

Abstract
In 1927, Guido Fanconi described a hereditary condition presenting panmyelopathy accompanied by short stature and hyperpigmentation, now better known as Fanconi Anemia (FA). With this discovery, the genetic and molecular basis underlying FA has emerged as a field of great interest. FA signaling is critical in the DNA damage response (DDR) to mediate the repair of damaged DNA. This has attracted a diverse range of investigators, especially those interested in aging and cancer. However, recent evidence suggests FA signaling also regulates functions outside the DDR, with implications for many other frontiers of research. Here, we discuss the characteristics of the FA gene fucnitons and expand upon current perspectives regarding the genetics of FA, conferring a myriad of molecular and cellular events.
Keywords: Fanconi anemia (FA), DNA damage response (DDR), Replication, Developmental defects, Cancer, and Aging
Introduction to Fanconi Anemia
Fanconi Anemia (FA) is a rare human genetic disease affecting approximately 1 out of every 136,000 newborns [1]. Clinically, FA contributes to numerous health complications (Box 1), including the early onset of aging, multi-organ congenital defects, bone marrow failure leading to pancytopenia, and a remarkably high predisposition to hematological and non-hematological malignancies [2]. Cells derived from FA patients display distinct patterns of chromosomal abnormalities. Additionally, these cells are characterized by hypersensitivity to DNA crosslinking agents, such as mitomycin C (MMC), diepoxybutane (DEB), and cisplatin [3].
Box 1. Clinical complications of FA and the FA complementation groups.
FA (Fanconi Anemia) is a rare human genetic disease, which occurs following germline mutations to any of the twenty-two FA genes. FA is characterized by an early onset of aging, cellular hypersensitivity to crosslinking agents, an extremely high predisposition to various cancers, and many other clinical problems, including skeletal defects, skin pigmentation, short stature, mental retardation, hearing loss, and other anatomic issues in the central nerve system, eye, heart, kidney, male sex organ, and gastrointestinal tract. Each group of FA patients results from a specific mutated FA gene. On the other hand, the non-mutated given gene (wild type), can complement the defect at the cellular level when introduced into each specific type of FA cells. Thereby, these corresponding FA groups are called FA complementation groups.
Currently, the DEB assay is a well-regarded tool used by physicians to diagnose FA, enabling the examination of tri- and quadri-radial figures in the chromosome spread of FA cells [4, 5]. With the recognition of new FA genes, gene therapy has arisen as a promising avenue by which to treat FA, as it avoids the major negative effects derived from hematopoietic stem cell transplantation (HCT), such as graft-versus-host-disease (GVHD), secondary cancers and endocrinopathies [6]. Despite the success of gene therapy in treating aplastic anemia, FA patients are still highly susceptible to cancers, with current technology unable to successfully target the entire somatic cell population [7]. Although considerable progress has been made in understanding FA and its clinical symptoms, the genetic and molecular mechanisms underpinning many of the developmental issues associated with FA require further elucidation. Further, the cellular and organic changes shown in FA patients suggest that the signal transduction pathway(s) underlying FA may regulate organ development. The defects that arise in FA patients could occur as early as the initiation of embryogenesis, and progress throughout the patient’s lifespan.
Long standing evidence suggests that a common signaling pathway acts to prevent the manifestation of FA. Comprised of at least twenty-two FA gene-encoded proteins (Table 1), the aforementioned signaling pathway has been coined the (canonical) FA pathway [8, 9]. Additionally, this pathway is commonly termed the FA-BRCA pathway, as several FA genes also encode breast cancer (BRCA) susceptibility gene products (Table 1). In trying to understand the nature of FA and its symptoms, many studies have shown that FA genes and pathways are perturbed. Hence, understanding both the canonical and noncanonical FA signaling transduction pathways, or FA pathway-dependent and independent signal transductions, has become an interesting subject of investigation, especially in relation to DNA damage [10–12]. The relevant FA research has also led to major breakthroughs in areas of molecular biology sitting outside DNA damage. Collectively, understanding of FA signaling, both FA pathway-dependent and independent, provides insight to study human aging/cancer and many of the other clinical complications displayed in FA patients.
Table 1.
Twenty-Two Fanconi Anemia Complementation Groups
| FANC- | Alias | FA patients (~ %) | Chr. Location | Protein Product (Kd) | Known Key Features of the Protein | Ub |
|---|---|---|---|---|---|---|
| A | FANCH | 64 | 16q24.3 | 163 | Core complex, Phosphorylated | + |
| B | 2 | Xp22.2 | 95 | Core complex | + | |
| C | 12 | 9q22.3 | 63 | Core complex | + | |
| D1 | BRCA2 | 2 | 13q12–13 | 380 | HR | − |
| D2 | 4 | 3p25.3 | 155, 162 | ID complex, monoubiquitinated, incision, TLS, HR, S phase arrest | + | |
| E | 1 | 6p21–22 | 60 | Core complex | + | |
| F | 2 | 11p15 | 42 | Core complex | + | |
| G | XRCC9 | 8 | 9p13 | 68 | Core complex | + |
| I | 1 | 15q25–26 | 150 | ID complex, phosphorylated, monoubiquitinated | + | |
| J | BACH1, BRIP1 | 2 | 17q22–24 | 130 | RecQ DEAH helicase family, HR, MMS, TLS, DSB repair | − |
| L | POG, PHF9 | 0.4 | 2p16.1 | 43 | Core complex, the ubiquitin ligase (E3) | + |
| M | 0.1 | 14q21.3 | 250 | DNA translocase activity, lesion recognition, core complex | + | |
| N | PALB2 | 0.7 | 16q12.1 | 130 | HR, DSB repair | − |
| O | RAD51C | 0.1 | 17q25.1 | 47 | RAD51 paralog, HR, | − |
| P | SLX4, BTBD12 | 0.5 | 16p13.3 | 200 | Scaffold protein, endonuclease, unhooking crosslink, TLS, Telomere maintenance | − |
| Q | ERCC4, XFP | 0.1 | 16p13.12 | 101 | Endonuclease, NER | − |
| R | RAD51 | 0.1 | 15q15.1 | 45 | HR | − |
| S | BRCA1 | 0.1 | 17q21.31 | 220 | HR | −/+ |
| T | UBE2T | <0.1 | 1q32.1 | 22.5 | Ubiquitin-conjugating enzyme (E2); NER | + |
| U | XRCC2 | <0.1 | 7q36.1 | 34 | Involved in HR, Resolving D-loop structure | − |
| V | REV7, MAD2L2 | <0.1 | 1p31 | 24 | Subunit DNA polymerase ζ involved in TLS | − |
| W | <0.1 | 16q23.1 | ~90 | The ubiquitin-protein ligase | −/+ |
Ub: Required for Monoubiquitination
In this review, we highlight how an understanding of the FA-associated genes and pathways can be utilized as a unique genetic model system to help explain various basic cellular processes, relevant to both FA and non-FA human cells. When discussing the role of FA genes in the FA signaling network, we acknowledge the distinction between the canonical FA-BRCA signaling pathway (Figure 1) and noncanonical (FA pathway-independent) signaling (Table 2). We further examine the contribution of FA signaling to the maintenance of cells with or without DNA damage, and discuss several emerging roles of FA signaling. By considering recent relevant studies, we are able to promote an innovative and insightful perspective regarding the understanding of FA signaling and the clinical/therapeutic implications of these findings toward treating FA. Finally, we discuss relevant prospective FA research, which we believe would provide an updated systemic grasp.
Figure 1. Schematic Representation of the FA Signaling Pathway.
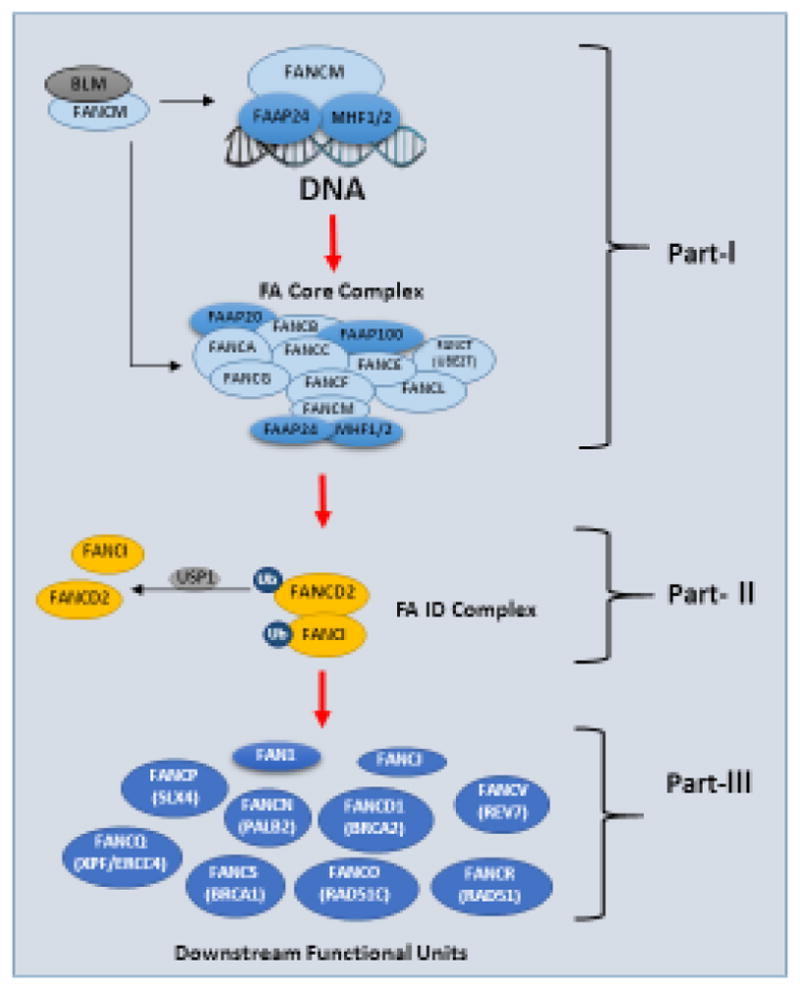
Part I) the FA proteins (FANCA, B, C, E, F, G, L, M, T and possibly I) along with FAAPs (FAAP 20/24/100 and MHF1/2) and other known proteins, assure the activity of ubiquitin E3 ligase for the monoubiquitination of FANCD2 and FANCI. Part II) FANCD2 and its paralog FANCI, as a central axis to connect or orchestrate the entire FA signaling pathway. Part III) downstream of Part-II, the remaining FA proteins. Monoubiquitinated FANCD2 and FANCI can be deubiquitinated by USP1, thereby inactivating the pathway. Red arrows indicate the canonical FA pathway. Ub indicates ubiquitination. Part I & III may include many others proteins, which have yet to be recognized. As such, the indefinite nature of FA signaling presents a particular challenge to translational studies utilizing the basic knowledge of FA to promote clinical understanding such as tumor resistance.
Table 2.
The FA Pathway-Independent RolesPlayed by FA proteins
| FA Proteins | Involved in DNA damage | Involved in other cellular processes |
|---|---|---|
| FANCA | CD40 signaling pathway; cell proliferation; inflammatory response; T cell differentiation; Sequence-specific DNA binding transcription factor activity | |
| FANCB | DNA damage repair (not entirely dependent on the FA core complex) | |
| FANCC | TP53 Regulation of DNA Repair Genes | Generic transcription pathway; Gene expression; Diabetes |
| FANCD1 | DNA damage repair, (not entirely dependent on the monoubiquitinated D2/I) | Cell cycle regulation; meiotic recombination; Presynaptic phase of homologous DNA pairing and strand exchange; Resolution of D-loop structures |
| FANCD2 | The HHR6 signaling pathway; The ATM signaling pathway; DNA damage repair; TP53 regulation of DNA Repair Genes; MiRNA regulation of DNA damage response | Replication: Replication-origin firing, Stalled replication forks; Mitochondria function; gene expression; |
| FANCE/F/G | DNA damage repair (not entirely depending on the FA core complex) | |
| FANCI | The ATR signaling pathway; TP53 regulation of DNA Repair Genes; DNA damage repair | Gene expression |
| FANCJ | DNA damage repair (not entirely dependent on the monoubiquitinated D2/I); G2/M DNA damage checkpoint | Cell cycle regulation; Cytosolic iron-sulfur cluster assembly; P53 activity; Presynaptic phase of homologous DNA pairing and strand exchange; Resolution of D-loop structures |
| FANCL | DNA damage repair (not entirely dependent on the FA core complex) | Ubiquitin mediated proteolysis |
| FANCM | ATR regulator, or a major sensor of the DDR | Stalled replication forks |
| FANCN | DNA damage repair (HR) (not entirely dependent on monoubiquitinated FANCD2) | Resolution of D-loop structures; Homologous DNA Pairing and Strand Exchange |
| FANCO | DNA damage repair (not entirely dependent on monoubiquitinated FANCD2) | Meiosis; Resolution of D-loop structures; Megakaryocyte development and platelet production; Cell cycle |
| FANCP | DNA damage repair (not entirely dependent on monoubiquitinated FANCD2/I) | Resolution of D-loop structures |
| FANCQ | DNA damage repair (DSB, NER) (not entirely dependent on monoubiquitinated FANCD2) | Transcription |
| FANCR | DNA damage repair (HR) (not entirely dependent on monoubiquitinated FANCD2/I); ATM signaling | Cell cycle; Meiosis; Rac1/Pak1/p38/MMP-2 pathway |
| FANCS | DNA damage repair (HR) (not entirely dependent on monoubiquitinated FANCD2/I); ATM signaling | Transcription (ATF-2, E2F, FOXA1 transcription factor networks); Androgen receptor signaling pathway; Aurora A signaling; Cell Cycle Checkpoints; Deubiquitinating; |
| FANCT | DNA damage repair (not entirely dependent on monoubiquitinated FANCD2/I); | Post-translational protein modification |
| FANCU | DNA damage repair (not entirely dependent on monoubiquitinated FANCD2/I) | Resolution of D-loop structures; Presynaptic phase of homologous DNA pairing and strand exchange |
| FANCV | TLS performed by POL1, POLK, REV1 or Zeta; post replication repair (not entirely dependent on monoubiquitinated FANCD2/I) | Cell cycle regulation; Shigellosis; Oocyte meiosis; Endoderm Differentiation |
| FANCW | Ubiquitination of RPA (not entirely for the activation of the FA pathway) | Ubiquitination; Mediation of p53 ubiquitination for its stability |
Resources: The Text and NCBI
The FA-BRCA Signaling Pathway
To date, twenty-two FA complementation groups (Table 1) have been identified [1, 13–15]. All of these groups have been identified as biallelic germline mutations that cause the FA phenotype, with the exception of FANCB and FANCR (Rad51) [16, 17]. Although the presentation of FA varies, dependent upon which FA gene(s) is mutated, the notion of a common signaling pathway involving the FA proteins is supported by the similarities in the clinical symptoms displayed throughout the FA-subtypes [1, 18]. Currently, in addition to the twenty-two FA proteins, the FA pathway consists of a number of FA associated proteins such as FAAP20/24/100, MHF1/2 (FAAP16/10) [19, 20], and several interacting partners including FAN1 [21–24], DNA polymerase eta [25] and REV1 [26].
The canonical FA signaling pathway is often dissected into three parts. Part I, comprises the FA core complex along with FANCT (ubiquitin conjugating enzme-E2) and upstream regulators. The core complex mainly acts as a ubiquitin ligase-E3, utilizing FANCL as the catalytic unit to monoubiquitinate FANCD2 and its paralog FANCI at K561 and K523, respectively [27]. Thus, Part I consists of nine known FA proteins, FAAPs and others [28, 29] (Figure 1). Part II, the FA ID complex is comprised of FANCD2 and FANCI (Figure 1). Part III, the functional units downstream of Part II, contains DNA repair proteins that act in coordination following the activation/monoubiquitination of FANCD2/FANCI (Figure 1). However, with the identification of new FA complementation groups, FA associated proteins and emerging functions, the categorization of the FA signaling pathway may need to be frequently revisited and refined.
Currently, it is not clear how many FA subtypes exist. Bioinformatics and technological advancements, especially in the various omics approaches (Box 2), such as proteomics and genomics, have led to the identification of seven new complementation groups (Q-W) in the last four years [14, 15, 17, 30–36]. Although the precise distribution of the FA-subtypes fluctuates, it is clear that over 80% of reported FA cases are associated with mutations, resulting in an inability to assemble the FA core complex and to monoubiquitinate FANCD2 (Table 1). This supports previous commentary that suggests FANCD2 is the focal point of the FA signaling pathway [37, 38]. FANCD2 monoubiquitination often represents the activation of the FA or FA-BRCA pathway [38].
Box 2.
‘‘Omics’’ is a general term used to describe the science of integrating the biological information of genes and proteins, and finding their interrelationships, aiming to understand & manipulate the regulatory mechanisms.
Genomics is the study of the overall structure, function and expression of the entire genome at DNA level. At the mRNA or protein level, the given study is termed Transcriptomics or Proteomics respectively. Additionally, Metabolomics studies all small-molecular-weight organic and inorganic compounds produced by the cell.
FA Signaling, Multifunctional Players & DNA Damage
Constant exposure to endogenous and exogenous genotoxic agents can compromise genome stability, when the DNA damage response (DDR) is compromised [39]. Checkpoint mechanisms serve a major regulatory function in governing the DDR and ensure the coordination of DNA repair proteins, which detect and repair DNA damage to protect cells from genome instability [39]. In these checkpoint systems, the activation of the ataxia telangiectasia-mutated (ATM) and ATM and Rad3-related (ATR) DNA repair pathways are well-recognized master responses to genotoxic stresses [40]. In the event of DNA damage, repair proteins perform various roles by sensing damaged DNA and repairing it. Alternatively, repair proteins initiate processes to eliminate damaged cells [41, 42]. The canonical FA pathway has been identified as an essential part of the DDR, and can be activated upon DNA damage [43–47]. The pathway is comprised of helicases (FANCM/J), nucleases or their collaborators (FANCQ/ or P), other enzymatic proteins (FANCL/T/V for E3, E2 and polymerase activities respectively), complex/scaffold proteins (FANCA/B/C/D2/E/F/G/I/P), as well as proteins involved in specific DNA damage repair processes (FANCD1/N/O/P/Q/R/S/U/V/W) (Table 1). Below, we describe the relevance of FA helicases, ligases, nucleases and scaffold proteins, as these activities are crucial in the DDR but under-recognized compared to those that were well known prior to being characterized as FA proteins.
Helicases
First discovered as an FA gene in 2005, FANCM gained considerable attention within the FA field due to its ability to directly interact with DNA [48]. Notably, FANCM is recognized as the only FA protein to have specific DNA-binding activity, as it possesses DNA translocase/helicase functions essential in sensing DNA damage [45, 49]. Together, FANCM, MHF1/2 and other associated proteins form a functional unit [50], which is stabilized by FAAP24 [51]. Furthermore, several independent studies have demonstrated that FANCM function is important for many processes such as, regulating crossovers during DNA double-strand break repair and the replicative bypass of DNA lesions [52–54], acting as an S-phase checkpoint protein, and promoting the recruitment of the FA core complex to sites of DNA damage [55]. Recent studies indicate association between FANCM mutations and high grade serous ovarian cancer susceptibility [56] as well as early-onset of familial breast cancer [57]. In contrast to its orthologs, which also possess the helicase domain structure, mammalian FANCM contains an additional domain(s) for interacting with MHF, RMI & others, with possible nuclease activity [55]. Currently, the importance of these domains is not clear. Nonetheless, future studies emphasizing individual domains will potentially provide in-depth understanding into the roles of FANCM as a multifunctional protein, advancing our understanding of FA signaling.
FANCJ (BRIP1) is a member of the RecQ DEAH helicase family and interacts with the BRCT repeats of BRCA1 [58]. Unlike other helicases, FANCJ functions specifically to unwind G-quadruplex (G4) structures that interfere with DNA replication, repair and RNA transcription [59, 60]. Therefore, the FANCJ helicase contributes to maintaining genomic integrity through these molecular events at G4-forming DNA regions, as well as supporting microsatellite maintenance [61]. Currently, FA helicase activity does not appear to be confined to a specific phase of the DDR. However, further research investigating the multifunctional nature of the helicase proteins within the FA signaling may provide additional insight into the intricacies of this signaling network.
Ligases
As of today, there are three FA proteins that are involved in the ligase activity. FANCL, as a catalytic subunit in the FA core complex- E3, is responsible for monoubiquitinating FANCD2 and its paralog FANCI [27]. Evidently, FANCL is a critical player as monoubiquitinated FANCD2 is the focal point and orchestrates the whole FA pathway. The newly identified FANCW, known as RFWD3 [14, 15], also performs the activity of the ubiquitin ligase-E3 in polyubiquitinating RPA, which is essential for ATR activation as well as the DSB repair [14, 15]. RFWD3 was also known to be an E3 ubiquitin ligase for p53 stability in the late response to DNA damage (Table 2). However, the relation of FANCW with effects of the p53 tumor suppressor activity in combination with the maintenance of genome stability needs in-depth study. FANCQ, previously known as XRCC4-XPF [62–65], however, promotes the ligase activity for DNA, instead of protein. XRCC4 is a key protein that enables the interaction of DNA Ligase IV with damaged DNA and therefore promotes ligation (of the ends) [66, 67]. To achieve this, some investigators believed that XRCC4 works in concert with Ku70/80 in non-homologous end-joining to repair double strand DNA breaks [68–70]. However, conflicting studies have produced opposing observations [71] in FANCS deficient FA cells, in which the related signaling was not required. This may result from the fact that FA gene products do not solely perform the same task, and further research is required. As a matter of fact, nearly all FA proteins possess both the FA pathway-dependent and independent roles (Figure 1 and Table 2).
Nucleases
FANCP (BTBD12/SLX4) [72], functions in regulating homologous recombination (HR). Although FANCP is not a nuclease itself, it acts as a multi-domain scaffold to facilitate the formation of various protein complexes, which are involved in executing the nuclease activity in humans and other mammals [73]. To this end, FANCP orchestrates part of the resolvase/nuclease activity [74], which cleaves the links between two homologous chromosomes that form during HR. On the basis of these known features, it appears that FANCP is an essential contributor to the DDR, which promotes transducing DNA signaling as well as repairing DNA damage.
Polymerases
FANCV is a recent FA protein to have been identified [35]. However, like many of the other FA proteins, it has been previously characterized as a DNA repair protein, REV7. Specifically, REV7 is one of two subunits of an error-prone DNA polymerase, namely DNA polymerase zeta (Pol zeta). Also, REV7 interacts with the REV1 polymerase, another error-prone DNA polymerase. Both pol zeta and REV1 are involved in translesion DNA synthesis, promoting post replication repair (PPR) [76]. The error-prone nature of FANCV suggests this type of enzymatic activities needs to be accurately regulated.
Scaffold Proteins
Although clear enzymatic activity has been shown for several FA proteins, many serve as important scaffold proteins, including FANCQ, FANCP (mentioned above) and FANCD2 (promoting/aiding distinct enzymatic activities of FAN1 [21–24], pol eta [25], Rev1 [26], and others). Although much effort has been dedicated to investigating the structural and functional features of FANCD2, recently an additional variant of the FANCD2 protein (namely FANCD2-V2) was found [37], alluding to the potential existence of further undiscovered information. Therefore, the FANCD2-V2 variant challenges the longstanding and thoroughly explored variant of the FANCD2 protein (namely, FANCD2-V1), which has been acknowledged to be the only variant to represent FANCD2 function within FA signaling. Due to structural and sequence similarity to FANCD2-V1, FANCD2-V2 was previously overlooked, differing by only 40 of 1471 AAs. Importantly, the ratio of FANCD2-V2/FANCD2-V1 expression is relatively higher in non-malignant cells/tissues and low stage tumors compared to their malignant counterparts. FANCD2-V2 thus appears to be more inclined to prohibiting the initiation of neoplastic transformation via its more potent tumor suppressive roles. Therefore, further investigation is required to understand the regulation of this differential expression and alternative functions as a scaffold protein and/or a protein with distinct enzymatic activities. Additionally, this recent discovery provides further molecular mechanisms, which promote the understanding of the genetics underlying FA. As many complex proteins are known to act as scaffolds, it is still unclear whether this property serves as either the sole and/or primary function, or whether scaffold proteins can function independent of protein complex formations.
In summary, the FA proteins promote an effective DDR as part of the FA signaling pathway. However, these proteins also have multifunctional roles, eluding to their FA pathway-independent (the noncanonical FA pathway) functions (Table 2). In the context of DNA damage repair, classifying the FA proteins as either sensors, transducers or effectors/executioners does not encapsulate the entirety of their function, and appears to be an obsolete exercise. As previously discussed, the FA gene products closely participate at multiple levels within the framework of the DDR. Helicase proteins largely function by unwinding DNA to support the DDR. This can occur either upstream of the DDR to initiate DNA damage sensing functions, or downstream as a repair effector/executioner to promote appropriate and accessible templates during repair. In addition, FA proteins carrying nuclease functions may act at the early phases of DNA repair, unhooking DNA crosslinks. However, these FA proteins may also perform scaffold functions essential in the formation of protein complexes for DNA damage repair. Nevertheless, within the FA field, it is still unclear whether scaffold proteins act solely as scaffolds, or if they also possess vehicle functions that recruit others to specific sites of DNA damage. All of these processes require further research.
FA signaling, DNA Repair-Independent & Replication
While a majority of the current FA literature has focused on the importance of FA signaling in the DDR, the roles of FA signaling in DNA replication has yet to be clearly defined. Studies in recent years have examined the close relationship of replicative helicases MCMs with FANCD2 [75–78]. This interaction has been demonstrated to be a key factor in the initiation of DNA replication to fire replication origins at a proper rate [75]. Furthermore, this demonstrated a role for the basal-level of FANCD2 monoubiquitination during the S phase of the cell cycle, under non-stressed conditions. In addition, this role of FANCD2 appears to be independent of its functions relating to the DDR.
Similar to DNA damage, the response to replicative stress is also known to promote FANCD2 monoubiquitination. Following this, FANCS/BRCA1 functions to protect stalled replication forks from degradation. This, however, can also be achieved by FANCO/Rad51 [79]. With the discovery of this redundant function, the potential role of FA signaling to regulate DNA replication independent of the DDR is now greater than previously thought. In addition, the relationship between BLM helicase and the FA proteins in replication also acts mostly independently of the known repair mechanisms. A molecular link was reported between FA and Bloom syndromes for more than a decade, with both diseases exhibiting overlapping phenotypes [80]. Indeed, studies have shown that the BLM and FA proteins interact through the formation of a super-molecular complex, which includes FANCS (BRCA1), FANCD2 and FANCA [81]. Studies investigating the functional interaction between FA signaling and BLM have recently demonstrated that BLM can promote the activation of FANCD2 and assist in FANCM recruitment upon stalled replication forks in a timely manner [82, 83].
During DNA replication, DNA unwinds at the origin and new strands are synthesized, leading to replication forks growing bi-directionally from the origin. These two distinct molecular events require effective regulation to faithfully duplicate genetic material, to which FA signaling plays a critical role. In addition, FANCD2 monoubiquitination and its interaction with numerous proteins are likely to be involved in other aspects of replication, such as elongation or termination. However, this hypothesis requires further investigation, which may in turn deliver novel insights into human aging, cancer, and the clinical symptoms associated with FA.
Activated/monoubiquitinated FANCD2 during replication appears not only to be independent from the DDR and repair functions that are commonly associated with FA signaling, but to also exist in a pathway-independent manner. In addition to FANCD2, it is widely acknowledged that FANCC, FANCA [84], FANCJ [61], FAAP24 [85] and FANCI [86] have all been reported to also possess pathway-independent roles (Table 2). Now with the discovery of over twenty further FA genes, the mechanisms underpinning FA display greater relevance than previously thought. New-found roles of the FA proteins highlight their continual significance within the DDR, evidenced by the recently identified FA protein, FANCV/REV7, and its ability to promote translesion DNA synthesis [36]. However, the unique roles of the FA signaling network, independent of DNA damage allude to additional unknown regulatory roles expanding well outside the DDR.
Emerging Roles of FA Signaling
Inhibition of FANCD2 monoubiquitination has been shown to deregulate cell proliferation/growth [87]. Following the impairment of FA signaling, the mechanistic consequences extend past deregulation in the DDR and aberrant replication. Indeed, the emerging roles of FA signaling may even encompass the M phase of the cell cycle [88–92], transcription [93, 94], mitochondrial function/metabolism [95, 96], telomere regulation [97] and more.
During the M phase of the cell cycle, FA signaling is highly regulated via the degradation of FANCM. This occurrence not only reduces FANCD2 monoubiquitination but also implements further regulatory roles within the cell cycle [88]. Additionally, in collaboration with BLM, FA signaling can promote proper chromosomal segregation at fragile sites [89, 90]. FANCD2 has been found to be essential for the protection of chromosomal integrity [91]. To achieve this, FANCD2 acts in concert with FANCI and BLM to survive mitosis with acentric chromosomes in a DDR-independent manner [91]. Furthermore, FANCP has also been reported to interact with Mus81 and others to promote appropriate chromosome segregation and to avoid mitotic catastrophe [92]. Moreover, crosstalk between FA signaling and other players expand the role of FA signaling in safeguarding chromosome stability during mitosis. In addition, the regulation of FA signaling by p21 (a cyclin-dependent kinase inhibitor) [98] and p53 [99] further supports the role of FA signaling in the regulation of cell proliferation. This possibly extends to all phases of the cell cycle, beyond the phases discussed. However, further research is required to validate this aspect of FA signaling as it currently remains unclear.
Recently, the FA signaling network has been implicated in a number of regulatory processes. The canonical FA pathway is most commonly characterized in DNA damage repair. By contrast, noncanonical FA (pathway-independent) signaling (Table 2) promotes both DNA damage repair as well as repair-independent roles. Although this mostly occurs at the DNA level, accumulated studies have demonstrated that its functional importance goes beyond the DNA level to safeguard genetic material. To correspond with this, FA signaling has recently been reported to regulate R-loops (Co-transcriptional RNA-DNA hybrids), which occur during transcription [93, 94]. Similarly, the FA proteins have also been found to be directly involved in mitochondria function [95, 96], where countless metabolic processes occur [100]. This suggests the potential involvement of FA signaling in the maintenance of cellular metabolomes (box 2). Interestingly, altered metabolite concentrations or deregulated metabolic flux may present another possible stimulus to FA signaling. Previously, FANCD2 has been demonstrated to protect cells from aldehydes; naturally occurring metabolic by-products. However, this appears to act more favorably in response to the DNA damaging and carcinogenic properties [101–103]. As such, acetaldehyde can activate the FA pathway, although it acts essentially as an inter-strand DNA-crosslinking agent [104].
A great deal of research has been dedicated to uncovering how FA signaling acts in relation to FANCD2 monoubiquitination. A recent study has demonstrated that in the event that the canonical FA pathway is inactivated, new FA signaling functions emerge [105]. In particular, when FANCD2 is not monoubiquitinated, it may exhibit Gain-of-Function (GOF) properties resulting in context specific gene expression. Currently, it is unknown whether this function is restricted to transcriptional regulation (Box 2). Nonetheless, this understanding adds an additional layer of complexity to the potential of FA signaling by continually displaying its ability to regulate numerous cellular processes and protect human cells from going awry from a variety of human diseases (Figure 2).
Figure 2. Deregulated FA signaling Gain of Function (GOF).

FA signaling is crucial to many cellular events in order to protect humans from diseases such aging, cancer, and severe bone marrow failure. Impaired FA signaling, however, not only loses the roles of the intact signaling pathway but also exhibits the GOF phenomenon, and promotes the clinical complications associated with FA.
Concluding Remarks and Perspectives
In this review, we have described recent progresses in the recognition of the molecular basis underlying the genetics of FA. Primarily, FA signaling is commonly characterized for its role in the DDR to maintain genome stability to prevent aging and cancer [106, 107]. Accordingly, genomic instability derived from compromised FA signaling is a likely commonality amongst many of the complications observed in FA patients. Nonetheless, in this review we have discussed an expanded role of FA signaling, accounting for the regulation of numerous cellular events, including DNA damage repair, DNA replication, and many other essential cellular processes. When aberrant, these confer aplastic anemia, cancer, aging and developmental defects. In conclusion, we believe that the regulation of normal biological functions carried out by FA signaling that acts in synergy with its DNA damage repair roles account for the clinical implications associated with FA. Despite the prevalence of multiple birth defects, defining a major characteristic of FA (Box 1), current research investigating the developmental biology of FA is extremely understudied. We believe that corresponding prospective research will uncover many unfamiliar roles hidden within the genetics of FA and provide insight pertaining the clinical complications of FA.
As described above, FA gene-encoded proteins may participate in cellular processes at multiple levels to critically sustain or maintain the flow of biological information (Figure 3). Currently, with metabolomics as the latest addition to the “omics” family, consisting of genomics, epigenomics, transcriptomics, and proteomics, the central dogma of molecular biology describes the flow of biological information ranging from DNA to the metabolic outputs (Figure 3). Therefore, metabolite profiling appears to be a potent approach to increasing our understanding of intracellular events governed by the genetics of FA. To demonstrate this, a recent metabolomics study [87] provides further support in understanding the role of FA signaling as a tumor suppressor (in non-FA patients) [108]. Thus, the integration of metabolite profiling with other omics-related approaches may enhance our understanding of the genetics of FA. In turn, this could validate an approach in determining commonality amongst the FA symptoms and further unexpected applications (See Outstanding Questions).
Figure 3. System view on the flow of biological information that can be influenced by FA signaling at multiple levels.

The flow of biological information in a system goes from DNA (genome) > RNA (transcriptome) > protein (proteome) > metabolite (metabolome). FA genes involved in the FA signaling network may be mutated and/or epigenetically modified. On the other hand, their normal transcripts can be abnormally processed or improperly translated into proteins, leading to a disordered metabolome, containing a variety of metabolites derived from multiple irregular cellular processes.
Outstanding Questions.
New FA cases exist, which cannot be assigned to any of the 21 complementation groups known thus far. How many FA complementation groups will be discovered?
Upon accumulated studies, there are more and more players identified as acting in the FA signaling network. What is the size of the entire FA signaling network?
FANCD2 has been observed to be expressed in two variants, both appearing to be important in the development of cancer and/or the early onset of aging. Will we discover this phenomenon in other FA proteins? What roles do the FA protein variants perform in human tumorigenesis or the premature aging?
Monoubiquitinated FANCD2 is a key representative for the activation of the FA signaling pathway. In the normal cycling cells, it only appears during DNA replication (the S phase of cell cycle), which is much less than the non-monoubiquitinated form. This non-monoubiquitinated form of FANCD2 has been shown to play important roles in the maintenance of cellular processes. Does FANCD2 function throughout all phases of cell cycle?
In terms of DNA-damaging therapeutic drugs, their efficacy might not look too optimistic, because there is always part of FA signaling to present in a manner of redundancy, overlap and/or the uncooperative, demanding constantly redefining the gained knowledge. In particular, are we able to thoroughly understand resistance derived from platinum, PARPi and/or others, upon which cells initiate DDR?
Highlights.
The Fanconi anemia genes are important for the maintenance of genome stability, encoding proteins crucial in the DNA damage response.
The FA core complex performs E3 ubiquitin ligase activity.
Monoubiquitination of FANCD2 and FANCI coordinates the activity of the downstream FA targets to repair damaged DNA.
FA signaling encompasses the complete cellular signaling network, initiated from any FA protein to function in both the canonical and non-canonical FA pathways, or to perform both FA pathway-dependent and -independent functions.
Inactivated FANCD2, i.e. FANCD2 that cannot be monoubiquitinated, can take on new roles (Gain of Function), resulting in the expression of genes not involved in the canonical FA pathway.
Acknowledgments
The referenced work from our own laboratory was supported in part by NIH grants (R01CA136532 & R01CA188251 to PF) and institutional supports from Mayo Clinic Foundation and University of Hawaii Cancer Center. We apologize for the absence of citations supporting the similar findings owing to the limitation of the reference number.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mamrak NE, Shimamura A, Howlett NG. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. 2016 doi: 10.1016/j.blre.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bogliolo M, Surralles J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev. 2015;33:32–40. doi: 10.1016/j.gde.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Taniguchi T, D'Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 2006;107(11):4223–33. doi: 10.1182/blood-2005-10-4240. [DOI] [PubMed] [Google Scholar]
- 4.Auerbach AD. Diagnosis of Fanconi anemia by diepoxybutane analysis. Curr Protoc Hum Genet. 2015;85:871–17. doi: 10.1002/0471142905.hg0807s85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oostra AB, et al. Diagnosis of fanconi anemia: chromosomal breakage analysis. Anemia. 2012;2012:238731. doi: 10.1155/2012/238731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tolar J, et al. Gene therapy for Fanconi anemia: one step closer to the clinic. Hum Gene Ther. 2012;23(2):141–4. doi: 10.1089/hum.2011.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alter BP. Cancer in Fanconi anemia, 1927–2001. Cancer. 2003;97(2):425–40. doi: 10.1002/cncr.11046. [DOI] [PubMed] [Google Scholar]
- 8.Moldovan GL, D'Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–49. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Higuera I, et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7(2):249–62. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 10.Kim H, D'Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26(13):1393–408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Longerich S, et al. Stress and DNA repair biology of the Fanconi anemia pathway. Blood. 2014;124(18):2812–9. doi: 10.1182/blood-2014-04-526293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys. 2014;43:257–78. doi: 10.1146/annurev-biophys-051013-022737. [DOI] [PubMed] [Google Scholar]
- 13.Ceccaldi R, Sarangi P, D'Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17(6):337–49. doi: 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- 14.Inano S, et al. RFWD3-Mediated Ubiquitination Promotes Timely Removal of Both RPA and RAD51 from DNA Damage Sites to Facilitate Homologous Recombination. Mol Cell. 2017;66(5):622–634.e8. doi: 10.1016/j.molcel.2017.04.022. [DOI] [PubMed] [Google Scholar]
- 15.Knies K, et al. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J Clin Invest. 2017;127(8):3013–3027. doi: 10.1172/JCI92069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meetei AR, et al. X-linked inheritance of Fanconi anemia complementation group B. Nat Genet. 2004;36(11):1219–24. doi: 10.1038/ng1458. [DOI] [PubMed] [Google Scholar]
- 17.Ameziane N, et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun. 2015;6:8829. doi: 10.1038/ncomms9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D'Andrea AD. Susceptibility pathways in Fanconi's anemia and breast cancer. N Engl J Med. 2010;362(20):1909–19. doi: 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang AT, Smogorzewska A. SnapShot: Fanconi anemia and associated proteins. Cell. 2015;160(1–2):354–354e1. doi: 10.1016/j.cell.2014.12.031. [DOI] [PubMed] [Google Scholar]
- 20.Singh TR, et al. MHF1-MHF2, a histone-fold-containing protein complex, participates in the Fanconi anemia pathway via FANCM. Mol Cell. 2010;37(6):879–86. doi: 10.1016/j.molcel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smogorzewska A, et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell. 2010;39(1):36–47. doi: 10.1016/j.molcel.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lachaud C, et al. Karyomegalic interstitial nephritis and DNA damage-induced polyploidy in Fan1 nuclease-defective knock-in mice. Genes Dev. 2016;30(6):639–44. doi: 10.1101/gad.276287.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pizzolato J, et al. FANCD2-associated nuclease 1, but not exonuclease 1 or flap endonuclease 1, is able to unhook DNA interstrand cross-links in vitro. J Biol Chem. 2015;290(37):22602–11. doi: 10.1074/jbc.M115.663666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu T, et al. FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science. 2010;329(5992):693–6. doi: 10.1126/science.1192656. [DOI] [PubMed] [Google Scholar]
- 25.Fu D, et al. Recruitment of DNA polymerase eta by FANCD2 in the early response to DNA damage. Cell Cycle. 2013;12(5):803–9. doi: 10.4161/cc.23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim H, et al. Regulation of Rev1 by the Fanconi anemia core complex. Nat Struct Mol Biol. 2012;19(2):164–70. doi: 10.1038/nsmb.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swuec P, et al. The FA Core Complex Contains a Homo-dimeric Catalytic Module for the Symmetric Mono-ubiquitination of FANCI-FANCD2. Cell Rep. 2017;18(3):611–623. doi: 10.1016/j.celrep.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meetei AR, et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet. 2003;35(2):165–70. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- 29.Rajendra E, et al. The genetic and biochemical basis of FANCD2 monoubiquitination. Mol Cell. 2014;54(5):858–69. doi: 10.1016/j.molcel.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bogliolo M, et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet. 2013;92(5):800–6. doi: 10.1016/j.ajhg.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang AT, et al. A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol Cell. 2015;59(3):478–90. doi: 10.1016/j.molcel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sawyer SL, et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015;5(2):135–42. doi: 10.1158/2159-8290.CD-14-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rickman KA, et al. Deficiency of UBE2T, the E2 Ubiquitin Ligase Necessary for FANCD2 and FANCI Ubiquitination, Causes FA-T Subtype of Fanconi Anemia. Cell Rep. 2015;12(1):35–41. doi: 10.1016/j.celrep.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hira A, et al. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am J Hum Genet. 2015;96(6):1001–7. doi: 10.1016/j.ajhg.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park JY, et al. Complementation of hypersensitivity to DNA interstrand crosslinking agents demonstrates that XRCC2 is a Fanconi anaemia gene. Journal of medical genetics. 2016 doi: 10.1136/jmedgenet-2016-103847. jmedgenet-2016–103847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bluteau D, et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest. 2016;126(9):3580–4. doi: 10.1172/JCI88010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han B, et al. Overlooked FANCD2 variant encodes a promising, portent tumor suppressor, and alternative polyadenylation contributes to its expression. Oncotarget. 2017;8(14):22490–22500. doi: 10.18632/oncotarget.14989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen Y, et al. Advances in the understanding of Fanconi Anemia Complementation Group D2 Protein (FANCD2) in human cancer. Cancer Cell Microenviron. 2015;2(4) doi: 10.14800/ccm.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 2013;5(9) doi: 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300(5625):1542–8. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 42.Sanchez Y, et al. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science. 1999;286(5442):1166–71. doi: 10.1126/science.286.5442.1166. [DOI] [PubMed] [Google Scholar]
- 43.Chen X, et al. A novel role for non-ubiquitinated FANCD2 in response to hydroxyurea-induced DNA damage. Oncogene. 2016;35(1):22–34. doi: 10.1038/onc.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song IY, et al. Rad18-mediated translesion synthesis of bulky DNA adducts is coupled to activation of the Fanconi anemia DNA repair pathway. J Biol Chem. 2010;285(41):31525–36. doi: 10.1074/jbc.M110.138206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8(10):735–48. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- 46.Fei P, Yin J, Wang W. New advances in the DNA damage response network of Fanconi anemia and BRCA proteins. FAAP95 replaces BRCA2 as the true FANCB protein. Cell Cycle. 2005;4(1):80–6. doi: 10.4161/cc.4.1.1358. [DOI] [PubMed] [Google Scholar]
- 47.Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493(7432):356–63. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meetei AR, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet. 2005;37(9):958–63. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang M, et al. The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Mol Cell. 2010;39(2):259–68. doi: 10.1016/j.molcel.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Q, et al. The MHF complex senses branched DNA by binding a pair of crossover DNA duplexes. Nat Commun. 2014;5:2987. doi: 10.1038/ncomms3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu S, et al. Crystal structures of isoorotate decarboxylases reveal a novel catalytic mechanism of 5-carboxyl-uracil decarboxylation and shed light on the search for DNA decarboxylase. Cell Res. 2013;23(11):1296–309. doi: 10.1038/cr.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Girard C, et al. AAA-ATPase FIDGETIN-LIKE 1 and Helicase FANCM Antagonize Meiotic Crossovers by Distinct Mechanisms. PLoS Genet. 2015;11(7):e1005369. doi: 10.1371/journal.pgen.1005369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Girard C, et al. FANCM-associated proteins MHF1 and MHF2, but not the other Fanconi anemia factors, limit meiotic crossovers. Nucleic Acids Res. 2014;42(14):9087–95. doi: 10.1093/nar/gku614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rohleder F, et al. FANCM interacts with PCNA to promote replication traverse of DNA interstrand crosslinks. Nucleic Acids Res. 2016;44(7):3219–32. doi: 10.1093/nar/gkw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xue X, Sung P, Zhao X. Functions and regulation of the multitasking FANCM family of DNA motor proteins. Genes Dev. 2015;29(17):1777–88. doi: 10.1101/gad.266593.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dicks E, et al. Germline whole exome sequencing and large-scale replication identifies FANCM as a likely high grade serous ovarian cancer susceptibility gene. Oncotarget. 2017 doi: 10.18632/oncotarget.15871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neidhardt G, et al. Association Between Loss-of-Function Mutations Within the FANCM Gene and Early-Onset Familial Breast Cancer. JAMA Oncol. 2016 doi: 10.1001/jamaoncol.2016.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cantor SB, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105(1):149–60. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- 59.Bharti SK, et al. Getting Ready for the Dance: FANCJ Irons Out DNA Wrinkles. Genes (Basel) 2016;7(7) doi: 10.3390/genes7070031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castillo Bosch P, et al. FANCJ promotes DNA synthesis through G-quadruplex structures. EMBO J. 2014;33(21):2521–33. doi: 10.15252/embj.201488663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matsuzaki K, et al. FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi anemia pathway. Genes Dev. 2015;29(24):2532–46. doi: 10.1101/gad.272740.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Osorio A, et al. Evaluation of rare variants in the new fanconi anemia gene ERCC4 (FANCQ) as familial breast/ovarian cancer susceptibility alleles. Hum Mutat. 2013;34(12):1615–8. doi: 10.1002/humu.22438. [DOI] [PubMed] [Google Scholar]
- 63.Lieber MR, et al. Flexibility in the order of action and in the enzymology of the nuclease, polymerases, and ligase of vertebrate non-homologous DNA end joining: relevance to cancer, aging, and the immune system. Cell Res. 2008;18(1):125–33. doi: 10.1038/cr.2007.108. [DOI] [PubMed] [Google Scholar]
- 64.Biedermann KA, et al. scid mutation in mice confers hypersensitivity to ionizing radiation and a deficiency in DNA double-strand break repair. Proc Natl Acad Sci U S A. 1991;88(4):1394–7. doi: 10.1073/pnas.88.4.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thompson LH, Jeggo PA. Nomenclature of human genes involved in ionizing radiation sensitivity. Mutat Res. 1995;337(2):131–4. doi: 10.1016/0921-8777(95)00018-f. [DOI] [PubMed] [Google Scholar]
- 66.Andres SN, et al. A human XRCC4-XLF complex bridges DNA. Nucleic Acids Res. 2012;40(4):1868–78. doi: 10.1093/nar/gks022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roy S, et al. XRCC4's interaction with XLF is required for coding (but not signal) end joining. Nucleic Acids Res. 2012;40(4):1684–94. doi: 10.1093/nar/gkr1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Z, et al. The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell. 1995;83(7):1079–89. doi: 10.1016/0092-8674(95)90135-3. [DOI] [PubMed] [Google Scholar]
- 69.Mansour WY, et al. Hierarchy of nonhomologous end-joining, single-strand annealing and gene conversion at site-directed DNA double-strand breaks. Nucleic Acids Res. 2008;36(12):4088–98. doi: 10.1093/nar/gkn347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schulte-Uentrop L, et al. Distinct roles of XRCC4 and Ku80 in non-homologous end-joining of endonuclease- and ionizing radiation-induced DNA double-strand breaks. Nucleic Acids Res. 2008;36(8):2561–9. doi: 10.1093/nar/gkn094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bunting SF, et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell. 2012;46(2):125–35. doi: 10.1016/j.molcel.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim Y, et al. Mutations of the SLX4 gene in Fanconi anemia. Nat Genet. 2011;43(2):142–6. doi: 10.1038/ng.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Klein HL, Symington LS. Breaking up just got easier to do. Cell. 2009;138(1):20–2. doi: 10.1016/j.cell.2009.06.039. [DOI] [PubMed] [Google Scholar]
- 74.Svendsen JM, et al. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009;138(1):63–77. doi: 10.1016/j.cell.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Panneerselvam J, et al. Basal level of FANCD2 monoubiquitination is required for the maintenance of a sufficient number of licensed-replication origins to fire at a normal rate. Oncotarget. 2014;5(5):1326–37. doi: 10.18632/oncotarget.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Panneerselvam J, et al. Fanconi Anemia Group D2 Protein Participates in Replication Origin Firing. Chemotherapy (Los Angel) 2016;5(3) doi: 10.4172/2167-7700.1000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Michl J, et al. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat Struct Mol Biol. 2016;23(8):755–7. doi: 10.1038/nsmb.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen YH, et al. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol Cell. 2015;58(2):323–38. doi: 10.1016/j.molcel.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22(1):106–16. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pichierri P, Franchitto A, Rosselli F. BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J. 2004;23(15):3154–63. doi: 10.1038/sj.emboj.7600277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meetei AR, et al. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol. 2003;23(10):3417–26. doi: 10.1128/MCB.23.10.3417-3426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Panneerselvam J, et al. BLM promotes the activation of Fanconi Anemia signaling pathway. Oncotarget. 2016;7(22):32351–61. doi: 10.18632/oncotarget.8707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ling C, et al. Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discov. 2016;2:16047. doi: 10.1038/celldisc.2016.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li J, et al. Fanconi anemia links reactive oxygen species to insulin resistance and obesity. Antioxid Redox Signal. 2012;17(8):1083–98. doi: 10.1089/ars.2011.4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Daschkey S, et al. Fatal Lymphoproliferative Disease in Two Siblings Lacking Functional FAAP24. J Clin Immunol. 2016;36(7):684–92. doi: 10.1007/s10875-016-0317-y. [DOI] [PubMed] [Google Scholar]
- 86.Zhang X, et al. FANCI is a negative regulator of Akt activation. Cell Cycle. 2016;15(8):1134–43. doi: 10.1080/15384101.2016.1158375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Panneerselvam J, et al. Distinct Metabolic Signature of Human Bladder Cancer Cells Carrying an Impaired Fanconi Anemia Tumor-Suppressor Signaling Pathway. J Proteome Res. 2016;15(4):1333–41. doi: 10.1021/acs.jproteome.6b00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kee Y, Kim JM, D'Andrea AD. Regulated degradation of FANCM in the Fanconi anemia pathway during mitosis. Genes Dev. 2009;23(5):555–60. doi: 10.1101/gad.1761309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Naim V, et al. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol. 2013;15(8):1008–15. doi: 10.1038/ncb2793. [DOI] [PubMed] [Google Scholar]
- 90.Chan KL, et al. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11(6):753–60. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 91.Bretscher HS, Fox DT. Proliferation of Double-Strand Break-Resistant Polyploid Cells Requires Drosophila FANCD2. Dev Cell. 2016;37(5):444–57. doi: 10.1016/j.devcel.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sarbajna S, Davies D, West SC. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014;28(10):1124–36. doi: 10.1101/gad.238303.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Garcia-Rubio ML, et al. The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet. 2015;11(11):e1005674. doi: 10.1371/journal.pgen.1005674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chatterjee N, Lin Y, Wilson JH. Fanconi anemia pathway regulates convergent transcription-induced cell death at trinucleotide repeats in human cells. Postdoc J. 2016;4(5):46–54. doi: 10.14304/surya.jpr.v4n5.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang T, et al. Fancd2 in vivo interaction network reveals a non-canonical role in mitochondrial function. Sci Rep. 2017;7:45626. doi: 10.1038/srep45626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jayabal P, et al. Involvement of FANCD2 in Energy Metabolism via ATP5alpha. Sci Rep. 2017;7(1):4921. doi: 10.1038/s41598-017-05150-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Root H, et al. FANCD2 limits BLM-dependent telomere instability in the alternative lengthening of telomeres pathway. Hum Mol Genet. 2016;25(15):3255–3268. doi: 10.1093/hmg/ddw175. [DOI] [PubMed] [Google Scholar]
- 98.Rego MA, et al. Regulation of the activation of the Fanconi anemia pathway by the p21 cyclin-dependent kinase inhibitor. Oncogene. 2012;31(3):366–75. doi: 10.1038/onc.2011.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jaber S, et al. p53 downregulates the Fanconi anaemia DNA repair pathway. Nat Commun. 2016;7:11091. doi: 10.1038/ncomms11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Golczak M, et al. LRAT-specific domain facilitates vitamin A metabolism by domain swapping in HRASLS3. Nat Chem Biol. 2015;11(1):26–32. doi: 10.1038/nchembio.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garaycoechea JI, et al. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489(7417):571–5. doi: 10.1038/nature11368. [DOI] [PubMed] [Google Scholar]
- 102.Langevin F, et al. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475(7354):53–8. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- 103.Pontel LB, et al. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol Cell. 2015;60(1):177–88. doi: 10.1016/j.molcel.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Joenje H. Metabolism: alcohol, DNA and disease. Nature. 2011;475(7354):45–6. doi: 10.1038/475045a. [DOI] [PubMed] [Google Scholar]
- 105.Panneerselvam J, et al. A Hidden Role of the Inactivated FANCD2: Upregulating DeltaNp63. Oncotarget. 2013 doi: 10.18632/oncotarget.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vijg J, Suh Y. Genome instability and aging. Annu Rev Physiol. 2013;75:645–68. doi: 10.1146/annurev-physiol-030212-183715. [DOI] [PubMed] [Google Scholar]
- 107.Yao Y, Dai W. Genomic Instability and Cancer. J Carcinog Mutagen. 2014;5 doi: 10.4172/2157-2518.1000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang J, et al. FAVL elevation in human tumors disrupts Fanconi anemia pathway signaling and promotes genomic instability and tumor growth. J Clin Invest. 2010;120(5):1524–34. doi: 10.1172/JCI40908. [DOI] [PMC free article] [PubMed] [Google Scholar]