

Abstract
Hydrogen exchange (HX) methods can reveal much about the structure, energetics, and dynamics of proteins. The addition of mass spectrometry (MS) to an earlier fragmentation-separation HX analysis now extends HX studies to larger proteins at high structural resolution and can provide information not available before. This chapter discusses experimental aspects of HX labeling, especially with respect to the use of MS and the analysis of MS data.
1. INTRODUCTION
Hydrogen–deuterium exchange mass spectrometry is increasingly being recognized as an important, information-rich, tool for protein studies (Englander, 2006; Kaltashov, Bobst, Nguyen, & Wang, 2013; Konermann, Tong, & Pan, 2008; Ling, Silva, Schriemer, & Schryvers, 2012; Pirrone, Iacob, & Engen, 2015; Zhang & Smith, 1993). The amide hydrogen (NH) of every residue (except proline) in every protein is available as a probe of its local environment. These probes can be used in many ways to inform us about the structural energetics and dynamics of proteins under native conditions, as well as how they respond to functional changes such as ligand binding and folding. Most hydrogen exchange experiments measure the exchange of H(1H) and D(2H) isotopes. This is reflected by the widespread use of the term hydrogen–deuterium exchange (HDX) rather than the more general term, hydrogen exchange (HX) that includes other isotopes. Mass spectrometry (MS) has taken an overwhelming lead since the late 1990s as the method of choice for HX measurements. While nuclear magnetic resonance (NMR) naturally achieves residue resolution, it requires ~104 times as much protein for each sample, is limited to fairly small proteins, <~200 residues, that are soluble near mM concentrations, and often requires isotopic enrichment with 15N.
This chapter will discuss HX-MS experiments in three sections: First HX labeling itself, beginning with exchange chemistry and the mechanisms of exchange in a protein. We then discuss the measurement of deuterated proteins by electrospray ionization MS. We will mostly deal with the fragment separation method (Englander & Englander, 1972; Englander & Kallenbach, 1983) in which labeled protein is enzymatically digested to yield peptide fragments that are in turn individually analyzed for D content based on the increase in mass (Δm) of deuterated peptides compared to unexchanged, all H, peptides. We finally discuss data processing, beginning with the identification of deuterated peptides and our method for moving toward residue resolution.
2. HX MECHANISM
2.1 Chemistry
The chemical exchange of a freely exposed amide NH is catalyzed by OH− and H+ and so is naturally a function of pH with a minimum rate at roughly pH 2.5. The rate is further influenced by inductive and steric effects from nearby amino acid side chains, temperature, and the isotopes involved. These effects are well calibrated and serve as reference rates when assessing structural protection (Bai, Milne, Mayne, & Englander, 1993; Connelly, Bai, Jeng, & Englander, 1993).
Careful control of pH is vital to successful HX measurements. Above pH 3 where proteins are usually studied, the rate increases by a factor of 10 with every pH unit. We take advantage of this pH dependence to control the rate of exchange for experimental convenience and most importantly to preserve the labeled state of the protein by quenching to the minimum rate. At pH 2.5 and 0 °C, the rate is near 0.01/min which allows time for the manipulations necessary for MS measurements.
2.2 HX Structural Physics
Stable hydrogen-bonded protein structure must open to expose the amide to solvent before it can exchange with its chemical rate as given by (Hvidt & Nielsen, 1966):
This scheme leads to a measured exchange rate, kex, given by:
| (1) |
here kch is the free peptide rate, a function of temperature, pH, and local sequence, and kop and kcl are the structural opening and closing rates that expose the amide.
For stable structure (kop ≪ kcl), this general scheme reduces to two limiting cases depending on the relative rates of reclosing (kcl) and the chemical exchange rate (kch).
When kcl ≪ kch, the protein opens and recloses many times before exchange occurs. The measured exchange rate then is kch during the fraction of time open. The equilibrium constant for opening, Kop, is given as kop/kcl, leading to a measured exchange rate as in Eq. (2).
| (2) |
In this case, referred to as EX2 or bimolecular exchange, exchange depends on kch and therefore on the pH. 1/Kop is often referred to as the protection factor. Protection is also often reported in free energy units as ΔGHX = −RT ln(Kop).
When kcl ≪ kch, exchange is limited by the opening rate, kop, and is independent of kch:
| (3) |
This case, known as EX1, sometimes referred to as correlated exchange, is most likely at high pH, where kch is fast, or low protein stability where kcl is slow.
When HX is measured by NMR, the exchange rate of each amide is measured individually and it is not easy to distinguish EX1 from EX2 in a single experiment. The most common test is to measure exchange at two or more pH values. If protein stability does not change with pH the exchange rate will change by a factor of 10 for each change of 1 pH unit under EX2 conditions and remain unaffected under EX1 conditions. If the exchange mechanism transitions between EX2 and EX1 as pH is raised, both the opening and closing rates can be measured by fitting the measured exchange rates as a function of pH to Eq. (1) allowing kop and kcl to float while using the known values of kch as a function of pH (Bedard, Mayne, Peterson, Wand, & Englander, 2008).
Measuring HX by MS can make the EX1, EX2 difference directly apparent and illustrates a significant advantage of using MS. MS measures exchange for groups of amino acids together whether peptides or the whole protein. If a segment of structure unfolds and stays unfolded for longer than the free peptide exchange rate before refolding, as in EX1 exchange, then all residues in that segment will fully exchange before it refolds. Under these conditions, at any point during the course of exchange the sample will contain peptides that are unexchanged (have not yet opened) and peptides that are fully exchanged (have opened at least once). As a result, it will display a bimodal isotopic profile. In EX2 exchange, individual amino acids are not correlated and so will always show a unimodal isotopic distribution that moves from starting to ending conditions (H to D or D to H) with longer exchange times.
3. EXPERIMENTAL CONSIDERATIONS—EXCHANGE LABELING
Figure 1 outlines the two major classes of HX labeling experiments and the two major methods used for reading the state of labeling by MS. Most experiments use continuous labeling followed by peptide level measurement. We will first discuss labeling protocols, then discuss MS methods for reading the label, and finally data analysis which includes choices depending on the nature of the labeling experiment.
Figure 1.

Classes of HX experiments.
3.1 Continuous Labeling (Native State)
Most HX experiments are some form of native state, continuous, labeling in which the exchange of the protein of interest is measured as a function of exchange time under conditions where the native state of the protein is stable. Experimental conditions are primarily determined by the protein being studied. The simplest way to begin exchange is to dilute a fully H protein sample into D2O under experimental conditions. It is generally desirable to make a large dilution so as to increase the dynamic range of the measurement, though this may be limited by protein solubility or other considerations.
Two dilutions are required. First, protein is diluted into exchange conditions and then after some time, quench buffer is added to bring the solution to pH ~2.5 accompanied by rapid cooling or freezing for later MS analysis. Commonly, each exchange time point is mixed separately in individual microcentrifuge tubes. This can be done either by hand or robotically. With effort, labeling times as short as 10 s can be achieved by either method.
It is important to consider the range of time points chosen for exchange samples. We would like to measure the rates of each amino acid in our protein but individual amino acids within a native protein may have exchange rates that vary by factors of 108 or more, from free peptide rates of less than a second to residues that exchange only via transient unfolding of the entire protein, more than a year for a stable protein (~12 kcal/mol). In order to capture this dynamic range and extract as much information as is available from the experiment, it is clear that we cannot be limited to exchange measured by simple dilution experiments over a single day, a dynamic range of only ~1000. At pH 7 and 20 °C, many positions will be fully exchanged by the first time point, 10–30 s, while many amides may not have measurably exchanged in 24 h. The measurement range can be extended for many proteins by moving the intrinsic HX rate into a more convenient timescale by adjusting the temperature, pH, or both. If the protein under study does not change its stability, a change of 1 pH unit up or down moves the intrinsic exchange rate up or down by a factor of 10, increasing the measurable range of protection (Coales et al., 2010). The exchange temperature can be similarly adjusted; a change of 10 °C yields a change in rate of almost a factor of 3. It is important to verify that protein stability does not change in response to the change in conditions by overlapping the timescales so that some residues are measured under different exchange conditions and show the same protection factors.
Faster exchange rates can also be accessed by use of a flow system to perform the labeling. Two examples are illustrated in Fig. 2. Exchange times are determined by flow rates and the volumes between mixers. Times as short as 10 ms can be measured. With our BioLogic SFM400, we are, in addition, able to stop and restart the flow allowing for exchange times of minutes in the same apparatus. Wang, Abzalimov, Bobst, and Kaltashov (2013) have used a flow system to spray directly without proteolysis or HPLC.
Figure 2.

(A) Flow apparatus used for short labeling times as described by Coales et al. (2010). Exchange begins when the sample is mixed with D2O buffer at mixing T1 pushed by pumps 1 and 2. After an exchange time, determined by the flow rate and the volume of the exchange loop, quench buffer is added. The sample flows through a pepsin column and onto a trap column. From the trap column, peptides are eluted to the MS by a gradient supplied by a separate pump. Not shown are valves that allow the reloading of the sample, D buffer, and quench loops, and that control flow through the trap column. (B) Our system using a BioLogic SFM400 stopped-flow mixer for labeling and a separate cooled chamber (Fig. 3) for proteolysis and chromatography. In the stopped-flow mixer, the speed of each syringe is separately controlled. For a simple exchange experiment, syringe 1 is not used. Unlabeled protein from syringe 2 is mixed with D2O buffer from syringe 3. After an exchange time determined by the volume of delay line 2 and the speed of flow exchange is quenched by syringe 4. The quenched sample flows to the injection loop of our proteolysis/HPLC system. When used for refolding experiments, we start with unfolded protein in syringe 1. Folding is started with a dilution into refolding conditions at mixer 1. From here, the labeling pulse is as above.
Once a sample has exchanged, the label must be preserved for MS analysis. The sample is quenched by lowering the temperature and pH before making the measurement as quickly as possible. It is important to verify that the pH of the quenched sample after the mix is as expected. We verify the pH of the mixture for each new batch of buffers. The optimum pH seems to be 2.5 for the initial quench where the ionic strength is 10’s or 100’s of mM and 2.3 for the HPLC buffers where the ionic strength is <20 mM (Walters, Ricciuti, Mayne, & Englander, 2012). We use 0.1% formic acid and 0.05% trifluoroacetic acid for the aqueous HPLC buffers and 0.1% formic acid for the acetonitrile buffer.
The quenched sample can be flash frozen and stored at −80 °C for later MS analysis. It is important that frozen samples be thawed without excess heating. This is aided by the addition of antifreeze in the form of 10% glycerol or some other molecule. With the freezing point depressed, thawing can be done fairly quickly (1–2 min) in a 0 °C bath.
As discussed above, exchange of H-bonded NH’s requires the transient opening of protecting structure. This can involve small-scale openings that only expose one amide NH at a time and larger scale openings that expose several amino acids together, all the way up to the global unfolding of the entire protein. Under EX2 conditions, the rate of a single residue will be determined by the fraction of time open, the sum of all opening reactions that expose that residue. Larger scale openings can be promoted by destabilizing conditions such as increased temperature or increasing concentrations of denaturant. This technique has been used to great effect by NMR for several proteins and has helped with the understanding of protein structural energetics and folding (Bai, Sosnick, Mayne, & Englander, 1995).
Some label will be unavoidably lost to back exchange during the workup for MS analysis. It is possible to measure the degree of back exchange with an “all D” control. In this experiment, a fully D protein sample subjected to the identical quench and analysis procedure used for exchange time points. The measured D occupancy for this sample can be used to calibrate the loss of label. The details of how back exchange is evaluated and corrected for depend on how the data are being analyzed and will be discussed in the analysis section.
Many people seem to have problems preparing all D samples. Given that some amides in stable proteins may be protected by 108 or more, simply waiting will not always work. We have had good success by heating a solution of protein in D2O, at the same D/H ratio that will be present during the exchange experiment, to the beginning of the thermal melt transition, usually ~5 °C less than the midpoint of the melt. The protein is always cycling back and forth between folded and unfolded states but at room temperature the unfolded fraction may be only 10−8 or less. At a temperature near the beginning of the melt, this fraction will approach 10−2, high enough to lower protection factors to only ~102–103 but not so high as to risk aggregation. In practice, we usually allow 10’s of minutes to be sure that the whole sample has cycled through the unfolded state.
3.1.1 Binding Sites and Epitope Mapping
HDX is frequently used to localize the binding site of a drug or antibody (epitope mapping) by measuring exchange rates for free, unbound protein and for protein bound to the ligand of interest. Exchange rates are expected to slow for those residues that are affected by the binding. This can occur by more than one mechanism. HX slowing can occur due to direct hydrogen bonding between the ligand and exposed NH’s on the protein. Binding interactions can stabilize a H-bond against exchange due to direct interaction of the ligand with either the H-bond donor or acceptor. Or interactions can stabilize H-bonded structure indirectly by stabilizing parts of the protein against larger scale openings (Mayne, Paterson, Cerasoli, & Englander, 1992; Paterson, Englander, & Roder, 1990; Wei et al., 2014).
It is of fundamental importance in binding studies to be sure that the protein binding site is saturated during the exchange time. The degree of saturation limits the extent to which HDX rate differences can be measured. If, for example, a ligand is bound only 90% of the time, then the maximal slowing will be a factor of 10 slower than for the unbound protein even in cases where exchange is completely stopped in the bound state. If this slowing affects only 1 or 2 residues in a peptide of 12 or more amino acids, the difference may be hard to distinguish.
Ligand bound experiments have to deal with the ligand in the MS analysis. The low pH quench will often unfold proteins and reduce binding. For small-molecule ligands, this alone may be enough if the ligand itself does not interfere with proteolysis or HPLC. In the case of protein–protein binding, e.g., antibody interactions, the ligand may be bigger than the protein being studied and is usually in molar excess. In this case, it is usually desirable to separate the protein from the antibody to avoid signal suppression and conflicts due to overlapping peptides. This is most easily accomplished by immobilizing the antibody on a column and binding the target protein to the antibody column. D2O buffer is infused into the column already saturated with bound protein to start exchange. The protein is eluted and exchange quenched in a single step by infusing low pH quench buffer through the column directly into the analysis system (Paterson et al., 1990).
3.2 Pulsed Labeling
Protein conformational kinetics, e.g., protein folding, can be followed by HX pulsed labeling methods. In this experiment, rather than measuring exchange as a function of time under steady-state conditions, we start a reaction and after some time take a snapshot of the hydrogen-bonded structure with a short pulse of high pH where exchange is fast enough to fully exchange all freely exposed amides in a few ms. After a labeling time of usually 10’s of ms, the pH is dropped to quench conditions and the sample analyzed.
While slower pulsed labeling experiments can be performed by manual mixing or by the same automated systems sometimes used for continuous labeling experiments, a flow system is needed for faster pulses. We use a BioLogic SFM400 stopped-flow mixer connected directly to the injection loop of our online proteolysis/HPLC system (Fig. 2). Three mixes are needed. The first, to start folding, will have to be large to dilute away denaturant. If exchange rates under folding conditions are slow relative to the folding time, then this dilution can also dilute the protein into D2O, or H2O if starting with unfolded D protein. The exchange pulse is started with an addition of high pH buffer. If the first dilution did not change the isotope (H to D or D to H), then this dilution will have to be large to move the protein to new isotopic solvent so that exchange can be measured. The pulse is quenched by the final addition of low pH buffer to bring the mix to pH 2.5.
There is an underappreciated feature of HX-MS in protein folding experiments. NMR measurements are done under native conditions. This is necessary in order to achieve the chemical shift dispersion necessary to resolve individual residues. In NMR-measured pulsed labeling experiments, the H/D pattern imposed by the pulse is “quenched” by native structural protection so only residues that are protected in the native protein will be measured, leaving open the possibility that nonnative structure could be present at the time of the pulse yet be unobserved if not protected by later native structure. MS-detected refolding experiments have so far not seen such non-native protection, instead only stepwise accumulation of native structure has been observed (Hu et al., 2013; Walters, Mayne, Hinshaw, Sosnick, & Englander, 2013).
4. EXPERIMENTAL CONSIDERATIONS—MS MEASUREMENT
4.1 Whole Protein
Whole protein analysis is done without proteolysis and with little or no chromatography. Volatile buffers such as ammonium acetate and the addition of organic solvent allow quenched protein to be sprayed directly. Otherwise it is necessary to exchange the buffer to something more suitable for electrospray ionization. We do this by trapping the protein on a small C4 column, washing away buffer salts, and then eluting with a step to ~75% acetonitrile. The eluted protein is sprayed directly (Fig. 3).
Figure 3.
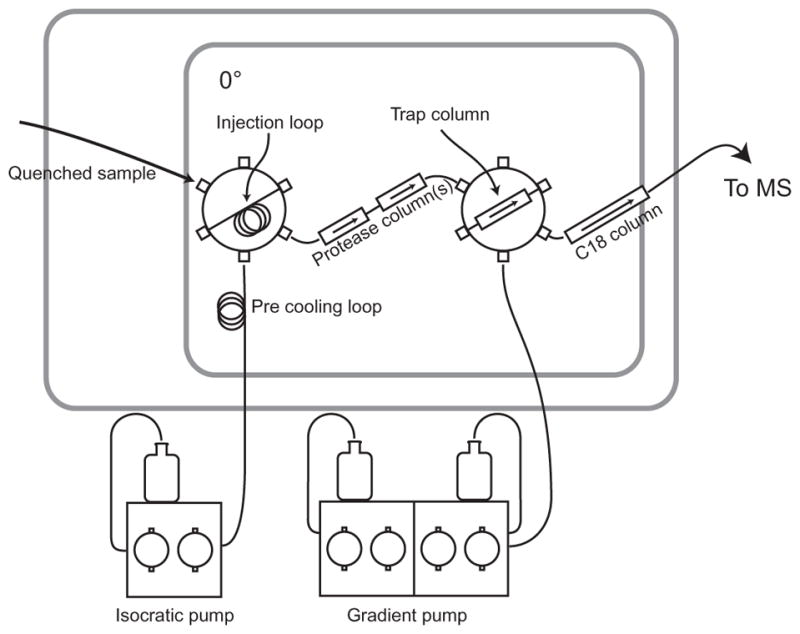
Our online digestion/HPLC system. The quenched sample is pushed through the protease column by a cold flow of pH 2.3 buffer (0.1% formic acid, 0.05% TFA). The produced peptides are bound by the small trap column and buffer salts are washed away. After switching the second valve peptides are eluted and separated by an H2O/acetonitrile gradient through a C18 analytical column. The output of the C18 column goes directly to the electrospray. Whole protein can be desalted for ESI by omitting the protease and analytical columns.
4.2 Peptide Resolved
In order to better locate the sites of exchange, the quenched protein is usually digested enzymatically into peptides that are then washed of buffer salts, and separated by rapid chromatography at 0 °C for electrospray ionization (Fig. 3) (Mayne et al., 2011). Quenched, labeled protein is loaded into the injection loop of a standard HPLC injection valve. A cold pH 2.3 aqueous buffer flow at 100–200 μl/min carries it through a protease column (sometimes more than one in tandem) and then via a second valve to a small reverse-phase “trap” column (typically C8 or C18, 1 mm×10 mm, 5-μm beads). The peptides produced bind to the trap column while buffer salts are washed away. Switching the second valve directs the flow from a gradient pump through the trap column and on through an analytical column to the electrospray. Variations of this basic plan with such features as reversing the flow during the protease wash or swapping protease columns are possible.
4.2.1 Proteolysis
Ideally, we would like to produce many sequentially overlapping peptides that redundantly cover the entire protein. Most workers have adopted the use of immobilized protease columns in preference to solution digestion. The speed of digestion and ease of incorporation into a flow system greatly favor the column system. While commercial protease columns are available, we produce our own. We couple protease, either pepsin or Fungal Protease type XIII from Sigma, to POROS 20 AL media from Applied Biosystems. We couple according to the manufacturer’s instructions at pH 4.4 with the addition of Na2SO4 (final concentration 750 mM) added over 2 h at room temperature. After several hours of additional reaction time, the beads are washed with pH 4.4 buffer for storage at 4 °C. A key step is to remove buffer salts from the commercial protease by gel filtration before beginning the reaction. Any amine containing buffer salts will compete with protein lysine side chains for binding sites. Once coupled, the beads are stable for months at 4 °C. We pack columns of either 1 or 2 mm diameter by 20 mm. We choose column sizes and flow rates according to the sizes and number of peptides we identify for each protein.
Digestion is often aided by the addition of denaturant to the quenched protein solution and is required for some proteins. We find our protease columns easily tolerate denaturant up to 2 M guanidinium chloride without damage. Addition of up to 200 mM TCEP (tris(2-carboxyethyl)phosphine) is included as necessary to reduce disulfides. Even with denaturant and TCEP, some proteins are resistant to proteolysis. There are some reports of successful electrochemical reduction of disulfide bonds (Mysling, Salbo, Ploug, & Jorgensen, 2014).
4.2.2 Chromatography
Peptides are eluted from the trap through an analytical HPLC column (C18, 0.3 mm×50 mm, 3-μm beads) to the electrospray. Gradients are roughly 10–45% acetonitrile over 12–15 min at ~10 μl/min. We find that a shorter gradient is not particularly useful in reducing back exchange as it only speeds the measurement of late eluting peptides while reducing HPLC separation. While the gradient is running, we can wash the protease column with injections of guanidinium chloride to wash any protein that might carry over.
Peptides are identified using standard MS/MS methods (Mayne et al., 2011). We are sure to include in our sequence database all potential contaminants including potential impurities, the proteases used, and other proteins analyzed using the same system. As an extra check on correct peptide ID’s, we confirm that sequentially overlapping peptides display consistent HD labeling.
5. MASS SPECTROMETER INSTRUMENT CONSIDERATIONS
HDX measurements can use many kinds of MS instruments. Everything we are discussing here assumes electrospray ionization. The combination of a large number of peptides, wide isotopic envelopes due to deuteration, and short HPLC gradients increases the probability of peptide overlap. High m/z resolution reduces the likelihood of individual isotopic peak overlap and high mass accuracy makes identification of peptides more certain. We use a Thermo Scientific LTQ Orbitrap XL usually operating at a resolution of 60,000 at m/z 400. Our data analysis program, ExMS, takes advantage of this higher resolution data to identify deuterated peptides.
Access to MS/MS capabilities is required for peptide identification but usually cannot be used to further localize D occupancy. Traditional collisional MS/MS fragmentation methods “scramble” amide D labels such that H’s and D’s equilibrate throughout the amides in a peptide before it fragments into daughter ions (Hamuro, Tomasso, & Coales, 2008). The increase in mass of daughter ions will reflect not the D occupancy levels of the daughter ion amino acids in the quenched protein but instead reflect the average fractional D occupancy of the peptide before fragmentation. Nonergodic fragmentation methods, electron-capture dissociation, and electron-transfer dissociation have been shown to cause minimal scrambling and provide the possibility of residue resolution of D occupancy. By analyzing the deuteration levels of a series of daughter ions differing by one amino acid at a time, the D occupancy of each new position can be determined by simple subtraction (Pan, Han, Borchers, & Konermann, 2009; Wang et al., 2013; Zehl, Rand, Jensen, & Jorgensen, 2008). In practice, enzymatic proteolysis may still be necessary for all but very short proteins to ensure coverage because not every possible site will fragment and the measurement of small mass differences in large fragments will limit accuracy. Care must still be taken to avoid scrambling in the spectrometer ion source. In some cases, parameters required to avoid ion scrambling severely reduce the signal.
6. DATA ANALYSIS
6.1 Whole Protein
Analysis of whole protein experiments is relatively straightforward. Continuous labeling EX2 experiments are characterized by the total D content as measured by the centroid shift. A back exchange correction can be applied by measuring the centroid of a fully D sample and correcting the measured Δm as discussed below.
Pulsed labeling experiments, EX1 exchange, or conformationally heterogeneous samples are expected to show multimodal isotopic profiles. If the m/z separation is sufficient, then the ratio of exchanged to unexchanged protein molecules can be readily measured by integrating the intensity of each envelope.
6.2 Peptide Analysis
Data analysis of HX experiments at the level of peptides can be an involved process depending on the nature of the exchange experiment and the peptide coverage. In all cases, it is important to remember that as the peptide bond is cut the N of the first amino acid of the resulting peptide becomes a free amine which exchanges very rapidly. In addition, the exchange rate of the second amino acid is greatly accelerated due to inductive effects on the chemical rate from the free amine (Bai et al., 1993). The result is that any D label present on the first amino acid is lost and, in general, label on the second residue is lost as well. In cases of very low back exchange if the first two side chains are isoleucine or valine, some label on the second residue might be retained.
6.2.1 Identification of Labeled Peptides
Identification of peptides with unknown levels of deuteration can be difficult. Several groups have developed software in an attempt to automate the identification and quantification of deuterated peptides, including (Pascal et al., 2012; Slysz et al., 2009) among others, as well as commercial software from Waters and Sierra Analytics. In general, one starts with a list of identified peptides and scans the MS chromatogram looking for each one while allowing for an unknown level of deuteration. The analysis usually starts with an all H sample where the m/z values are known in order to accurately measure the retention time window for each peptide. Once the retention time is accurately known, then deuterated samples are analyzed. A search over a narrow range of retention times simplifies the search over a larger range of m/z values.
We have developed the program ExMS (Kan, Mayne, Sevaugan Chetty, & Englander, 2011) which uses a list, with retention times, of peptides previously identified by MS/MS to identify deuterated peptides from MS data. In short, for each peptide on its list ExMS uses high-resolution accurate mass data to identify sets of isotopically resolved peaks that are consistent with deuterated samples of that peptide. A series of tests are applied to confirm the quality of the identification. The extracted data for each peptide are output as a table containing the retention time, the intensity of each isotopic peak, the overall intensity, and the centroid value for each peptide as well as information about each test.
At this point, various options are available depending on the nature of the experiment. One can simply plot average D occupancy, the increased centroid above the all H value, as a function of time as increased weight in Daltons or as percent exchanged. Percent D should be calculated as:
where Δm is the centroid mass above the centroid of the unlabeled peptide, frac D is the fraction D present during the exchange period, N is the number of amino acids in the peptide, and P is the number of proline residues in the peptide beyond the first two amino acids. As discussed above, no label is expected to be retained on the first two amino acids.
Whichever scale is used, it is helpful to indicate the size of the peptide involved so the reader can convert between the two scales. The use of a D scale rather than percent more readily relates to structural protection which operates on a residue level.
Protection provided by H-bonded structure directly influences the rate of exchange under EX2 conditions. Therefore, in order to measure the energetics of opening reactions that allow exchange, it is most useful to pay attention not to simple deuteration levels but instead to exchange rates (Fig. 4).
Figure 4.

EX2 exchange. (A, B) Mass spectra of a peptide produced by pepsin digest (RVALTEDRLPRL) as a function of exchange time. The top spectra are for an unexchanged, all H, sample and the bottom are from a fully exchanged, all D, sample used to calibrate back exchange. Intermediate time points show the progress of exchange for (A) the protein free in solution and (B) the protein bound to an antibody. The increase in centroid above the all H sample uncorrected for back exchange is plotted in (C), corrected for back exchange in (D). In the absence of back exchange, we could expect nine deuterons for an all D sample. The measured centroid increase for the all D sample is 7 D’s indicating 78% recovery. It can be seen that antibody binding apparently slows exchange for at least six positions by a factor of 1000 or more.
In pulsed labeling experiments or under EX1 conditions, analysis can involve a simple centroid shift measurement of average D occupancy. Alternatively, more information is available if multiple labeled populations are resolvable as in Fig. 5. In this case, the relative intensities of differently labeled populations yield information about the number of sites labeled (m/z axis) and the fraction of molecules in each resolved labeled population (relative intensities of each population).
Figure 5.

Example data from a pulsed labeling experiment following the folding of maltose binding protein (MBP) showing bimodal isotopic distributions. Three peptides are shown. Residues 21–43 (A), 76–89 (B), and 346–370 (C). Folding time before the labeling pulse is indicated. In this case, starting with all D unfolded protein, the heavy fraction reflects protein molecules that were protected from exchange during a 43-ms, pH 9 pulse. Different parts of the protein acquire protecting structure at different times after folding starts showing the progressive formation of the native structure. The data shown in gray are from unfolded and native control experiments and show that the protected and unprotected fractions behave like fully folded and unfolded protein. The fits are with binomials. A concerted EX1 exchange mechanism will yield similar data. From Walters et al. (2013).
6.2.2 Back Exchange Correction
The measured level of back exchange is an important measure of data quality. D recovery can be measured on a per-peptide basis by subjecting an all D sample to the same proteolysis, HPLC, MS workflow used for experimental samples. The D recovery can then be calculated as:
where Δm(D, measured) is the measured shift for an “all D” sample. Δm(D, expected) is the expected shift for an all D sample in the absence of back exchange calculated as:
here frac(D) is the fraction D present when the all D sample was prepared, N is the number of amino acids in the peptide, and P is the number of proline residues in the peptide beyond the first two amino acids.
A simple back exchange correction can be applied as:
It should be stressed that all back exchange corrections are at best approximate. The exchange correction suggested above implicitly assumes that labeled residues in the H/D experiment back exchange at the average exchange rate of the entire peptide as measured from the all D experiment. In reality, for a given peptide only some residues may have high D occupancy and may have exchange rates higher or lower than the average. A somewhat more sophisticated way of dealing with back exchange is discussed below in the context of moving toward residue-resolved analysis. In general, it is far better to reduce back exchange rather than try to correct for it. Even in experiments where back exchange seems like it can be ignored in analysis (ligand binding, EX1, pulsed labeling), the presence of back exchange still reduces the quality of the collected data, reducing the dynamic range available for resolving differences in exchange with and without ligand or for resolving folded and unfolded populations.
6.3 Toward Residue Resolution
For EX2 experiments, many groups have developed analysis methods that attempt to localize D occupancy beyond the peptide level. All such methods seek to use the information available from multiple sequentially overlapping peptides or fragments to localize D occupancy as much as possible given the cut sites. If nonergodic fragmentation methods are used or proteolysis provides sets of peptides with a common end differing by single residue steps at the other end, then simple subtraction can be used (Wang et al., 2013). We will not attempt to review the various published algorithms designed to deal with more general cases but will briefly describe the method we have developed, HDsite (Kan, Walters, Mayne, & Englander, 2013).
The HDsite algorithm takes advantage of not just the average, centroid, D occupancy for each peptide but also uses the information available in the shape of the isotopic distribution. Many different D occupancy patterns for a partially exchanged peptide can give rise to the same centroid value with different isotopic envelopes (Fig. 6). HDsite uses the set of D occupancies for each residue as adjustable parameters to simultaneously fit the measured isotopic envelopes for a set of overlapping peptides. While D sites cannot be localized beyond the information available from the cut sites in a set of overlapping peptides HDsite is able to resolve within “switchable” residues variable D occupancy levels without being able to assign which D level goes with which residue. The algorithm has been tested against NMR-measured rates for staphylococcal nuclease.
Figure 6.

Toward residue resolution. (A) Bars represent the nearly 300 overlapping peptides obtained for staphylococcal nuclease. The staggered ends of overlapping peptides can in principle locate D occupancy to higher resolution than the peptide level. Given this set of peptides, the potential resolution is indicated by the dot and dashed line along the bottom. Small spots indicate residues that should be resolvable. Bars represent groups or two or more residues that always appear together and cannot be resolved. The HDsite algorithm can provide D occupancy information for each of these “switchable” residues but cannot assign D values to a particular residue in the group. (B–D) The basis for the HDsite algorithm. Simulated isotopic distributions for three different D distributions, all with the same centroid, yield different MS isotopic distributions. (B) All eight sites 50% D. (C) Four sites 100% D, four sites 0% D. (D) Four sites 10% D, four sites 90% D. Adapted from Kan et al. (2013).
Back exchange can be dealt with as part of the fitting process. An effective back exchange time is measured for each peptide based on the measured centroid of an all D sample using the known free peptide rates under quench conditions for the residues of that peptide. For each iteration in the fitting procedure, the D occupancy levels are adjusted according to the effective back exchange time for that peptide. These adjusted D occupancies are used to calculate the envelope that is compared to experimental data.
We have since developed a version of the program that fits the entire exchange time course with a set of exchange rates, one for each residue rather than fitting D occupancy values for each time point. While computationally more intensive, this version improves the quality of the fit by reducing the number of adjustable parameters from one D occupancy per residue for each time point to one rate per residue for the whole time course.
6.4 Analysis of Pulsed Exchange Data
Data from pulsed labeling experiments, conformationally heterogeneous samples, and EX1 experiments are all expected to give rise to multimodal MS profiles. In these experiments, one is often more interested in the relative magnitude of the heavy and light populations than the degree of exchange (centroid) of each population. Analysis is greatly dependent on the quality of the MS data, particularly the separation of the labeled populations on the m/z axis. In favorable cases, the populations are separated well enough so that integration of the intensity of each is straightforward as is the calculation of the centroid of each. In other cases, separation is not sufficient to fully separate the isotopic envelopes of two populations. In this case, it is necessary to do some sort of curve fitting to extract the relative intensities and centroids of the labeled populations. This is not as simple a problem as it might seem because the shape of each isotopic distribution is not necessarily known. In practice, Gaussians or binomials can give good results except in cases where the overlap is particularly severe. The best quality data come from peptides with well-resolved protected and unprotected envelopes. Generally, this means peptides that are long enough so that 13C envelopes are narrower than mass differences due to D but not so long that a given peptide spans more than one protein folding unit. We find that peptides of ~10–15 amino acids give good results. A high dynamic range of H/D during the pulse (ideally 0% D protein exchanging in 100% D or vice versa) and very low back exchange also improve separation of differentially labeled fractions.
7. CONCLUSION
HX methods continue to extend our understanding of protein structure, folding, and dynamics. HX methods are being applied to an increasing number of protein systems and sample handling techniques continue to advance. The use of MS has greatly extended the range of proteins amenable to HX analysis while vastly reducing the amount of protein required. As mass spectrometers with ever higher resolution and sensitivity become available combined with advances in data processing software, we can expect to extract more and more detailed information from HDX experiments.
References
- Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins: Structure, Function, and Genetics. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Sosnick TR, Mayne L, Englander SW. Protein folding intermediates: Native-state hydrogen exchange. Science. 1995;269(5221):192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard S, Mayne LC, Peterson RW, Wand AJ, Englander SW. The foldon substructure of staphylococcal nuclease. Journal of Molecular Biology. 2008;376(4):1142–1154. doi: 10.1016/j.jmb.2007.12.020. http://dx.doi.org/10.1016/j.jmb.2007.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coales SJ, Sook Yen E, Lee JE, Ma A, Morrow JA, Hamuro Y. Expansion of time window for mass spectrometric measurement of amide hydrogen/deuterium exchange reactions. Rapid Communications in Mass Spectrometry. 2010;24(24):3585–3592. doi: 10.1002/rcm.4814. http://dx.doi.org/10.1002/rcm.4814. [DOI] [PubMed] [Google Scholar]
- Connelly GP, Bai Y, Jeng MF, Englander SW. Isotope effects in peptide group hydrogen exchange. Proteins. 1993;17(1):87–92. doi: 10.1002/prot.340170111. [DOI] [PubMed] [Google Scholar]
- Englander SW. Hydrogen exchange and mass spectrometry: A historical perspective. Journal of the American Society for Mass Spectrometry. 2006;17(11):1481–1489. doi: 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander SW, Englander JJ. Hydrogen tritium exchange methods. Methods in Enzymology. 1972;26c:406–413. doi: 10.1016/s0076-6879(72)26021-9. [DOI] [PubMed] [Google Scholar]
- Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic-acids. Quarterly Reviews of Biophysics. 1983;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- Hamuro Y, Tomasso JC, Coales SJ. A simple test to detect hydrogen/ deuterium scrambling during gas-phase peptide fragmentation. Analytical Chemistry. 2008;80(17):6785–6790. doi: 10.1021/ac800645f. http://dx.doi.org/10.1021/ac800645f. [DOI] [PubMed] [Google Scholar]
- Hu W, Walters BT, Kan ZY, Mayne L, Rosen LE, Marqusee S, et al. Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(19):7684–7689. doi: 10.1073/pnas.1305887110. http://dx.doi.org/10.1073/pnas.1305887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hvidt A, Nielsen SO. Hydrogen exchange in proteins. Advances in Protein Chemistry. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- Kaltashov IA, Bobst CE, Nguyen SN, Wang S. Emerging mass spectrometry-based approaches to probe protein-receptor interactions: Focus on overcoming physiological barriers. Advanced Drug Delivery Reviews. 2013;65(8):1020–1030. doi: 10.1016/j.addr.2013.04.014. http://dx.doi.org/10.1016/j.addr.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan ZY, Mayne L, Sevaugan Chetty P, Englander SW. ExMS: Data analysis for HX-MS experiments. Journal of the American Society for Mass Spectrometry. 2011;22(11):1906–1915. doi: 10.1007/s13361-011-0236-3. http://dx.doi.org/10.1007/s13361-011-0236-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan ZY, Walters BT, Mayne L, Englander SW. Protein hydrogen exchange at residue resolution by proteolytic fragmentation mass spectrometry analysis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(41):16438–16443. doi: 10.1073/pnas.1315532110. http://dx.doi.org/10.1073/pnas.1315532110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L, Tong X, Pan Y. Protein structure and dynamics studied by mass spectrometry: H/D exchange, hydroxyl radical labeling, and related approaches. Journal of Mass Spectrometry. 2008;43(8):1021–1036. doi: 10.1002/jms.1435. http://dx.doi.org/10.1002/jms.1435. [DOI] [PubMed] [Google Scholar]
- Ling JM, Silva L, Schriemer DC, Schryvers AB. Hydrogen-deuterium exchange coupled to mass spectrometry to investigate ligand-receptor interactions. Methods in Molecular Biology. 2012;799:237–252. doi: 10.1007/978-1-61779-346-2_15. http://dx.doi.org/10.1007/978-1-61779-346-2_15. [DOI] [PubMed] [Google Scholar]
- Mayne L, Kan ZY, Sevugan Chetty P, Ricciuti A, Walters BT, Englander SW. Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method. Journal of the American Society for Mass Spectrometry. 2011;22(11):1898–1905. doi: 10.1007/s13361-011-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne L, Paterson Y, Cerasoli D, Englander SW. Effect of antibody binding on protein motions studied by hydrogen-exchange labeling and two-dimensional NMR. Biochemistry. 1992;31(44):10678–10685. doi: 10.1021/bi00159a006. [DOI] [PubMed] [Google Scholar]
- Mysling S, Salbo R, Ploug M, Jorgensen TJ. Electrochemical reduction of disulfide-containing proteins for hydrogen/deuterium exchange monitored by mass spectrometry. Analytical Chemistry. 2014;86(1):340–345. doi: 10.1021/ac403269a. http://dx.doi.org/10.1021/ac403269a. [DOI] [PubMed] [Google Scholar]
- Pan J, Han J, Borchers CH, Konermann L. Hydrogen/deuterium exchange mass spectrometry with top-down electron capture dissociation for characterizing structural transitions of a 17 kDa protein. Journal of the American Chemical Society. 2009;131(35):12801–12808. doi: 10.1021/ja904379w. http://dx.doi.org/10.1021/ja904379w. [DOI] [PubMed] [Google Scholar]
- Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, et al. HDX workbench: Software for the analysis of H/D exchange MS data. Journal of the American Society for Mass Spectrometry. 2012;23(9):1512–1521. doi: 10.1007/s13361-012-0419-6. http://dx.doi.org/10.1007/s13361-012-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson Y, Englander SW, Roder H. An antibody binding site on cytochrome c defined by hydrogen exchange and two-dimensional NMR. Science. 1990;249(4970):755–759. doi: 10.1126/science.1697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrone GF, Iacob RE, Engen JR. Applications of hydrogen/deuterium exchange MS from 2012 to 2014. Analytical Chemistry. 2015;87(1):99–118. doi: 10.1021/ac5040242. http://dx.doi.org/10.1021/ac5040242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slysz GW, Baker CA, Bozsa BM, Dang A, Percy AJ, Bennett M, et al. Hydra: Software for tailored processing of H/D exchange data from MS or tandem MS analyses. BMC Bioinformatics. 2009;10:162. doi: 10.1186/1471-2105-10-162. http://dx.doi.org/10.1186/1471-2105-10-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters BT, Mayne L, Hinshaw JR, Sosnick TR, Englander SW. Folding of a large protein at high structural resolution. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(47):18898–18903. doi: 10.1073/pnas.1319482110. http://dx.doi.org/10.1073/pnas.1319482110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters BT, Ricciuti A, Mayne L, Englander SW. Minimizing back exchange in the hydrogen exchange—Mass spectrometry experiment. Journal of the American Society for Mass Spectrometry. 2012;23:2132–2139. doi: 10.1007/s13361-012-0476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Abzalimov RR, Bobst CE, Kaltashov IA. Conformer-specific characterization of nonnative protein states using hydrogen exchange and top-down mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(50):20087–20092. doi: 10.1073/pnas.1315029110. http://dx.doi.org/10.1073/pnas.1315029110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Mo J, Tao L, Russell RJ, Tymiak AA, Chen G, et al. Hydrogen/ deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: Methodology and applications. Drug Discovery Today. 2014;19(1):95–102. doi: 10.1016/j.drudis.2013.07.019. http://dx.doi.org/10.1016/j.drudis.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehl M, Rand KD, Jensen ON, Jorgensen TJD. Electron transfer dissociation facilitates the measurement of deuterium incorporation into selectively labeled peptides with single residue resolution. Journal of the American Chemical Society. 2008;130(51):17453–17459. doi: 10.1021/ja805573h. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Science. 1993;2(4):522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]