

Abstract
Objective
To evaluate 18F-AV-1451 tau PET binding among microtubule-associated protein tau (MAPT) mutation carriers.
Methods
Using a case-control study, we quantitatively and qualitatively compared tau PET scans in 10 symptomatic and 3 asymptomatic MAPT mutation carriers (n = 13, age range 42–67 years) with clinically normal (CN) participants (n = 241, age range 42–67 years) and an Alzheimer disease (AD) dementia cohort (n = 30, age range 52–67 years). Eight participants had MAPT mutations that involved exon 10 (N279K n = 5, S305N n = 2, P301L n = 1) and tend to form 4R tau pathology, and 5 had mutations outside exon 10 (V337M n = 2, R406W n = 3) and tend to form mixed 3R/4R tau pathology.
Results
Tau PET signal was qualitatively and quantitatively different between participants with AD, CN participants, and MAPT mutation carriers, with the greatest signal intensity in those with AD and minimal regional signal in MAPT mutation carries with mutations in exon 10. However, MAPT mutation carriers with mutations outside exon 10 had uptake levels within the AD range, which was significantly higher than both MAPT mutation carriers with mutations in exon 10 and controls.
Conclusions
Tau PET shows higher magnitude of binding in MAPT mutation carriers who harbor mutations that are more likely to produce AD-like tau pathology (e.g., in our series, the non–exon 10 families tend to accumulate mixed 3R/4R aggregates). Exon 10 splicing determines the balance of 3R and 4R tau isoforms, with some mutations involving exon 10 predisposing to a greater proportion of 4R aggregates and consequently a lower level of AV-1451 binding, as seen in this case series, thus supporting the notion that this tau PET ligand has specific binding properties for AD-like tau pathology.
Many neurodegenerative diseases are characterized by the presence of tau protein pathology.1 In these disorders, the tau protein is abnormally hyperphosphorylated before forming various types of intracellular aggregates (e.g., paired helical filaments, ribbons, and/or straight filaments). There are 6 isoforms of the tau protein encoded by the microtubule-associated protein tau (MAPT) gene, with half of these isoforms including 3 conserved microtubule-binding domain sequence repeats (3R tau) and the other half containing 4 repeats (4R tau). The inclusion of exon 10 in gene transcripts determines whether the protein product will be 3R or 4R tau. MAPT gene mutations that alter the splicing of exon 10 lead to excess 4R tau relative to 3R tau.2 The aggregate-forming filaments come in at least 3 distinct types: homogeneous 3R, homogeneous 4R, and heterogeneous 3R/4R.3,4 The broad spectrum of heterogeneous filaments formed with mixtures of 3R/4R tau allows diverse physical and biological properties, with more restricted properties for homogeneous 3R and 4R filaments. The tau aggregates in Alzheimer disease (AD) are paired helical filaments composed of 3R/4R mixtures of all 6 tau isoforms5; in progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD), they are composed of 4R aggregates, and in Pick disease, they are composed of 3R aggregates. A striking feature of the pathology observed in MAPT mutations is that they may resemble the inclusions seen in any of these tauopathies (AD, PSP, CBD, or Pick disease).6
A major advance in the study of these tau-associated neurodegenerative diseases has been the development of PET ligands that bind to tau protein aggregates. The most widely used ligand currently, 18F-AV-1451 or flortaucipir (tau PET), formally called T807, was developed to bind to AD-type tau aggregates.7 While 18F-AV-1451 strongly binds tau in AD dementia, the unique physical properties present in 3R/4R tau mixtures that led to this strong binding may not extend to homogeneous 3R and 4R tau aggregates.8–12 However, in vivo tau PET binding has been reported in patients with PSP,13,14 those with CBD,15,16 and MAPT mutation carriers.17–19 The current literature has not reached a consensus regarding in vivo 18F-AV-1451 binding in non-Alzheimer tauopathies. Given the heterogeneous nature of the tau pathology observed in MAPT mutations, they allow the in vivo evaluation of tau PET as a biomarker for various forms of tau pathology within a similar patient population. Therefore, we compared tau PET binding between MAPT mutation carriers, with heterogeneous tau aggregates, to participants with AD dementia with 3R/4R tau and clinically normal (CN) participants without significant tau pathology.
Methods
Standard protocol approvals, registrations, and patient consents
All participants provided written consent with approval of the Mayo Clinic Foundation and Olmsted Medical Center Institutional Review boards.
Participants
Participants were part of the Mayo Clinic Study of Aging or Mayo Clinic Alzheimer's Disease Research Center as described previously,20,21 as well as the Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (https://clinicaltrials.gov/show/NCT02372773) and Advancement of Research and Treatment in Frontotemporal Lobar Degeneration (https://clinicaltrials.gov/show/NCT02365922) protocols. All participants or their designees provided written consent with approval of the Olmsted Medical Center and/or Mayo Clinic Institutional Review boards. This study included 10 symptomatic and 3 asymptomatic MAPT mutation carriers (n = 13, age range 42–67 years) compared with amyloid-negative CN participants (n = 241, age range 42–67 years) and an amyloid-positive AD dementia cohort (n = 30, age range 52–67 years). Eight participants had MAPT mutations that involved exon 10 (N279K n = 5, S305N n = 2, P301L n = 1) and tend to form 4R tau pathology, and 5 had mutations outside exon 10 (V337M n = 2, R406W n = 3) and tend to form mixed 3R/4R tau pathology. Table provides a description of the MAPT participants. For an illustrative comparison, we also included tau PET imaging from 1 symptomatic participant with a progranulin (PGRN) gene mutation. All gene mutation carriers (MAPT and PGRN) were amyloid-PET negative.
Table.
MAPT mutation carrier characteristics
Multimodality neuroimaging
For tau PET, participants were injected with 370 MBq (range 333–407 MBq) 18F-AV-1451 before imaging, and imaging was performed as a 20-minute PET acquisition between 80 and 100 minutes after injection. PET images were acquired with 1 of 2 PET/CT scanners (DRX; GE Healthcare, Chicago, IL) operating in 3-dimensional mode (septa removed). A CT scan was obtained for attenuation correction. PET sinograms were iteratively reconstructed into a 256-mm field of view (FOV). The pixel size was 1.0 mm and the slice thickness was 3.3 mm. Standard corrections were applied. All participants underwent MRI scanning at 3T with a standardized protocol that included a 3-dimensional magnetization-prepared rapid gradient echo sequence. Parameters were as follows: repetition time/echo time/T1 time 2,300/3/900 milliseconds, flip angle 8°, FOV 26 cm, in-plane matrix 256 × 256 with a phase FOV of 0.94, and slice thickness 1.2 mm. Scans were performed on 1 of 2 scanners from the same manufacturer.
Image analysis
Tau PET cortical uptake was assessed with regions of interest (ROIs) defined by an in-house–modified version of the AAL atlas (47 bilateral ROIs) as previously described.22 Individual tau PET ROI median uptake values were normalized to cerebellar crus to calculate regionally specific standardized uptake value ratios (SUVRs). Data with and without partial volume correction were evaluated with the previously described method.22
Statistical methods
To compare tau PET SUVR in controls, MAPT mutation carriers, and those with AD, both visual and analytic methods were used. The age range of the comparison cohorts used was limited to 42 to 67 years, which is the age range of those with the MAPT mutation. For a visual comparison, boxplots of the raw tau PET SUVR values were created for each ROI. A linear model with a 3-level group variable (CN, MAPT, AD) predicting tau PET SUVR adjusted for age was run within the brain ROI for MAPT mutations (i.e., the temporal pole). An identical exploratory analysis of every ROI is also reported in the supplementary material. Because tau PET SUVR in the temporal pole is skewed right, a log transformation was used in the model. A t test was then performed to compare the least-squares means of log tau PET SUVR from the model for each of the 3 groups. To assess the validity of the linear models, a QQ-plot of the residuals was used. For the temporal pole region, the residuals were satisfactorily normally distributed. Linear models were similarly used to examine the relationship between imaging variables and clinical severity.
Results
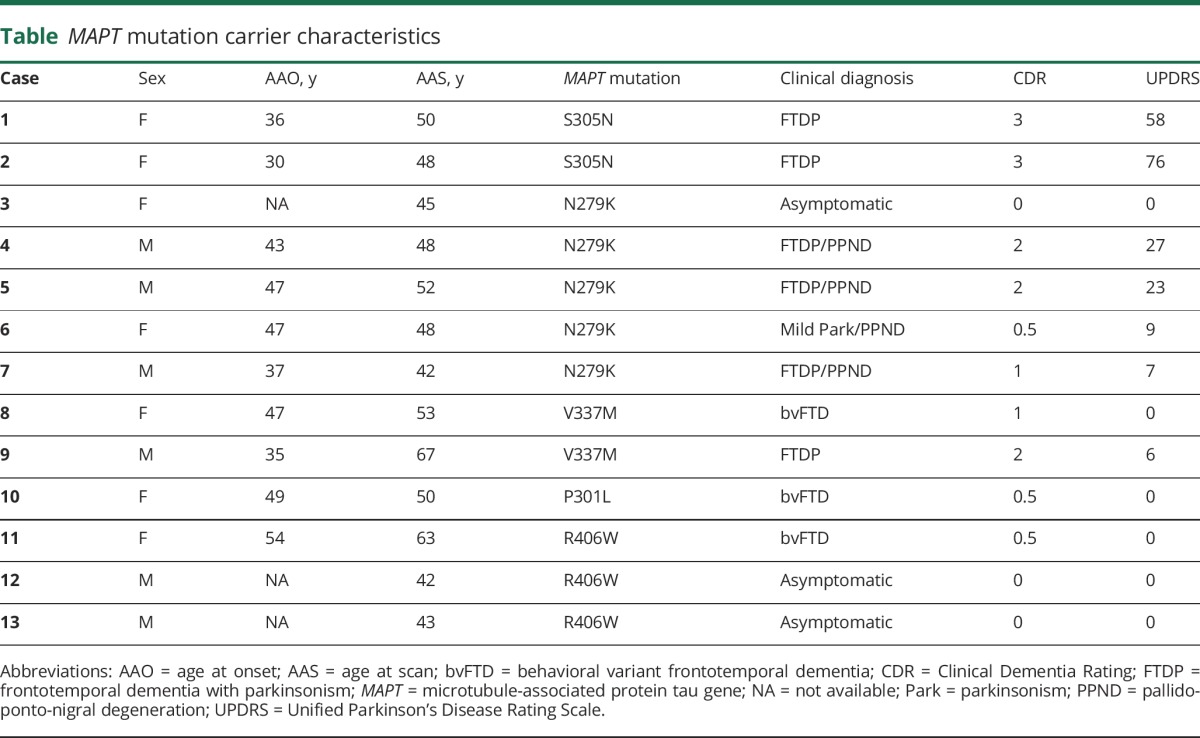
On visual (figure 1) and quantitative (figure 2) assessment, there was a higher tau PET SUVR level in the temporal pole in participants with MAPT relative to CN individuals, but these values were lower than levels seen in participants with AD (figure e-1, links.lww.com/WNL/A232, for a comparison of all regions). However, the participants with MAPT mutations outside exon 10 (i.e., V337M and R406W) that are expected to have AD-like tau (mixed 3R/4R) pathology showed qualitatively (figure 1) and quantitatively (figure 2) higher magnitude of tau PET signal that is closer to the level seen in cases with AD dementia. The participants with MAPT with mutations outside exon 10 had higher tau PET SUVRs in the temporal pole than participants with mutations inside exon 10 (p = 0.035, figure 2B) and CN (p < 0.001) but did not differ from participants with AD (p = 0.363). However, the participants with mutations inside exon 10 had lower levels of tau PET SUVR than participants with AD (p < 0.001), but they did not differ significantly from controls (p = 0.177). Given the small sample sizes, these statistical comparisons by exon 10 status should be interpreted cautiously.
Figure 1. Qualitative comparison of AV-1451 among MAPT mutation carriers.

Each participant's AV-1451 tau PET scan is overlaid on their structural MRI in native space. The dynamic range for the color map is the same for each case, and reference examples of a control and a patient with AD are included for comparison in the same dynamic range (top left corner). Case number and mutation location are listed for each participant. AD = Alzheimer disease; CN = clinically normal; MAPT = microtubule-associated protein tau; SUVR = standardized uptake value ratio.
Figure 2. Quantitative comparison of temporal pole AV-1451 among MAPT mutation carriers relative to controls and participants with AD dementia.
(A) Temporal pole tau PET SUVR for each participant is plotted separated by group (MAPT, CN, and AD; CN vs AD p < 0.001, CN vs MAPT p < 0.001, AD vs MAPT p < 0.001). MAPT mutation and clinical status are encoded in the marker type as per the figure legend for that group. Given that the asymptomatic participant with the N279K mutation is difficult to distinguish from the symptomatic participants, that participant is colored red in this plot; figures e-1 and e-2 (links.lww.com/WNL/A232) contain this same information for every ROI. The p value for the pairwise comparison of each group controlling for age is displayed above the plot. (B) The same information is plotted for the MAPT group separated by mutation location (inside vs outside exon 10 p = 0.035). The temporal pole SUVR was significantly greater for participants with mutations outside exon 10 relative to inside exon 10. See the main text for a description of the comparison to AD and CN by mutation location. AD = Alzheimer disease; CN = clinically normal; MAPT = microtubule-associated protein tau gene; ROI = region of interest; SUVR = standardized uptake value ratio.
The overall pattern of tau PET signal in participants with MAPT clearly distinguished them from those with AD dementia in that the signal is concentrated in the temporal poles rather than more widespread as is seen in AD dementia (figures 1 and 3 and table e-1, links.lww.com/WNL/A232). In the MAPT carriers with mutations outside exon 10, the temporal pole signal was also more concentrated in the gray matter, which is in contrast to the relatively greater white matter involvement in participants with MAPT mutation involving exon 10. Correcting for partial volume averaging effects did not alter these patterns (figure e-2 ). Figure 4 provides representative examples of the AV-1451 signal for each MAPT mutation site.
Figure 3. Representative renderings of tau PET uptake in MAPT mutations relative to AD.
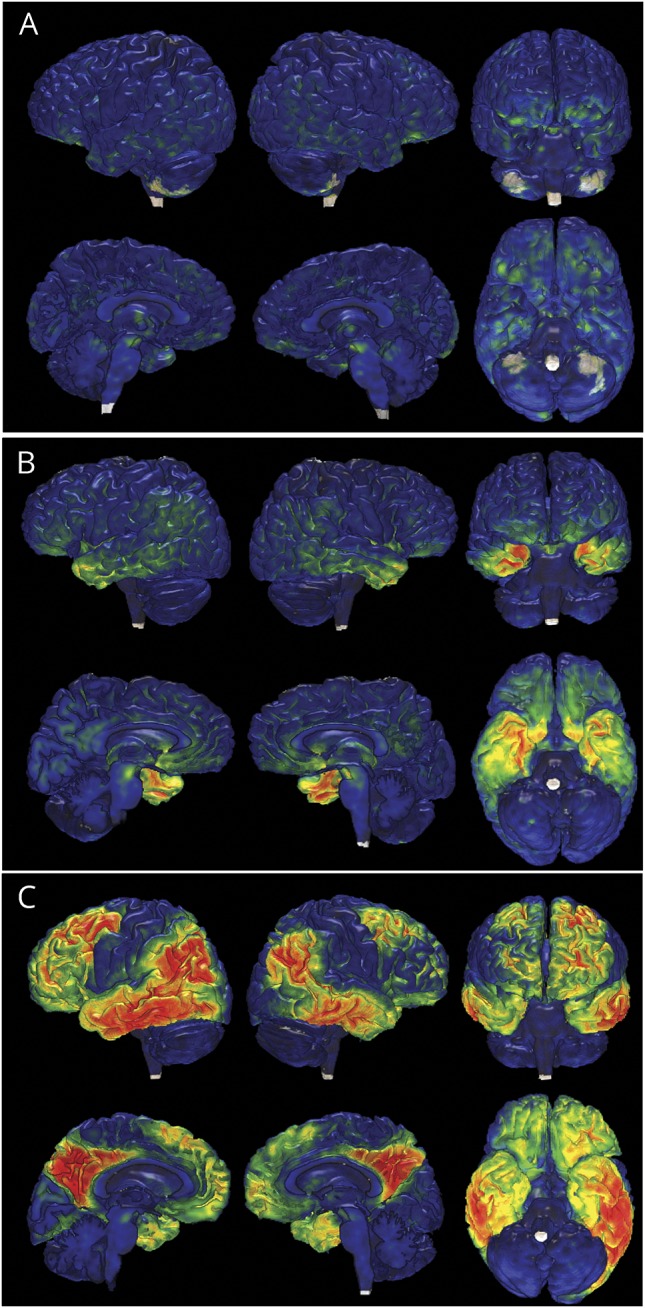
Tau PET global distributions are displayed on individual participant's brain renderings for 3 participants: (A) case 5, representative of participants with 4R MAPT with marginally elevated levels of tau PET signal; (B) case 11, representative of participants with mixed 3R/4R MAPT, and (C) a representative patient with AD dementia. The color maps contain the same dynamic range as figure 1. AD = Alzheimer disease; 4R = 4 repeat; MAPT = microtubule-associated protein tau gene; 3R = 3 repeat.
Figure 4. Example AV-1451 scans for each MAPT mutation location.

Schematic of the MAPT gene, with exons 1 and 9 through 13 displayed, and the approximate locations of each of the mutations included in this study. Axial sections of AV-1451 uptake are displayed for representative case examples for each mutation. MAPT = microtubule-associated protein tau gene.
The low levels of tau PET signal that can be seen in MAPT mutation carriers with expected 4R pathology (figure e-3A, links.lww.com/WNL/A232) can follow a regional distribution similar to that seen in MAPT mutations carriers with expected mixed 3R/4R pathology (figure e-3B); however, it is of a much lower magnitude and is more concentrated in white matter. It is uncertain whether this low-level signal is related to tau PET binding to 4R tau pathology because similar regionally specific uptake patterns are present in patients with frontotemporal dementia (FTD) who are not expected to have tau pathology at all such as amyloid-PET–negative PGRN mutation carriers that typically have TDP-43 pathology (figure e-3C). In addition, low levels of uptake seen in a 45-year-old asymptomatic N279K mutation carrier fell within the range of symptomatic N279K mutations carriers (compare the red asterisk relative to purple circles in figure 2 and figure e-1). However, this was not true for asymptomatic participants with R406W mutation (compare the brown asterisk relative to green circles in figure 2 and figure e-1). One of these participants had little to no signal (in the same range as participants with N279K mutation), but the other asymptotic participant with R406W mutation had signal in the AD range. Furthermore, among the participants with MAPT mutation, there was no discernible relationship between disease severity and AV-1451 SUVR, but there was between disease severity and temporal pole thickness (figure e-4).
Discussion
This study clearly demonstrates that AV-1451 has unique binding properties for AD-like tau pathology in vivo among MAPT mutation carriers. Despite the similar clinical and structural imaging features in these MAPT mutation carriers, tau PET imaging with AV-1451 demonstrated different binding intensities for MAPT carriers with mutations known to form primarily pathologic aggregates composed of 4R tau isoforms relative to mutations that are known to produce AD-like mixed 3R/4R isoform aggregates. The participants with MAPT mutation with suspected AD-like aggregates had a higher-magnitude tau PET signal than controls and MAPT mutation carriers who tend to produce 4R aggregates. However, the regional distribution of the marginally elevated tau PET signal seen in cases with suspected 4R followed the expected distribution of the underlying tau pathology in that it was seen in the same frontotemporal brain regions as in the MAPT carriers with suspected mixed 3R/4R tau.
This marginally elevated binding in participants with suspected 4R may not be related primarily to differences in density of tau pathology, partly because cases with suspected 4R with severe atrophy never reach the level of SUVR magnitude seen in cases with 3R/4R MAPT. In fact, the intensity appeared not to rise proportionally with more clinically advanced cases of 4R-associated neurodegeneration (see the red asterisk in figure 2 and figures e-1 and e-4, links.lww.com/WNL/A232), in contrast to those with AD dementia and MAPT mutation carriers who produce AD-like tau and develop relatively greater SUVRs when they are at more advanced disease stages (figure 2). However, longitudinal imaging would be necessary to fully evaluate the utility of this regionally specific, marginally elevated tau PET signal in an individual participant. Such studies would be able to evaluate whether this signal tracks with clinical progression in a meaningfully different way than other imaging modalities that do not directly measure tau pathology (e.g., glucose-PET and structural MRI).12 Given that participants with suspected 4R at advanced stages of neurodegeneration did not display more elevated tau PET signal, it is possible that either binding may peak during a period of maximal active neurodegeneration and then decline in later disease stages when the rate of neurodegeneration slows or there is less residual brain parenchyma with abnormal tau protein deposition to which the ligand can bind. However, significant brain parenchyma loss does not seem to significantly impair tau PET binding in cases with 3R/4R tau pathology. This, however, remains speculative in the absence of several years of longitudinal imaging across an individual's disease course.
Alternative explanations for marginally elevated levels of tau PET signal in the white matter of cases with 4R tauopathy may include 2 different but related etiologies: nonspecific changes related to neurodegeneration and specific signal related to 4R tau pathology. The regionally specific nature of the tau PET signal in cases with 4R MAPT may suggest a direct relationship to 4R tau pathology, but this is not necessarily the case. This low-level binding may not be related to tau pathology binding because marginally elevated levels of tau PET signal occur in a regionally specific manner in neurodegenerative cases that are not expected to be tauopathies at all (e.g., semantic dementia and familial FTD with PGRN mutations, figure e-3, links.lww.com/WNL/A232). Therefore, one cannot assume that regionally specific signal indicates tau pathology binding in vivo. Our interpretation that this in vivo marginally elevated signal may not be directly related to binding of 4R tau pathologic aggregates is in line with ex vivo studies of the binding properties of this ligand8–12 and with the fact that it does not appear to be associated with clinical severity. There may be some proportion of the regionally specific signal that is related to direct binding to 4R tau aggregates; however, this signal is unlikely to be a useful biomarker of this pathology given the known potential for this ligand to have nonspecific binding of a similar, or possibly greater, magnitude in affected brain regions in nontauopathies and its lack of association with clinical severity. Some additional explanations that any true 4R binding might not be associated with clinical severity may be that this marginally elevated signal appears in the preclinical disease phase and does not rise in the clinical phase or there is regional heterogeneity in the associations of clinical severity across different phenotypes. Combined in vivo and ex vivo studies are required to determine the etiology of the nonspecific binding in areas of active neurodegeneration so that these pitfalls can be better understood in the evaluation of alternative PET ligands that are more sensitive to aggregates composed of 4R isoforms.
The majority of MAPT mutations involve exon 10 and its splicing and lead to an imbalance in 3R and 4R tau isoforms.23 The participants in this study with MAPT mutations involving exon 10 all tend to produce pathologic aggregates composed primarily of 4R tau isoforms and to display marginally elevated levels of AV-1451 binding with a relative predominance for the white matter of brain regions affected by neurodegeneration. This is in contrast to the participants who carry mutations outside exon 10 that tend to produce mixed 3R/4R pathology and have high levels of AV-1451 signal with a relatively greater involvement of the gray matter (figure 4). However, it should be emphasized that MAPT mutations outside exon 10 do not always produce mixed 3R/4R pathology. Similarly, it should also be emphasized that not all exon 10–associated mutations produce only 4R tau pathology. Instead of illustrating the effect of exon 10 mutations, we believe that these phenotypically similar cases of familial FTD are illustrating the unique selectivity of AV-1451 for mixed 3R/4R tau pathology. Paired helical filaments are uniquely abnormal pathologic aggregates in which the microtubule-binding regions of both 3R and 4R tau form the core structure,5 and they also display unique binding properties with AV-1451 ex vivo8–12 and in vivo, as demonstrated here. However, a limitation of our in vivo study is the lack of pathologic evaluation of the participants who were scanned for this study. We therefore were obliged to extrapolate from previous pathologic evaluations of affected family members from these kindreds or other kindreds with the same mutations.3,18,19,24–29 It should also be noted that all MAPT N279K mutation carriers included in this study (n = 5) are from the same extended kindred known as the pallido-ponto-nigral degeneration family.25
This study helps to reconcile some discrepancies in the existing literature regarding the biomarker potential of AV-1451 for non-AD tauopathies. Some studies have shown high binding in 3R/4R MAPT18,19 and 4R MAPT mutations17 but have not directly compared among MAPT mutations, as was done in this study. In addition, some studies have reported good biomarker potential for AV-1451 in 4R tauopathies related to PSP13 and CBD,15,16 while others report poor12 or uncertain14 biomarker potential in 4R tauopathies. In this context, the current study sheds a unique light on the potential clinical utility of AV-1451 as a biomarker of neurodegenerative disease. We have observed that elevated tau PET signal is in a bitemporal predominant pattern in some MAPT mutation carriers, but the magnitude of this signal is low and predominately in the white matter for 4R MAPT and elevated in the AD range for all mixed 3R/4R MAPT. However, the regional distribution in 3R/4R MAPT is distinct from that seen in AD (figures 1 and 3). Therefore, tau PET appears to provide a specific biomarker signature for AD-related neurodegeneration. In contrast, the heterogeneity in tau pathologic isoforms associated with MAPT mutations and the uncertainty in the biological meaning of in vivo AV-1451 binding in 4R tau cases when it is present make this tool a less useful biomarker for these non-AD tauopathies.
Acknowledgment
The authors thank all the dedicated and caring research participants and their families for supporting this work. They also thank AVID Radiopharmaceuticals, Inc, for its support in supplying 18F-AV-1451 precursor, chemistry production advice and oversight, and Food and Drug Administration regulatory cross-filing permission and documentation needed for this work.
Glossary
- AD
Alzheimer disease
- CBD
corticobasal degeneration
- CN
clinically normal
- FOV
field of view
- 4R
4 repeats
- FTD
frontotemporal dementia
- MAPT
microtubule-associated protein tau
- PGRN
progranulin
- PSP
progressive supranuclear palsy
- ROI
region of interest
- SUVR
standardized uptake value ratio
- 3R
3 repeats
Footnotes
Editorial, page 495
Author contributions
D.J.: conception and design of the study, acquisition and analysis of data, and drafting the manuscript. D.K. and J.G.: acquisition and analysis of data, drafting the manuscript. J.S.: acquisition and analysis of data, statistical analyses, drafting the manuscript, figures, and tables. M.S., C.S., C.D., Z.W., R.R., K.K., and R.P.: acquisition and analysis of data. C.J. and V.L.: conception and design of the study, acquisition and analysis of data. B.B.: conception and design of the study, acquisition and analysis of data, and drafting the manuscript.
Study funding
This research was supported by NIH grants U01 AG045390, U54 NS092089, R01 AG011378, R01 AG041581, U01 AG006786, P50 AG016574, and P50 NS072187; the Liston Family Foundation; the GHR Foundation; Foundation Dr. Corinne Schuler; the Mayo Foundation; and the Robert H. and Clarice Smith and Abigail van Buren Alzheimer's Disease Research Program.
Disclosure
D. Jones receives research support from the NIH and the MN Partnership for Biotechnology and Medical Genomics. D. Knopman receives research support from the NIH and the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation. He serves on a Data Safety Monitoring Board for Lundbeck Pharmaceuticals and for the DIAN study and is an investigator in clinical trials sponsored by Biogen, TauRX Pharmaceuticals, Lilly Pharmaceuticals, and the Alzheimer's Disease Treatment and Research Institute, University of Southern California. J. Graff-Radford is partially supported by the NIH/National Institute on Aging (NIA) K76AG057015. J. Syrjanen, M. Senjem, C. Schwarz, and C. Dheel report no disclosures relevant to the manuscript. Z. Wszolek is partially supported by the NIH/National Institute of Neurological Disorders and Stroke (NINDS) P50 NS072187, NIH/NIA (primary) and NIH/NINDS (secondary) 1U01AG045390-01A1, Mayo Clinic Center for Regenerative Medicine, Mayo Clinic Center for Individualized Medicine, Mayo Clinic Neuroscience Focused Research Team (Cecilia and Dan Carmichael Family Foundation and the James C. and Sarah K. Kennedy Fund for Neurodegenerative Disease Research at Mayo Clinic in Florida), a gift from Carl Edward Bolch, Jr., and Susan Bass Bolch, and the Sol Goldman Charitable Trust. He serves as co-editor-in-chief of Parkinsonism and Related Disorders and as associate editor of the European Journal of Neurology. R. Rademakers received honoraria for lectures or educational activities not funded by industry; she serves on the Medical Advisory Board of the Association for Frontotemporal Degeneration and on the Board of Directors of the International Society for Frontotemporal Dementia. K. Kantarci serves on the Data Safety Monitoring Board for Takeda Global Research & Development Center, Inc and data monitoring boards of Pfizer and Janssen Alzheimer Immunotherapy and is funded by the NIH (R01 AG040042 [principal investigator (PI)], R21 NS066147 [PI], P50 AG44170/Project 2 [PI], P50 AG16574/Project 1 [PI], and R01 AG11378 [coinvestigator]). R. Petersen works as a consultant for Merck Inc, Roche Inc, Biogen Inc, Eli Lily and Company, and Genentech Inc; receives publishing royalties for Mild Cognitive Impairment (Oxford University Press, 2003); and receives research support from the NIH and the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation. C. Jack receives research funding from the NIH and the Alexander Family Alzheimer's Disease Research Professorship at Mayo Clinic. V. Lowe is a consultant for Bayer Schering Pharma, Merck Research, and Piramal Imaging Inc and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals, the NIH (NIA, National Cancer Institute), the Elsie and Marvin Dekelboum Family Foundation, the Liston Family Foundation, and the MN Partnership for Biotechnology and Medical Genomics. B. Boeve has served as an investigator for clinical trials sponsored by GE Healthcare and FORUM Pharmaceuticals. He receives royalties from the publication Behavioral Neurology of Dementia (Cambridge Medicine, 2009). He serves on the Scientific Advisory Board of the Tau Consortium. He has consulted for Isis Pharmaceuticals. He receives research support from the NIH, the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation, and the Mangurian Foundation. Go to Neurology.org/N for full disclosures.
References
- 1.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci 2001;24:1121–1159. [DOI] [PubMed] [Google Scholar]
- 2.McCarthy A, Lonergan R, Olszewska DA, et al. Closing the tau loop: the missing tau mutation. Brain 2015;138:3100–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol 2015;41:24–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siddiqua A, Margittai M. Three- and four-repeat tau coassemble into heterogeneous filaments. J Biol Chem 2010;285:37920–37926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hasegawa M, Watanabe S, Kondo H, et al. 3R and 4R tau isoforms in paired helical filaments in Alzheimer's disease. Acta Neuropathol 2014;127:303–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cairns NJ, Lee VM, Trojanowski JQ. The cytoskeleton in neurodegenerative diseases. J Pathol 2004;204:438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia CF, Arteaga J, Chen G, et al. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer's disease. Alzheimers Dement 2013;9:666–676. [DOI] [PubMed] [Google Scholar]
- 8.Marquie M, Normandin MD, Vanderburg CR, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol 2015;78:787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowe VJ, Curran G, Fang P, et al. An autoradiographic evaluation of AV-1451 tau PET in dementia. Acta Neuropathol Commun 2016;4:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coakeley S, Cho SS, Koshimori Y, et al. Positron emission tomography imaging of tau pathology in progressive supranuclear palsy. J Cereb Blood Flow Metab 2016:37:3150–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marquie M, Normandin MD, Meltzer AC, et al. Pathological correlations of [F-18]-AV-1451 imaging in non-Alzheimer tauopathies. Ann Neurol 2017;81:117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith R, Scholl M, Honer M, Nilsson CF, Englund E, Hansson O. Tau neuropathology correlates with FDG-PET, but not AV-1451-PET, in progressive supranuclear palsy. Acta Neuropathol 2017;133:149–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitwell JL, Lowe VJ, Tosakulwong N, et al. [18 F]AV-1451 tau positron emission tomography in progressive supranuclear palsy. Mov Disord 2017;32:124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Passamonti L, Vazquez Rodriguez P, Hong YT, et al. 18F-AV-1451 positron emission tomography in Alzheimer's disease and progressive supranuclear palsy. Brain 2017;140:781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Josephs KA, Whitwell JL, Tacik P, et al. [18F]AV-1451 tau-PET uptake does correlate with quantitatively measured 4R-tau burden in autopsy-confirmed corticobasal degeneration. Acta Neuropathol 2016;132:931–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McMillan CT, Irwin DJ, Nasrallah I, et al. Multimodal evaluation demonstrates in vivo 18F-AV-1451 uptake in autopsy-confirmed corticobasal degeneration. Acta Neuropathol 2016;132:935–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones WRB, Cope TE, Passamonti L, et al. [F-18]AV-1451 PET in behavioral variant frontotemporal dementia due to MAPT mutation. Ann Clin Transl Neur 2016;3:940–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith R, Puschmann A, Scholl M, et al. F-18-AV-1451 tau PET imaging correlates strongly with tau neuropathology in MAPT mutation carriers. Brain 2016;139:2372–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spina S, Schonhaut DR, Boeve BF, et al. Frontotemporal dementia with the V337M MAPT mutation: tau-PET and pathology correlations. Neurology 2017;88:758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kantarci K, Petersen RC, Przybelski SA, et al. Hippocampal volumes, proton magnetic resonance spectroscopy metabolites, and cerebrovascular disease in mild cognitive impairment subtypes. Arch Neurol 2008;65:1621–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts RO, Geda YE, Knopman DS, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 2008;30:58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowe VJ, Kemp BJ, Jack CR, Jr, et al. Comparison of 18F-FDG and PiB PET in cognitive impairment. J Nucl Med 2009;50:878–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu F, Gong CX. Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener 2008;3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reed LA, Schmidt ML, Wszolek ZK, et al. The neuropathology of a chromosome 17-linked autosomal dominant parkinsonism and dementia (“pallido-ponto-nigral degeneration”). J Neuropathol Exp Neurol 1998;57:588–601. [DOI] [PubMed] [Google Scholar]
- 25.Wszolek ZK, Pfeiffer RF, Bhatt MH, et al. Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann Neurol 1992;32:312–320. [DOI] [PubMed] [Google Scholar]
- 26.Sumi SM, Bird TD, Nochlin D, Raskind MA. Familial presenile dementia with psychosis associated with cortical neurofibrillary tangles and degeneration of the amygdala. Neurology 1992;42:120–127. [DOI] [PubMed] [Google Scholar]
- 27.Lindquist SG, Holm IE, Schwartz M, et al. Alzheimer disease-like clinical phenotype in a family with FTDP-17 caused by a MAPT R406W mutation. Eur J Neurol 2008;15:377–385. [DOI] [PubMed] [Google Scholar]
- 28.Mirra SS, Murrell JR, Gearing M, et al. Tau pathology in a family with dementia and a P301L mutation in tau. J Neuropathol Exp Neurol 1999;58:335–345. [DOI] [PubMed] [Google Scholar]
- 29.Iijima M, Tabira T, Poorkaj P, et al. A distinct familial presenile dementia with a novel missense mutation in the tau gene. Neuroreport 1999;10:497–501. [DOI] [PubMed] [Google Scholar]