

ABSTRACT
In this study, the authors present a sample of 71 patients with hereditary optic neuropathy and negative genetic test results for OPA1/OPA3/LHON. All of these patients later underwent genetic testing to rule out WFS. As a result, 53 patients (74.7%) were negative and 18 patients (25.3%) were positive for some type of mutation or variation in the WFS gene. The authors believe that this study is interesting because it shows that a sizeable percentage (25.3%) of patients with hereditary optic 25 neuropathy and negative genetic test results for OPA1/OPA3/LHON had WFS mutations or variants.
KEYWORDS: Central diabetes insipidus, diabetes mellitus, hereditary optic neuropathies, neurosensory deafness, optic atrophy, Wolfram syndrome
Introduction
The most common hereditary optic neuropathies are dominant optic atrophy (DOA) and Leber’s hereditary optic neuropathy (LHON).1 However, other conditions, such as Wolfram syndrome (WFS), must be taken into account in the differential diagnosis of hereditary optic neuropathies.
WFS is a rare recessive autosomal disease characterised by the presence of diabetes mellitus (DM) and optic atrophy (OA) with early onset, typically before the age of 15 years. In addition, WFS may be associated with neurosensory deafness, central diabetes insipidus (DI), and progressive neurological disorders (such as ataxia, peripheral neuropathy, cognitive impairment, and psychiatric manifestations).2,3 The ophthalmological manifestations of WFS, in addition to OA, include the presence of cataracts, nystagmus, and, in certain cases, sensory strabismus.4
WFS may be caused by mutations in the WFS1 (Online Mendelian Inheritance in Man [OMIM] 222300) or CDGSH iron sulphur domain 2 (CISD2; OMIM 604928) gene. WFS1 is located on chromosome 4p16.1 and encodes the protein wolframin, which is located in the membrane of the endoplasmic reticulum (ER). CISD2 is located on chromosome 4q22 and encodes ER inter-membrane small protein (ERIS).2,3
Mutations in the CISD2 gene are responsible for the WFS type 2 (WFS2). This rare syndrome produces the same manifestations as classic WFS, with one exception: DI is absent.2 WFS2-related diseases are rare and have been described in Jordanian families in the literature. They are caused by mutations in the CISD2 gene, located on chromosome 4q22.2
The diagnosis of WFS is based on the presence of characteristic symptoms and clinical signs and is confirmed by a genetic test. Given that in WFS, the clinical manifestations progress with age, it is difficult to make a diagnosis at a young age.
In 2011, Yu-Wai-Man et al. published a series of 188 patients who were phenotypically compatible with DOA. In this group of patients, genetic testing was performed for OPA1/OPA3 and was positive in 14.4% of cases.5
In the present study, we present a sample of 71 patients with hereditary optic neuropathy and negative genetic test results for OPA1/OPA3/LHON. All of these patients underwent genetic testing to rule out WFS, regardless of whether they met the clinical criteria for WFS.
Methodology
Patients
This is a retrospective study of a sample of 71 patients with hereditary optic neuropathy who were referred to the King Khaled Eye Specialist Hospital in Riyadh from different geographical areas of Saudi Arabia from 2010 to 2014.
The inclusion criteria were as follows: a visual acuity (VA) decrease in the first two decades of life, temporal or diffuse pallor of the optic disc, and an absence of other neurological and ophthalmological diseases that could explain the optic neuropathy (e.g., demyelinating disease, perinatal hypoxia, previous trauma). Patients with nystagmus or strabismus were included. Medical and family histories were recorded for all patients; however, for certain patients, these data could not be reliably obtained.
All patients were given a full neuro-ophthalmological examination, including VA analysis, colour vision assessment using Ishihara plates, biomicroscopy using a slit lamp, a description of ocular motility, and fundus appearance assessment. For patients who exhibited a sufficient degree of cooperation, campimetry (Goldmann or Humphrey) and spectral domain optical coherence tomography (OCT) (Heidelberg Engineering, Inc., Heidelberg, Germany) of the optic nerve were conducted.
All patients underwent genetic testing for OPA1, OPA3, LHON, and WFS.
Genetic testing
Blood samples from all 71 patients were used for genetic testing for OPA1/OPA3/LHON. Subsequently, genetic testing was performed for WFS1 and WFS2.
The 30 coding exons and the exon-intron boundaries of OPA1 on chromosome 3q28–29 (OMIM 605290) were amplified by polymerase chain reaction (PCR) and sequenced directly. The resulting sequence data were compared with the reference sequence NM_130837.2.
We also conducted a deletion analysis of OPA1 (OMIM 605290) with the material obtained by multiplex ligation-dependent probe amplification (MLPA) using the SALSA MLPA P229-B1 OPA1 kit (Schouten, xx, xx). All coding exons of OPA1 and the adjacent regions were screened for deletions/duplications.
To identify OPA3 mutations, the two coding exons and the exon-intron boundaries of the OPA3 gene on chromosome 19q13.32 (OMIM 606580) were amplified by PCR and sequenced directly. The resulting sequence data were compared with the reference sequence NM_025136.3.
Additionally, given the clinical overlap between patients with DOA and LHON, the three most frequent genetic LHON mutations, at mitochondrial DNA (mtDNA) positions 11,778, 3460, and 14,484, were excluded in the analyses of all patients. For these analyses, mtDNA was obtained from peripheral blood leukocytes. Portions of the ND-4, ND-1, and ND-6 genes were amplified using the PCR-based amplification-refractory mutation system.
Finally, genomic DNA was screened for mutations in the WFS1 gene and CISD2 gene. The coding exons of known WFS genes were enriched using Roche/NimbleGen sequence capture technology (Madison, WI, USA) and sequenced on an Illumina HiSeq 1500 system (next-generation sequencing [NGS]; San Diego, CA, USA) Genomic DNA was fragmented, and the coding exons of the analysed genes as well as the corresponding exon-intron boundaries were enriched using the Roche/NimbleGen sequence capture approach, amplified, and sequenced simultaneously by Illumina technology (NGS) using an Illumina HiSeq 1500 system. The target regions were sequenced with an average coverage of 678-fold. For more than 99% of the regions of interest, a 20-fold coverage was obtained.
NGS data analysis was performed using bioinformatics analysis tools as well as JSI Medical Systems software (version 4.1.2; Boston, MA, USA). Identified variants and indels were filtered against external and internal databases and filtered depending on their allele frequency, focusing on rare variants with a minor allele frequency (MAF) of 1% or less. Nonsense frameshift and canonical splice site variants were primarily considered likely pathogenic. Assessment of pathogenicity of identified non-synonymous variants was performed using bioinformatics prediction programmes such as Mutation Taster, Polyphen-2, Mutation Assessor, FATHMM, etc. Only those variants that were predicted probably damaging by the majority of the used algorithms were considered likely pathogenic. Putatively pathogenic differences between the wild-type sequence (human reference genome according to University of California Santa Cruz [UCSC] Genome Browser: hg19, GRCh37) and the patient’s resulting sequence data for the WFS1 gene (OMIM 606201; locus chromosome 4p16.1) were compared with the reference sequence NM_006005.3.
Limitations of the NGS method are as follows: Mutations in insufficiently covered regions and rare alteration in other parts of the genes (e.g., regulatory regions), low-level mosaics, or deep intronic splice mutations cannot be excluded with this analysis. In principle, high coverage of NGS data enables copy number variation (CNV) analysis indication of deletions or duplications in the analysed genomic regions, but not completely ruling out such structural variants. Positive CNV results are validated by independent approaches such as MLPA.
Results
Our case series consisted of a total of 71 patients with negative genetic test results for OPA1/OPA3/LHON. Subsequently, these patients underwent genetic testing for WFS, with 53 patients negative and 18 patients positive for some type of mutation or variation in the WFS gene. We will separately describe the clinical characteristics of the patients who were negative for WFS and those who were positive for WFS.
Group of patients who were negative for WFS (WFS−) (53 patients)
The sample of 53 patients with no mutation was composed of 35 men (66%) and 18 women (34%), with a mean age of 21.2 years (range: 7–57 years). In all, 24 patients (45.3%) had a positive family history of optic nerve disease and 41 (77.4%) had consanguineous parents. The mean age at which the onset of visual symptoms occurred was 4.40 years (range: 0–20).
VAs ranged from 20/20 to 20/400 (mean: 20/80). Using Ishihara colour plates, colour vision ranged from 0/15 to 15/15 plates read correctly (mean: 3/15). In total, 41 patients had temporal optic disc pallor (77.4%) and 12 (22.6%) had diffuse pallor. Visual campimetry showed that most patients exhibited caecocentral scotomas (48.4%), followed by an enlarged blind spot (16.1%), arcuate defects (12.9%), and generalised decreased sensitivity (16.1%), and that 6.5% had normal visual fields.
Thirteen patients (24.5%) presented with nystagmus, and nine patients had strabismus (17%). The range of eye movements was normal in all these patients, and the nystagmus was horizontal and pendular. Eight patients (15.1%) had hearing loss, and two patients (3.8%) had ataxia. None of the patients in this group had DM or DI.
Retinal nerve fibre layer (RNFL) analysis by OCT of the optic nerve showed a mean value of 66.06 microns (range: 45–94). Cranial magnetic resonance (MR) imaging was normal (except for thinning of the optic nerve and/or optic chiasm) in all patients in this group.
In this group of 53 patients, genetic tests for OPA1/OPA3/LHON/WFS were all negative.
Group of patients who were positive for WFS (WFS+) (18 patients)
Table 1 summarises the general clinical characteristics of these 18 patients (dividing them into 2 groups: homozygous-heterozygous), including their mean age, the existence of consanguinity, the presence of a prior family history, the existence of DM and the age of onset, the presence of OA and the age of onset of visual symptoms, the presence of DI and the age of onset, the existence of auditory loss, and the coexistence of neurological symptoms. The table also describes the significant findings of the cranial MR imaging evaluations performed.
Table 1.
Clinical characteristics of the patients (N = 18).
| Characteristic | Homozygous (5/18) | Heterozygous (13/18) |
|---|---|---|
| Age | Mean: 20.4; range: 8–45 | Mean: 19.1; range: 6–35 |
| Consanguinity | 5/5 (100%) | 13/13 (100%) |
| Family history | 2/5 (40%) | 7/13 (53.8%) |
| DM | 3/5 (60%) | 3/13 (23%) |
| Age of onset of DM | Mean: 16.3; range: 4–30 | Mean: 4.6; range: 2–6 |
| Optic atrophy | 5/5 (100%) | 13/13 (100%) |
| Age of onset of loss of vision | Mean: 13.4; range: 6–35 | Mean: 7.1; range: 5–9 |
| DI | 1/5 (20%) | 3/13 (23%) |
| Age of onset of DI | Mean: 25 | Mean: 17; range: 10–30 |
| Hearing loss | 2/5 (40%) | 2/13 (15.3%) |
| Neurological symptoms | 0/5 (0%) | 5/13 (38.4%) Ataxia (3/5), tremor (1/5), plantar dorsiflexionweakness (1/5), mental delay (1/5) |
| Brain MRI | Thinning of the optic nerve 3/5 (60%) | Thinning of the optic nerve 7/13 (53.8%) Cerebellar atrophy 1/13 (7.69%) |
Note. DM = diabetes mellitus; DI = diabetes insipidus; MRI = magnetic resonance imaging.
The mean age in this group was 19.5 years. In total, 50% had a positive family history, and 100% presented consanguinity between parents. DM was present in 33.3% of this group, with a mean onset age of 10.5 years. DI and auditory loss were present in 22.2% of the patients, and 27.7% presented neurological symptoms, of which ataxia was the most frequent. With regard to the cranial MR evaluations, the most frequently found characteristic in this group was thinning of the optic nerve (55.5%).
Table 2 summarises the most important ophthalmological findings in the WFS+ patients, including the mean VA, the colour vision results, the most frequent findings from visual campimetry and OCT, and the fundus appearance results, as well as data regarding the existence of cataracts, nystagmus, and strabismus.
Table 2.
Ophthalmologic characteristics.
| Visual acuity | Mean: 20/100 | Range: 20/25–3/200 |
|---|---|---|
| Colour vision | Mean: 4/15 | Range: 0/15–8/15 |
| Visual perimetry | Normal 1/18 (8.3%) | Central scotoma 4/12 (33.3%) |
| Cecocentral scotoma 4/12 (33.3%) | ||
| Arcuate scotoma 2/12 (16.6%) | ||
| Enlarged blind spot 1/12 (8.3%) | ||
| OCT (RNFL in microns) | Mean: 68.7 | Range: 44–95 |
| OCT (localisation of the defect) | 4 quadrants 17/26 (65.3%) | |
| Temporal quadrant 7/26 (26.92%) | ||
| Superior/nasal 2/26 (7.69%) | ||
| Fundoscopy | Temporal pallor 29/36 (80.5%) | |
| Global pallor 7/36 (19.4%) | ||
| Optic nerve cupping 4/18 (22.2%) | ||
| Presence of cataract | 0/36 | 0% |
| Nystagmus | 4/36 (11.1%) | Jerk side 2/4 (50%) |
| Pendular 1/4 (25%) | ||
| Elliptical 1/4 (25%) | ||
| Strabismus | 7/36 (19.4%) | Exotropia 6/7 (85.7%) |
| Esophoria 1/7 (14.2%) |
Note. OCT = optic coherence tomography.
The specific contents of Table 2 are as follows. The mean VA was 20/100, with a range between 20/25 and 3/200. Visual campimetry was conducted in 12 patients but was impossible in the remaining 6 patients. For these 12 patients, the most frequent defects were central scotoma (33.3%) and caecocentral scotoma (33.3%). The mean thickness of the RNFL was 68.7 microns, and the most frequent defect was thinning of the four quadrants (65.3%). Fundoscopy showed temporal disc pallor in 80.5% of patients. Diabetic retinopathy was not present in any of the 18 patients. Similarly, cataracts were not found in any of these patients, and 11.1% of the patients presented nystagmus in the examination; the most frequent type was jerk side-beat nystagmus. Finally, 19.4% of these patients had strabismus, and exotropia was the most frequent finding (85.7%).
Table 3 details the WFS mutations or genetic variants in this group of 18 patients. The table demonstrates that all of the genetic mutations were located in exon 8 of the WFS1 gene. The most frequently found type was missense mutations (50%). Of the mutations, 61.1% were heterozygous. In this study, we identified seven new mutations or variations not previously described in the scientific literature. Table 3 also shows how many patients had a prior family history compatible with hereditary optic neuropathy. Specifically, 13 patients had heterozygous mutations, 7 of whom had a positive family history. Although one of the limitations of this study is the lack of a complex genetic segregation analysis, the fact that certain heterozygous patients had a family history reinforces the hypothesis that the mutations were not simple genetic polymorphisms or variations, at least in a subset of the patients.
Table 3.
Genetic mutations.
| Patient | Family history | Gene | Mutation | Phenotype | State | Comments |
|---|---|---|---|---|---|---|
| Patient 1 | − |
WFS1 Exon 8 |
Missense variant c.1495C→T |
OA + | Heterozygous | Most of the bioinformatics programs predict that the variant is a benign polymorphism |
| Patient 2 | − |
WFS1 Exon 8 |
Missense mutation c.1831C→T |
OA + Tremor |
Heterozygous | New mutation in the literature |
| Patient 3 | + (sister) |
WFS1 Exon 8 |
Missense mutation c.2620G→A |
OA + Drop foot |
Heterozygous | New mutation in the literature |
| Patient 4 | − |
WFS1 Exon 8 |
Missense mutation c.2020G→A |
OA + DM + HL + |
Homozygous | Mutation already described in the literature |
| Patient 5 | + (brother) |
WFS1 Exon 8 |
Indel mutation c.1046_1047delinsAG | OA + DM + HL + Ataxia |
Heterozygous | New mutation in the literature Leads to an amino acid exchange in position 349 of the protein sequence |
| Patient 6 | + (2 brothers) |
WFS1 Exon 8 |
Deletion c.2343_2644delCT |
OA + DM + DI + HL + |
Homozygous | Premature stop codon Truncated WFS1 protein Mutation already described in the literature |
| Patient 7 | + (1 uncle) |
WFS1 Exon 8 |
Run-on mutation c.2673A→C | OA + | Homozygous | Mutation leading to the elimination of the normal stop codon and degradation of themRNA (nonsense-mediated decay) New mutation in the literature |
| Patient 8 | − |
WFS1 Exon 8 |
Missense mutation c.2104G→A |
OA + | Homozygous | New mutation in the literature |
| Patient 9 | − |
WFS1 Exon 8 |
Non-stop mutation c.2373A→C |
OA + DM + |
Homozygous | Mutation leading to the elimination of the normal stop codon and degradation of themRNA (nonsense-mediated decay) New mutation in the literature |
| Patient 10 | − |
WFS1 Exon 8 |
Missense variant c.2452C→T |
OA + DI + |
Heterozygous | This variant has been described in the homozygous state as a disease-causing WFS mutationin one family. However, the heterozygous state of our patient would not sufficiently explain the phenotype |
| Patient 11 | − |
WFS1 Exon 8 |
Missense mutation c.1633G→A |
OA + DI + Ataxia |
Heterozygous | New mutation in the literature |
| Patient 12 | − |
WFS1 Exon 8 |
Missense mutation c.577A→C |
OA + | Heterozygous | Most of the bioinformatics programs predict that the variant is a benign polymorphism |
| Patient 13 | − |
WFS1 Exon 8 |
Missense mutation c.2620G→A | OA + | Heterozygous | New mutation in the literature |
| Patient 14 | + (sister) |
WFS1 Exon 8 |
Missense mutation c.2620G→A | OA + DI + HL + Ataxia |
Heterozygous | New mutation in the literature |
| Patient 15 | + (sister, below) |
WFS1 Exon 8 |
Missense mutation c.1123C→T | OA + | Heterozygous | New mutation in the literature |
| Patient 16 | + (sister, above) |
WFS1 Exon 8 |
Missense mutation c.1123C→T | OA + | Heterozygous | New mutation in the literature |
| Patient 17 | + (brother, below) |
WFS1 Exon 8 |
Nonsense mutation c.2034G→A | OA + DM + |
Heterozygous | Premature stop codon Truncated WFS1 protein New mutation in the literature |
| Patient 18 | + (brother, above) |
WFS1 Exon 8 |
Nonsense mutation c.2034G→A | OA + DM + |
Heterozygous | Premature stop codon Truncated WFS1 protein New mutation in the literature |
Note. The table shows the genetic mutations found in WFS+ patients.
Figure 1A–C show the appearance of the optic nerve of three patients with positive genetic test results for WFS mutations (respectively Patients 3, 4, and 12 in Table 1). Patient 3 exhibited optic nerve pallor localised in the temporal sector, which occurred in 80.5% of our patients. Patient 2 exhibited global pallor of the optic nerve, which occurred in 19.4% of our patients. Finally, Patient 12 showed pallor and excavation, which occurred in 22.2% of our patients.
Figure 1.
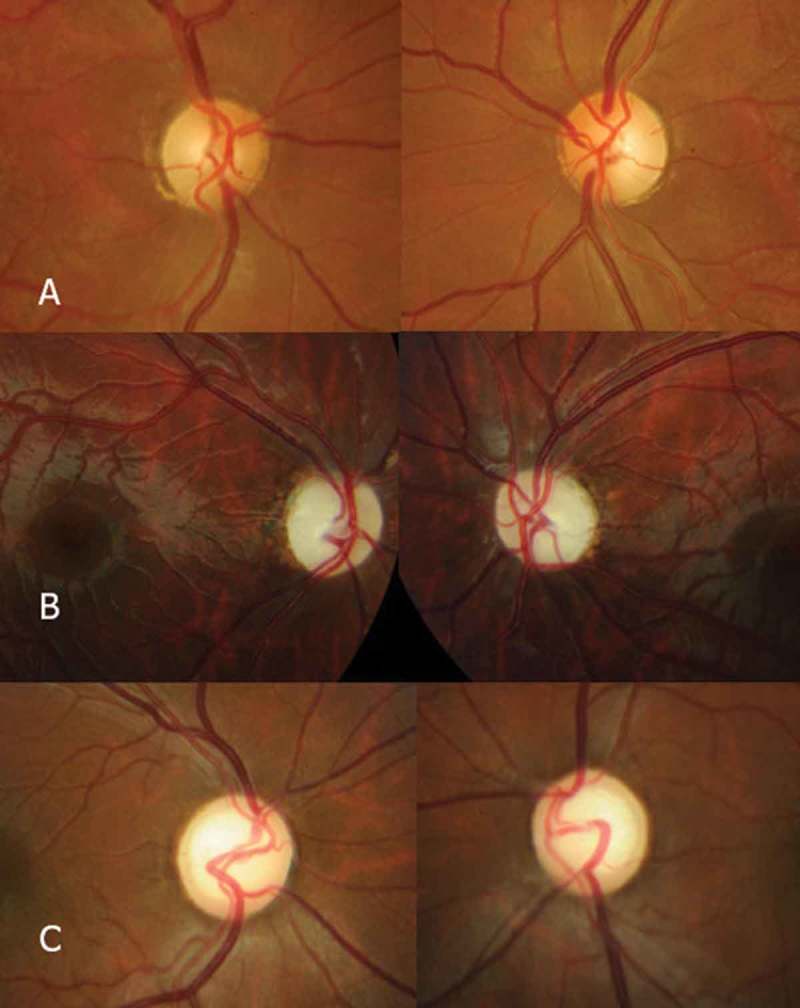
Photocomposition showing the appearance of the optic nerves of Patients 3 (A), 4 (B), and 12 (C) among the WFS+ patients. Patient 3 exhibited optic nerve pallor localised in the temporal sector, which occurred in 80.5% of the patients. Patient 2 showed global pallor of the optic nerve, which occurred in 19.4% of the patients. Finally, Patient 12 exhibited pallor and excavation, which occurred in 22.2% of the patients.
Figure 2 shows two examples of OCT corresponding to two patients positive for WFS mutations, with RNFL loss predominantly in the temporal sector (Patient 3) and globally in the four quadrants (Patient 2).
Figure 2.
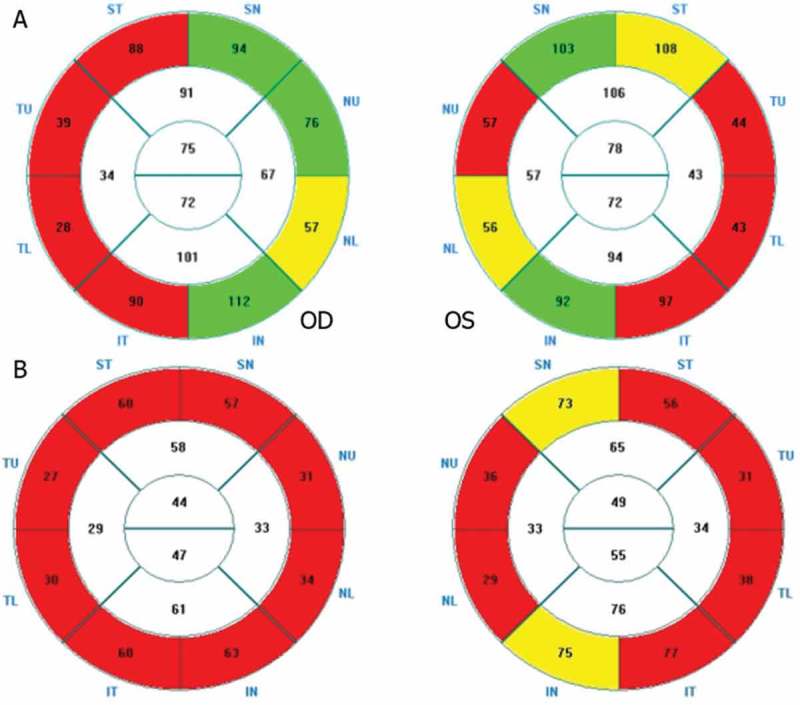
OCT showing RNFL loss in two WFS+ patients. (A) RNFL predominantly in the temporal sector (Patient 3). (B) Global RNFL loss in the four quadrants (Patient 2).
Discussion
In this study, we present a sample of 71 patients with hereditary optic neuropathy and negative genetic test results for OPA1/OPA3/LHON. All of these patients underwent genetic testing to rule out WFS. As a result, 18 (25.3%) were identified as patient carriers with mutations or variations in the WFS gene. We will now focus on the clinical and diagnostic significance of the presence of these mutations.
Currently, there are no clear data in the scientific literature proving the utility of genetic testing for WFS in patients with hereditary optic neuropathy and negative genetic test results for OPA1/OPA3/LHON, independently of whether they meet the clinical criteria for WFS. We believe that this study may shed some light on whether to adopt this strategy.
WFS is a progressive disease that begins with the onset of DM and OA before the age of 10 years. It is classically accepted that these two characteristics define the disease in 100% of patients. However, in the present study, only approximately 20% of the patients presented with DM. DM tends to have a more benign course than classic DM that is not associated with WFS, with a lower prevalence of diabetic retinopathy.2,6 Nevertheless, severe hyperglycaemia is more frequently present.7
Neurosensory deafness is present in 60% of individuals, with a mean age of onset of 12.5 years.2,8 This condition varies from congenital deafness to very mild auditory loss. Neurological abnormalities are present in 62% of individuals, with a mean onset age of 30 years. The main manifestations include truncal or gait ataxia, episodes of apnoea, cognitive decline, neurogenic bladder, and psychiatric manifestations.2,6
Central DI occurs in 72% of patients, with a mean onset age of 15.5 years.2,8
The mean age of death is 30 years due to respiratory failure secondary to brain stem atrophy.2,8
This brief summary of the clinical manifestations of WFS may help to indicate that since WFS is a progressive disease, the diagnostic criteria will depend on the disease stage of the patient. Currently, the most reliable diagnostic criterion continues to be the presence of juvenile DM and OA.8 In addition, the majority of patients have a family history of DM or deafness.3
In our study, we separately examined the clinical and ophthalmological characteristics of one group of patients with negative genetic test results for WFS (WFS−) (53 patients) and another group of patients with positive genetic test results for WFS (WFS+) (18 patients). The main difference between the two groups was the absence of DM in the WFS− group, compared with a rate of 33.3% in the WFS+ patients. DI was not detected in any WFS− patients, whereas it occurred in 22.2% of WFS+ patients. Hearing loss was present in 15.1% of WFS− patients, compared with 27.7% of WFS+ patients. Finally, neurological symptoms (mainly ataxia) were present in 3.8% of WFS− patients, compared with 27.7% of the WFS+ group. These differences can be readily understood, since most of these events (except ataxia and hearing loss, which can be found in certain patients with OPA1+ syndrome) are specific to WFS.
If we specifically compare the clinical data of the WFS+ group in our study with data in other studies in the literature conducted on patients diagnosed with WFS, we find that the variability of clinical expression in our sample is quite different compared with what has been previously published. For example, a study conducted by Barrett et al.6 showed that different clinical manifestations were present in WFS patients at the following rates: DM and OA, 100%; DI, 72%; auditory loss, 60%; and neurological symptoms, 62%. However, as has been previously stated, it should be kept in mind that WFS is a clinically progressive disease. As a result, the degree of clinical expression depends on the age of the selected patients.
As for the mutations or genetic variations present in the WFS+ group in our sample, we must acknowledge that their presence does not prove that these patients actually had true WFS. For example, Patients 1 and 12 most likely had benign variations of the WFS1 gene, given that the majority of bioinformatics programmes predict that these variants are benign polymorphisms. Another example is Patient 10, who had a heterozygous missense c.2452C→T mutation. This variant has been described as a disease-causing WFS mutation in the homozygous state in one family. However, the heterozygous state would not sufficiently explain the phenotype of our patient.
However, these three patients presented optic nerve pallor that is difficult to justify as stemming from other causes (OPA1, OPA3, and LHON gene mutations were all negative). In any case, we must recognise that the only aspect of the clinical spectrum of WFS that these patients presented was OA (Patients 1 and 12) or OA and DI (Patient 10). Therefore, the presence of these benign polymorphisms was very likely an incidental finding in these patients.
However, we cannot exclude the possibility that in these patients, there may have been another mutation in an intronic regulatory region, which would not have been detected by sequencing all exons. This possibility may be further investigated by sequencing all introns, which was not undertaken in the present study.
The pathogenetic value of the mutations found in the remaining WFS+ patients is supported by the following findings: negative genetic test results for OPA1/OPA3/LHON in all these patients, no other reasons for the OA, the presence of a positive family history in 50% of the patients and of consanguinity in 100%, and the presence of compatible clinical manifestations in a proportion of these patients (compared with its absence in WFS− patients).
Other relevant findings are, for example, that Patients 15–16 and 17–18 were two pairs of siblings and therefore presented the same mutation responsible for WFS. Other, unrelated patients in our sample also had the same mutation, such as Patients 3, 13, and 14; these patients presented the same heterozygous missense mutation, namely, c.2620G→A (p.Ala874Thr).
With respect to genetic mutations reported in different studies on patients with a clinical diagnosis of WFS, until 2008, 71 missense, nonsense, and splice site mutations; 37 deletions; and 14 insertions had been reported. The majority of these mutations were truncating, compatible with a pattern of haploinsufficiency. Nevertheless, a small number of non-truncating missense mutations have been reported.2,9 In our sample, the most frequent mutation type was a missense mutation, which was present in 50% of our patients.
Mutations in the WFS1 gene are usually located in exon 8, although they have also been reported in exons 3, 4, 5, and 6.2,9 In accordance with these data, 100% of our patients presented a mutation in exon 8 of the WFS1 gene.
The phenotypic-genotypic correlation is difficult to establish. However, several authors2,10,11 have hypothesised that patients who present inactivating mutations in both alleles of WFS1 (such as the presence of a deletion that leads to a premature stop codon) experience more severe clinical expression than individuals with non-inactivating mutations (such as missense mutations). However, one of our patients with more severe clinical symptoms and greater neurological symptomatology (ataxia, nystagmus) possessed a missense mutation, which contradicts the hypothesis presented above.
In the literature, patients with de novo mutations have also been described,2,12 and in certain patients, mutations in mtDNA have been identified.13 However, this last point has recently been discounted by other authors.6,14
One of the main limitations of the present study is the absence of complex genetic segregation analysis. This is an important point, and because of this absence, we recognise that several of the mutations that we observed might have been false-positive results. The absence of this analysis is due to the fact that in many cases, the relatives of the patients refused to undergo genetic study because their visual health was normal. Another explanation is that many patients were living in remote areas of the country, making it difficult for other family members to travel with them to undergo genetic testing.
However, despite reasonable doubts about the true pathogenetic value of several of the mutations present in WFS+ patients, we believe that this study is interesting because it shows that a sizeable percentage (25.3%) of patients with optic neuropathy and negative genetic test results for OPA1/OPA3/LHON presented WFS mutations or variants in this sample. Certainly, this does not mean that patients can be exclusively diagnosed with WFS using genetic testing. Nevertheless, the results of genetic testing may at least allow us to identify those patients in need of close monitoring to identify the onset of clinical symptoms compatible with WFS.
Conclusions
This study presents a cohort of patients with a clinical phenotype compatible with hereditary optic neuropathy and negative genetic test results for OPA1/OPA3/LHON gene mutations. When genetic testing was later performed for WFS, 25.3% of the patients were identified as patient carriers with mutations or variants in the WFS gene. Although certain results are likely to represent only benign polymorphisms, we believe that it would be advisable to complete genetic testing for WFS mutations (regardless of whether patients meet the clinical criteria for WFS) in patients with negative genetic test results for classical OPA1/OPA3/LHON mutations.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
References
- [1].Yu-Wai-Man P, Griffiths PG, Chinnery PF.. Mitochondrial optic neuropathies—disease mechanisms and therapeutic strategies. Prog Retin Eye Res 2011;30:81–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tranebjærg L, Barrett T, Rendtorff ND. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, eds. WFS1-Related Disorders. GeneReviews®. Seattle, WA: University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK1116/.Accessed September 16, 2016. [Google Scholar]
- [3].Marshall BA, Permutt MA, Paciorkowski AR, Hoekel J, Karzon R, Wasson J, Viehover A, White NH, Shimony JS, Manwaring L, Austin P, Hullar TE, Hershey T; Washington University Wolfram Study Group. Phenotypic characteristics of early Wolfram syndrome. Orphanet J Rare Dis 2013;8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hoekel J, Chisholm SA, Al-Lozi A, Hershey T, Tychsen L; Washington University Wolfram Study Group. Ophthalmologic correlates of disease severity in children and adolescents with Wolfram syndrome. J AAPOS 2014;18:461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yu-Wai-Man P, Shankar SP, Biousse V, Miller NR, Bean LJ, Coffee B, Hegde M, Newman NJ.. Genetic screening for OPA1 and OPA3 mutations in patients with suspected inherited optic neuropathies. Ophthalmology 2011;118:558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Barrett TG, Bundey SE.. Wolfram (DIDMOAD) syndrome. J Med Genet 1997;34:838–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rohayem J, Ehlers C, Wiedemann B, Holl R, Oexle K, Kordonouri O, Salzano G, Meissner T, Burger W, Schober E, Huebner A, Lee-Kirsch MA; Wolfram Syndrome Diabetes Writing Group. Diabetes and neurodegeneration in Wolfram syndrome: a multicenter study of phenotype and genotype. Diabetes Care 2011;34:1503–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Barrett TG. Differential diagnosis of type 1 diabetes: which genetic syndromes need to be considered? Pediatr Diabetes 2007;8(Suppl 6):15–23. [DOI] [PubMed] [Google Scholar]
- [9].Cryns K, Sivakumaran TA, Van den Ouweland JM, Pennings RJ, Cremers CW, Flothmann K, Young TL, Smith RJ, Lesperance MM, Van Camp G.. Mutational spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Hum Mutat 2003;22:275–287. [DOI] [PubMed] [Google Scholar]
- [10].d’Annunzio G, Minuto N, D’Amato E, de Toni T, Lombardo F, Pasquali L, Lorini R.. Wolfram syndrome (diabetes insipidus, diabetes, optic atrophy, and deafness): clinical and genetic study Diabetes Care 2008;31:1743–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cano A, Rouzier C, Monnot S, Chabrol B, Conrath J, Lecomte P, Delobel B, Boileau P, Valero R, Procaccio V, Paquis-Flucklinger V; French Group of Wolfram Syndrome, Vialettes B. Identification of novel mutations in WFS1 and genotype-phenotype correlation in Wolfram syndrome. Am J Med Genet A 2007;143A:1605–1612. [DOI] [PubMed] [Google Scholar]
- [12].Hansen L, Eiberg H, Barrett T, Bek T, Kjaersgaard P, Tranebjaerg L, Rosenberg T.. Mutation analysis of the WFS1 gene in seven Danish Wolfram syndrome families; four new mutations identified. Eur J Hum Genet 2005;13:1275–1284. [DOI] [PubMed] [Google Scholar]
- [13].Rötig A, Cormier V, Chatelain P, Francois R, Saudubray JM, Rustin P, Munnich A.. Deletion of mitochondrial DNA in a case of early-onset diabetes mellitus, optic atrophy, and deafness (Wolfram syndrome, MIM 222300). J Clin Invest 1993;91:1095–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Barrett TG, Scott-Brown M, Seller A, Bednarz A, Poulton K, Poulton J.. The mitochondrial genome in Wolfram syndrome. J Med Genet 2000;37:463–466. [DOI] [PMC free article] [PubMed] [Google Scholar]