

Abstract
The sigma 1 receptor (σ1R) is a structurally unique transmembrane protein that functions as a molecular chaperone in the endoplasmic reticulum (ER), and has been implicated in cancer, neuropathic pain, and psychostimulant abuse. Despite physiological and pharmacological significance, mechanistic underpinnings of structure-function relationships of σ1R are poorly understood, and molecular interactions of selective ligands with σ1R have not been elucidated. The recent crystallographic determination of σ1R as a homo-trimer provides the foundation for mechanistic elucidation at the molecular level. Here we report novel bioluminescence resonance energy transfer (BRET) assays that enable analyses of ligand-induced multimerization of σ1R and its interaction with BiP. Haloperidol, PD144418, and 4-PPBP enhanced σ1R homomer BRET signals in a dose dependent manner, suggesting their significant effects in stabilizing σ1R multimerization, whereas (+)-pentazocine and several other ligands do not. In non-denaturing gels, (+)-pentazocine significantly decreased whereas haloperidol increased the fraction of σ1R multimers, consistent with the results from the homomer BRET assay. Further, BRET assays examining heteromeric σ1R-BiP interaction revealed that (+)-pentazocine and haloperidol induced opposite trends of signals. From molecular modeling and simulations of σ1R in complex with the tested ligands, we identified initial clues that may lead to the differed responses of σ1R upon binding of structurally diverse ligands. By combining multiple in vitro pharmacological and in silico molecular biophysical methods, we propose a novel integrative approach to analyze σ1R-ligand binding and its impact on interaction of σ1R with client proteins.
Keywords: sigma 1 receptor, BRET, assay development, (+)-pentazocine, haloperidol, BiP
1. Introduction
The sigma 1 receptor (σ1R) is an intriguing transmembrane protein that does not share sequence homology to any known eukaryotic protein family, except for a fungal sterol isomerase (Hanner et al., 1996). It has been characterized as a molecular chaperone in the endoplasmic reticulum (ER) (Hayashi and Su, 2007), and may be a promising therapeutic target for several neuropsychiatric disorders (Kourrich et al., 2012; Maurice and Su, 2009). In addition, σ1R has been shown to be involved in pain (Romero et al., 2016; Sanchez-Fernandez et al., 2017), psychostimulant abuse (Katz et al., 2017; Sabino et al., 2017), and neurodegenerative diseases (Maurice and Goguadze, 2017; Nguyen et al., 2017) among others (Albayrak and Hashimoto, 2017; Soriani and Rapetti-Mauss, 2017; Wang et al., 2017). Depending on the physiological readout, σ1R ligands have been described as “agonists” or “antagonists”, as for G-protein coupled receptors (GPCRs). For instance, σ1R “antagonists” demonstrate efficacy in counteracting neuropathic pain (Romero et al., 2016) and drug seeking behavior in stimulant abuse (Katz et al., 2017), whereas σ1R “agonists” display favorable effects in depression (Fishback et al., 2010). However, it is worth noting that the efficacy distinction between “agonist” and “antagonist” has not reached consensus in a therapeutic context. For instance, in the development of antipsychotics both an “antagonist” (Ferris et al., 1986; Gilmore et al., 2004) and an “agonist” (Albayrak and Hashimoto, 2017) have been found to be beneficial. Furthermore, the underlying molecular mechanistic differences between agonists and antagonists have not been well characterized across different pathological contexts (Katz et al., 2016; Merlos et al., 2017), and it must be noted that some of the observations and interpretations are not monolithic (Katz et al., 2017). For example, the σ1R ligand BMY 14802 was characterized as both an antagonist and an agonist (Schoenwald et al., 1995; Taylor et al., 1993). This discrepancy can be attributed to readouts and interpretations at different downstream effector levels. Indeed only a few studies have evaluated the efficacy of σ1R ligands specifically at the level of σ1R-σ1R interaction (Gomez-Soler et al., 2014; Gromek et al., 2014; Mishra et al., 2015).
σ1R has a multitude of client proteins and the interactions with them may influence the nature of ligand action and signaling outcome (Su et al., 2016). Thus, client protein coupling and subsequent effector activation or inactivation have been reported as the major biological function of σ1R (Su et al., 2016). Among the different client proteins, in particular, binding immunoglobulin protein (BiP), also known as heat shock 70 kDa protein 5, has been well-characterized (Ha et al., 2014; Miki et al., 2015; Ono et al., 2013; Penas et al., 2011) and shown to regulate ER-originated events such as calcium release and receptor trafficking (Hayashi and Su, 2007).
To understand the molecular mechanism of ligands on σ1R, simplified signaling-independent methods are expected to better categorize the ligands, which may require going beyond the canonical agonist/antagonist definitions. In light of recent studies on σ1R homomerization in response to a variety of ligands (Gromek et al., 2014; Mishra et al., 2015), ligand-induced changes on receptor multimerization state may provide at least qualitative criterion for functional categorization. Recent crystal structures of ligand-bound σ1R, which are solved in homo-trimers, have revealed its transmembrane topology as well as the ligand binding site (Schmidt et al., 2016). Even though discrepancies with previous reports with regard to the structural topology (Ortega-Roldan et al., 2015; Ossa et al., 2017) remain, the high-resolution structural information revealed by the crystal structures sets the path to fundamental understanding of biophysical and pharmacological properties of σ1R at the molecular level. Specifically, the identification of the homomerization interface provides a framework to design the constructs that are feasible to study the ligand-induced changes of multimerization state with pharmacological assays.
The bioluminescence resonance energy transfer (BRET) assay is a reliable protein-protein proximity assay. The methodology is capable of uncovering both distance changes between two proteins (Pfleger et al., 2006), and large conformational rearrangements within a protein such as ligand-induced conformational changes of GPCRs (Lohse et al., 2012). Importantly, BRET is suited for real-time kinetic tracking of the movements of labeled proteins. Here we develop novel BRET assays to characterize the ligand-induced changes in the homomerization of σ1R and its interaction with BiP. The findings in σ1R homomer BRET assay were validated by a biochemical assay, and were further shown to correlate with the results from computational modeling. Herein, we report the pharmacological characterizations of 8 known σ1R ligands in a signaling-independent manner.
2. Materials and methods
2.1. σ1R radioligand binding in guinea pig cortex
Male Hartley guinea pig cortices were dissected from freshly harvested brains (shipped cold in phosphate-buffered saline (PBS) buffer from BioReclamation IVT) and frozen at -80 °C for future use. On test day, thawed guinea pig cortices were suspended and homogenized in 20 volumes (w/v) (10 mM Tris.HCl, 0.32M Sucrose, pH 7.4 at 25 °C) with a glass-teflon apparatus and centrifuged (∼1,200 rpm) for 10 min at 4 °C. The supernatant was collected in a clean tube and the pellet re-suspended in 10 ml of cold buffer and centrifuged again (∼1,200 rpm) for 10 min at 4 °C. The supernatants were pooled together and centrifuged (20,000 rpm) for 15 min at 4 °C. The final pellet was suspended in ice-cold binding buffer at 50 mg/ml concentration (original wet weight). A Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration present in the tissue preparation (1.25 mg/ml). All test compounds were freshly dissolved in 30% DMSO and 70% H2O to a stock concentration of 1 mM or 100 μM. To assist the solubilization of free-base compounds, 10 μl of glacial acetic acid was added along with the DMSO (in place of 10 μl final H2O volume). Each test compound was then diluted into 10 half-log serial dilutions using 30% DMSO as the vehicle. Radioligand competition experiments were conducted in 96-well plates containing 300 μl fresh binding buffer, 50 μl of diluted test compound, 100 μl of tissue preparation (125 μg/well total protein amount), and 50 μl of radioligand diluted in binding buffer ([3H]-(+)-pentazocine: 3 nM final concentration, ARC, Saint Louis, MO). Nonspecific binding was determined using 10 μM PRE-084 and total binding was determined with 30% DMSO vehicle (3% DMSO final concentration). All compound dilutions were tested in triplicate and the competition reactions started with the addition of the tissue preparation and incubated for 120 min at room temperature. The reaction was terminated by filtration through Perkin Elmer Uni-Filter-96 GF/B, presoaked for 120 min in 0.05% polyethylenimine, using a Brandel 96-Well Plates Harvester Manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed 3 times with 3 ml (3 × 1 ml/well) of ice cold binding buffer. 65 μL Perkin Elmer MicroScint 20 Scintillation Cocktail was added to each well and filters were counted using a Perkin Elmer MicroBeta Microplate Counter (calculated efficiency: 31%). IC50 values for each compound were determined from inhibition curves and Ki values were calculated using the Cheng-Prusoff equation; Kd values for [3H]-(+)-pentazocine (σ1R: 5.18 nM) and Bmax (1091 fmol/mg) were determined via separate homologous competitive binding experiments. Ki values were determined from at least 3 independent experiments and are reported as mean ± SEM.
2.2. σ1R radioligand binding in HEK293 cell membranes
HEK293T cells were grown as described below in section 2.4. Upon reaching 80-90% confluence, non-transfected HEK293T cells were harvested using pre-mixed Earle's Balanced Salt Solution (EBSS) with 5 mM EDTA (Life Technologies) and centrifuged at 3,000 rpm for 10 min at 21 °C. The supernatant was removed and the pellet was resuspended in 10 mL hypotonic lysis buffer (5 mM MgCl2, 5 mM Tris, pH 7.4 at 4 °C) and centrifuged at 20,000 rpm for 30 min at 4 °C. The pellet was then resuspended in fresh EBSS binding buffer made from 8.7 g/L Earle's Balanced Salts without phenol red (US Biological, Salem, MA) and 2.2 g/L sodium bicarbonate, pH to 7.4. A Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration. On test day, the experiments were conducted in 96-well plates containing 300 μL fresh binding buffer, 50 μL of diluted test compound, 100 μL of membranes (100 μg/well total protein), and 50 μL of radioligand diluted in binding buffer ([3H]-(+)-pentazocine: 3 nM final concentration, [3H]-haloperidol: 2 nM final concentration, Saint Louis, MO). Nonspecific binding was determined using 10 μM (+)-pentazocine or 10 μM haloperidol, respectively (final concentrations), and total binding was determined with 30% DMSO vehicle (3% DMSO final concentration). All compound dilutions were tested in triplicate and the reactions incubated for 120 min at room temperature. The reactions were terminated by filtration through Perkin Elmer Uni-Filter-96 GF/B, presoaked for 120 min in 0.5% polyethylenimine, and the filters counted as described above (calculated efficiency 31% and 29% for [3H]-(+)-pentazocine and [3H]-haloperidol, respectively). Kd (σ1R: 7.76 nM) and Bmax (2080 fmol/mg) for [3H]-(+)-pentazocine, as well as Kd (σ1R: 9.25 nM) and Bmax (5784 fmol/mg) for [3H]-haloperidol, were determined via separate homologous competitive binding experiments. Ki values were determined from at least 3 independent experiments and are reported as mean ± SEM.
2.3. [3H]-(+)-Pentazocine association kinetics and competition kinetics by other σ1R ligands
Membranes from non-transfected HEK293T were collected as described above. HEK293T cells, either intact or transfected with σ1R-Venus and σ1R-NanoLuciferase, were harvested using pre-mixed Earle's Balanced Salt Solution (EBSS) with 5 mM EDTA (Life Technologies) and centrifuged at 1,200 rpm for 5 min at 21 °C to prevent cell lysis. The supernatant was removed and the pellet was then resuspended in fresh EBSS binding buffer made from 8.7 g/L Earle's Balanced Salts without phenol red (US Biological, Salem, MA) and 2.2 g/L sodium bicarbonate, pH to 7.4. A Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration. The experiments were conducted in 96-well plates containing 300 μl fresh binding buffer, 50 μl of 30% DMSO vehicle (3% DMSO final concentration for total binding) or 10 μM (+)-pentazocine (final concentration for non-specific binding), 100 μl of membranes or whole cell preparation (100 μg/well total protein), and 50 μl of radioligand diluted in binding buffer ([3H]-(+)-pentazocine: 3 nM final concentration). The reactions were incubated at room temperature and terminated by filtration through Perkin Elmer Uni-Filter-96 GF/B (presoaked in 0.5% polyethylenimine) at different time points. Non-specific binding was subtracted from the total binding at each time point and the obtained specific binding values were normalized to the maximum specific binding achieved at 120 min. T1/2 values represent the time necessary to obtain 50% of the radioligand bound. Bmax values (membranes: 871 fmol/mg; whole non-transfected cells: 1041 fmol/mg and BRET-transfected cells: 4289 fmol/mg) were calculated as well from the association kinetic experiments. All the values were determined from at least 3 independent experiments performed in triplicate and are reported as mean ± SEM. For time dependent specific competition of σ1R ligands in different cell preparations versus [3H]-(+)-pentazocine, HEK293 membranes, whole non-transfected cells and whole BRET-transfected cells were collected as described above. Each test compound was tested for its ability to displace the total amount of radioligand bound at different time points. The concentration used for each compound was determined as the minimum concentration necessary to fully compete with the radioligand at the equilibrium (120 min). The experiments were conducted in 96-well plates containing 300 μl fresh binding buffer (8.7 g/L Earle's Balanced Salts without phenol red and 2.2 g/L sodium bicarbonate, pH to 7.4.), 50 μl of 30% DMSO vehicle (3% DMSO final concentration for total binding) or the drug dilution at the desired concentration, 100 μl of membranes or whole cell preparation (100 μg/well total protein), and 50 μl of radioligand diluted in binding buffer ([3H]-(+)-pentazocine: 3 nM final concentration). The reactions were incubated at room temperature and terminated by filtration through Perkin Elmer Uni-Filter-96 GF/B (presoaked in 0.5% polyethylenimine) at different time points. For each time point the amount of specific binding was normalized to 100% (total specific binding in presence of the vehicle) and 0% (residual radioligand bound in presence of 500 nM (+)-pentazocine). The data represent the average of two independent experiments with triplicate determinations for each drug concentration at the different time points. The multiple statistical comparison was obtained by two-way ANOVA followed by post-hoc Tukey analysis.
2.4. DNA constructs, transfection, and cell culture
Human σ1R is fused in pcDNA3.1 plasmid to the NanoLuciferase fragments, full length NanoLuciferase (NL), or mVenus (VN; monomeric YFP variant) N-terminally or C-terminally by introduction of restriction enzyme site to generate the following constructs. The N-terminal fragment (NL1) or C-terminal fragment (NL2) of NanoLuciferase is subcloned as follows: KpnI-NL1-BamHI-σ1R, KpnI-NL2-BamHI-σ1R, σ1R-BamHI-NL1-XhoI, σ1R-BamHI-NL2-XhoI. Full length NanoLuciferase or mVenus is subcloned as follows: KpnI-NL-BspEI-σ1R, NheI-VN-HindIII-σ1R, σ1R-XhoI-VN-XbaI, σ1R-XhoI-NL-XbaI. N-terminal fusion of FLAG-2×His8 to σ1R is made as previously described (Hong et al., 2017). Human BiP is fused in pcDNA3.1 plasmid to venus as follows: NheI-VN-HindIII-BiP, BiP-XhoI-VN-XbaI. All the constructs were confirmed by sequence analysis.
For luciferase complementation, a constant amount of total plasmid (2 μg) was transfected in human embryonic kidney cells 293T (HEK 293T) using polyethylenimine (PEI; Sigma-Aldrich, St. Louis, MO) in a 1 to 2 ratio in 6-well plates. For acceptor saturating BRET, using PEI, a constant amount of total plasmid cDNA (5 μg) in varied donor:acceptor ratios was transfected in HEK 293T in 6-well plates. For drug induced BRET, a constant amount of total plasmid cDNA (15 μg) in 1:24 (= donor:acceptor ratio) was transfected using PEI. Cells were maintained in culture with Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and kept in an incubator at 37 °C and 5% CO2. Experiments were performed approximately 48 h post-transfection.
2.5. Luciferase complementation and bioluminescence resonance energy transfer (BRET)
Luciferase complementation is conducted as follows. Cells were harvested, washed, and resuspended in PBS. Approximately 200,000 cells/well were distributed in 96-well plates. Luminescence was measured at 485 nm using a Mithras LB940 reader (Berthold Technologies, Bad Wildbad, Germany).
Acceptor saturating BRET is performed as described previously (Sohy et al., 2009). Briefly, after cell resuspension and distribution in 96-well plates as described in luciferase complementation, expression of venus fusion proteins was estimated by measuring fluorescence at 535 nm following excitation at 485 nm. Expression of NL fusion proteins was estimated by measuring the luminescence of the cells after incubation with 5 μM coelenterazine h. In parallel, BRET was measured as a ratio between measurements at 535 nm (fluorescence) and at 485 nm (luminescence) using a Mithras LB940 reader (Berthold Technologies, Bad Wildbad, Germany). Results are plotted as fluorescence over luminescence vs. basal-subtracted BRET ratio.
Drug induced BRET is conducted as reported previously (Urizar et al., 2011). Cells were prepared in 96-well plates as in acceptor saturating BRET. 5 μM coelenterazine h (substrate for BRET) was added to each well. Three minutes after addition of coelenterazine h, ligands [(+)-pentazocine (Sigma), PRE-084 (Tocris), NE100 (Tocris), JHW007 (Newman lab), cocaine (NIDA), PD144418 (Tocris), haloperidol (Tocris), 4PPBP (Tocris)] in series of dilution were added to each well. BRET was measured as in acceptor saturating BRET. Results are calculated for the BRET change (BRET ratio for the corresponding drug minus BRET ratio in the absence of the drug). Emax values are expressed as the basal subtracted BRET change in the dose-response graphs.
2.6. Non-denaturing gel assay
As previously described (Hong et al., 2017), HEK293 cells were stably transfected with N-terminal FLAG-2×His8 tagged σ1R (FH-σ1R). Confluent cells in 24-well plates were first incubated with haloperidol or vehicle at 37 °C for 0.5 h in culture medium, followed by addition of (+)-pentazocine (if indicated) and incubation at 37 °C for 1 h. Cells were then washed with cold PBS with 1 mM MgCl2 and 0.1 mM CaCl2 (PBSCM), harvested and lysed with lysis buffer (0.1% glyco-diosgenin, NaCl 150 mM, EDTA 1 mM, Tris 50 mM, pH 7.5, and protease inhibitors) for 2 h at 4 °C. Supernatants after centrifugation (20,000 g, 10 min) were mixed with 2× sample buffer (8% perfluorooctanoic acid (PFO), 40% glycerol, bromophenol blue 0.005%, Tris 100 mM, pH 7.5) to a final concentration of 4% PFO, and heated at 37 °C for 10 min. Samples were run in 5-15% polyacrylamide Tris-glycine gels (running buffer: 0.1% PFO, 25 mM Tris, 192 mM glycine, pH 8.3). Proteins were transferred to PVDF membranes and immunoblotted with rat monoclonal anti-Flag L5 antibody (Biolegend, San Diego, CA).
2.7. Data analysis
All experiments were repeated at least three times. Regression and other statistical analyses were performed using GraphPad Prism version 7.0 (GraphPad Software, San Diego, CA).
2.8. Molecular docking and simulations
The crystal structure of σ1R in complex with PD144418 (σ1R/PD144418, PDB ID 5HK1 (Schmidt et al., 2016)) was used as the starting point for our modeling studies. Except for PD144418, all other compounds described in this study were docked into the binding site revealed by the crystal structure using the induced-fit docking (IFD) protocol (Sherman et al., 2006) implemented in the Schrodinger suite (release 2016-4). Since all of these compounds, except for PRE-084, possess a nitrogen that should be protonated at pH 7, they are expected to form a salt bridge with Glu172 in the binding site. The pKa of the nitrogen on PRE-084 was calculated to be 5.82±1.47 at pH 7 (which is consistent with the experimentally measured value of 6.2, see text), using the Epik protocol in the Schrodinger suite (release 2016-4). However, PRE-084 was still prepared as the protonated form since the protonation is likely to happen in the micro-environment of the binding site (see Supplementary Method and Supplementary Results). Therefore, the formation of the salt bridge was used as a filter for the docking poses. We considered all stereoisomers for each ligand, including those for the phenylcyclohexane ring of PRE-084, and those resulting from two possible ways in protonating the aforementioned nitrogen for haloperidol, 4-PPBP, cocaine, (+)-pentazocine, and JHW007. Several poses for each ligand or stereoisomer with significantly different orientations in the binding site were further evaluated by binding pose metadynamics simulations (Clark et al., 2016) in which the ligand RMSD deviation from the starting pose was set to be the collective variable. Poses that are clearly unfavored in the metadynamics simulations, i.e., with higher RMSD fluctuations and lower hydrogen bond persistency, were discarded, while others were selected for the molecular dynamics (MD) simulations. In the end, one to three poses were selected for each compound for the following MD simulations (Table S1).
MD simulations of the σ1R/ligand complexes were performed in an explicit water and 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) lipid bilayer environment using Desmond MD System (version 4.9; D. E. Shaw Research, New York, NY) with the OPLS3 force field for protein, ligand, and lipids (Harder et al., 2016) and with the SPC water model. The missing residues at the N-terminus of σ1R in the crystal structure of σ1R/PD144418 were constructed using MODELLER (John and Sali, 2003). Each of the σ1R/ligand complexes were then placed into the POPC lipid bilayer using the orientation of σ1R/PD144418 retrieved from the Orientation of Proteins in Membranes database (Lomize et al., 2006). The system charges were neutralized, and 150 mM NaCl was added. Simulations were performed with a cutoff distance of 12 Å for the nonbonded interactions; and the particle mesh Ewald method was used to evaluate long-range electrostatic effects. In the isothermal-isobaric (NPT) ensemble, constant temperature (310 K) and 1 atm constant pressure were maintained with Langevin dynamics on an anisotropic flexible periodic cell, with a constant-ratio constraint applied on the lipid bilayer in the X-Y plane. Each system was first minimized and then equilibrated with restraints on the ligand heavy atoms and protein backbone atoms, followed by production runs with all atoms unrestrained, as described previously (Michino et al., 2017; Michino et al., 2015). For each complex, we collected multiple trajectories (Table S1).
2.9. Ligand pose analysis
The stability of each ligand in the MD simulations were evaluated by calculating i) the RMSD of the ligands using the ligand pose in the last frame of each trajectory as the reference, and ii) the distance of the hydrogen on the charged N to the carboxyl group of Glu172. For the following analysis, we chose the most stable pose of each ligand that had the ligand RMSD below 1.5 Å for at least 300 ns with the persistent formation of the salt bridge – whereas most of the compounds had only one pose that was clearly better than the others, the two isomers of (+)-pentazocine were similarly stable in the MD simulations and both were included in the subsequent studies.
To classify the σ1R ligands based on their binding modes, we calculated the interaction frequencies of these ligands with the binding site residues. For the reasons described in the Discussion (section 4), we did not include cocaine in this analysis. Two poses of (+)-pentazocine were combined so the following analysis was not biased. A protein residue was considered to form an interaction with the ligand if any heavy atom-heavy atom distance between them was within 4 Å. To obtain the mean interaction frequency, we randomly sampled 100 frames from each of the MD trajectories of the stable poses of each complex, which was repeated for 30 times and the mean contact frequency values were plotted as the heat map. Then, a hierarchical clustering was performed to classify the σ1R ligands based on the identities of contacting residues and their interaction frequencies with the ligands. To prevent the big difference caused by some individual residues, e.g., those with high contact frequency, we used Canberra distance to calculate the pairwise distances between two ligands ( , in which pi, qi represent the contact frequency between the ligand p or q and the residue i; while n represents the total number of interaction residues being considered; if pi = qi = 0, then di = 0). Then, Ward's method was applied to merge the clusters.
3. Results
3.1. The endogenous σ1R in HEK 293T cells has similar binding affinities for σ1R ligands asthose in guinea pig cortex
Several cellular models have been used for the functional characterization of σ1R. While differentiated cells may have advantages in addressing signaling effects of σ1R, transfectable heterologous cells are amenable for addressing biophysical and pharmacological aspects of σ1R due to their ability to robustly express membrane proteins. In order to pursue σ1R pharmacology in the latter strategy, we first investigated expression levels of endogenous σ1R in HEK 293T cells so that later the levels of heterologous expression could be derived (see section 3.3). Thus, we measured σ1R expression levels (Bmax) and σ1R ligand binding affinities (Kd) with [3H]-(+)-pentazocine (a previously reported σ1R “agonist” (Maurice and Su, 2009; Rousseaux and Greene, 2015)) in the membrane preparation of HEK 293T cells, and found they were comparable to those determined in guinea pig cortex (Table 1, Kd values were determined by homologous competitive binding as described in the Methods section), which has been routinely used in pharmacological studies of σ1R ligands (Su, 1982; Tam and Cook, 1984). Note the Kd value of (+)-pentazocine for guinea pig cortex was similar to that reported previously (Bowen et al., 1993).
Table 1. Radioligand binding properties for [3H]-haloperidol and [3H]-(+)-pentazocine.
Kd and Bmax are determined using [3H]-haloperidol and [3H]-(+)-pentazocine in HEK 293T cell (second column) and guinea pig cortex (third column) membrane preparations.
| Compound | HEK 293T membrane | Guinea pig cortex | ||
|---|---|---|---|---|
| [3H]-haloperidol | Kd ± SEM (nM) | n | NA | |
| 7.87 ± 3.47 | 5 | |||
| Bmax ± SEM (fmol/mg) | n | |||
| 5847 ± 2221 | 5 | |||
| [3H]-(+)-pentazocine | Kd ± SEM (nM) | n | Kd ± SEM (nM) | n |
| 7.76 ± 0.52 | 3 | 5.18 ± 0.75 | 3 | |
| Bmax ± SEM (fmol/mg) | n | Bmax ± SEM (fmol/mg) | n | |
| 2080 ± 163 | 3 | 1091 ± 79 | 3 | |
Next, based on wide usage reported in the literature, eight structurally diverse σ1R ligands (Figure 1) were selected for the HEK 293T cell-based pharmacological characterizations. As expected, in the competition assays with [3H]-(+)-pentazocine, the Ki values for these eight compounds showed a consistent match between the HEK 293T cells and guinea pig cortex (Table 2, representative binding curves are shown in Supplementary Figure 1), indicating the binding affinities characterized by the cell-based assay can capture those in the tissue, the latter of which is in a more physiological context. We further assessed σ1R affinities using another σ1R radioligand, [3H]-haloperidol (a previously reported σ1R “antagonist” (Rousseaux and Greene, 2015)) (Table 1). The Ki values were generally higher (lower affinity) across all the tested compounds using [3H]-haloperidol compared to [3H]-(+)-pentazocine (Table 2). Nonetheless, the relative differences of the Ki values among the tested compounds were similar between [3H]-(+)-pentazocine and [3H]-haloperidol.
Figure 1.
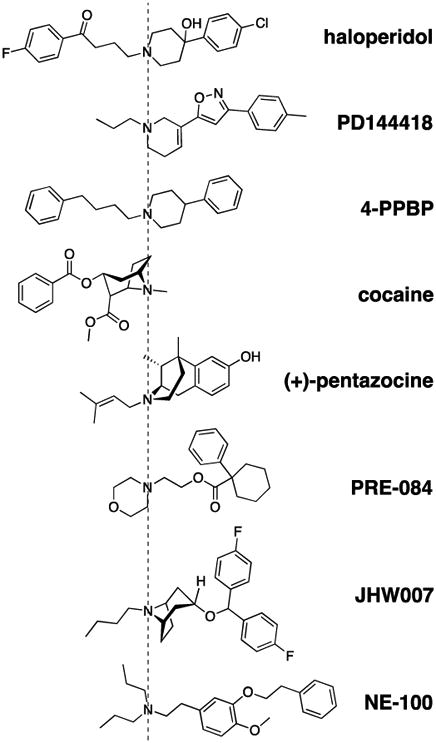
Chemical structures of the eight compounds aligned by their overall binding orientations. The compounds are oriented based on the resulting poses in the ligand binding pocket of σ1R from our molecular modeling and simulation results, and are aligned by the charged N atom.
Table 2. Radioligand competition binding with [3H]-haloperidol or [3H]-(+)-pentazocine.
Competition results using [3H]-haloperidol (second column) or [3H]-(+)-pentazocine (third and fourth columns) are shown. For [3H]-(+)-pentazocine binding, results for membrane preparation of guinea pig cortex (third column) in addition to HEK 293T (fourth column) are shown.
| Compound | Competition vs. 2 nM [3H]-haloperidol | Competition vs. 3 nM | [3 H]-(+)-pentazocine | |||
|---|---|---|---|---|---|---|
| HEK 293T membrane | Guinea pig cortex | HEK 293T membrane | ||||
| Ki ± SEM (nM) | n | Ki ± SEM (nM) | n | Ki ± SEM (nM) | n | |
| haloperidol | 8.24 ± 4.11 | 3 | 0.835 ± 0.315 | 3 | 0.965 ± 0.016 | 3 |
| (+)-pentazocine | 174 ± 48.4 | 3 | 5.11 ± 0.514 | 3 | 7.63 ± 0.318 | 3 |
| cocaine | 32100 ± 9110 | 3 | 3710 ± 1170 | 3 | 4660 ± 1920 | 3 |
| PD144418 | 2.67 ± 0.578 | 4 | 0.842 ± 0.307 | 3 | 0.894 ± 0.110 | 3 |
| JHW007 | 1.85 ± 0.0579 | 3 | 1.48 ± 0.289 | 4 | 0.554 ± 0.172 | 3 |
| PRE-084 | 208 ± 43.2 | 3 | 11.0 ± 0.640 | 3 | 21.0 ± 5.93 | 3 |
| NE-100 | 16.3 ± 4.43 | 4 | 1.14 ± 0.283 | 4 | 1.75 ± 0.382 | 6 |
| 4-PPBP | 2.28 ± 0.558 | 3 | 0.857 ± 0.153 | 3 | 1.16 ± 0.219 | 3 |
3.2. Topology analysis by proximity assays indicate N- and C-termini of σ1R are on differentsides of membranes
A few different topological orientations of N- and C-termini relative to the membrane have been proposed for σ1R, based on the hydrophobicity analysis and truncation mutation studies (Brune et al., 2013; Kourrich et al., 2012). Such topological insight of σ1R is key to understanding its homomeric interaction as well as its interactions with client proteins. Thus, guided by the recent homotrimeric crystal structures, we studied the relative topology between N- and C-termini by both bimolecular complementation and BRET approaches (Figure 2).
Figure 2.
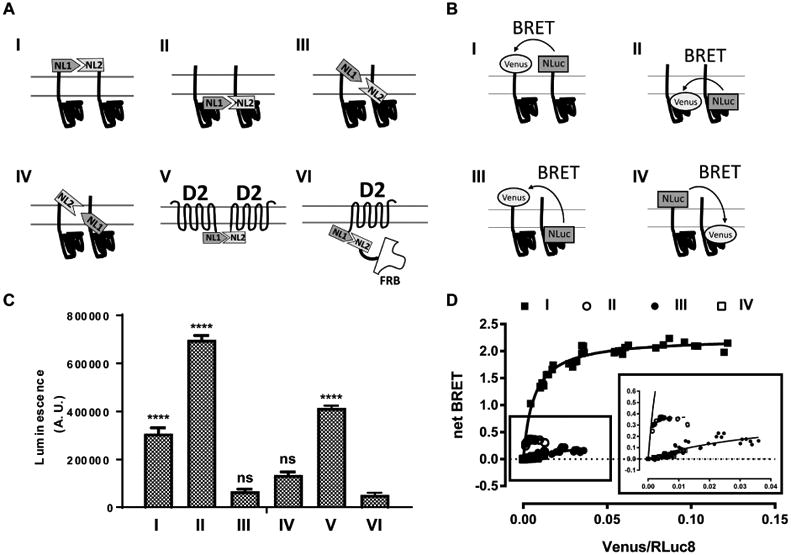
N- and C-termini of σ1R are not close to each other in the proximity assays. (A) Schematic representation for NanoLuciferase complementation assay. (B) Schematic representation for BRET assay. (C) NanoLuciferase complementation is performed between σ1R and σ1R with four different configurations (I-IV; NL1-σ1:NL2-σ1, σ1-NL1:σ1-NL2, NL1-σ1:σ1-NL2, σ1-NL1:NL2-σ1), between D2R-NL1 and D2R-NL2 (V), or between D2R-NL1 and FRB-NL2 (VI). One way ANOVA followed by posthoc Dunnett's test against D2R-NL1:FRB-NL2 (VI) shows significance (p < 0.0001) for NL1-σ1:NL2-σ1 (I), σ1-NL1:σ1-NL2 (II), and D2R-NL1:D2R-NL2 (V). Data represents mean ± S.E.M. (n = 3 or more). (D) Acceptor-saturating BRET between σ1R and σ1R using N- and C-terminal fusion constructs to address the relative transmembrane topology: NL-σ1 and VN-σ1 (I, solid square), σ1-NL and σ1-VN (II, open circle), σ1-NL and VN-σ1 (III, solid circle), and NL-σ1 and σ1-VN (IV, open square). Inset magnifies the curves for σ1-NL and σ1-VN (II), σ1-NL and VN-σ1 (III), and NL-σ1 and σ1-VN (IV). The results of both assays are consistent with the crystal structure showing N- and C-termini on opposite sides of membrane.
By fusing recently reported NanoLuciferase split fragments (i.e. NL1 and NL2) (Yano et al.) to proteins, the level of complementation between NL1 and NL2 is assessed by luminescence (Figure 2A), from which we can deduce the protein-protein proximity. As a positive control, we used dopamine D2 receptor (D2R) C-terminal fusions of NL1 and NL2 (D2R-NL1:D2R-NL2), a pair of constructs that are known to readily complement (Figure 2A). As a negative control, we used D2R-NL1 and a non-membrane associated cytosolic protein FRB fused to NL2 (D2R-NL1:FRB-NL2), because they approach to each other only randomly (Figure 2A). For σ1R, NL1 and NL2 are fused not only to the same ends, i.e., either both to the N-terminus, NL1-σ1R:NL2-σ1R (NL1-σ1:NL2-σ1), or both to the C-terminus, σ1R-NL1:σ1R-NL2 (σ1-NL1:σ1-NL2), but also to the opposite ends, i.e., NL1-σ1:σ1-NL2 or NL2-σ1:σ1-NL1 (Figure 2A). Consistent with the topology revealed by the crystal structures that N- and C-termini are on two sides of the membrane, only the same-end fusion σ1R pairs and the D2R-NL1:D2R-NL2 pair showed a significantly higher luminescence level above the D2R-NL1:FRB-NL2 pair (Figure 2C).
We then used an acceptor saturating BRET method, a commonly-used ratiometric assay that has the advantage of withstanding off-target ligand interaction with a luciferase. As previously reported (Marullo and Bouvier, 2007), a saturating profile and higher net BRET values are the signature of specific interactions between the donor and acceptor (i.e., NL and VN), whereas linear or low net BRET values indicate a random collision between the donor and acceptor and thus not likely to form a complex between the two fused-proteins. Consistent with the findings from the above bimolecular NanoLuciferase complementation, only the same-end fusion construct pairs, the N-terminal pair NL-σ1R:VN-σ1R (NL-σ1:VN-σ1) and the C-terminal pair σ1-NL:σ1-VN, satisfied the high and saturable BRET signals, whereas the opposite-end fusion construct pairs (NL-σ1:σ1-VN and σ1-NL:VN-σ1) exhibited a low linear BRET profile (Figure 2B,D).
3.3. PRE-084 shows unique kinetics in entering the ligand binding site of σ1R
Photoaffinity labeling experiments (Fontanilla et al., 2008) and the recently solved σ1R crystal structures (Schmidt et al., 2016) demonstrate that σ1R ligand binding site is on the same side of the membrane as its C-terminus. However, the locale of the binding site with regard to the membrane topology is much less certain. The C-terminus (and therefore the binding site) has been variously asserted to be located within the ER lumen (Hayashi and Su, 2007), in the cytoplasm (Aydar et al., 2002), or outside the cell (Balasuriya et al., 2013). More recently, based on the crystal structures, Schmidt et al. have asserted that the σ1R ligand binding site is in the cytoplasm (Schmidt et al., 2016). In this configuration, the binding site consistently remains within the cell, regardless of σ1R being at the ER or translocated to the cell membrane. Such a locale would require ligands to permeate through the plasma membrane first in order to access the binding site, which may result in different binding kinetics in intact whole cells compared to membrane preparations. To explore this possibility, we examined ligand binding kinetics in both preparations.
First, in both the intact whole cell and membrane preparations, we studied the displacement of [3H]-(+)-pentazocine by the σ1R ligands listed in Table 1, for which the concentration was determined based on their Ki values (Supplementary Figure 2A,B). Normalized to total binding in the absence of cold ligand and complete displacement by 500 nM cold (+)-pentazocine as 100% and 0%, respectively, haloperidol, JHW007, and NE100 showed immediate displacement even at 15 min of incubation for both membrane preparation and whole cells (note the negative values indicate that the tested cold ligand may reach the binding sites faster than (+)-pentazocine). However, PRE-084 showed significantly slower kinetics than the others in the whole cell assay, whereby it only displaced 60.6% [3H]-(+)-pentazocine at 15 min and its displacement remained below 76.1% at 120 min. In contrast, at these timepoints it displaced 92.4–96.1% [3H]-(+)-pentazocine in the membrane preparation. The experimentally determined pKa (6.2 for PRE-084 and 9.2-9.3 for (+)-pentazocine) suggests that PRE-084 is not charged in the solutions (pH 7.4), whereas (+)-pentazocine is. Although this difference indicates that PRE-084 is more plasma-membrane permeable at a physiological pH, other properties may play a role in entering the ligand binding site, thus yielding the slower access of PRE-084 to the site.
Next, HEK 293T cells transfected with the C-terminal BRET construct pair σ1-NL:σ1-VN were characterized by binding kinetics analysis (Supplementary Figure 2C). We found all the tested ligands, including PRE-084, displaced [3H]-(+)-pentazocine in these cells as fast as in the membrane preparation of the non-transfected cells, indicating that the transiently transfected plasma membrane is permeable to σ1R ligands, possibly due to compromised membrane integrity (Grandinetti et al., 2012).
From the binding kinetics, we calculated Bmax and Kd for intact whole cells (1041 ± 540 fmol/mg and 1.7 ± 0.1 nM, respectively), BRET-transfected whole cells (4289 ± 2745 fmol/mg and 2.0 ± 0.3 nM), and membrane preparation (871 ± 403 fmol/mg and 3.3 ± 1.4 nM). As expected, BRET-transfected whole cells showed ∼5-fold higher Bmax than intact cells, confirming that transient transfection adds binding sites and the transfected fusion σ1R constructs retain the functional integrity of the protein. Binding kinetic analysis of (+)-pentazocine association time (T1/2, the time to bind 50% of the available binding sites) among intact whole cells, BRET-transfected whole cells, and membrane preparation yielded near identical values (21.4 ± 3.86, 21.3 ± 4.54, and 21.1 ± 4.44 min, respectively) and suggested fast permeability of (+)-pentazocine (Supplementary Figure 2D).
Taken together, while other tested ligands exhibited cell permeability comparable to [3H]-(+)-pentazocine in intact cells, PRE-084 showed slower kinetics. The lack of slower kinetics of PRE-084 in the membrane preparation and the BRET-transfected cells suggests that the process of permeation through the intact membrane may play a role in its kinetics. However, whereas the involvement of the plasma membrane permeation is consistent with the ligand binding site of σ1R being on the cytoplasmic side, pKa values suggest that PRE-084 is more permeable than (+)-pentazocine. Thus, the locale of the binding site with regard to membrane topology requires further investigation.
3.4. σ1R homomer BRET defines two categories of σ1R ligands
Next, we used the C-terminal σ1-NL:σ1-VN BRET construct pair to evaluate whether the ligand-induced BRET signal changes have any applicability in differentiating the σ1R ligands in a dose dependent manner. In all four time points tested, there were clear dose-response effects by haloperidol, 4-PPBP, and PD144418, but weaker or almost no effects by (+)-pentazocine, PRE-084, NE-100, JHW007, and cocaine (Figure 3A-D). The results at later time points (10, 30, and 60 min) showed more prominent increases in the induced BRET signals for haloperidol, 4PPBP, and PD144418, but very little by the others (Figure 3A-D). The differences in potencies among haloperidol, 4PPBP, and PD144418 correlated with their different binding affinities (Table 2), although the potencies are consistently lower than the Ki values. We categorized haloperidol, 4PPBP, and PD144418 to be active, whereas the other compounds were inactive in this assay. Even though there could be a spectrum of Emax values depending on the σ1R ligands, we set the threshold by two-way ANOVA Dunnett test at the level of PD144418 Emax (∼0.02) resulting in the classification of ligands into two groups: haloperidol, 4PPBP, and PD144418 that showed positive dose-dependent induction of BRET signaling, and (+)-pentazocine, PRE-084, NE100, JHW007, and cocaine that produced no detectable effects on BRET signaling.
Figure 3.
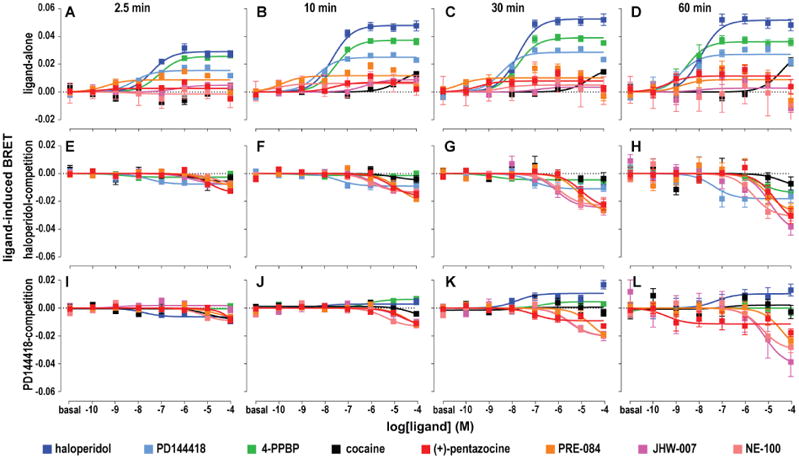
Drug induced σ1R homomer BRET changes. Drug induced BRET between C-terminally fused σ1-NL and σ1-VN is detected at 2.5, 10, 30, or 60 min for ligand alone (A-D), ligand in the presence of 1 μM haloperidol (E-H), or 1 μM PD144418 (I-L). σ1R ligands are represented in different colors – (+)-pentazocine (red), PRE-084 (orange), NE100 (pink), JHW007 (magenta), cocaine (black), haloperidol (blue), PD144418 (cyan), and 4PPBP (green). Data represents mean ± S.E.M. (n = 5 or more).
To demonstrate that the lack of BRET signals in the single-ligand mode was not due to the inability of (+)-pentazocine, PRE-084, NE-100, JHW007, or cocaine to bind to σ1R, we used a competition mode of the assay by adding 1 μM haloperidol (Figure 3E-H) or 1 μM PD144418 (Figure 3I-L) prior to addition of the tested ligands (see Methods). We found all five compounds showed dose-dependent effects in the competition mode – the negative values in Figure 3E-L indicate direct competition, as the BRET signals induced by the pre-added haloperidol or PD144418 were subtracted to 0 in the analysis. The inhibition potencies for these drugs were correlated with their relative differences in binding affinities as well, though the overall potency values were more right-shifted (less potent) than the Ki values. Further, when the lower efficacy (Emax shown in the single-ligand mode) ligand PD144418 was displaced by haloperidol or 4PPBP, both of which have higher Emax values, positive BRET signals were induced as compared to the negative signal observed for the non-efficacious ligands (Figure 3I-L).
Of note, when we carried out the same assays using the N-terminal NL-σ1:VN-σ1 construct pair (Supplementary Figure 3), the data could not be deciphered by conventional regression analysis. Thus, even though the BRET ratios for the N-terminal pair were higher than the C-termini pair, it was more feasible to study the structure-activity relationships (SAR) of these σ1R ligands in the σ1R homomerization with the C-terminal fusion constructs.
3.5. Haloperidol promotes the formation of higher order σ1R homomers in a non-denaturinggel assay
Because haloperidol increased σ1R homomeric interaction in our BRET assays, we further explored ligand effects on σ1R multimerization using a biochemical method described recently (Hong et al., 2017). In non-denaturing gels, we observed low molecular weight (MW) bands likely corresponding to monomer and dimer of σ1R, and high MW bands indicative of σ1R multimers. Concentrations of 0.1 or 1 μM haloperidol dramatically increased the multimer band intensities and decreased the low MW band intensities, in a dose dependent manner. Consistent with enhanced BRET signals by haloperidol in Figure 2, this result showed that haloperidol promoted or stabilized σ1R multimerization. On the other hand, 10 μM (+)-pentazocine decreased the density of multimer bands while increasing those of low MW bands (Figure 4). In addition, we evaluated the capability of (+)-pentazocine to inhibit the multimer-promoting effect by haloperidol, using two ratios of haloperidol and (+)-pentazocine, 0.1:10 μM (1:100) and 1:10 μM (1:10). Results showed that (+)-pentazocine inhibited haloperidol's effect in a dose dependent manner (Figure 4).
Figure 4.
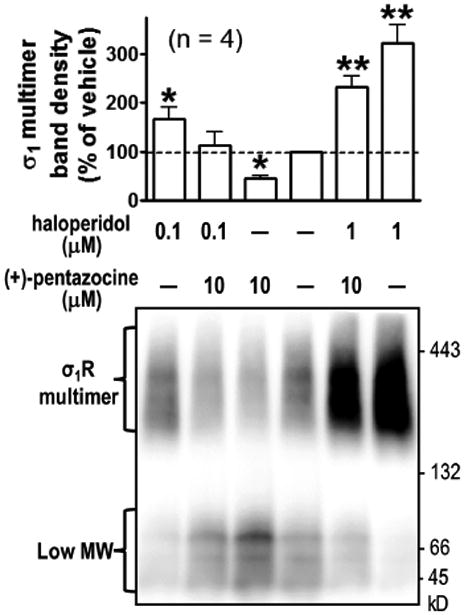
Non-denaturing gel analysis of drug-induced σ1R-σ1R interaction. Cells expressing FH-σ1R (MW: 32 kDa) were incubated with haloperidol in culture medium at 37 °C for 0.5 h, followed by incubation of (+)-pentazocine for 1 h. Cells were lysed with GDN lysis buffer and subjected to PFO-PAGE. Flag antibodies detected high-MW multimeric and low-MW (likely monomer and dimer) bands, based on their apparent MW. Quantified results of multimeric band signals (mean ± SEM, n = 4 experiments), with a representative blot. * P < 0.05, ** P < 0.01, one way ANOVA and post-hoc Dunnett's test, compared with vehicle. Faint signals (∼ 50 kDa) are likely due to partial degradation of dimeric σ1R.
These results suggest that the well-characterized σ1R agonist (+)-pentazocine and antagonist haloperidol had distinct effects on the multimerization state of σ1R. Whereas haloperidol promotes the formation of higher order homomers, (+)-pentazocine shifts the equilibrium in the opposite direction.
3.6. Computational study reveals a structural basis for ligand categorization
The crystal structures of σ1R/PD144418 and σ1R/4-IBP (PDB ID 5HK1 and 5HK2) (Schmidt et al., 2016) reveal an occluded and elongated binding cavity in a cupin-like β-barrel, with the highly conserved Glu172 located near the center of the cavity, forming a salt bridge with the ligands.
Because the crystal structures have revealed an unexpected topology for the number of transmembrane segments and the locations of the N- and C-termini, we first evaluated the stability of these structures in the lipid bilayer environment. Thus, we immersed the trimers of σ1R/PD144418 complex and the apo state of σ1R (starting from the structure with PDB ID 5HK1) in the explicit lipid-bilayer (see section 2.8) and carried out prolonged MD simulations (Table S1). We found the integrities of the σ1R structure and the trimer interface were well retained in both conditions. Thus, whereas the functional dynamics of the homomer states of σ1R remains to be fully characterized, these studies support the crystal structures as providing a reliable framework to investigate the SAR of structurally diverse σ1R ligands.
We then systematically characterized the binding modes of all the σ1R ligands in Figure 1. We carried out molecular docking of these compounds in all their possible stereoisomeric forms and selected several potential binding poses to be further evaluated by both metadynamics and prolonged conventional MD simulations (see section 2.7). From the simulation results, we identified the most stable modes for each ligand (see section 2.8, and Supplementary Figure 5). Whereas we could narrow down the most stable pose for the other ligands to one, we found two isoforms of (+)-pentazocine that are protonated differently on the charged N atom were similarly stable, and thus we have included the results of both forms in our following interacting-residue analysis.
Based on the MD simulation results, we carried out a hierarchical clustering analysis of σ1R ligands according to their interacting patterns with the binding site residues (see section 2.9). When the number of clusters is reduced to two, we found that haloperidol, PD144418 and 4-PPBP form one cluster, while the rest compounds are in the other cluster, the results of which are correlated well with those from the homomer BRET assay. Interestingly, a fluorophenyl ring of JHW007 protruded into a small pocket that was not occupied by any of haloperidol, PD144418 and 4-PPBP (Figure 5C) and formed a polar interaction with Thr189 (Figure 5D). Similarly, PRE-084 and one of the (+)-pentazocine isomers could also occupy this pocket, with the latter stably forming a hydrogen bond (H-bond) with Thr189 (Figure 5D). This pocket is encircled by Val84, Asn85, Ala86, Leu186, Asp188, Thr189, Thr198, and Thr202, and a water molecule is found in this pocket in the crystal structure of σ1R/PD144418. Among these residues, Asn85 and Asp188 face the central tunnel formed by the trimer, while Thr198 forms a H-bond with Asp195, which interacts with His116 and Arg119 of another monomer. Thus, this small pocket appears to be associated with multimerization, and the different interactions with this pocket between the two groups of ligands are potentially correlated with their divergent propensities in promoting or stabilizing the formation of σ1R multimer.
Figure 5.
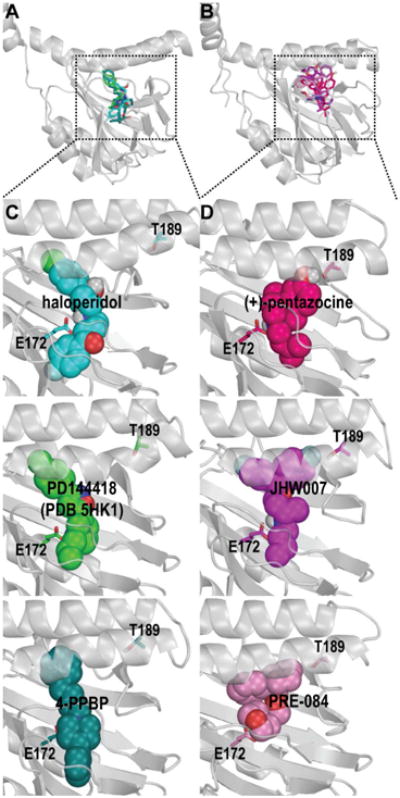
Molecular modeling and simulations of σ1R in complex with its ligands reveal distinct interaction patterns. The stable poses of ligands with multimer-promoting effect (haloperidol, PD144418, and 4-PPBP) and those without ((+)-pentazocine, PRE-084, JHW007, and NE-100) are shown in panels A and B, respectively. The zoom-in views of the representative ligands from each group are shown in panels C and D. Note JHW007, (+)-pentazocine, and PRE-084 protrude into a pocket that is not occupied by the multimer-promoting ligands.
3.7. Haloperidol and (+)-pentazocine have opposite impacts on σ1R-BiP coupling
To demonstrate the applicability of the BRET approach in investigating the functional coupling between σ1R and its client proteins, we focused on characterizing the interaction between σ1R and BiP. First, the acceptor-saturating BRET between σ1R and BiP showed robust increases in BRET signals in two out of four tested BRET construct pairs, i.e., NL-σ1:VN-BiP and σ1-NL:VN-BiP, with the saturability indicating specific interactions between the two proteins (Supplementary Figure 4A,B). BiP-VN C-terminal fusion does not seem to be a good sensor for BRET since the C-terminal domain plays a role in substrate binding for BiP (Yang et al., 2015). To be consistent with the σ1R homomer BRET, we used the σ1-NL:VN-BiP pair to evaluate the impact of σ1R ligand binding on this coupling. Interestingly, (+)-pentazocine showed the bimodal dose response effect at 2.5 and 10 min time points, whereas haloperidol showed a negative induced BRET signal at 60 min (Figure 6). In the competition mode of the assay, the presence of 1 μM (+)-pentazocine diminished the negative BRET signal induced by haloperidol. These results suggest two temporally distinct events: one immediately after the ligand addition and one at a later time point. A similar trend was observed in the N-terminal fusion pair (NL-σ1R:VN-BiP; see Supplementary Results and Supplementary Figure 4C).
Figure 6.
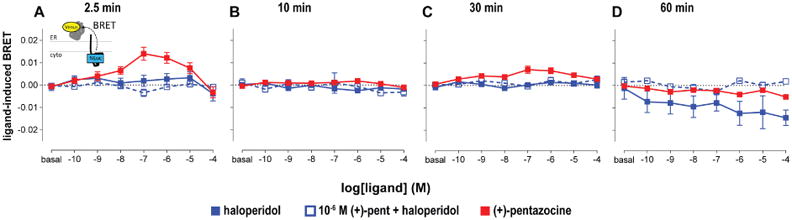
Drug induced σ1R-BiP BRET changes. Dose response change in BRET is detected between C-terminally fused σ1-NL and VN-BiP at 2.5 (A), 10 (B), 30 (C), 60 (D) min for (+)-pentazocine (red), haloperidol (blue solid), haloperidol in the presence of 1 μM (+)-pentazocine (blue open). Data represents mean ± S.E.M. (n = 5 or more).
4. Discussion
In our binding experiments, the Bmax determined by [3H]-haloperidol showed a statistically nonsignificant trend to be higher than that determined by [3H]-(+)-pentazocine (Table 1), suggesting the presence of off-target binding site(s) for haloperidol, such as the σ2 receptor (σ2R) and aminergic GPCRs. However, the curve fitting could not detect or differentiate more than one binding site. Nevertheless, our results are consistent with σ2R contributing to the Bmax in [3H]-haloperidol but not in [3H]-(+)-pentazocine binding – in a recent study, the σ2R was found to have a Kd of 54.1 nM for haloperidol, which is similar to that of σ1R and cannot be resolved in slope analysis; in comparison, σ2R has a Kd of 2.47 μM for (+)-pentazocine, which is a much higher value than the Kd at σ1R (Alon et al., 2017).
Employing a novel σ1R homomer BRET assay that detects ligand-induced changes between σ1R monomers, we are able to identify ligands that produced a positive dose-dependent increase of σ1R-σ1R homomer BRET signals, such as haloperidol, 4-PPBP, and PD144418. Although (+)-pentazocine and other ligands did not produce clear response in this assay, it is possible that a higher sensitivity is necessary to discern subtle BRET signals in transiently transfected cells with abundantly expressed BRET sensor proteins. However, they attenuated the increase in BRET signals elicited by haloperidol, PD144418 or 4-PPBP. The fact that the displacement of PD144418 by haloperidol or 4-PPBP resulted in a higher Emax value is consistent with graded effects among the haloperidol-like ligands (Figure 3K,L).
Because σ1R homomer BRET assays did not directly measure functional outcomes after σ1R ligand binding, we do not use “agonist” and “antagonist” in the interpretation of our results. While traditionally σ1R agonists and antagonists are defined by various functional assays such as anti-nociception, the molecular mechanisms that delineate σ1R agonists and antagonists remain to be elucidated. Indeed, in label-free dynamic mass redistribution assays, we did not see significant changes by all tested σ1R ligands, indicating that their σ1R-dependent signaling events are very different from GPCR-dependent ones (Schroder et al., 2010) and perhaps not as robust near the plasma membrane (Supplementary Results and Supplementary Figure 6). Although some ligands exhibited unexpected results in the BRET assays, we did observe similar phenotypes in “agonists” ((+)-pentazocine and PRE-084) and “antagonists” (haloperidol and PD144418), consistent with their characterization in the literature (Maurice and Su, 2009). Nonetheless, the unexpected phenotypes of NE-100, and 4-PPBP are intriguing. It is plausible that such discrepancies could be attributed to subtle responses by these ligands that are not detected by our BRET assays, or masked by their off-target effects, or likely due to extrinsic factors such as client proteins. In addition, the inactivity of cocaine in our σ1R homomer BRET assay is worth noting. Although the low potency of cocaine in the single-ligand homomer BRET assays renders it difficult to interpret its phenotype, based on the results at 30 and 60 min time points (Figure 3C,D), it is tempting to speculate that cocaine is more like haloperidol in this aspect. Interestingly, JHW007 has been found to counter-act the stimulant and reinforcing effects of cocaine (Hiranita et al., 2017), and is different from haloperidol in our BRET assays. Indeed, JHW007 is a dual acting ligand that binds to both σ1R and the dopamine transporter (DAT) with high affinities (Agoston et al., 1997; Hiranita et al., 2017; Katz et al., 2004). JHW007 stabilizes DAT in a more inward-facing conformation from that stabilized by cocaine (Abramyan et al., 2017; Beuming et al., 2008; Bisgaard et al., 2011; Loland et al., 2008). Thus, if cocaine and JHW007 belong to different categories of σ1R ligands, we can envision that JHW007 antagonizes the actions of cocaine at both DAT and σ1R, two important targets that have been found to interact dynamically at or near the plasma membrane (Hong et al., 2017; Sambo et al., 2017). Interestingly, to the extent of our simulations in this study, we could identify a stable pose of cocaine that does not protrude into the small pocket occupied only by JHW007, (+)-pentazocine, and PRE-084.
Homomer BRET does not provide information directly on how many monomers constitute the σ1R complex as the BRET ratio is not quantitative for a donor:acceptor stoichiometric ratio. However, it is worth noting that the Emax are saturable (e.g., haloperidol, PD144418, and 4PPBP) indicating that the equilibrated donor:acceptor ratio is capped at a certain level within the BRET-participating populations. Although we may not know the stoichiometric ratio, it is interesting to observe graded saturation levels of Emax among different σ1R ligands, which may indicate different higher order homomers.
To evaluate the ligand effects on the homomerization state of σ1R directly, we employed a biochemical assay described recently (Hong et al., 2017). By comparing the results from these non-denaturing gels and BRET assays, we conclude that the increase of the BRET signal is associated with the formation of higher-order homomers, based on the following two common trends: first, the observation that haloperidol promotes the multimerization and (+)-pentazocine competes against that effect in gel assays is in line with the differences observed between these two ligands in the homomer BRET assay; second, the multimer band is made of different populations and distinctly heavier than a trimer, indicating the possibility of several high-order multimers, which may be reflected by different strengths of BRET signals elicited by the haloperidol-like ligands; third, the density changes for the multimer are inversely correlated with those of low-MW populations (monomer and dimer), consistent with the observed displacement of haloperidol by (+)-pentazocine in the competition BRET assay.
σ1R has been shown to regulate chaperone activities of BiP in the ER via σ1R-BiP interaction (Hayashi and Su, 2007; Ortega-Roldan et al., 2013). The opposite effects of (+)-pentazocine and haloperidol demonstrated by our σ1R-BiP BRET assay, along with their effects in the homomer BRET assay, argue that the σ1R-BiP interaction is modulated differently by these σ1R ligands. Indeed, they also induced distinct effects on σ1R homomerization in biochemical assays using non-denaturing gels. Interestingly, the opposite pharmacological profiles between the homomer BRET and σ1R-BiP BRET can be interpreted as that the promotion of homomerization by haloperidol diminishes σ1R-BiP interaction, whereas unchanged or diminished homomerization by (+)-pentazocine may facilitate σ1R interacting with BiP. We propose that the physiological significance of σ1R homomerization is tightly linked to the interaction with its client protein BiP. Such a scenario closely resembles the hypothesis proposed on the interaction between σ1R and DAT in a recent study (Hong et al., 2017). Determining whether similar changes occur during the interaction of σ1R with other client proteins will require further investigations. Although there may be different interpretations of ligand effects on the σ1R-BiP interaction, both current and previous studies (Hayashi and Su, 2007) suggest that σ1R ligands dynamically regulate σ1R-BiP interactions. In BRET assays, we were able to examine time-course and dose-response of σ1R ligand in live cells, which was not achieved by previous co-immunoprecipitation methods. Thus, the bimodal dose response and time dependent change of the (+)-pentazocine effect (Figure 6) may indicate that the ligand-induced cellular trafficking of σ1R affects its interaction with ER-resident BiP. Indeed, a (+)-pentazocine-induced decrease in σ1R-BiP interaction at later time points in our study here coincides with a previous report of 30 min (+)-pentazocine incubation time point (Su et al., 2016).
Note, however, since BRET appears to happen across the membrane in our HEK 293T cell model, our BRET results on σ1R-BiP interaction are consistent with, but do not definitively prove the orientation of σ1R relative to the ER lumen and cytoplasm depicted in the cartoons in Figure 6 and Supplementary Figure 4, i.e., with the N terminus facing the ER lumen (or the extracellular side). Interestingly, recent work by the Yang group (Mavylutov et al., 2017) suggests that the C terminus of σ1R may face the ER lumen.
In our MD simulations, we found that the trimer models of the σ1R/PD144418 complex and its apo form embedded in lipid bilayers are highly stable in terms of the integrity of the ligand binding site and the trimer interface. This finding supports that the crystal structure from which these models were derived is in a valid conformation, thereby the N- and C-termini of the protein are on two different sides of the membrane. The results of our topological analysis of σ1R by the BRET assay resonate with this conclusion as well. Based on these consistent conclusions from different approaches, we carried out a σ1R structure-based SAR study to investigate the structural basis for the different BRET profiles of the two groups of σ1R ligands. In particular, we found JHW007, (+)-pentazocine, and PRE-084 protrude into a small pocket near the trimer interface that was not occupied by haloperidol, 4PPBP, and PD144418. Thus, our computational results described herein reveal the initial clues of the SAR profile for the tested ligands.
Whereas our work establishes a novel molecular biophysical approach to classify σ1R ligands, it is important to point out that the resulting classifications of the ligands are not necessarily in contradiction to previous characterizations of these ligands, many of which are based on functional readouts downstream of the initial σ1R-ligand interactions. Such readouts are assumed to have integrated their impact from the interactions between σ1R and its client proteins, and the off-target effects of ligands. Thus, our BRET approach sheds light on the ligand-induced conformational changes of σ1R, in combination and correlation with the σ1R structure-based SAR studies, it would provide a framework at the atomistic level to investigate the structure-function relationship of σ1R.
5. Conclusions
We established a novel σ1R homomer BRET assay to profile the pharmacological properties of σ1R ligands based on induced changes in σ1R homomerization, and identified multimer-promoting and non-promoting ligands, the conclusion of which is substantiated by a biochemical assay at the protein-protein interaction level. The results of a complementary BRET assay on the σ1R-BiP interaction reflected the reciprocal pharmacological behavior seen in σ1R homomerization. In characterizing the binding modes of the experimentally tested compounds at σ1R with extensive MD simulations, we revealed initial clues for the structural basis that may be responsible for stabilization of the homomer.
Supplementary Material
Highlights.
Both our experimental and computational results support the topology revealed by the crystal structures of σ1R.
Novel BRET assays are capable of examining σ1R homomeric and σ1R-BiP heteromeric interactions upon ligand binding.
Our findings shed light on the ligand-induced conformational changes of σ1R.
Acknowledgments
This study was supported by the Intramural Research Program of the National Institutes of Health, National Institute on Drug Abuse. WCH was supported by Butler University faculty startup fund. The authors would like to thank Dr. Sergi Ferré for generously sharing equipment. We thank Dr. Tsung-Ping Su for insightful discussions, and Dr. Ning-Sheng Cai for her technical expertise.
Footnotes
Author contribution: HY and LS designed the study. AB performed the binding assays; HY carried out the BRET assays; MX and ADF performed molecular docking and dynamics simulations; WCH and SNS designed and performed non-denaturing gel assays; DAG performed the pKa titrations; MX, ADF, AMA, and LS analyzed the modeling results, HY, AB, MX, WCH, AHN and LS interpreted the experimental results; HY and LS wrote the initial draft, with all authors participating in revising the manuscripts.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abramyan AM, Stolzenberg S, Li Z, Loland CJ, Noe F, Shi L. The Isomeric Preference of an Atypical Dopamine Transporter Inhibitor Contributes to Its Selection of the Transporter Conformation. ACS Chem Neurosci. 2017 doi: 10.1021/acschemneuro.7b00094. [DOI] [PubMed] [Google Scholar]
- Agoston GE, Wu JH, Izenwasser S, George C, Katz J, Kline RH, Newman AH. Novel N-substituted 3 alpha-[bis(4′-fluorophenyl)methoxy]tropane analogues: selective ligands for the dopamine transporter. J Med Chem. 1997;40:4329–4339. doi: 10.1021/jm970525a. [DOI] [PubMed] [Google Scholar]
- Albayrak Y, Hashimoto K. Sigma-1 Receptor Agonists and Their Clinical Implications in Neuropsychiatric Disorders. Adv Exp Med Biol. 2017;964:153–161. doi: 10.1007/978-3-319-50174-1_11. [DOI] [PubMed] [Google Scholar]
- Alon A, Schmidt HR, Wood MD, Sahn JJ, Martin SF, Kruse AC. Identification of the gene that codes for the sigma2 receptor. Proc Natl Acad Sci U S A. 2017;114:7160–7165. doi: 10.1073/pnas.1705154114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydar E, Palmer CP, Klyachko VA, Jackson MB. The sigma receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron. 2002;34:399–410. doi: 10.1016/s0896-6273(02)00677-3. [DOI] [PubMed] [Google Scholar]
- Balasuriya D, Stewart AP, Edwardson JM. The sigma-1 receptor interacts directly with GluN1 but not GluN2A in the GluN1/GluN2A NMDA receptor. J Neurosci. 2013;33:18219–18224. doi: 10.1523/JNEUROSCI.3360-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat Neurosci. 2008;11:780–789. doi: 10.1038/nn.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgaard H, Larsen MA, Mazier S, Beuming T, Newman AH, Weinstein H, Shi L, Loland CJ, Gether U. The binding sites for benztropines and dopamine in the dopamine transporter overlap. Neuropharmacology. 2011;60:182–190. doi: 10.1016/j.neuropharm.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen WD, De Costa BR, Hellewell SB, Walker JM, Rice KC. [3H]-(+)-Pentazocine: A potent and highly selective benzomorphan-based probe for sigma-1 receptors. Molecular Neuropharmacology. 1993;3:117–126. [Google Scholar]
- Brune S, Pricl S, Wunsch B. Structure of the sigma1 receptor and its ligand binding site. J Med Chem. 2013;56:9809–9819. doi: 10.1021/jm400660u. [DOI] [PubMed] [Google Scholar]
- Clark AJ, Tiwary P, Borrelli K, Feng S, Miller EB, Abel R, Friesner RA, Berne BJ. Prediction of Protein-Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations. J Chem Theory Comput. 2016;12:2990–2998. doi: 10.1021/acs.jctc.6b00201. [DOI] [PubMed] [Google Scholar]
- Ferris RM, Tang FL, Chang KJ, Russell A. Evidence that the potential antipsychotic agent rimcazole (BW 234U) is a specific, competitive antagonist of sigma sites in brain. Life Sci. 1986;38:2329–2337. doi: 10.1016/0024-3205(86)90640-5. [DOI] [PubMed] [Google Scholar]
- Fishback JA, Robson MJ, Xu YT, Matsumoto RR. Sigma receptors: potential targets for a new class of antidepressant drug. Pharmacol Ther. 2010;127:271–282. doi: 10.1016/j.pharmthera.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanilla D, Hajipour AR, Pal A, Chu UB, Arbabian M, Ruoho AE. Probing the steroid binding domain-like I (SBDLI) of the sigma-1 receptor binding site using N-substituted photoaffinity labels. Biochemistry. 2008;47:7205–7217. doi: 10.1021/bi800564j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore DL, Liu Y, Matsumoto RR. Review of the pharmacological and clinical profile of rimcazole. CNS Drug Rev. 2004;10:1–22. doi: 10.1111/j.1527-3458.2004.tb00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Soler M, Fernandez-Duenas V, Portillo-Salido E, Perez P, Zamanillo D, Vela JM, Burgueno J, Ciruela F. Predicting the antinociceptive efficacy of sigma(1) receptor ligands by a novel receptor fluorescence resonance energy transfer (FRET) based biosensor. J Med Chem. 2014;57:238–242. doi: 10.1021/jm401529t. [DOI] [PubMed] [Google Scholar]
- Grandinetti G, Smith AE, Reineke TM. Membrane and nuclear permeabilization by polymeric pDNA vehicles: efficient method for gene delivery or mechanism of cytotoxicity? Mol Pharm. 2012;9:523–538. doi: 10.1021/mp200368p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromek KA, Suchy FP, Meddaugh HR, Wrobel RL, LaPointe LM, Chu UB, Primm JG, Ruoho AE, Senes A, Fox BG. The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J Biol Chem. 2014;289:20333–20344. doi: 10.1074/jbc.M113.537993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha Y, Shanmugam AK, Markand S, Zorrilla E, Ganapathy V, Smith SB. Sigma receptor 1 modulates ER stress and Bcl2 in murine retina. Cell Tissue Res. 2014;356:15–27. doi: 10.1007/s00441-013-1774-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E, Glossmann H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci U S A. 1996;93:8072–8077. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J Chem Theory Comput. 2016;12:281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Hiranita T, Hong WC, Kopajtic T, Katz JL. sigma Receptor Effects of N-Substituted Benztropine Analogs: Implications for Antagonism of Cocaine Self-Administration. J Pharmacol Exp Ther. 2017;362:2–13. doi: 10.1124/jpet.117.241109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong WC, Yano H, Hiranita T, Chin FT, McCurdy CR, Su TP, Amara SG, Katz JL. The sigma-1 receptor modulates dopamine transporter conformation and cocaine binding and may thereby potentiate cocaine self-administration in rats. J Biol Chem. 2017;292:11250–11261. doi: 10.1074/jbc.M116.774075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B, Sali A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003;31:3982–3992. doi: 10.1093/nar/gkg460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz JL, Hiranita T, Hong WC, Job MO, McCurdy CR. A Role for Sigma Receptors in Stimulant Self-Administration and Addiction. Handb Exp Pharmacol. 2017 doi: 10.1007/164_2016_94. [DOI] [PubMed] [Google Scholar]
- Katz JL, Hong WC, Hiranita T, Su TP. A role for sigma receptors in stimulant self-administration and addiction. Behav Pharmacol. 2016;27:100–115. doi: 10.1097/FBP.0000000000000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz JL, Kopajtic TA, Agoston GE, Newman AH. Effects of N-substituted analogs of benztropine: diminished cocaine-like effects in dopamine transporter ligands. J Pharmacol Exp Ther. 2004;309:650–660. doi: 10.1124/jpet.103.060525. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Su TP, Fujimoto M, Bonci A. The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci. 2012;35:762–771. doi: 10.1016/j.tins.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Nuber S, Hoffmann C. Fluorescence/bioluminescence resonance energy transfer techniques to study G-protein-coupled receptor activation and signaling. Pharmacol Rev. 2012;64:299–336. doi: 10.1124/pr.110.004309. [DOI] [PubMed] [Google Scholar]
- Loland CJ, Desai RI, Zou MF, Cao J, Grundt P, Gerstbrein K, Sitte HH, Newman AH, Katz JL, Gether U. Relationship between conformational changes in the dopamine transporter and cocaine-like subjective effects of uptake inhibitors. Mol Pharmacol. 2008;73:813–823. doi: 10.1124/mol.107.039800. [DOI] [PubMed] [Google Scholar]
- Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- Marullo S, Bouvier M. Resonance energy transfer approaches in molecular pharmacology and beyond. Trends Pharmacol Sci. 2007;28:362–365. doi: 10.1016/j.tips.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Maurice T, Goguadze N. Sigma-1 (sigma1) Receptor in Memory and Neurodegenerative Diseases. Handb Exp Pharmacol. 2017 doi: 10.1007/164_2017_15. [DOI] [PubMed] [Google Scholar]
- Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavylutov T, Chen X, Guo L, Yang J. APEX2- tagging of Sigma 1-receptor indicates subcellular protein topology with cytosolic N-terminus and ER luminal C-terminus. Protein & Cell. 2017 doi: 10.1007/s13238-017-0468-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlos M, Burgueno J, Portillo-Salido E, Plata-Salaman CR, Vela JM. Pharmacological Modulation of the Sigma 1 Receptor and the Treatment of Pain. Adv Exp Med Biol. 2017;964:85–107. doi: 10.1007/978-3-319-50174-1_8. [DOI] [PubMed] [Google Scholar]
- Michino M, Boateng CA, Donthamsetti P, Yano H, Bakare OM, Bonifazi A, Ellenberger MP, Keck TM, Kumar V, Zhu C, Verma R, Deschamps JR, Javitch JA, Newman AH, Shi L. Toward Understanding the Structural Basis of Partial Agonism at the Dopamine D3 Receptor. J Med Chem. 2017;60:580–593. doi: 10.1021/acs.jmedchem.6b01148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michino M, Free RB, Doyle TB, Sibley DR, Shi L. Structural basis for Na(+)-sensitivity in dopamine D2 and D3 receptors. Chem Commun (Camb) 2015;51:8618–8621. doi: 10.1039/c5cc02204e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y, Tanji K, Mori F, Wakabayashi K. Sigma-1 receptor is involved in degradation of intranuclear inclusions in a cellular model of Huntington's disease. Neurobiol Dis. 2015;74:25–31. doi: 10.1016/j.nbd.2014.11.005. [DOI] [PubMed] [Google Scholar]
- Mishra AK, Mavlyutov T, Singh DR, Biener G, Yang J, Oliver JA, Ruoho A, Raicu V. The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem J. 2015;466:263–271. doi: 10.1042/BJ20141321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L, Lucke-Wold BP, Mookerjee S, Kaushal N, Matsumoto RR. Sigma-1 Receptors and Neurodegenerative Diseases: Towards a Hypothesis of Sigma-1 Receptors as Amplifiers of Neurodegeneration and Neuroprotection. Adv Exp Med Biol. 2017;964:133–152. doi: 10.1007/978-3-319-50174-1_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono Y, Tanaka H, Tsuruma K, Shimazawa M, Hara H. A sigma-1 receptor antagonist (NE-100) prevents tunicamycin-induced cell death via GRP78 induction in hippocampal cells. Biochem Biophys Res Commun. 2013;434:904–909. doi: 10.1016/j.bbrc.2013.04.055. [DOI] [PubMed] [Google Scholar]
- Ortega-Roldan JL, Ossa F, Amin NT, Schnell JR. Solution NMR studies reveal the location of the second transmembrane domain of the human sigma-1 receptor. FEBS Lett. 2015;589:659–665. doi: 10.1016/j.febslet.2015.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Roldan JL, Ossa F, Schnell JR. Characterization of the human sigma-1 receptor chaperone domain structure and binding immunoglobulin protein (BiP) interactions. J Biol Chem. 2013;288:21448–21457. doi: 10.1074/jbc.M113.450379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossa F, Schnell JR, Ortega-Roldan JL. A Review of the Human Sigma-1 Receptor Structure. Adv Exp Med Biol. 2017;964:15–29. doi: 10.1007/978-3-319-50174-1_3. [DOI] [PubMed] [Google Scholar]
- Penas C, Pascual-Font A, Mancuso R, Fores J, Casas C, Navarro X. Sigma receptor agonist 2-(4-morpholinethyl)1 phenylcyclohexanecarboxylate (Pre084) increases GDNF and BiP expression and promotes neuroprotection after root avulsion injury. J Neurotrauma. 2011;28:831–840. doi: 10.1089/neu.2010.1674. [DOI] [PubMed] [Google Scholar]
- Pfleger KD, Seeber RM, Eidne KA. Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein-protein interactions. Nat Protoc. 2006;1:337–345. doi: 10.1038/nprot.2006.52. [DOI] [PubMed] [Google Scholar]
- Romero L, Merlos M, Vela JM. Antinociception by Sigma-1 Receptor Antagonists: Central and Peripheral Effects. Adv Pharmacol. 2016;75:179–215. doi: 10.1016/bs.apha.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Rousseaux CG, Greene SF. Sigma receptors [sigmaRs]: biology in normal and diseased states. J Recept Signal Transduct Res. 2015:1–62. doi: 10.3109/10799893.2015.1015737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabino V, Hicks C, Cottone P. Sigma Receptors and Substance Use Disorders. Adv Exp Med Biol. 2017;964:177–199. doi: 10.1007/978-3-319-50174-1_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambo DO, Lin M, Owens A, Lebowitz JJ, Richardson B, Jagnarine DA, Shetty M, Rodriquez M, Alonge T, Ali M, Katz J, Yan L, Febo M, Henry LK, Bruijnzeel AW, Daws L, Khoshbouei H. The sigma-1 receptor modulates methamphetamine dysregulation of dopamine neurotransmission. Nat Commun. 2017;8:2228. doi: 10.1038/s41467-017-02087-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Fernandez C, Entrena JM, Baeyens JM, Cobos EJ. Sigma-1 Receptor Antagonists: A New Class of Neuromodulatory Analgesics. Adv Exp Med Biol. 2017;964:109–132. doi: 10.1007/978-3-319-50174-1_9. [DOI] [PubMed] [Google Scholar]
- Schmidt HR, Zheng S, Gurpinar E, Koehl A, Manglik A, Kruse AC. Crystal structure of the human sigma1 receptor. Nature. 2016;532:527–530. doi: 10.1038/nature17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenwald RD, Barfknecht CF, Shirolkar S, Xia E. The effects of sigma ligands on protein release from lacrimal acinar cells: a potential agonist/antagonist assay. Life Sci. 1995;56:1275–1285. doi: 10.1016/0024-3205(95)00073-9. [DOI] [PubMed] [Google Scholar]
- Schroder R, Janssen N, Schmidt J, Kebig A, Merten N, Hennen S, Muller A, Blattermann S, Mohr-Andra M, Zahn S, Wenzel J, Smith NJ, Gomeza J, Drewke C, Milligan G, Mohr K, Kostenis E. Deconvolution of complex G protein-coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nat Biotechnol. 2010;28:943–949. doi: 10.1038/nbt.1671. [DOI] [PubMed] [Google Scholar]
- Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Sohy D, Yano H, de Nadai P, Urizar E, Guillabert A, Javitch JA, Parmentier M, Springael JY. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the Protean Effects of “Selective” Antagonists. Journal of Biological Chemistry. 2009;284:31270–31279. doi: 10.1074/jbc.M109.054809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriani O, Rapetti-Mauss R. Sigma 1 Receptor and Ion Channel Dynamics in Cancer. Adv Exp Med Biol. 2017;964:63–77. doi: 10.1007/978-3-319-50174-1_6. [DOI] [PubMed] [Google Scholar]
- Su TP. Evidence for sigma opioid receptor: binding of [3H]SKF-10047 to etorphine-inaccessible sites in guinea-pig brain. J Pharmacol Exp Ther. 1982;223:284–290. [PubMed] [Google Scholar]
- Su TP, Su TC, Nakamura Y, Tsai SY. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharmacol Sci. 2016;37:262–278. doi: 10.1016/j.tips.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam SW, Cook L. Sigma opiates and certain antipsychotic drugs mutually inhibit (+)-[3H] SKF 10,047 and [3H]haloperidol binding in guinea pig brain membranes. Proc Natl Acad Sci U S A. 1984;81:5618–5621. doi: 10.1073/pnas.81.17.5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DP, Eison MS, Moon SL, Schlemmer RF, Jr, Shukla UA, VanderMaelen CP, Yocca FD, Gallant DJ, Behling SH, Boissard CG, et al. A role for sigma binding in the antipsychotic profile of BMY 14802? NIDA Res Monogr. 1993;133:125–157. [PubMed] [Google Scholar]
- Urizar E, Yano H, Kolster R, Galés C, Lambert N, Javitch JA. CODA-RET reveals functional selectivity as a result of GPCR heteromerization. Nat Chem Biol. 2011;7:624–630. doi: 10.1038/nchembio.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Cui X, Roon P, Saul A, Smith SB. The Role of Sigma1R in Mammalian Retina. Adv Exp Med Biol. 2017;964:267–284. doi: 10.1007/978-3-319-50174-1_18. [DOI] [PubMed] [Google Scholar]
- Yang J, Nune M, Zong Y, Zhou L, Liu Q. Close and Allosteric Opening of the Polypeptide-Binding Site in a Human Hsp70 Chaperone BiP. Structure. 2015;23:2191–2203. doi: 10.1016/j.str.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, Cai N, Javitch JA, Ferre S. Luciferase complementation based detection of G protein coupled receptor activity. doi: 10.2144/btn-2018-0039. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.