structure factors for the RIT-bound WT, opt-WT, and water-, RIT-, and BEC-bound Cys-less CYP3A4 were deposited as Protein Data Bank entries 5VCO, 5VCC, 5VCD, 5VCE, and 5VCG, respectively.
RESULTS AND DISCUSSION
Mutagenesis Screening and Production of Cys-less CYP3A4
Previous mutagenesis studies indicated that substitution of multiple cysteines decreases CYP3A4 stability and expression levels.7,17 De novo incorporation of the modifiable surface cysteines, needed for conformational studies, could further jeopardize production of the properly folded and functional enzyme. Therefore, a codon-optimized gene was synthesized to boost the level of CYP3A4 expression in E. coli (Figure S2). Compared to the native WT Δ3–22 CYP3A4 (1 mg/g of cells), the level of expression of opt-WT was 3-fold higher (∼3 vs 1 mg/g of cells).
The synthetic gene was used as a template for screening mutagenesis. During the first round, each non-heme-ligating cysteine was replaced with the small Ala and isosteric Ser residue. As shown in Figure 1, the level of production of CYP3A4 was not significantly affected by the C58A/S mutations but increased by 1.5/1.8- and 1.4/2.7-fold for the Ala/Ser variants of Cys377 and Cys468, respectively. In contrast, there was a 40–70% decrease in the level of C98A/S mutant expression and 90% for C239A/S. Elimination of Cys64 had the most deleterious effect, as both the C64S and C64A variants were expressed at negligible levels (<1% compared to opt-WT CYP3A4).
Figure 1.
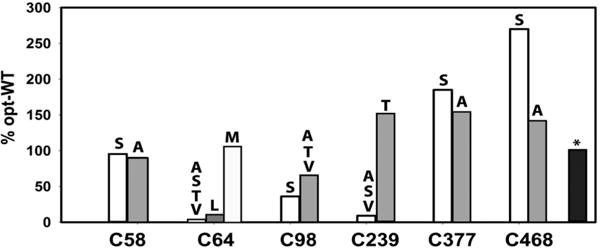
Effect of single-cysteine mutations on the expression of codon-optimized CYP3A4 in E. coli. The CYP3A4 content was determined in the bacterial lysate by measuring the ferrous P450–CO adduct. All values were normalized and shown as a percentage of the expression level of opt-WT. The standard deviation from the mean values did not exceed 10%. A black bar with an asterisk corresponds to the C58A/C64M/C98A/C239T/C377A/C468S variant.
To find more tolerable replacements, cysteines 64, 98, and 239 were replaced with the small chain Thr and Val residues. No improvement in the expression level was observed for the C64T/V, C98T/V, or C239V mutants (Figure 1). The C239T variant, however, was expressed 1.6-fold higher than opt-WT CYP3A4. Since Cys64 is part of the hydrophobic cluster (described in detail below), this residue was next mutated to Leu and Met. There was a slight increase (∼10%) in the C64L mutant expression, but the C64M variant was produced as robustly as opt-WT.
Based on these findings, a well-expressed C64M/C239T/C468S variant was then cloned and used as a template for introduction of C58A/S, C98A/T/V, and C377S/A mutations to identify the most optimal combination for Cys-less CYP3A4. The C58A/C64M/C98A/C239T/C377A/C468S variant was chosen for biochemical and structural characterization because it was expressed at the highest level close to that for opt-WT CYP3A4 (Figure 1).
Spectral Properties and Equilibrium Ligand Binding
Optical spectra of opt-WT and Cys-less CYP3A4 were similar but not identical to that of the original WT (Figure 2A). The latter protein has a 416 nm Soret band and equal α- and β-bands, whereas both opt-WT and Cys-less CYP3A4 have the absorption maximum at 417–418 nm, an elevated shoulder at 370 nm, and a lower β-band. These subtle type II spectral changes may indicate that opt-WT and Cys-less CYP3A4 are partially bound to a heteroatom of a ligand, the source and chemical nature of which are yet to be determined.
Figure 2.
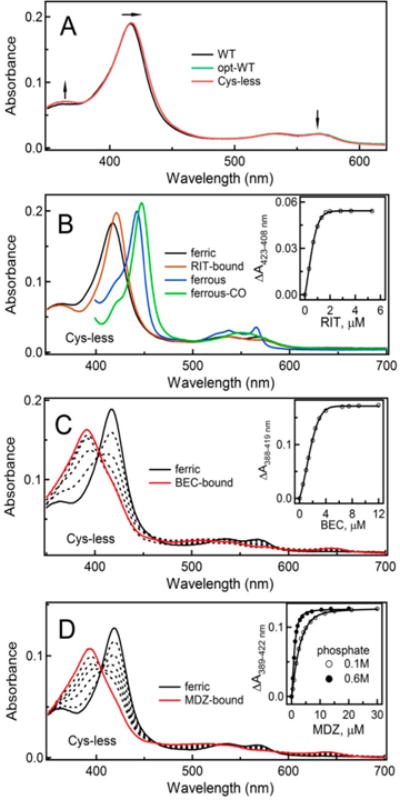
(A) Absorbance spectra of WT, opt-WT, and Cys-less CYP3A4. Arrows point to spectral differences indicative of a partial type II spectral shift in opt-WT and Cys-less CYP3A4. (B) Spectral changes induced by RIT in Cys-less CYP3A4. (C and D) Equilibrium titration of Cys-less CYP3A4 with BEC and MDZ, respectively. Insets are titration plots and quadratic or hyperbolic fittings. The derived Ks values are listed in Table 1.
Other dissimilarities were observed during equilibrium titrations with the inhibitor RIT and substrates BEC and MDZ. The ligand-induced spectral changes in opt-WT and Cys-less CYP3A4 were similar, and those for Cys-less CYP3A4 are shown in Figure 2. As expected,4,24 RIT induced a red shift in the Soret band, and the ferrous RIT-bound form had a characteristic 442 nm absorption peak (Figure 2B). Type I ligands BEC and MDZ, on the other hand, led to a nearly complete high-spin shift (Figure 2C,D). Compared to WT, titration of opt-WT and Cys-less CYP3A4 with either ligand required a longer time for completion. Judging from the dissociation constants [Ks (Table 1)], both proteins bind RIT as tightly as WT but the affinity for BEC is 1.5–2-fold lower. As observed for WT,3 an increase in the phosphate concentration from 0.1 to 0.6 M led to a 3-fold decrease in Ks for MDZ. Interestingly, regardless of the ionic strength, the Ks for the Cys-less CYP3A4–MDZ complex was severalfold lower than the respective values for WT and opt-WT (Table 1). This suggests that some Cys mutations might assist in the structural reorganization required for the association of MDZ.
Table 1.
Equilibrium Titration and Kinetic Parameters for the Ligand Binding to WT, Opt-WT, and Cys-less CYP3A4
| WT | opt-WT | Cys-less | |
|---|---|---|---|
| Equilibrium Titrations, Ks (μM) | |||
| RIT | 0.05a | 0.045 ± 0.008 | 0.047 ± 0.012 |
| BEC | 0.3b | 0.77 ± 0.14 | 0.45 ± 0.06 |
| MDZ | |||
| 0.1 M phosphate | 4.8c | 3.6 ± 0.1 | 0.73 ± 0.09 |
| 0.6 M phosphate | 1.8c | 1.6 ± 0.1 | 0.22 ± 0.04 |
| Ligand Binding Kinetics | |||
| RIT | |||
| kfast(s−1) | 1.24 ± 0.05 (70%)d | 0.68 ± 0.02 (33%) | 0.31 ± 0.03 (33%) |
| kslow(s−1) | 0.26 ± 0.02 (30%) | 0.033 ± 0.003 (67%) | 0.037 ± 0.005 (67%) |
| BEC | |||
| kfast(s−1) | 0.17 ± 0.02 (52%) | 0.016 ± 0.003 (49%) | 0.007 ± 0.001 (54%) |
| kslow(s−1) | 0.0089 ± 0.0014 (48%) | 0.0016 ± 0.0004 (51%) | 0.0013 ± 0.003 (46%) |
| MDZ, 0.1 M phosphate | |||
| kfast(s−1) | 1.50 ± 0.05 (70%) | 0.19 ± 0.03 (43%) | 0.15 ± 0.02 (40%) |
| kslow(s−1) | 0.022 ± 0.004 (30%) | 0.021 ± 0.003 (57%) | 0.022 ± 0.03 (60%) |
| MDZ, 0.6 M phosphate | |||
| kfast(s−1) | 3.4 ± 0.3 (82%) | 0.26 ± 0.03 (49%) | 0.27 ± 0.02 (43%) |
| kslow(s−1) | 0.23 ± 0.04 (18%) | 0.027 ± 0.004 (51%) | 0.036 ± 0.003 (57%) |
| First Electron Transfer from CPR | |||
| kfast(min−1) | 1.20 ± 0.05 (65%) | 0.72 ± 0.02 (85%) | 1.22 ± 0.03 (64%) |
| kslow(min−1) | 0.26 ± 0.02 (35%) | 0.030 ± 0.003 (15%) | 0.22 ± 0.02 (36%) |
Kinetics of Ligand Binding
Kinetics of RIT, BEC, and MDZ binding to WT, opt-WT, and Cys-less CYP3A4 were measured by monitoring the appearance or disappearance of the low-spin form on a stopped flow spectrophotometer (Figures 3 and 4). All reactions were biphasic and, regardless of the ligand type, the rate constants for the fast and slow phase (kfast and kslow, respectively) were the highest for WT (Table 1). The largest difference in the kinetic parameters was observed for the BEC binding reaction, whereas the amplitude of the absorbance change varied the most during RIT association because of a large difference in the percentage of the fast kinetic phase: 70% for WT versus 33% for opt-WT and Cys-less CYP3A4. Binding of MDZ to all proteins proceeded faster at a high ionic strength (Figure 4A,B), but the stimulating effect was more pronounced for WT (Table 1). A smaller 420 nm absorbance change observed during formation of the WT CYP3A4–MDZ complex (Figure 4A,B) can be explained by the shift in the Soret maximum and incompletion of the high-spin conversion. Together, the equilibrium binding and kinetic data indicate that opt-WT and Cys-less CYP3A4 interact with the type I and type II ligands in a similar manner but considerably slower than WT and have a lower affinity for BEC.
Figure 3.
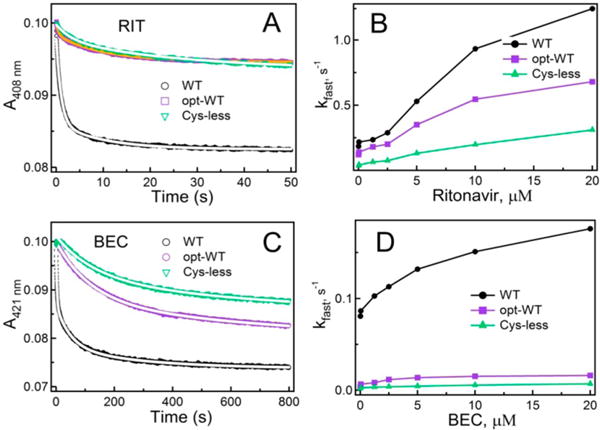
Kinetics of RIT and BEC binding to WT, opt-WT, and Cysless CYP3A4 measured at ambient temperature in 0.1 M phosphate buffer (pH 7.4). (A and C) Kinetic traces recorded in a stopped flow spectrophotometer after mixing 2 μM CYP3A4 with 40 μM RIT and BEC, respectively. Reactions were biphasic under all studied conditions. Fitting curves are shown as white solid lines. (B and D) Dependence of the observed rate constant for the fast phase (kfast) on the concentration of RIT and BEC, respectively. The kfast and kslow values measured at the maximal ligand concentration are listed in Table 1.
Figure 4.
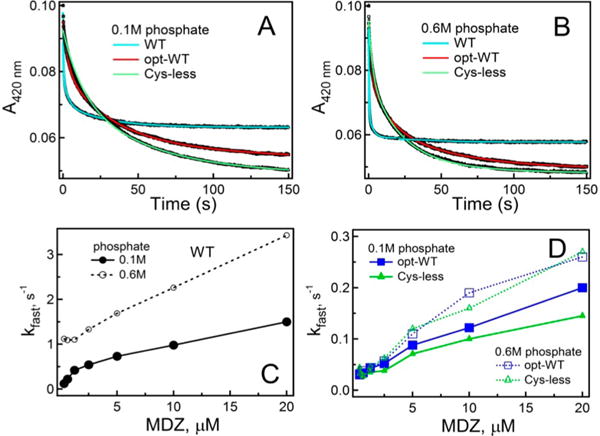
Kinetics of MDZ binding to WT, opt-WT, and Cys-less CYP3A4. (A and B) Kinetic traces obtained after mixing 2 μM proteins and 40 μM MDZ in 0.1 and 0.6 M phosphate buffer (pH 7.4), respectively. Kinetics were biphasic for the entire MDZ concentration range. Solid cyan, red, and green lines are fitting curves. (C) Dependence of kfast on MDZ concentration derived for WT CYP3A4. (D) Plots of kfast vs MDZ concentration for opt-WT and Cys-less CYP3A4. The kfast and kslow values measured at the maximal MDZ concentration are listed in Table 1.
BFC Debenzylase Activity and Electron Transfer from CPR
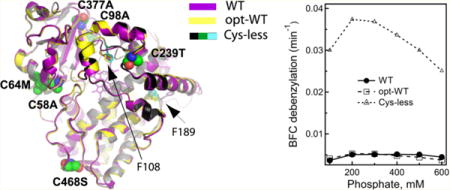
To investigate if/how cysteine depletion affects the catalytic function of CYP3A4, the BFC debenzylase activity was measured in a reconstituted system with the redox partner CPR. BFC is a model substrate that forms an easily detectable fluorescent product after O-debenzylation. At high phosphate concentrations, BFC causes a small high-spin shift in WT and none in opt-WT or Cys-less CYP3A4 (Figure S3). Strikingly, under all studied conditions, Cys-less CYP3A4 oxidized BFC 6–8-fold faster than WT and opt-WT (Figure 5A). A similar trend was observed for the C98S/C239S/C468G mutant of Δ3–12 CYP3A4, reported to possess a 1.5-fold higher activity than WT.17
Figure 5.
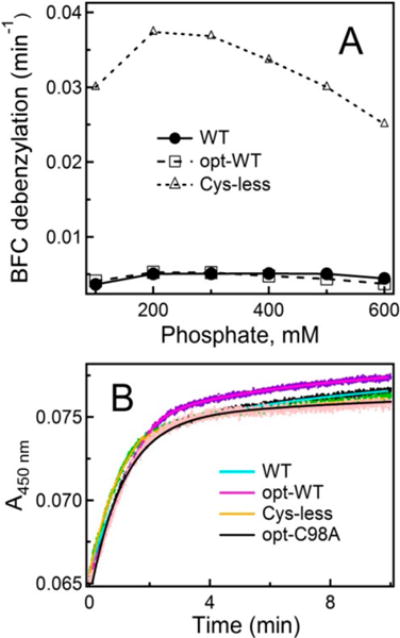
(A) Effect of phosphate concentration on the BFC debenzylation activity of WT, opt-WT, and Cys-less CYP3A4. The standard deviation from the mean values did not exceed 10%. (B) Kinetics of the first electron transfer from CPR to various forms of CYP3A4 measured under anaerobic conditions in CO-saturated 0.1 M phosphate buffer (pH 7.4) supplemented with 30 μM MDZ. Under these conditions, all proteins were in the high-spin form. The C98A mutant of opt-WT CYP3A4 (opt-C98A) was used as a control. Fitting curves are shown as solid lines; the derived rate constants are listed in Table 1.
One factor that could contribute to this phenomenon is an increased level of electron flow from CPR, as one of the mutated residues (Cys98) modulates the affinity of the CYP3A4–CPR complex.25 To test this possibility, we determined the rate constant for the first electron transfer from CPR by monitoring formation of the ferrous CYP3A4– CO adduct. The C98A mutant, which has no effect on the CYP3A4–CPR interaction,25 was included in this experiment as a control. Since the heme reduction rate depends on the high-spin content, 30 μM MDZ was added to the reaction mixture to convert the heme iron to the high-spin form. Within the studied time interval, the electron transfer reactions for all CYP3A4 forms were biphasic and the derived rate constants were in the same range (Figure 5B and Table 1). The kfast and kslow values for the C98A mutant were also similar: 0.90 ± 0.07 and 0.06 ± 0.01 min−1, respectively (82% reduction in the fast phase). These results suggest that factors other than the interprotein electron transfer must be responsible for a drastic increase in the catalytic activity of Cys-less CYP3A4.
Crystal Structure of Opt-WT CYP3A4
Because cysteine mutations could have a structural impact, crystal structures of opt-WT and Cys-less CYP3A4 were determined and compared. The X-ray model of the water-coordinated opt-WT was determined to 1.7 Å, the highest resolution achieved so far for CYP3A4, and refined to the lowest R factors [R and Rfree values of 16.6 and 20.4%, respectively (Table S1)]. Overall, this and the previously reported 2.05 Å 1TQN model of the ligand-free WT CYP3A4 were very similar, with a root-mean-square deviation (rmsd) between the ordered Cα atoms of 0.49 Å (Figure 6A). The higher resolution and better quality of the opt-WT structure allowed the identification of additional solvent networks, alternative side chain conformations, and multiple glycerol and ethylene glycol binding sites. One glycerol and two ethylene glycol molecules were present in the active site, forming a H-bonding network with the hemeligating water molecule and the Arg212 and Ser119 side chains (Figure 6C).
Figure 6.
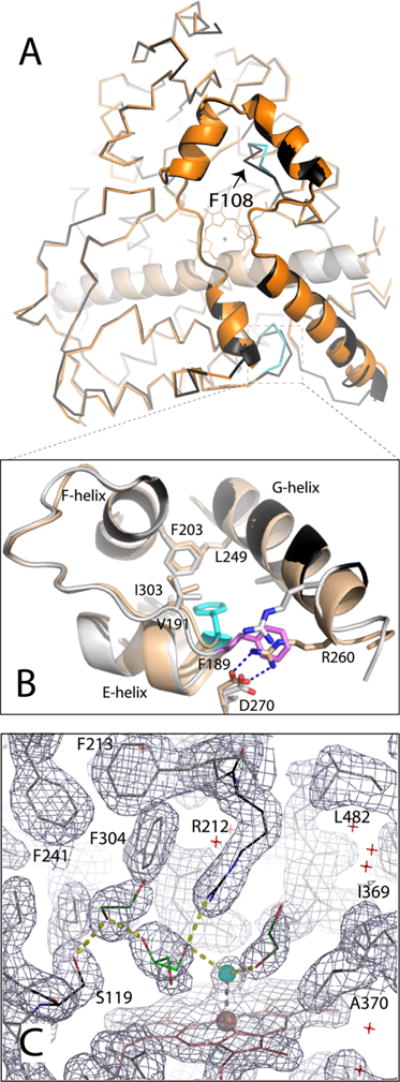
Crystal structure of opt-WT CYP3A4. (A) Structural superposition of the WT (1TQN, beige and orange) and opt-WT CYP3A4 (gray and black). The differently folded fragments in opt-WT are colored cyan; Phe108 is denoted with an arrow. (B) Magnified view of the region surrounding Phe189, whose side chain is oriented toward and away from the protein core in opt-WT and WT CYP3A4 (colored cyan and pink, respectively). The “inward” conformation allows Phe189 to interact with the hydrophobic cluster formed by Val191, Phe203, Leu249, and Ile303 and allows formation of the Arg260–Asp270 salt bridge linking the G and H helices. (C) Active site of opt-WT CYP3A4. The heme-bound water is colored cyan; other solvent molecules are depicted as red crosses. Glycerol and two ethylene glycol molecules (shown as light and dark green sticks, respectively) are interlinked with the water ligand and the Ser119 and Arg212 side chains through H-bonding interactions (shown as yellow dotted lines). The gray mesh is the 2Fo – Fc electron density map contoured at 1σ.
Comparison of the 1TQN and opt-WT structures showed also that, besides small positional deviations in the flexible surface loops, there were notable folding differences in the regions surrounding Phe108 and Phe189 (Figure 6A,B). Phe189 is located at the end of the E helix and, in opt-WT, is directed toward the protein core and engaged in hydrophobic interactions with Val191, Phe203, Leu249, and Ile303. Because of the tighter turn at the end of the G helix, Arg260 can approach and form a strong double salt bridge with Asp270, thereby locking Phe189 in an “inward” conformation and strengthening its interactions with the hydrophobic cluster. In the 1TQN structure, Phe189 points toward the solvent channel, which prevents formation of the Arg260–Asp270 salt bridge and weakens crosstalk between the E and G helices (Figure 6B). This could affect conformational dynamics in the F–G fragment and, consequently, the ligand binding rates.
Two other ligand-free structures of WT CYP3A4 (PDB entries 1W0E and 4I3Q) were determined to a lower resolution (2.8 and 2.6 Å, respectively). Comparative analysis of the Phe189 sites showed that the 4I3Q structure resembles 1TQN, whereas the 1W0E model is more similar to 5VCC (Figure S4). Asp270 is disordered in the 1W0E structure, and it is unclear whether the Arg260–Asp270 salt bridge can be established. In any case, naturally occurring conformational variations in the Phe189 region could complicate the substrate binding and catalytic behavior of CYP3A4. The impact of structural heterogeneity at the Phe108 site will be discussed in detail below.
Crystal Structure of Cys-less CYP3A4
The 1.95 Å structure of Cys-less CYP3A4 is nearly identical to that of opt-WT [rmsd between the ordered Cα atoms of 0.27 Å (Figure 7A)], and its active site glycerol is oriented and connected to Ser119, Arg212, and the heme-ligating water in a similar manner (compare Figures 6C and 7B). This is in contrast to the 1TQN structure of WT CYP3A4, where no such glycerol was observed despite the fact that it was present in excess in the purification and crystallization buffer.26 Therefore, it is tempting to speculate that the active site glycerol could contribute to an altered ligand binding and catalytic behavior of opt-WT and Cys-less CYP3A4 (Figures 3–5A and Table 1). Previous studies showed that glycerol may act as both a competitive inhibitor and an activator of mammalian CYPs.27,28 The X-ray data provide the first insights into the possible mechanism and suggest that the tightly bound glycerol molecule could modulate ligand binding in several ways: by crowding and reducing access to the active site, by interlocking the active site residues critical for ligand binding, and/or by decreasing the flexibility of the F–G fragment. Since the catalytic center remains accessible in the glycerol-bound form, the smaller volume and higher rigidity of the active site could promote metabolism of small compounds, such as BFC.
Figure 7.
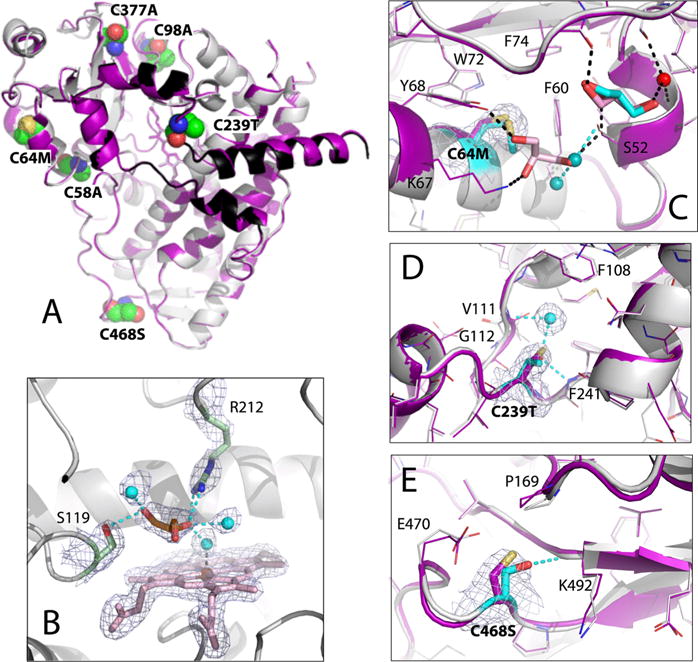
Crystal structure of Cys-less CYP3A4. (A) Superposition of Cys-less (gray and black) and opt-WT CYP3A4 (purple). The mutated residues are in CPK representation and labeled. (B) View of the active site where glycerol (orange sticks) forms H-bonds with the Ser119 and Arg212 side groups and three water molecules (cyan spheres). The 2Fo – Fc electron density map contoured at 1σ is shown as gray mesh. (C–E) Structural changes at the C64M, C239T, and C468S mutational sites, respectively. The superimposed Cys-less and and opt-WT structures are rendered in gray/cyan and purple, respectively. In Cys-less CYP3A4, the newly recruited water molecules and glycerol are shown as cyan spheres and cyan sticks, respectively; cyan dotted lines are the newly formed H-bonds. Water, glycerol, and H-bonds observed in opt-WT CYP3A4 are represented in the same manner but colored red, pink, and black, respectively. The 2Fo – Fc electron density maps at the mutation sites are shown as gray mesh contoured at 1σ.
Glycerol also is a well-known protein stabilizing agent29 that permitted isolation of the first functionally active mammalian P450 five decades ago.30 Nowadays, 10–20% glycerol is added to the purification and storage solutions to prevent the conversion of CYPs to the inactive P420 form. In the Cysless CYP3A4 structure, 11 glycerol molecules are bound to the outer surface. Four of these peripheral sites coincide, and two others are replaced with ethylene glycol in opt-WT (Figure S5). Notably, in both structures, four glycerol molecules are clustered near the A′ and A helices, through which CYP3A4 is thought to associate with the microsomal membrane.31–33 The extensive polyol-mediated inter-residue bridges observed in the X-ray structure may improve stability and prevent inactivation and proteolytic degradation of CYP3A4.
Structural Differences between Opt-WT and Cys-less CYP3A4
Comparison of the opt-WT and Cys-less CYP3A4 structures revealed conformational variations at the C64M, C239T, and C468A mutational sites and in the Phe108-containing fragment.
C64M Site
Cys64 is adjacent to the aromatic cluster comprised of Tyr53, Phe54, Phe60, Tyr68, Trp72, and Phe74 (Figure 7C) and forms S–π interactions with the Phe60 ring. The cluster is also stabilized by two glycerol molecules that shield the hydrophobic surface from the solvent and mediate a H-bonding network with the main and side chains of the 67–68 and 51–52 fragments and Thr42 (Figure 7C). Due to steric clashing, Met in position 64 expels the neighboring glycerol and the site becomes more solvated. However, the methionine sulfur is positioned suitably for the S–π contacts with Phe60. The latter interaction seems to be important for protein stability and/or folding, as five other investigated substitutes of Cys64 profoundly diminish CYP3A4 expression (Figure 1).
C239T Site
Cys239 is located in the G′–G connecting loop, part of the F–G fragment. The C239T replacement promotes formation of two new H-bonding contacts: with the Phe241 amide nitrogen and the newly recruited water molecule that is also connected to the Val111 main chain (Figure 7D). The 111–112 peptide bond flip is required to allow the latter interaction. Since Val111 resides in the B–C loop, directly involved in the ligand binding process, the Thr239-triggered changes could lower the degree of motional freedom of both the B–C and F–G fragments and alter the ligand binding kinetics. It is puzzling why the C239S mutation has such a negative impact on CYP3A4 expression (Figure 1) because serine could similarly mediate the H-bonding interactions and is isosteric to cysteine.
C468S Site
Cys468 is positioned at the very start of the C-terminal loop, the upper part of which lines the wall of the substrate access channel. Introduction of polar Ser468 promotes the H-bond formation with the amide nitrogen of Lys492 from the opposite side of the loop (Figure 7E). This newly established connection does not appear to impose any restraints on the C-terminal loop or nearby residues and secondary structure elements. Thus, the C468S mutation could affect conformational stability but is unlikely to feasibly influence the ligand binding properties of CYP3A4.
Phe108 Site
The 105–111 fragment is part of the B–C loop that comprises the opposite wall of the active site cavity. Two of its residues, Phe108 and Val111, are within the van der Waals distance from Ile120, Phe213, Phe220, Val240, and Phe241 that, together with the I-helix Phe304, form a hydrophobic cluster adjacent to the active site (Figure 8A). In Cys-less CYP3A4, the 105–111 fragment adapts a single conformation, and Phe108 is well-ordered and turned sideways toward the hydrophobic pocket [“sideway” rotamer (Figure 8A)]. This is in contrast to the 1TQN structure of WT CYP3A4, where the 107–109 main chain is 3 Å farther away and a distinct Phe108 rotamer points toward the cluster (“inward” orientation). In opt-WT, both conformations are observed but the “sideway” orientation is less favorable [44% occupancy (Figure 8B)]. It should be emphasized that these variations are not crystal-induced artifacts, as all three proteins were crystallized in the same I222 space group where no packing constraints are imposed on the 105–111 fragment. Second, the outlined conformational differences are important because they could modulate the strength of the hydrophobic inter-residue contacts and, as a consequence, the rigidity of the active site that could increase or decrease when Phe108 is in a “sideway” or “inward” orientation, respectively. Since reorganization of the aforementioned cluster is required for the productive orientation of MDZ,3 conformational heterogeneity in the Phe108 area might, in part, be responsible for the observed differences in the MDZ binding reaction (Figure 4 and Table 1).
Figure 8.
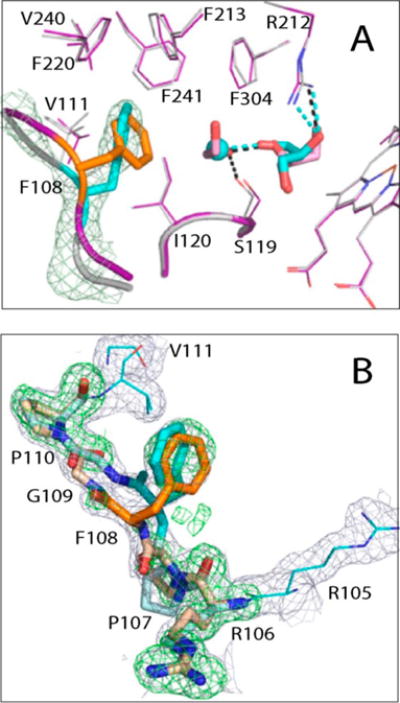
Conformational heterogeneity at the Phe108 site. (A) Superposition of Cys-less (gray and cyan) and opt-WT CYP3A4 (purple, orange, and pink). In Cys-less CYP3A4, the 105–111 fragment adapts a single conformation where Phe108 is well-ordered and approaches the hydrophobic cluster adjacent to the active site (residues Ile120, Phe213, Phe220, Val240, Phe241, and Phe304) in a “sideway” orientation. In opt-WT, an “inward” orientation of Phe108 is more preferable (orange), and the water molecule linked to glycerol is replaced with ethylene glycol. (B) Conformational heterogeneity of the 105–1111 fragment in opt-WT CYP3A4. The 2Fo – Fc and simulated annealing Fo – Fc omit electron density maps are contoured at 1σ and 3σ and shown as gray and green mesh, respectively.
It is unclear at the moment how the folding of the Phe108-containing segment is controlled. The two closest mutations, C239T and C98A, are 11–19 Å away, and the latter has no structural impact. One regulating factor could be the Val111-Gly112 peptide flip triggered by the C239T substitution (Figure 7D). The resulting Val111 shift (∼0.7 Å) could give enough space for the “sideway” Phe108 orientation. Additionally, in Cys-less CYP3A4, the active site glycerol is linked to a water rather than ethylene glycol molecule (Figure 8A), which could provide extra space for structural readjustments. Finally, glycerol itself could be a key player as it locks Arg212 and the nearby Phe213 in a fixed conformation, which not only strengthens interactions at the Phe108 site but also decreases the flexibility of the F–G fragment.
Crystal Structure of the RIT-Bound Cys-less CYP3A4
To elucidate if/how Cys mutations affect the ligand binding mode, crystal structures of the RIT- and BEC-bound forms of Cys-less CYP3A4 were determined (Table S1). Also, for more accurate identification of the RIT-dependent changes, the WT CYP3A4–RIT complex was crystallized in the I222 space group. The major differences between the RIT-bound forms were observed in the F–G and C-terminal fragments (Figure 9A). Both alterations are likely triggered by the C58A substitution because it leads to repositioning of the 55–59 segment and the nearby Met371 side chain (Figure 9B). To avoid steric hindrance with the new Phe57 conformer, the RIT terminal group swings aside and its isopropyl thiazole becomes disordered. The F′–G′ connecting loop (residues 211–217) follows the movement and penetrates deeper into the active site to fill the emptied space and optimize the protein–ligand interactions. The C-terminal loop, in turn, moves farther away to prevent clashing. Importantly, the RIT side groups, largely contributing to the binding affinity,34 are positioned similarly in WT and Cys-less CYP3A4 (Figure 9B). This could explain why Cys-less CYP3A4 binds RIT tightly but slower than WT (Figure 3 and Table 1). On the other hand, comparison of the 1NXU and 5VCO structures of RIT-bound WT CYP3A4 showed that the manner of crystal packing could affect the RIT backbone conformation, carbonyl oxygen orientation, and placement of the Phe-2 side group (Figure S5).
Figure 9.
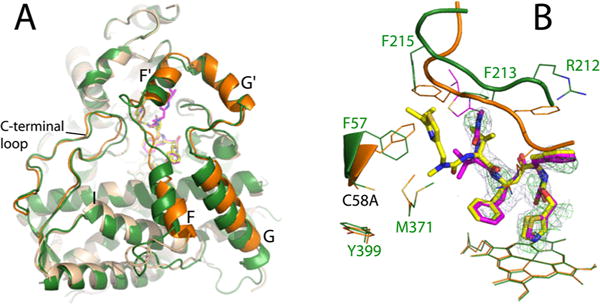
Crystal structure of the RIT-bound Cys-less CYP3A4. (A) Superposition of RIT-bound Cys-less (beige and orange) and WT CYP3A4 (green) shows conformational differences in the F–G fragment and C-terminal loop. (B) RIT orientation in Cys-less (magenta) and WT CYP3A4 (yellow). In the mutant, the disordered isopropyl thiazole portion of RIT is depicted in thin lines. The C58A substitution triggers repositioning of the Phe57 and Met371 side chains, due to which the RIT terminal group and the F′–G′ loop (residues 211–217) accommodate distinct conformations to avoid steric clashing. The 2Fo – Fc and simulated annealing Fo – Fc omit maps around RIT in Cys-less CYP3A4 are shown as gray and green mesh contoured at 1σ and 3σ, respectively.
Crystal Structure of the BEC-Bound Cys-less CYP3A4
The BEC-bound form was crystallized in the same space group as the WT CYP3A4–BEC complex (PDB entry 3UA1) and determined to a similar resolution (Table S1), which allowed unbiased comparison of the substrate binding modes. There was no mutational impact on the BEC orientation. Similar to the 3UA1 structure, BEC binds to Cys-less CYP3A4 in an extended and productive conformation (Figure 10A), with the cyclic tripeptide moiety approaching the heme and the lysergic group sandwiched between the walls of the substrate channel. The primary oxidation sites, the C8′ and C9′ atoms of the proline ring,35,36 are within 3.6 Å of the heme iron. The latter distance is slightly shorter than in the 3UA1 structure because the tripeptide group is pulled deeper into the active site and H-bonded to the Ser119 and Arg212 side chains. The lysergic moiety, in turn, moves farther from Thr224 and loses a H-bond connection. This leads to a higher level of ligand disorder (an average B factor of 100 Å2 vs 62 Å2 in the 3UA1 structure) and discontinuity in the electron density (Figure 10A). The latter feature is not the result of partial ligand binding because BEC, a high-affinity substrate, nearly fully saturates the binding site (occupancy of ∼0.9).
Figure 10.
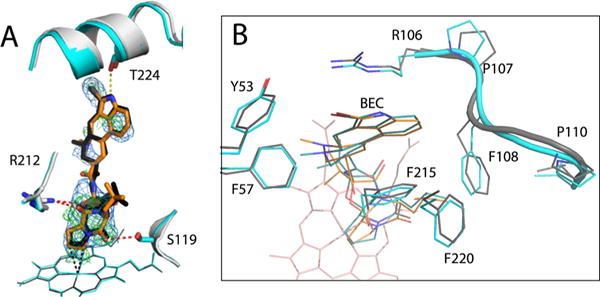
Crystal structure of the BEC-bound Cys-less CYP3A4. (A) Structural superposition of the BEC-bound forms of Cys-less (gray and orange) and WT CYP3A4 (PDB entry 3UA1, cyan and black). In both structures, BEC binds in an extended and productive conformation. However, in Cys-less CYP3A4, the cyclic tripeptide moiety is closer to the heme and H-bonded to Ser119 and Arg212 (red dotted lines), whereas the lysergic group is farther from Thr224 and cannot form the H-bond observed in the 3UA1 structure (yellow dotted line). The 2Fo – Fc and simulated annealing Fo – Fc omit maps around BEC in Cys-less CYP3A4 are contoured at 1σ and 3σ and shown as blue and green mesh, respectively. (B) Top view of the active site showing residues that flank the lysergic moiety of BEC. In Cys-less CYP3A4, the 105–111 fragment is folded differently and Phe108 clashes with BEC’s lysergic group, forcing it to move aside and lose a H-bond connection with Thr224.
One reason for the BEC binding mode variations in Cys-less CYP3A4 could be the differently folded 105–111 peptide, flanking BEC’s lysergic group. In the “sideway” orientation, the Phe108 Cβ atom is much closer to the lysergic group (3.1 vs 4.5 Å for the “inward” rotamer in the 3UA1 structure (Figure 10B)]. To minimize steric hindrance, the latter moiety shifts aside and its interaction with Thr224 is weakened. Considering that Cys-less CYP3A4 has a lower affinity and interacts with BEC considerably slower than WT (Figure 3 and Table 1), it can be concluded that the newly formed H-bonds with the tripeptide group cannot compensate for the loss of the BEC– Thr224 interaction, which likely initiates or drives association of the ligand with CYP3A4. Certainly, further mechanistic and molecular dynamics studies are required to fully understand the cause of structural and functional differences in WT, opt-WT, and Cys-less CYP3A4.
CONCLUSIONS
Utilization of the optimized gene of human CYP3A4 as a template for screening mutagenesis enabled the preparation of a well-expressed, stable, and functional C58A/C64M/C98A/C239T/C377A/C468S variant that could be used in biochemical and biophysical studies. The largest negative impact on the CYP3A4 expression level was observed upon elimination of cysteines 64 and 239, suggesting their importance for protein integrity and/or folding.
Codon optimization not only boosted the level of production but also affected the conformational and ligand binding properties of CYP3A4. Structural differences were observed in the Phe108 and Phe189 regions, as well as at the C64M, C239T, and C468S mutational sites. Unlike WT, both opt-WT and Cys-less CYP3A4 contained the active site glycerol that interlocked Ser119 and Arg212, directly involved in ligand binding, and the hydrophobic cluster adjacent to Phe108. Crowding and decreased flexibility of the active site and the F– G fragment might contribute to the distinct ligand binding ability of opt-WT and Cys-less CYP3A4. Moreover, if naturally occurring in vivo, association of the active site glycerol and structural variations in the Phe108 and Phe189 regions could increase conformational heterogeneity and complicate the substrate binding and catalytic behavior of microsomal CYP3A4.
The high-resolution structure of opt-WT CYP3A4 provided a clearer view of the protein architecture, side chain arrangement, and solvent networks and could serve as a more accurate starting model for determination of novel CYP3A4 structures. Structural comparison of RIT-bound WT CYP3A4 crystallized in different space groups, on the other hand, showed that the ligand binding mode can be notably affected by crystal packing, which must be taken into account in the structure-based drug design. Finally, the non-heme-ligating cysteines were found to contribute to the conformational and functional integrity of CYP3A4, and therefore, in the chemical modification studies, the respective impacts of cysteine mutations need to be determined prior to the label attachment in order to better understand and more accurately interpret the experimental results.
Supplementary Material
Acknowledgments
This work involves research performed at the Stanford Synchrotron Radiation Lightsource and the Advanced Light Source. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including Grant P41GM103393). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH11231. The author thanks Dr. T. L. Poulos for the critical reading of the manuscript.
Funding
This work was supported by National Institutes of Health Grant ES025767.
ABBREVIATIONS
- BEC
bromoergocryptine
- BFC
7-benzyloxy-(4-trifluoromethyl)coumarin
- CYP3A4
cytochrome P450 3A4
- CPR
cytochrome P450 reductase
- Cys-less and opt-WT CYP3A4
C58A/C64M/C98A/C239T/C377A/C468S variant and wild-type CYP3A4 produced in E. coli using a codon-optimized synthetic gene, respectively
- MDZ
midazolam
- RIT
ritonavir
- WT
wild-type CYP3A4 produced in E. coli using nonoptimized human cDNA
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00334.
Location of non-heme-ligating cysteines in CYP3A4, a nucleotide sequence of the codon-optimized CYP3A4 gene, BFC-dependent spectral changes in various CYP3A4 forms, comparison of the Phe189 sites in the ligand-free CYP3A4 structures, location of the glycerol and ethylene glycol molecules on the surface of opt-WT and Cys-less CYP3A4, packing of RIT-bound WT CYP3A4 in different crystal forms and its effect on the RIT binding mode, and X-ray data collection and refinement statistics (PDF)
ORCID
Irina F. Sevrioukova: 0000-0002-4498-6057
Notes
The author declares no competing financial interest.
References
- 1.Guengerich FP. Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Rendic S, Di Carlo FJ. Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab Rev. 1997;29:413–580. doi: 10.3109/03602539709037591. [DOI] [PubMed] [Google Scholar]
- 3.Sevrioukova IF, Poulos TL. Structural basis for regiospecific midazolam oxidation by human cytochrome P450 3A4. Proc Natl Acad Sci U S A. 2017;114:486–491. doi: 10.1073/pnas.1616198114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sevrioukova IF, Poulos TL. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc Natl Acad Sci U S A. 2010;107:18422–18427. doi: 10.1073/pnas.1010693107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ekroos M, Sjogren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc Natl Acad Sci U S A. 2006;103:13682–13687. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davydov DR, Rumfeldt JAO, Sineva EV, Fernando H, Davydova NY, Halpert JR. Peripheral ligand binding site in cytochrome P450 3A4 located with fluorescence resonance energy transfer (FRET) J Biol Chem. 2012;287:6797–6809. doi: 10.1074/jbc.M111.325654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sineva EV, Rumfeldt JA, Halpert JR, Davydov DR. A large-scale allosteric transition in cytochrome P450 3A4 revealed by luminescence resonance energy transfer (LRET) PLoS One. 2013;8:e83898. doi: 10.1371/journal.pone.0083898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davydov DR, Davydova NY, Sineva EV, Kufareva I, Halpert JR. Pivotal role of P450-P450 interactions in CYP3A4 allostery: the case of alpha-naphthoflavone. Biochem J. 2013;453:219–230. doi: 10.1042/BJ20130398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsalkova TN, Davydova NY, Halpert JR, Davydov DR. Mechanism of interactions of alpha-naphthoflavone with cytochrome P450 3A4 explored with an engineered enzyme bearing a fluorescent probe. Biochemistry. 2007;46:106–119. doi: 10.1021/bi061944p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davydov DR, Davydova NY, Tsalkova TN, Halpert JR. Effect of glutathione on homo- and heterotropic cooperativity in cytochrome P450 3A4. Arch Biochem Biophys. 2008;471:134–145. doi: 10.1016/j.abb.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davydov DR, Sineva EV, Sistla S, Davydova NY, Frank DJ, Sligar SG, Halpert JR. Electron transfer in the complex of membrane-bound human cytochrome P450 3A4 with the flavin domain of P450BM-3: the effect of oligomerization of the heme protein and intermittent modulation of the spin equilibrium. Biochim Biophys Acta, Bioenerg. 2010;1797:378–390. doi: 10.1016/j.bbabio.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davydov DR, Davydova NY, Sineva EV, Halpert JR. Interactions among cytochromes P450 in microsomal membranes: oligomerization of cytochromes P450 3A4, 3A5 and 2E1 and its functional consequences. J Biol Chem. 2015;290:3850–3864. doi: 10.1074/jbc.M114.615443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davydov DR, Yang Z, Davydova N, Halpert JR, Hubbell WL. Conformational mobility in cytochrome P450 3A4 explored by pressure-perturbation EPR spectroscopy. Biophys J. 2016;110:1485–1498. doi: 10.1016/j.bpj.2016.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baer BR, Wienkers LC, Rock DA. Time-dependent inactivation of P450 3A4 by raloxifene: identification of Cys239 as the site of apoprotein alkylation. Chem Res Toxicol. 2007;20:954–964. doi: 10.1021/tx700037e. [DOI] [PubMed] [Google Scholar]
- 15.Pearson JT, Wahlstrom JL, Dickmann LJ, Kumar S, Halpert JR, Wienkers LC, Foti RS, Rock DA. Differential time-dependent inactivation of P450 3A4 and P450 3A5 by raloxifene: a key role for C239 in quenching reactive intermediates. Chem Res Toxicol. 2007;20:1778–1786. doi: 10.1021/tx700207u. [DOI] [PubMed] [Google Scholar]
- 16.Wen B, Doneanu CE, Gartner CA, Roberts AG, Atkins WM, Nelson SD. Fluorescent photoaffinity labeling of cytochrome P450 3A4 by lapachenole: identification of modification sites by mass spectrometry. Biochemistry. 2005;44:1833–1845. doi: 10.1021/bi048228c. [DOI] [PubMed] [Google Scholar]
- 17.Menard A, Huang Y, Karam P, Cosa G, Auclair K. Site-specific fluorescent labeling and oriented immobilization of a triple mutant of CYP3A4 via C64. Bioconjugate Chem. 2012;23:826–836. doi: 10.1021/bc200672s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Polic V, Auclair K. Allosteric activation of cytochrome P450 3A4 via progesterone bioconjugation. Bioconjugate Chem. 2017;28:885–889. doi: 10.1021/acs.bioconjchem.6b00604. [DOI] [PubMed] [Google Scholar]
- 19.Sevrioukova IF, Poulos TL. Anion-dependent stimulation of CYP3A4 monooxygenase. Biochemistry. 2015;54:4083–4096. doi: 10.1021/acs.biochem.5b00510. [DOI] [PubMed] [Google Scholar]
- 20.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of COOT. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 23.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–321. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sevrioukova IF, Poulos TL. Structural and mechanistic insights into the interaction of cytochrome P4503A4 with bromoergocryptine, a type I ligand. J Biol Chem. 2012;287:3510–3517. doi: 10.1074/jbc.M111.317081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen B, Lampe JN, Roberts AG, Atkins WM, David Rodrigues A, Nelson SD. Cysteine 98 in CYP3A4 contributes to conformational integrity required for P450 interaction with CYP reductase. Arch Biochem Biophys. 2006;454:42–54. doi: 10.1016/j.abb.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J Biol Chem. 2004;279:38091–38094. doi: 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- 27.Yoo JS, Cheung RJ, Patten CJ, Wade D, Yang CS. Nature of N-nitrosodimethylamine demethylase and its inhibitors. Cancer Res. 1987;47:3378–3383. [PubMed] [Google Scholar]
- 28.Yang MX, Cederbaum AI. Glycerol increases content and activity of human cytochrome P-4502E1 in a transduced HepG2 cell line by protein stabilization. Alcohol: Clin Exp Res. 1997;21:340–347. [PubMed] [Google Scholar]
- 29.Gekko K, Timasheff SN. Mechanism of protein stabilization by glycerol: preferential hydration in glycerol-water mixtures. Biochemistry. 1981;20:4667–4676. doi: 10.1021/bi00519a023. [DOI] [PubMed] [Google Scholar]
- 30.Ichikawa Y, Yamano T. Reconversion of detergent- and sulfhydryl reagent-produced P-420 to P-450 by polyols and glutathione. Biochim Biophys Acta, Bioenerg. 1967;131:490–497. doi: 10.1016/0005-2728(67)90008-4. [DOI] [PubMed] [Google Scholar]
- 31.Denisov IG, Shih AY, Sligar SG. Structural differences between soluble and membrane bound cytochrome P450s. J Inorg Biochem. 2012;108:150–158. doi: 10.1016/j.jinorgbio.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baylon JL, Lenov IL, Sligar SG, Tajkhorshid E. Characterizing the membrane-bound state of cytochrome P450 3A4: structure, depth of insertion, and orientation. J Am Chem Soc. 2013;135:8542–8551. doi: 10.1021/ja4003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Treuheit NA, Redhair M, Kwon H, McClary WD, Guttman M, Sumida JP, Atkins WM. Membrane interactions, ligand-dependent dynamics, and dtability of cytochrome P4503A4 in lipid nanodiscs. Biochemistry. 2016;55:1058–1069. doi: 10.1021/acs.biochem.5b01313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sevrioukova IF, Poulos TL. Ritonavir analogues as a probe for deciphering the cytochrome P450 3A4 inhibitory mechanism. Curr Top Med Chem. 2014;14:1348–1355. doi: 10.2174/1568026614666140506120647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maurer G, Schreier E, Delaborde S, Loosli HR, Nufer R, Shukla AP. Fate and disposition of bromocriptine in animals and man. I: Structure elucidation of the metabolites. Eur J Drug Metab Pharmacokinet. 1982;7:281–292. doi: 10.1007/BF03189631. [DOI] [PubMed] [Google Scholar]
- 36.Maurer G, Schreier E, Delaborde S, Nufer R, Shukla AP. Fate and disposition of bromocriptine in animals and man. II: Absorption, elimination and metabolism. Eur J Drug Metab Pharmacokinet. 1983;8:51–62. doi: 10.1007/BF03189581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.