

Abstract
Objective
To describe the phenomenon of tau-negative amnestic dementia mimicking Alzheimer disease (AD) clinically and radiologically and to highlight the importance of biomarkers in AD research.
Methods
Eight participants with amnestic mild cognitive impairment or AD dementia were evaluated by a behavioral neurologist and had a standardized neuropsychological battery performed. All participants completed structural (MRI) and molecular (amyloid and tau PET) imaging. AD-signature thickness and adjusted hippocampal volume served as structural biomarkers, while standardized uptake value ratios (SUVRs) from validated regions of interest for amyloid and tau PET were used to determine molecular biomarker status.
Results
All participants were thought to have AD as the primary driver of their symptoms before any PET imaging. All participants had hippocampal atrophy, and 2 participants fell below the AD-signature thickness cutoff for elderly controls (2.57), with a further 3 falling below the more stringent cutoff based on young controls (2.67). Four participants were amyloid positive (SUVR >1.42), and all were tau negative (SUVR <1.33).
Conclusions
The participants presented here were clinically impaired, with structural imaging evidence of neurodegeneration, in the absence of any significant tau accumulation. Therefore, AD is unlikely as a cause of their clinical presentation and neurodegenerative imaging findings. Several implications are discussed, including the need to establish amyloid and tau positivity in N+ participants before enrolling them in trials of disease-modifying therapy agents for AD.
The availability first of amyloid PET imaging and now of tau PET imaging has fundamentally changed our approach to the antemortem diagnosis of dementia due to Alzheimer disease (AD).1,2 While it has long been known from neuropathologic studies that the clinical diagnosis of what was previously called probable AD in the 1984 criteria was only about 80% sensitive and specific at best, the neuropathologic basis of amnestic mild cognitive impairment (MCI) shows an even weaker relationship to neuropathologic AD.3 Recent studies using amyloid PET imaging revealed similar inaccuracies in the clinical diagnoses of MCI or AD dementia.4,5 With the availability of tau PET, further experience has been gained in the clinical-biomarker associations with pathophysiologic AD that will also lead to more refined diagnoses of what is and is not AD.1,6
Recently, the A/T/N biomarker classification scheme was proposed,7 allowing independent classification of amyloid, tau, and neurodegeneration status. Patients who are A+ are thought to be on the AD pathway, with A− participants either being normal (A−/T−/N−) or harboring some non-AD pathology (A−/T−/N+, A−/T+/N+, A−/T+/N−).1 Crucially, however, amyloid positivity does not imply that neurodegeneration is due to AD: T−/N+ cases likely have another pathology driving their degeneration regardless of amyloid status.1 With the recent addition of tau PET, this group can be identified and studied more readily.2 If A+/T−/N+ participants can mimic AD clinically, this group may inadvertently be included in AD trials, with serious implications. Here, we report 8 T−/N+ cases that were clinically diagnosed with AD dementia or amnestic MCI (aMCI) due to AD.
Methods
Clinical assessment
All 8 participants were evaluated in the Mayo Clinic Alzheimer's Disease Research Center. A consensus diagnosis was given at a multidisciplinary meeting based on established criteria.8 All participants completed a comprehensive neuropsychological battery as part of the Uniform Data Set designed by the National Alzheimer's Coordinating Center. Test scores were then converted to z scores controlling for age, sex, and education (https://www.alz.washington.edu/WEB/npsych_means.html). Because conversion is not available for the Rey-Osteroth Complex Figure, the raw score was converted to a standard score based on Mayo's Older Americans Normative Studies.
Standard protocol approvals, registrations, and patient consents
The Mayo Clinic Institutional Review Board reviewed and approved this study. Written informed consent was obtained from all participants or their guardians.
Imaging protocol
Details of PET imaging and analysis have been described elsewhere.1 Briefly, Pittsburgh compound B was used for amyloid PET, and flortaucipir (AV-1451) was used for tau PET. An amyloid standardized uptake value ratio (SUVR) was derived from the voxel-weighted median uptake in the prefrontal, orbitofrontal, parietal, temporal, anterior and posterior cingulate, and precuneus regions of interest.1 The tau SUVR was based on the voxel-weighted median uptake in the entorhinal, amygdala, parahippocampal, fusiform, inferior temporal, and middle temporal regions of interest.1 MRI was performed on 1 of 3 compatible 3T GE systems (GE Healthcare, Chicago, IL), and the AD-signature thickness (AD-sig) and hippocampal volume adjusted for intracranial volume were calculated. Previously established cut points were used.1
Results
Clinical findings
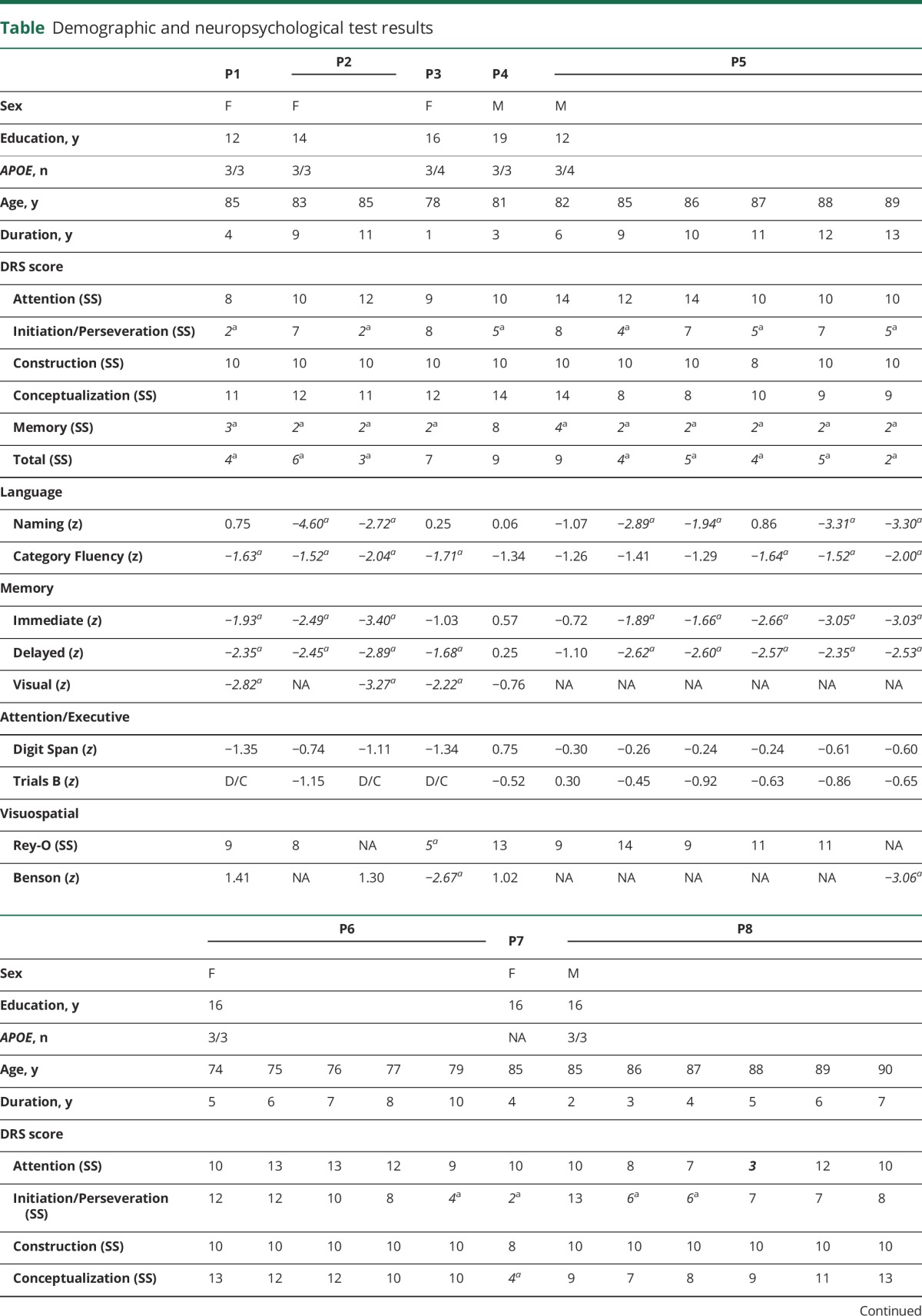
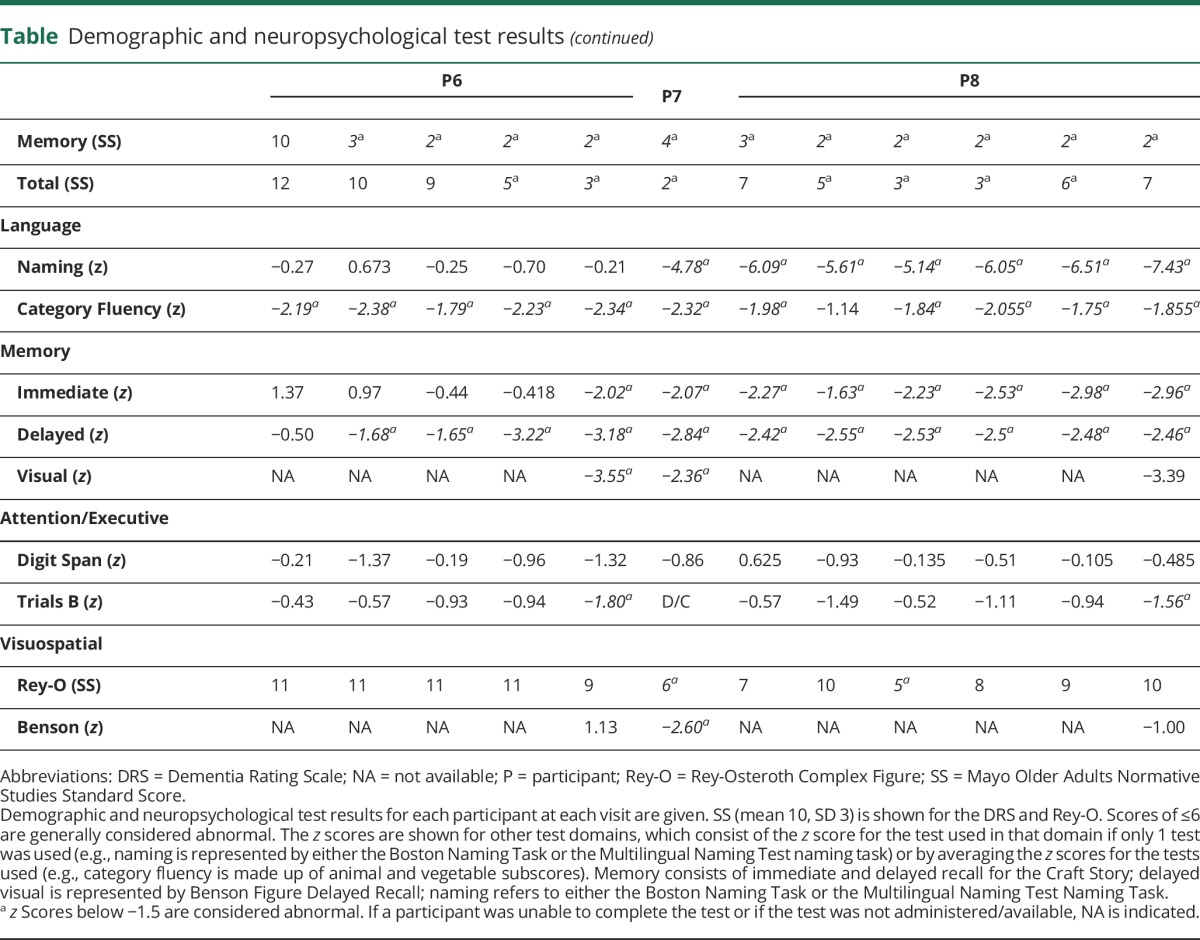
All participants were thought to have AD as the primary driver of their symptoms before any PET imaging and met the criteria for AD dementia (patients 1, 2, 5–8) or aMCI (patients 3 and 4). Neuropsychological results are summarized in the table. Both participants 1 and 2 had experienced several years of progressive short-term memory (STM) loss before presenting for evaluation, by which time some language impairment was evident in participant 2. Participants 3 and 4 had STM loss, corroborated by informants, but with minimal impairment of their activities of daily living. Participant 5 had an initial course of ≈6 years of progressive STM loss before other cognitive domains became affected. Participant 6 passed through a phase of aMCI before being diagnosed with dementia at the last visit. Participant 7 presented with STM loss for a few years before developing more general cognitive impairment by the time of initial assessment. Finally, participant 8 presented with STM loss, which remained the main cause of impairment in activities of daily living throughout follow-up despite poor test scores for nonmemory domains.
Table.
Demographic and neuropsychological test results
Imaging findings
Imaging results are shown in the figure. A cognitively normal and biomarker-negative (A−/T−/N−) control and a participant with typical AD dementia (A+/T+/N+) are shown for reference. All participants had hippocampal atrophy (hippocampal volume adjusted for intracranial volume <−1.15), and 3 participants fell below the AD-sig cutoff for elderly controls (2.57). Four participants were amyloid positive (SUVR >1.42), and all were tau negative (SUVR <1.33).
Figure. Results of structural and molecular imaging.
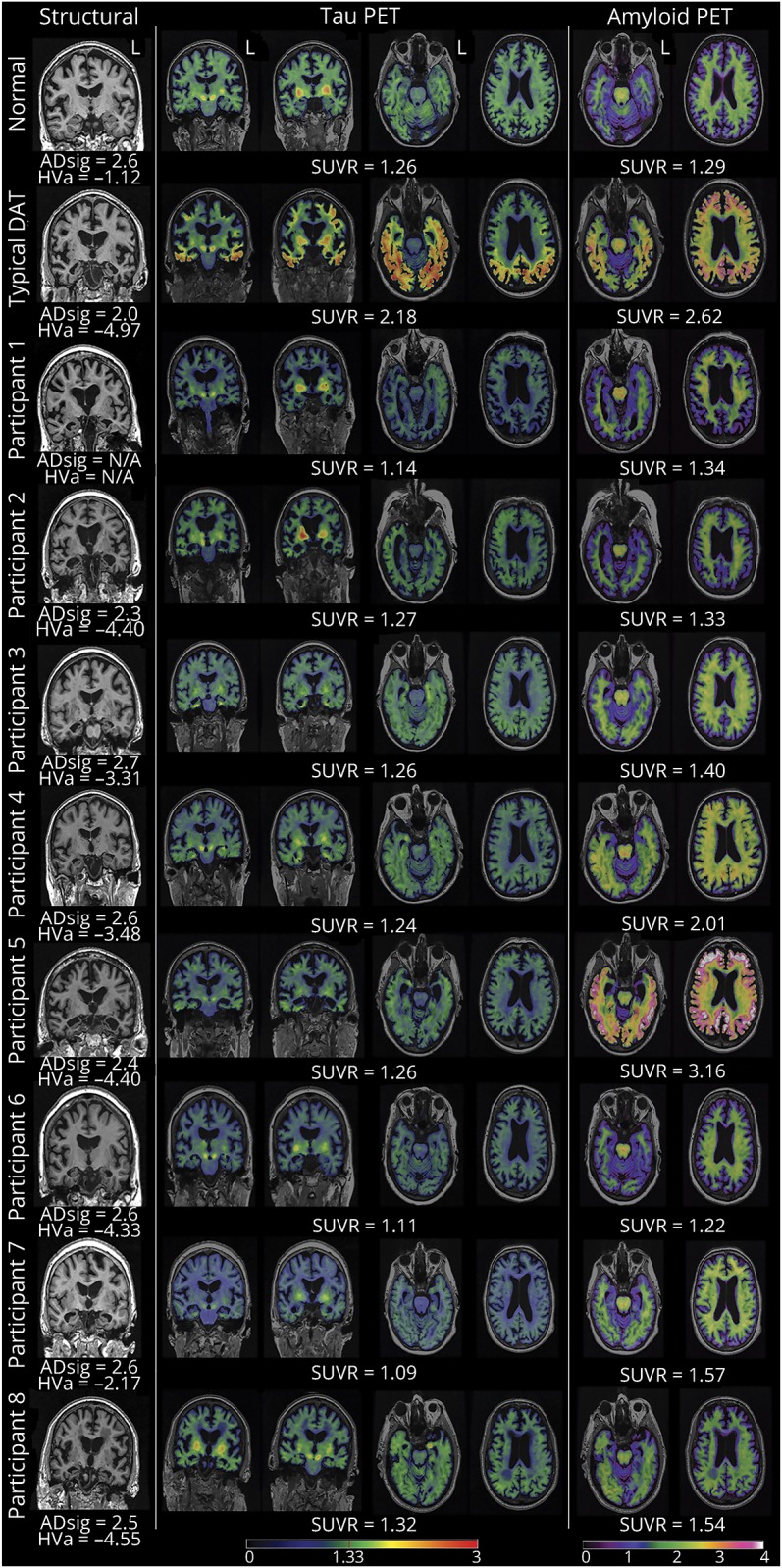
Representative images from coronal MRI (left), tau PET imaging (middle), and amyloid PET imaging (right) for each participant, including a control and a typical case with biomarker-positive AD dementia. The AD-sig value and SUVRs for tau and amyloid PET are shown below each participant's scans. AD-sig and HVa processing for participant 1 were not available. AD = Alzheimer disease; AD-sig = Alzheimer disease signature thickness; DAT = dementia of the Alzheimer type; HVa = hippocampal volume adjusted for intracranial volume; SUVR = standardized uptake value ratio.
Discussion
The participants presented here were clinically impaired with structural imaging evidence of neurodegeneration in the absence of any significant tau accumulation. Therefore, AD is unlikely to be a cause of their clinical presentation and neurodegenerative imaging findings. This highlights several important points about AD and the clinical and imaging metrics used to diagnose it. The lack of specificity of clinical AD criteria is evident, consistent with the fact that a significant proportion of cases with clinically suspected AD dementia lack AD pathology or biomarker evidence for AD, while a large proportion of clinically normal individuals have AD pathology or biomarker evidence supporting AD. Our findings plus prior reports have made it highly likely that many participants enrolled in AD dementia trials over the last few decades may not have had AD pathophysiology driving their clinical syndrome.4,5,9 In addition, the presence of hippocampal atrophy in all participants and volume loss in other characteristic AD cortical areas in some emphasize the limitations of structural imaging, a problem of etiologic nonspecificity that increases with age as a greater proportion of cognitively normal participants fall below cutoff values.9 However, the 4 participants who were amyloid positive emphasize the need for biomarker models that incorporate both amyloid and tau.6
With the ATN model, the 8 participants would correctly be labeled as A−/T−/N+ (participants 1–3 and 6) or A+/T−/N+ (participants 2, 5, 7 and 8), 2 patterns that are inconsistent with current models of pure AD given the absence of tau in the setting of neurodegeneration.1,7 We have previously postulated that neurodegeneration in the A−/T−/N+ group is the result of a heterogeneous group of non-AD pathologies that increase in prevalence with age such as hippocampal sclerosis, argyrophilic grain disease, and cerebrovascular disease.9 Similarly, for the group of A+/T−/N+ participants, we suspect that a non-AD process is driving the neurodegeneration, with amyloidosis representing an early and asymptomatic phase of AD. Among cognitively normal participants, the prevalence of the A−/T−/N+ and A+/T−/N+ groups increases above the age of 75 years and peaks at ≈18% to 22% and 17% to 18%, respectively.9 From the data presented here and other data from our center, we estimate that ≈27.5% of aMCI or dementia thought to be due to AD in those >75 years of age might be tau-PET negative, with half of these being amyloid-PET negative (appendix e-1, http://links.lww.com/WNL/A239). This is consistent with prior reports that ≈50% of aMCI in the >80-year-old cohort is caused by non-AD pathologies, most notably hippocampal sclerosis and argyrophilic grain disease.3 The age and the focal medial temporal and hippocampal atrophy in our participants would be consistent with hippocampal sclerosis,3,10 a suspicion that was recently confirmed in 1 participant (participant 2) who had TDP-43–positive hippocampal sclerosis at autopsy and minimal evidence for AD (appendix e-1).
A notable limitation of our study is the fact that biomarker status was assessed with PET only, which may lack sensitivity to low Thal and Braak stages of the disease, so early AD may have been missed in our biomarker-negative cases. However, this would not account for their prominent amnesia and neurodegeneration, which would have to have an additional non-AD etiology. Similarly, the lack of pathologic confirmation can be seen as a weakness, but the delay between clinical presentation and pathologic examination complicates clinic-pathologic correlations. We have compelling in vivo evidence that our participants lacked significant tau deposition, which would have been suspected if their degeneration was due to AD, again implicating non-AD pathologies.
The cases reported here highlight the importance of establishing amyloid and tau positivity in N+ participants before enrolling them in trials of disease-modifying therapy agents for AD.
Acknowledgment
The authors are grateful to the participants and their families for participating in aging and dementia research. They also thank AVID Radiopharmaceuticals, Inc, for its support in supplying AV-1451 precursor, chemistry production advice and oversight, and Food and Drug Administration regulatory cross-filing permission and documentation needed for this work.
Glossary
- AD
Alzheimer disease
- AD-sig
Alzheimer disease–signature thickness
- aMCI
amnestic mild cognitive impairment
- MCI
mild cognitive impairment
- STM
short-term memory
- SUVR
standardized uptake value ratio
Author contributions
Hugo Botha and William G. Mantyh: acquisition of data, analysis and interpretation of data, manuscript preparation. Jonathan Graff-Radford: acquisition of data, analysis and interpretation of data, critical revision of manuscript for intellectual content. Mary M. Machulda: acquisition of data, interpretation of data, critical revision of manuscript for intellectual content. Scott A. Przybelski and Heather J. Wiste: analysis and interpretation of data. Matthew L. Senjem and Joseph E. Parisi: interpretation of data. Bradley F. Boeve: analysis and interpretation of data, critical revision of manuscript for intellectual content. Ronald C. Petersen and Melissa E. Murray: analysis and interpretation of data. Val J. Lowe: acquisition of data, analysis and interpretation of data, study supervision, critical revision of manuscript for intellectual content. David S. Knopman: acquisition of data, analysis and interpretation of data. Clifford R. Jack: analysis and interpretation of data, study supervision, critical revision of manuscript for intellectual content. David T. Jones: acquisition of data, analysis and interpretation of data, manuscript preparation.
Study funding
Study funded by P50 AG016574, and AG011378 and AG041851 from the National Institute on Aging and the Robert H. and Clarice Smith and Abigail van Buren Alzheimer's Disease Research Program.
Disclosure
H. Botha, W. Mantyh, and J. Graff-Radford report no disclosures relevant to the manuscript. M. Machulda receives research support from the NIH and the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation. S. Przybelski, H. Wiste, M. Senjem, and J. Parisi report no disclosures relevant to the manuscript. R. Petersen works as a consultant for Merck Inc, Roche Inc, Biogen Inc, Eli Lily and Company, and Genentech Inc; receives publishing royalties for Mild Cognitive Impairment (Oxford University Press, 2003); and receives research support from the NIH and the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation. M. Murray has served on the editorial boards of BMC Neurology and Frontiers in Neurology and has been a consultant for Avid Radiopharmaceuticals. B. Boeve has served as an investigator for clinical trials sponsored by GE Healthcare and FORUM Pharmaceuticals; receives royalties from the publication of a book titled Behavioral Neurology of Dementia (Cambridge Medicine, 2009); serves on the Scientific Advisory Board of the Tau Consortium; has consulted for Isis Pharmaceuticals; and receives research support from the NIH, the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation, and the Mangurian Foundation. V. Lowe is a consultant for Bayer Schering Pharma, Merck Research, and Piramal Imaging Inc and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals, the NIH (National Institute on Aging, National Cancer Institute), the Elsie and Marvin Dekelboum Family Foundation, the Liston Family Foundation, and the MN Partnership for Biotechnology and Medical Genomics. D. Knopman receives research support from the NIH and the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation. He serves on a Data Safety Monitoring Board for Lundbeck Pharmaceuticals and for the Dominantly Inherited Alzheimer Network study and is an investigator in clinical trials sponsored by Biogen, TauRX Pharmaceuticals, Lilly Pharmaceuticals, and the Alzheimer's Disease Treatment and Research Institute, University of Southern California. C. Jack receives research funding from the NIH and the Alexander Family Alzheimer's Disease Research Professorship at Mayo Clinic. D. Jones reports no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Jack CR, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement 2017;13:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwarz AJ, Yu P, Miller BB, et al. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 2016;139:1539–1550. [DOI] [PubMed] [Google Scholar]
- 3.Petersen RC, Parisi JE, Dickson DW, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol 2006;63:665–672. [DOI] [PubMed] [Google Scholar]
- 4.Lowe VJ, Peller PJ, Weigand SD, et al. Application of the National Institute on Aging-Alzheimer's Association AD criteria to ADNI. Neurology 2013;80:2130–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N Engl J Med 2014;370:322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jack CR, Knopman DS, Jagust WJ, Petersen RC. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jack CR Jr, Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016;87:539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jack CR, Wiste HJ, Weigand SD, et al. Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50-95 years: a cross-sectional study. Lancet Neurol 2017;16:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson PT, Smith CD, Abner EL, et al. Hippocampal sclerosis of aging, a prevalent and high-morbidity brain disease. Acta Neuropathol 2013;126:161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]