

Abstract
A 17-year-old male presented with acute hemolysis with stomatocytosis, elevated mean corpuscular hemoglobin concentration (MCHC), and osmotic gradient ektacytometry consistent with marked erythrocyte dehydration. Erythrocytes from both parents also demonstrated evidence of dehydration with elevated MCHC and abnormal ektacytometry, but neither to the degree of the patient. Genetic studies revealed the patient had hereditary xerocytosis (HX) due to a novel PIEZO1 mutation inherited from his mother and hemoglobin C (HbC) trait inherited from his father. HbC trait accentuated the erythrocyte dehydration of HX. Coinheritance of interrelated disorders and/or modifier alleles should be considered whenever severe erythrocyte dehydration is observed.
Keywords: dehydration, erythrocyte, genetic modifier, hemoglobin C, xerocytosis
1 | INTRODUCTION
Inheritance of modifier alleles is known to influence disease phenotype. One of the best characterized examples is coinheritance of a mutant G6PD allele in glucose-6-phosphate deficiency with a mutant UGT1A1 Gilbert’s disease allele, increasing severity of neonatal hyperbilirubinemia.1 We describe a young man with hereditary xerocytosis (HX) due to a PIEZO1 mutation whose erythrocyte dehydration and clinical severity were accentuated by coinheritance of hemoglobin C (HbC) trait. Modifier alleles are expected to ameliorate or worsen the erythrocyte dehydration seen in inherited and acquired disorders of the erythrocyte including HX, sickle cell disease, beta-thalassemia, and hereditary spherocytosis.
2 | METHODS AND RESULTS
2.1 | Kindred
A 17-year-old boy with HbC trait presented with bleeding, petechiae, and severe thrombocytopenia thought to be idiopathic thrombocytopenia in the setting of Epstein–Barr virus infection and was treated with intravenous immune globulin. Several days later, he developed acute Coombs-negative hemolytic anemia with abundant spherocytes and stomatocytes on peripheral smear. After resolution of the thrombocytopenic and hemolytic episode, few stomatocytes persisted, his hemoglobin normalized but his erythrocytes exhibited elevated mean corpuscular hemoglobin concentration (MCHC) (Table 1). Additional evaluation revealed cholelithiasis. Erythrocytes from his mother and father also showed elevated MCHC. Osmotic gradient ektacytometry (LoRRca MaxSis) revealed a pattern consistent with erythrocyte dehydration in the proband and both parents, with the proband’s erythrocytes more severely affected than either parent (Fig. 1).2
TABLE 1.
Hematologic parameters
| Mother | Father | Proband | Control | |
|---|---|---|---|---|
| Hemoglobin (g/dl) | 14 | 16 | 13 | |
| Mean corpuscular hemoglobin concentration (MCHC) | 35.3 | 35.9 | 36.8 | |
| Mean corpuscular hemoglobin (pg) | 29.0 | 28.5 | 28.7 | |
| Mean corpuscular volume (fl) | 82 | 79 | 78 | |
| Ektacytometry: Omin | 0.144 | 0.139 | 0.136 | 0.156 |
| Ektacytometry: Ohyper | 469 | 459 | 412 | 490 |
| PIEZO1 R1955C | Yes | No | Yes | |
| Hb C trait | No | Yes | Yes |
FIGURE 1.
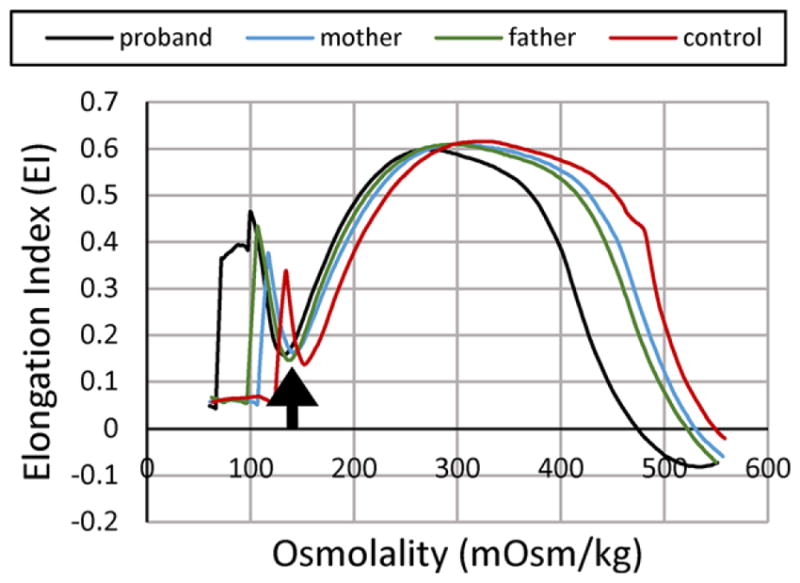
Osmotic gradient ektacytometry. Characteristics of erythrocyte dehydration in the proband and his parents include reduced deformability index with a leftward shift of the minimal osmolality point (Omin, the osmolality yielding release of 50% of hemoglobin reflecting the surface area to volume ratio of erythrocytes, arrow) and a leftward shift of the high osmolality region (which contains Ohyper or O′)
2.2 | Genetic studies
To determine the etiology of the proband’s anemia, whole exome sequencing was performed on peripheral blood derived genomic DNA as described.3 Whole exome sequencing data were aligned to the human genome (hg19) and analyzed. Mean target coverage was high, 67.6, and >95.3% of all targeted bases were read more than 10×. Genotypes for single nucleotide and indel variants were called using the GATK Haplotype Caller and submitted to the Annovar annotation pipeline.
A variant was identified in the PIEZO1 gene, with abnormal DNA sequences in the alignment of multiple short sequencing reads in exon 5. The corresponding DNA sequence revealed a heterozygous G to A substitution (nucleotide position chr16:8878 6879) changing Arg (CGC) to Cys (TGC) at amino acid 1955 (NM_001142864:c.C5863T:p.R1955C). The PIEZO1 missense mutation was predicted to be pathogenic with a CADD phred score of 14.9, and damaging by SIFT as well as several other mutation prediction algorithms. The mutation is in a highly conserved residue, with a highly significant vertebrate phastCons score of 1.0 and a highly significant PhyloP conservation score of 2.9. The PIEZO1 R1955 amino acid is highly conserved across vertebrate species, including the clades of placental mammals, the extant Eutherians, birds, reptiles and frogs. This variant is very rare. It is not present in the NHLBI Exome Sequencing Project database. It is present at frequency of 0.0004 in both the 1000 Genomes and ExAC (>60,000 exomes) databases. In addition, a heterozygous HbC allele was confirmed at the beta-globin gene (HBB) locus (chr11:5248233 C to T; NM_000518:c.G19A:p.E7K).
The variants in the PIEZO1 and the HBB genes identified by exome sequencing were validated by conventional Sanger sequencing (not shown). No other deleterious variants were detected in the coding regions of the PIEZO1, KCNN4, and SLC4A1 genes. Sequencing genomic DNA from the parents revealed that the proband inherited the PIEZO1 variant from his Caucasian mother and the HbC allele from his African American father. These studies were approved by the Yale Institutional Review Board (#12377).
3 | DISCUSSION
HX (Greek ξηρο, xero—dry) and related disorders are characterized by dehydrated erythrocytes with decreased water and solute content.4,5 These dominantly inherited disorders are associated with well-compensated anemia, with complications including hydrops fetalis, hemolytic and aplastic episodes, thromboses, gallstones, and propensity for iron overload as an adult.6 Laboratory findings include compensated anemia, reticulocytosis, elevated MCHC, rare target cells, dessicytes, and stomatocytes on peripheral blood smear, decreased erythrocyte osmotic fragility, and a characteristic pattern on osmotic gradient ektacytometry.7 Borderline platelet counts have been noted and exercise-induced hemolysis has also been reported. Most cases are due to mutations in PIEZO1.5,8
The clinical, laboratory, and genetic findings in this kindred were supportive of the diagnosis of HX, with the proband first coming to medical attention during a viral illness. His mother, who also has HX, was unaware that she had an inherited anemia. Several unexpected observations were made when performing diagnostic studies: (i) Erythrocytes from the proband and his mother had evidence of dehydration, with the dehydration of the proband’s erythrocytes much more severe than his mother’s, even though both have the same PIEZO1 mutation. (ii) Erythrocytes from the proband’s father also exhibited dehydration (elevated MCHC, leftward shifted ektacytometry), and while he does not have a PIEZO1 mutation, he has hemoglobin AC.2,9 The proband has both HX and hemoglobin AC contributing to the marked erythrocyte dehydration phenotype.
HbC is a variant hemoglobin due to a glutamic acid to lysine substitution at codon 6 of the HBB gene. Its primary clinical significance is when it is coinherited in trans with sickle cell disease or beta-thalassemia. In HbC carriers, ~28–44% of total hemoglobin is HbC and there is typically no anemia even though erythrocyte survival may be shortened.10,11 Erythrocytes of HbC carriers are dehydrated, thus red blood cell indices demonstrate elevated MCHC.12,13 Although there have been conflicting studies, HbC appears to confer resistance to malaria in both heterozygotes and homozygotes.14
Numerous disorders are associated with erythrocyte dehydration. Primary disorders include HX and related syndromes, whereas dehydration is a secondary phenomenon in sickle cell disease, beta-thalassemia, HbC, and hereditary spherocytosis.4 Thus, coinheritance of these disorders may influence erythrocyte dehydration and clinical severity, even when the inheritance of one entity is believed to be benign, such as HbC. For example, in a mouse model of sickle cell disease, a genetic variant that activates KCl cotransport worsens erythrocyte dehydration and clinical manifestations of sickle cell disease.15 Similarly, a patient with hemoglobin SC and hereditary spherocytosis suffered from recurrent sequestration crisis with altered erythrocyte rheology that improved after splenectomy.16 In our proband, coinheritance of two variants associated with erythrocyte dehydration, a PIEZO1 variant and HbC trait, led to more severe erythrocyte dehydration, aggravating the clinical phenotype of HX.
Genome-wide association studies (GWAS) have demonstrated that a significant component of erythrocyte hydration is genetically determined.17,18 In normal humans, variation in indices of erythrocyte hydration, including cell volume and hemoglobin, is strongly influenced by genetic factors.19,20 Because several pathways regulate hydration in the erythrocyte, it follows that any of the proteins involved in maintaining erythrocyte volume homeostasis, transporters, channels, their regulatory kinases and phosphatases, and other regulatory proteins may serve as modifiers of erythrocyte hydration.21
Thus, both coinheritance of two interrelated disorders and/or modifier alleles, which by themselves could be asymptomatic, should be considered whenever more severe erythrocyte dehydration and clinical severity are observed in a patient with primary or secondary abnormalities of erythrocyte hydration.
Acknowledgments
Grant sponsor: Doris Duke Charitable Foundation; Grant sponsor: NIDDK; Grant number: RO1DK104046.
Abbreviations
- HbC
hemoglobin C
- HX
hereditary xerocytosis
- MCHC
mean corpuscular hemoglobin concentration
Footnotes
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
References
- 1.Kaplan M, Renbaum P, Levy-Lahad E, et al. Gilbert syndrome and glucose-6-phosphate dehydrogenase deficiency: A dose-dependent genetic interaction crucial to neonatal hyperbilirubinemia. Proc Natl Acad Sci USA. 1997;94:12128–12132. doi: 10.1073/pnas.94.22.12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Da Costa L, Suner L, Galimand J, et al. Diagnostic tool for red blood cell membrane disorders: Assessment of a new generation ektacytometer. Blood Cells Mol Dis. 2016;56:9–22. doi: 10.1016/j.bcmd.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glogowska E, Lezon-Geyda K, Maksimova Y, Schulz VP, Gallagher PG. Mutations in the Gardos channel (KCNN4) are associated with hereditary xerocytosis. Blood. 2015;126:1281–1284. doi: 10.1182/blood-2015-07-657957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallagher PG. Disorders of red cell volume regulation. Curr Opin Hematol. 2013;20:201–207. doi: 10.1097/MOH.0b013e32835f6870. [DOI] [PubMed] [Google Scholar]
- 5.Andolfo I, Alper SL, De Franceschi L, et al. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood. 2013;121:3925–3935. S1–S12. doi: 10.1182/blood-2013-02-482489. [DOI] [PubMed] [Google Scholar]
- 6.Archer NM, Shmukler BE, Andolfo I, et al. Hereditary xerocytosis revisited. Am J Hematol. 2014;89:1142–1146. doi: 10.1002/ajh.23799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glogowska E, Gallagher PG. Disorders of erythrocyte volume homeostasis. Int J Lab Hematol. 2015;37(1):85–91. doi: 10.1111/ijlh.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zarychanski R, Schulz VP, Houston BL, et al. Mutations in the mechanotransduction protein PIEZO1 are associated with hereditary xerocytosis. Blood. 2012;120:1908–1915. doi: 10.1182/blood-2012-04-422253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson RM, Ravindranath Y. Osmotic scan ektacytometry in clinical diagnosis. J Pediatr Hematol Oncol. 1996;18:122–129. doi: 10.1097/00043426-199605000-00005. [DOI] [PubMed] [Google Scholar]
- 10.Ballas SK, Larner J, Smith ED, Surrey S, Schwartz E, Rappaport EF. The xerocytosis of Hb SC disease. Blood. 1987;69:124–130. [PubMed] [Google Scholar]
- 11.Prindle KH, Jr, McCurdy PR. Red cell lifespan in hemoglobin C disorders (with special reference to hemoglobin C trait) Blood. 1970;36:14–19. [PubMed] [Google Scholar]
- 12.Brugnara C, Kopin AS, Bunn HF, Tosteson DC. Regulation of cation content and cell volume in hemoglobin erythrocytes from patients with homozygous hemoglobin C disease. J Clin Invest. 1985;75:1608–1617. doi: 10.1172/JCI111867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hinchliffe RF, Norcliffe D, Farrar LM, Lilleyman JS. Mean cell haemoglobin concentration in subjects with haemoglobin C, D, E and S traits. Clin Lab Haematol. 1996;18:245–248. [PubMed] [Google Scholar]
- 14.Travassos MA, Coulibaly D, Laurens MB, et al. Hemoglobin C trait provides protection from clinical falciparum malaria in malian children. J Infect Dis. 2015;212:1778–1786. doi: 10.1093/infdis/jiv308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown FC, Conway AJ, Cerruti L, et al. Activation of the erythroid K-Cl cotransporter Kcc1 enhances sickle cell disease pathology in a humanized mouse model. Blood. 2015;126:2863–2870. doi: 10.1182/blood-2014-10-609362. [DOI] [PubMed] [Google Scholar]
- 16.Warkentin TE, Barr RD, Ali MA, Mohandas N. Recurrent acute spleni sequestration crisis due to interacting genetic defects: Hemoglobin SC disease and hereditary spherocytosis. Blood. 1990;75:266–270. [PubMed] [Google Scholar]
- 17.Ganesh SK, Zakai NA, van Rooij FJ, et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat Genet. 2009;41:1191–1198. doi: 10.1038/ng.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamatani Y, Matsuda K, Okada Y, et al. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat Genet. 2010;42:210–215. doi: 10.1038/ng.531. [DOI] [PubMed] [Google Scholar]
- 19.Soranzo N, Spector TD, Mangino M, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet. 2009;41:1182–1190. doi: 10.1038/ng.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferreira MA, Hottenga JJ, Warrington NM, et al. Sequence variants in three loci influence monocyte counts and erythrocyte volume. Am J Hum Genet. 2009;85:745–749. doi: 10.1016/j.ajhg.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallagher PG. Transporting down the road to dehydration. Blood. 2015;126:2775–2776. doi: 10.1182/blood-2015-10-675488. [DOI] [PubMed] [Google Scholar]