

Abstract
The shortage of molecular information for taxol-producing fungi has greatly impeded the understanding of fungal taxol biosynthesis mechanism. In this study, the transcriptome of one taxol-producing endophytic fungus Cladosporium cladosporioides MD2 was sequenced for the first time. About 1.77 Gbp clean reads were generated and further assembled into 16,603 unigenes with an average length of 1110 bp. All of the unigenes were annotated against seven public databases to present the transcriptome characteristics of C. cladosporioides MD2. A total of 12,479 unigenes could be annotated with at least one database, and 1593 unigenes could be annotated in all queried databases. In total, 8425 and 3350 unigenes were categorized into 57 GO functional groups and 262 KEGG pathways, respectively, exhibiting the dominant GO terms and metabolic pathways in the C. cladosporioides MD2 transcriptome. One potential and partial taxol biosynthetic pathway was speculated including 9 unigenes related to terpenoid backbone biosynthesis and 40 unigenes involved in the biosynthetic steps from geranylgeranyl diphosphate to 10-deacetylbaccatin III. These results provided valuable information for the molecular mechanism research of taxol biosynthesis in C. cladosporioides MD2.
Electronic supplementary material
The online version of this article (10.1186/s13568-018-0567-6) contains supplementary material, which is available to authorized users.
Keywords: Endophytic fungus, Cladosporium cladosporioides, Transcriptome, Taxol
Introduction
Taxol (also named as Paclitaxel) originally isolated from Taxus brevifolia (Wani et al. 1971) was well-known as an anti-cancer drug used in clinic. Fermentation processes using taxol-producing microorganisms were considered as an efficient and sustainable way to produce taxol (Gond et al. 2014; Choi et al. 2016). Since one taxol-producing endophytic fungus Taxomyce andreanae isolated from the bark of T. brevifolia was firstly reported (Stierle et al. 1993), more than 40 fungal genera about 200 endophytic fungi had been reported to produce taxol (Flores-Bustamante et al. 2010; Kusari et al. 2014). However, low and un-stable taxol productivity in fungi restricted their industrial application (Gond et al. 2014; Kusari et al. 2014). Targeted genetic engineering of taxol-producing fungi was an effective way to exploit their taxol productivity, however there were difficult challenges due to the almost complete lack of molecular information on the exact pathway, key rate-limiting steps, and availability of a complete set of genes involved in the taxol biosynthesis of fungi (Flores-Bustamante et al. 2010; Gond et al. 2014). Thus, a prerequisite was to discover taxol-related genes and uncover the molecular mechanism of taxol biosynthesis in fungi (Flores-Bustamante et al. 2010; Kusari et al. 2014).
High-throughput RNA-seq technology has been extensively used for transcriptome analysis of many organisms without reference genomes to discover novel and interesting genes (Grabherr et al. 2011; Zhang et al. 2013). Some of representative examples were that several transcriptomes of Taxus were investigated with RNA-seq technology to comprehensively reveal candidate genes involved in taxol biosynthesis. da Hao et al. (2011) used RNA-seq technology to sequence the leaf transcriptome of Taxus mairei, and obtained 36,493 unigenes, including hundreds of taxane biosynthetic genes and some genes involved in tissue specific functions. Li et al. (2012) used RNA-seq technology to investigate the transcriptional profile of Taxus chinensis cells in response to methyl jasmonate (MeJA) elicitation, obtained 46,581 unigenes, and detected 13,469 differentially expressed genes, including all of the known jasmonate (JA) biosynthesis/JA signaling pathway genes and taxol-related genes. Sun et al. (2013) used RNA-seq technology to investigate the transcriptomes of MeJA-treated and un-treated Taxus x media cells, obtained 40,348 unigenes, detected all the 29 known genes involved in the biosynthesis of terpenoid backbone and taxol, and found multiple candidates for the unknown steps in taxol biosynthesis. Additionally, Qiao et al. (2014) used RNA-seq technology to sequence the leaf transcriptome of Cephalotaxus hainanensis, obtained 39,416 unigenes, and detected a number of putative candidate genes involved in taxol biosynthesis. More importantly, most of taxol-related genes in Taxus have been cloned and characterized in vitro by functional expression in Escherichia coli or yeast using ‘surrogate’ substrates, and one taxol biosynthesis pathway in Taxus consisting of about 20 biosynthetic enzymes was depicted (Jennewein and Croteau 2001; Walker and Croteau 2001; Croteau et al. 2006; Guo et al. 2006; Liu et al. 2016). These fruitful results gave us a relatively comprehensive knowledge of the taxol biosynthesis in Taxus. However, little was known about the fungal taxol biosynthesis pathway and taxol-related genes in fungi.
Recently, Yang et al. (2014) sequenced the genome of one taxol-producing fungus Penicillium aurantiogriseum NRRL 62431 isolated from Corylus avellana L., identified candidate genes involved in taxol biosynthesis, and found low sequence identities between the taxol biosynthetic candidate genes of P. aurantiogriseum NRRL 62431 and that of its host hazel as well as that of Taxus baccata. However, the molecular information on the taxol biosynthesis of Cladosporium cladosporioides MD2, one taxol-producing fungus isolated from T. x media (Zhang et al. 2009), was almost completely lacking. In this study, the transcriptome of C. cladosporioides MD2 was firstly sequenced by Illumina RNA-seq technology to generate 19,714,114 clean reads, that were de novo assembled with Trinity program (Grabherr et al. 2011) into 16,603 unigenes. All of the unigenes were annotated and analyzed by using BLAST against various protein and nucleotide databases, and the biological functions and metabolic pathways of these unigenes were further characterized through Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation. One potential and partial taxol biosynthesis pathway in C. cladosporioides MD2 was speculated, and dozens of unigenes were annotated to be involved in taxol biosynthesis. These results provided a valuable source for further gene discovery, functional genomic research and comparative transcriptome analysis of C. cladosporioides MD2.
Materials and methods
Fungal material and RNA extraction
Cladosporium cladosporioides MD2 was identified by China Center for Type Culture Collection (CCTCC) and deposited under CCTCC no. 207063 (Zhang et al. 2009). One milliliter of the spore suspension (about 108 spores) of C. cladosporioides MD2 was cultivated in 100 mL of potato dextrose liquid medium at 28 °C and 150 rpm for 132 h (equal to five and a half days), and the mycelium pellets were collected by centrifugation for RNA extraction. According to the manufacturer’s instruction, total RNA was extracted by using TRIzol® Reagent (Invitrogen, California, USA), treated with DNase I (Invitrogen, California, USA), dissolved in RNase-free water, and stored in a − 80 °C freezer. The quality and integrity of total RNA were detected by using Nanodrop2000 spectrophotometer (Thermo Fisher Scientific, California, USA) and Bioanalyzer 2100 (Agilent, California, USA), respectively. The purity of total RNA was assessed by both A260/280 and A260/230 ratios.
cDNA synthesis and library sequencing
About 60 μg of total RNA at a concentration ≥ 600 ng/μL, OD260/280 = 1.8 ~ 2.2 and RNA integrity number (RIN) ≥ 7.0 were used for construction of a cDNA sequencing library using a TruSeqTM RNA Sample Preparation Kit (Illumina, San Diego, California, USA). Briefly, poly-A mRNA samples were isolated from the total RNA using oligo (dT) attached magnetic beads and then interrupted into 200–500 bp fragments. Subsequently, the fragmented mRNAs as well as random hexamer primers and reverse transcriptase were used to synthesize first-strand cDNAs, that were used to synthesize second-strand cDNAs with DNA polymerase I and RNase H. The cDNA products were purified with QIAquick PCR Purification Kit (Qiagen, California, USA), end-repaired and A-tailed using End Repair Mix (Qiagen, California, USA), and then ligated with sequencing index adapters (Illumina, San Diego, California, USA). The ligated products were isolated by gel electrophoresis to select suitable fragments and were subsequently amplified to generate the final cDNA libraries, that were sequenced using Illumina HiSeq™ 2500 sequencing platform at Wuhan Bio-Broad Biotechnology Co., Ltd. (Wuhan, China).
De novo assembly by trinity
The raw reads were filtered to generate clean reads by removal of adaptors, ambiguous reads with unknown nucleotides larger than 10%, and low-quality reads where more than half of the bases’ qualities were less than five (Quality score < 5). The clean reads were deposited in NCBI Sequence Read Archive (SRA) Sequence Database with accession number SRR6147052. Subsequently, the clean reads were de novo assembled by Trinity program (Grabherr et al. 2011) to generate non-redundant unigenes. In brief, all the clean reads were assembled into contigs with different k-mer values to assess optimal assembly range. The assembled transcripts were as reference sequences for transcript annotation and re-validated by aligning clean reads back to transcripts (Miller et al. 2012). Each of clean reads was re-mapped to reference sequences using Bowtie (Langmead 2010) with default parameters. The longest transcripts within each operationally assembled contig were defined as unigenes.
Transcriptome annotation
The annotation of unigenes were performed by using BLAST algorithm against NCBI non-redundant protein sequence (Nr) database, NCBI non-redundant nucleotide sequence (Nt) database, Swiss-Prot database, Protein family (Pfam) database with an E-value threshold of 1.0E−5, and the best aligning results were used to annotate unigenes and decide the sequence direction of each unigene. Additionally, some unigenes that had no alignment hit in the above databases were further estimated by using ESTScan (3.0.3) software (Iseli et al. 1999) to predict their protein coding sequences (CDS). With Nr and Swiss-Prot annotation, Blast2GO software (Conesa et al. 2005) was used to perform gene ontology (GO) annotation of unigenes to obtain cellular component, molecular function and biological process terms. After retrieving the associated GO terms, the possible functions of unigenes were predicted against eukaryotic Orthologous Group (KOG) database. Additionally, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways annotation of unigenes was performed by mapping the sequences obtained from Blast2GO to the contents of KEGG database (Kanehisa and Goto 2000).
Analysis of potential fungal taxol biosynthesis pathway
The peptide sequences of unigenes were searched on Carbohydrate-Active Enzymes (CAZy) database (Park et al. 2010) to find the enzymes involved in taxol biosynthesis pathway. In parallel, the reported gene sequences involved in the taxol biosynthesis from Taxus or other organisms were retrieved from GenBank. All the sequences were then manually aligned by BLAST search against the assembled contigs to predict potential genes involved in the taxol biosynthesis. All the hits with an E-value threshold of 1.0E−10 were used as queries to search KEGG database again for further identification, and the genes were kept if their subjects were annotated as the enzymes involved in the taxol biosynthesis.
Reverse-transcribed PCR validation of unigenes
Total RNA from C. cladosporioides MD2 was reverse-transcribed by using SuperScript III Reverse Transcriptase (Invitrogen, California, USA) and oligo(dT)18. Ten of assembled unigenes were randomly selected for reverse-transcribed PCR (RT-PCR) validation, and the primer sequences of these assembled unigenes were listed in Additional file 1: Table S1.
Results
Transcriptome sequencing and assembly
The transcriptome of C. cladosporioides MD2 was sequenced with Illumina sequencing technology to generate 19,776,638 raw reads consisting of 1,779,897,420 nucleotides (nt). After removing the adaptor sequences, ambiguous reads and low-quality reads, a total of 19,714,114 clean reads (approximate 1.77 Gbp) were obtained (GenBank Accession Number: SRR6147052), and were further de novo assembled into 16,603 unigenes with an average length of 1110 bp and an N50 of 1894 bp (Table 1). Among them, 6976 unigenes, 2942 unigenes and 3819 unigenes were within the ranges of 200–500, 500–1000 and 1000–2000 bp, respectively, and 2866 unigenes were longer than or equal to 2000 bp (Fig. 1a). Additionally, ten of the assembled unigenes of C. cladosporioides MD2 were randomly selected for RT-PCR validation, and all of the 10 assembled unigenes got right amplifications (Additional file 1: Figure S1). These results indicated the suitability of mRNA-seq for extensive and accurate assembly of the C. cladosporioides MD2 transcriptome.
Table 1.
Assembly summary of C. cladosporioides MD2 unigenes
| Assembly | Number |
|---|---|
| Number of raw reads | 19,776,638 |
| Number of clean reads | 19,714,114 |
| Total unigenes | 16,603 |
| N50 length (bp) | 1894 |
| Mean length (bp) | 1110 |
| Min length (bp) | 201 |
| Max length (bp) | 10,055 |
| Unigenes with 200 ~ 500 bp | 6976 |
| Unigenes with 500 ~ 1000 bp | 2942 |
| Unigenes with 1000 ~ 2000 bp | 3819 |
| Unigenes with ≥ 2000 bp | 2866 |
Fig. 1.
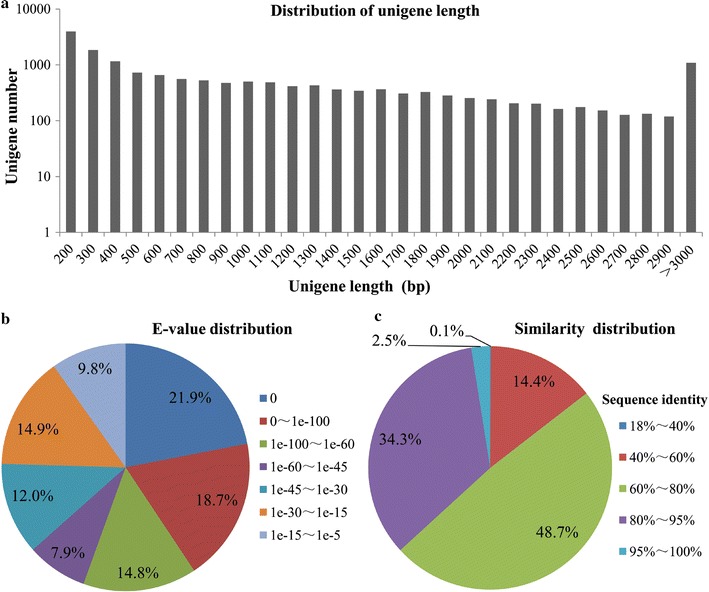
mRNA-seq and de novo assembly of C. cladosporioides MD2 transcriptome. a Distribution of unigene length. b E-value distribution of BLAST hits for each unique sequence against the Nr database. c Similarity distribution of the top BLAST hits for each sequence against the Nr database
Transcriptome annotation
All of the 16,603 unigenes were annotated by BLAST alignment against different protein and nucleotide databases. A total of 11,670 unigenes (70.29%) had homologous sequences in the Nr database (Table 2), suggesting that most of the unigenes could be translated into proteins. Based on the annotation results of Nr database, E-value distribution analysis showed that 75.30 and 63.30% of the matched sequences had strong homology with E-value < 1.0E−30 and E-value < 1.0E−45, respectively, and whereas only 24.5% of the matched sequences had high similarity with E-value from 1.0E−30 to 1.0E−5 (Fig. 1b). Similarity distribution analysis showed that 36.80% of the mapped sequences had a similarity higher than 80%, whereas 63.2% of the mapped sequences had a similarity ranging from 18 to 80% (Fig. 1c). Additionally, species distribution analysis revealed that C. cladosporioides MD2 had a number of homologous sequences with several species, and the genes from Baudoinia compniacensis had the highest similarity (19.90%) with that from C. cladosporioides MD2, followed by Dothistroma septosporum (14.50%), Pseudocercospora fijiensis (10.90%), Zymoseptoria brevis (8.40%), and Zymoseptoria tritici (7.10%). The low sequence similarity of the genes from C. cladosporioides MD2 to the genes from these above species suggested the impossibility of using the genomes of these relative species as references for the transcriptome analysis of C. cladosporioides MD2.
Table 2.
Summary of C. cladosporioides MD2 unigene annotations
| Databases | Number of mapped unigenes | Mapping rates (%) |
|---|---|---|
| Nr | 11,670 | 70.29 |
| Nt | 3048 | 18.35 |
| KEGG | 3350 | 20.17 |
| SwissProt | 7211 | 43.43 |
| Pfam | 8431 | 50.77 |
| GO | 8425 | 50.74 |
| KOG | 4904 | 29.53 |
| Annotated by all seven databases | 1593 | 9.59 |
| Annotated by at least one database | 12,479 | 75.16 |
Additionally, 3048 unigenes (18.35%), 8431 unigenes (50.78%), 7211 unigenes (43.43%), 8425 unigenes (50.74%), 4904 unigenes (29.53%) and 3350 unigenes (20.17%) were annotated in the databases of Nt, Pfam, Swiss-Prot, GO, KOG and KEGG, respectively, and the mapping rates were from 18 to 71% (Table 2). In total, 12,479 unigenes (75.16%) could be annotated with at least one database, and 1593 unigenes (9.59%) could be annotated in all of seven queried databases, with relatively well defined functional annotations (Table 2). These annotations gave us a valuable resource for investigating specific pathways and function genes in C. cladosporioides MD2. Furthermore, all of the unigenes were aligned by BLASTX (E-value ≤ 1.0E−5) to all of seven queried databases to predict their coding sequences (CDS), and the correct reading frames of the nucleotide sequences of unigenes (5′-3′ direction) were defined by the highest rank in the BLAST results. In addition, the CDS of some unigenes, that could not be aligned to the above databases, were predicted by ESTScan software. In total, 16,578 unigenes were identified to harbor their CDS, where the CDS of 11,975 unigenes were identified by BLAST alignment and that of 4603 were estimated by the ESTScan.
GO and KOG classification of unigenes
To classify the predicted function of all unigenes, Blast2GO software was employed to search against GO database and KOG database. A total of 8425 unigenes were categorized into 57 functional groups under three GO main categories, including ‘Biological Process’, ‘Cellular Component’ and ‘Molecular Function’. The ‘Biological Process’ category occupied the largest proportion (25 terms), followed by ‘Cellular Component’ (18 terms) category and ‘Molecular Function’ category (14 terms) (Fig. 2; Additional file 1: Table S2). In the ‘Biological Process’ category, the major GO terms included ‘cellular process’ (4944 unigenes), ‘metabolic process’ (4861 unigenes), ‘single-organism process’ (4090 unigenes), ‘biological regulation’ (1627 unigenes), ‘localization’ (1583 unigenes) and ‘regulation of biological process’ (1496 unigenes). In the ‘Cellular Component’ category, the representative terms included ‘cell’ (2707 unigenes), ‘cell part’ (2707 unigenes), ‘organelle’ (1870 unigenes), ‘macromolecular complex’ (1575 unigenes), ‘membrane’ (1512 unigenes) and ‘membrane-part’ (1429 unigenes). In the ‘Molecular Function’ category, the dominant GO terms were ‘binding’ (4519 unigenes), ‘catalytic activity’ (4156 unigenes), ‘transporter activity’ (726 unigenes) and ‘nucleic acid binding transcription factor activity’ (415 unigenes). A high percentage of genes (more than 4000 unigenes) were categorized into ‘cellular process’, ‘metabolic process’, ‘single-organism process’, ‘binding’ and “catalytic activity”, demonstrating the dominated biological activities being taking place in the C. cladosporioides MD2 transcriptome. In addition, 4904 unigenes were categorized (E-value ≤ 1.0E−5) in 25 functional KOG clusters (Fig. 3), and the five largest categories were ‘general function predictions only’ (827 unigenes), ‘post-translational modification, protein turnover, chaperones’ (492 unigenes), ‘signal transduction mechanisms’ (368 unigenes), ‘translation, ribosomal structure and biogenesis’ (329 unigenes) and ‘energy production and conversion’ (327 unigenes). These results demonstrated the major biological functions of the genes expressed in the C. cladosporioides MD2 transcriptome.
Fig. 2.
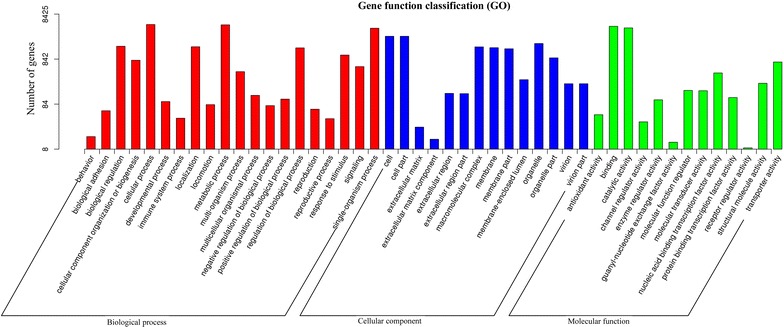
GO annotation of the C. cladosporioides MD2 unigenes. The annotated unigenes were classified into cellular component, molecular function and biological process categories
Fig. 3.
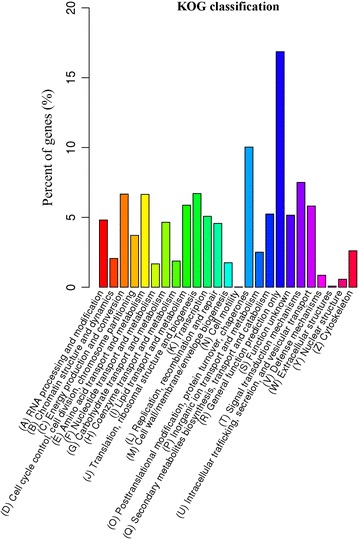
KOG classification of the C. cladosporioides MD2 unigenes. The left y-axis indicated the percent of genes in a category. The dominating categories included ‘general function predictions only’ (827 unigenes), ‘post-translational modification, protein turnover, chaperones’ (492 unigenes), ‘signal transduction mechanisms’ (368 unigenes), ‘translation, ribosomal structure and biogenesis’ (329 unigenes), and ‘energy production and conversion’ (327 unigenes)
KEGG analysis of unigenes
To better understand the active genes involved in the biological pathways of C. cladosporioides MD2, all of the unigenes were mapped to KEGG database. A total of 3350 unigenes were assigned to 262 KEGG pathways, and the top five pathways hierarchy (ranked by unigene numbers) were ‘Carbohydrate metabolism’ (388 unigenes), ‘Amino acid metabolism’ (307 unigenes), ‘Overview’ (287 unigenes), ‘Translation’ (280 unigenes) and ‘Signal transduction’ (222 unigenes) (Fig. 4). The dominant KEGG pathways included ‘biosynthesis of amino acids’ (157 unigenes, ko01230), followed in sequence by ‘carbon metabolism’ (150 unigenes, ko01200), ‘ribosome’ (104 unigenes, ko03010), ‘spliceosome’ (97 unigenes, ko03040), ‘purine metabolism’ (79 unigenes, ko00230), ‘cell cycle-yeast’ (78 unigenes, ko04111) and ‘starch and sucrose metabolism’ (78 unigenes, ko00500) (Additional file 1: Table S3), suggesting that most of the genes expressed in the C. cladosporioides MD2 transcriptome were involved in basic metabolic pathways.
Fig. 4.
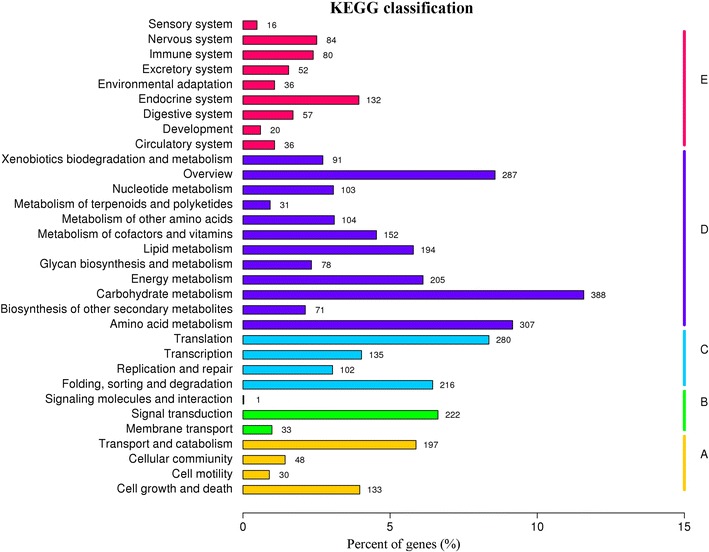
KEGG classification of the C. cladosporioides MD2 unigenes. a Cellular processes; b environmental information processing; c genetic information processing; d metabolism; e organismal systems. The numbers on the column represented the estimation of unigenes within the pathway
Dozens of unigenes were detected to be involved in ‘diterpenoid biosynthesis’. Among them, 9 unigenes were involved in terpenoid backbone biosynthesis (Fig. 5; Additional file 1: Table S4). Unigenes of c12899_g1 and c8934_g1 were homologous to an acetyl-CoA C-acetyltransferase (AACT) gene. Unigenes of c13511_g1, c14615_g1, c12988_g1 and c10355_g1 were homologous to the genes for hydroxymethylglutaryl-CoA synthase (HMGS), hydroxymethylglutaryl-CoA reductase (HMGR), mevalonate kinase (MK), diphosphomevalonate decarboxylase (MDD), respectively. The AACT, HMGS, HMGR, MK and MDD were key enzymes for the biosynthesis of isopentenyl diphosphate (IPP) in the mevalonate pathway (Miziorko 2011). Isopentenyl-diphosphate delta-isomerase (IPI), farnesyl diphosphate synthase (FDPS) and geranylgeranyl diphosphate synthase (GGPPs) regulated the biosynthesis flux of geranylgeranyl diphosphate (Geranygeranyl-PP, GGPP), a universal precursor for diterpene biosynthesis. Unigenes of c11713_g1, c10023_g1 and c1886_g1 were homologous to the IPI gene, FDPS gene and GGPPs gene, respectively. Additionally, 40 unigenes were annotated to be involved in the biosynthetic steps from GGPP to taxol within ‘diterpenoid biosynthesis’ pathway.
Fig. 5.
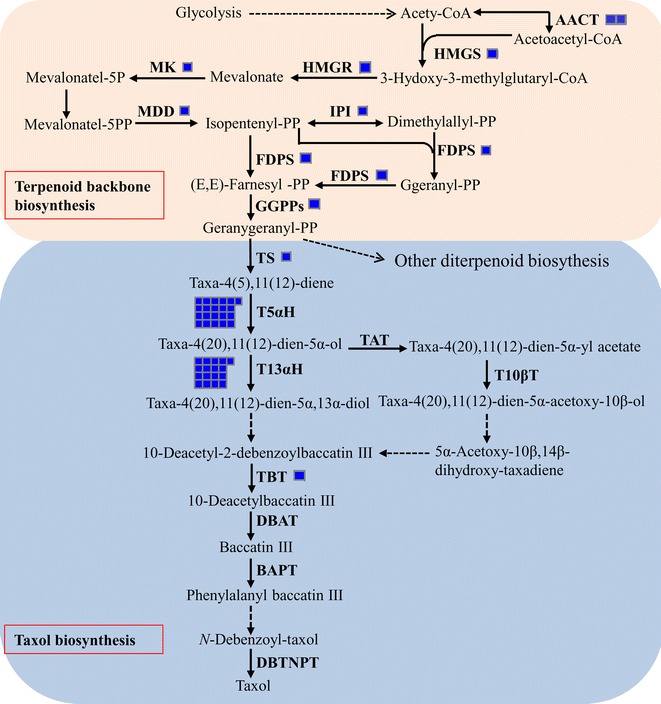
Overview of the potential fungal taxol biosynthesis pathway of C. cladosporioides MD2. The numbers of genes in families with at least one gene were indicated by the numbers of blue rectangles. Identified enzymes included: AACT acetyl-CoA C-acetyltransferase, HMGS hydroxymethylglutaryl-CoA synthase, HMGR hydroxymethylglutaryl-CoA reductase, MK mevalonate kinase, MDD diphosphomevalonate decarboxylase, IPI isopentenyl-diphosphate delta-isomerase, FDPS farnesyl diphosphate synthaseisopentenyl-diphosphate delta-isomerase, GGPPs geranylgeranyl diphosphate synthaseisopentenyl-diphosphate delta-isomerase, TS taxadiene synthaseisopentenyl-diphosphate delta-isomerase, T5αH taxadiene 5-alpha-hydroxylaseisopentenyl-diphosphate delta-isomerase, T13αH taxane 13-alpha-hydroxylaseisopentenyl-diphosphate delta-isomerase, TBT 2-alpha-hydroxytaxane 2-O-benzoyltransferase
Prediction of one fungal taxol biosynthesis pathway
Among the 40 unigenes involved in the taxol biosynthesis of C. cladosporioides MD2 (Fig. 5; Additional file 1: Table S4), unigene c11040_g1 was homologous to a gene for taxadiene synthase (TS), that catalyzed the cyclization of GGPP into taxa-4(5),11(12)-diene (Wildung and Croteau 1996), one committed step in the construction of the unique taxane skeleton for the biosynthesis of taxol and related taxoids (Walker and Croteau 2001). Twenty-one unigenes were homologous to a gene for taxadiene 5-alpha-hydroxylase (T5αH), that catalyzed the hydroxylation of taxadiene C5 to taxadienol (Jennewein et al. 2004). Seventeen unigenes were homologous to a gene for taxane 13-alpha-hydroxylase (T13αH), that catalyzed the hydroxylation of taxane C13 (Jennewein et al. 2001). Unigene c5600_g1 was homologous to a gene for 2-alpha-hydroxytaxane 2-O-benzoyltransferase (TBT), that catalyzed the conversion of a 2-debenzoyl ‘taxoid-type’ intermediate to 10-deacetylbaccatin III (10-DAB), a late-stage acylation step in the taxol biosynthetic pathway (Walker and Croteau 2000a).
Correspondingly, a potential and partial taxol biosynthetic pathway from GGPP to 10-DAB in the C. cladosporioides MD2 transcriptome was speculated (Fig. 5). The GGPP was cyclized by TS into taxa-4(5),11(12)-diene, and further catalyzed by T5αH into taxa-4(20),11(12)-dien-5α-ol. Then, the taxa-4(20),11(12)-dien-5α-ol was catalyzed by T13αH into taxa-4(20),11(12)-dien-5α,13α-diol, and followed to synthesize 10-deacetyl-2-debenzoylbaccatin III by several undefined steps, which might be a novel and potential biosynthesis branch of 10-deacetyl-2-debenzoylbaccatin III in C. cladosporioides MD2. Finally, the 10-deacetyl-2-debenzoylbaccatin III was catalyzed by TBT into 10-DAB. These unigenes homologous to the genes for TS, T5αH, T13αH and TBT provided candidates for further verification of the fungal taxol biosynthesis pathway in C. cladosporioides MD2.
Discussion
A fact was that successive replating of most of taxol-producing fungi in semisynthetic medium resulted in a decrease in taxol production (Flores-Bustamante et al. 2010; Venugopalan and Srivastava 2015). The original taxol yield and 10-DAB yield of C. cladosporioides MD2 were about 800 and 120 µg/L in 2009 (Zhang et al. 2009), however they were dramatically decreased to about 5–7 and 50 µg/L in 2014, respectively. Afterwards, the taxol yield and 10-DAB yield of C. cladosporioides MD2 basically remained unchanged up to the final test in July, 2016. This biological phenomenon of loss in taxol productivity with subculture was certainly related to the taxol biosynthesis mechanism of C. cladosporioides MD2. Here, the transcriptome of the successively subcultured C. cladosporioides MD2 with taxol production drawdown was investigated for the first time with a RNA-seq technology to explore gene-expressed characteristics in this situation. About 1.77 Gbp clean reads were generated from the transcriptome of C. cladosporioides MD2 and further assembled into 16,603 unigenes, that were annotated with seven protein and nucleic databases. Based on the KEGG analysis, 40 unigenes in the C. cladosporioides MD2 transcriptome were annotated to be related to the fungal taxol biosynthesis, and were homologous to the genes for TS, T5αH, T13αH, and TBT, respectively.
According to the proved taxol biosynthesis pathway in Taxus (Jennewein and Croteau 2001; Walker and Croteau 2001; Croteau et al. 2006; Guo et al. 2006; Liu et al. 2016), GGPP was cyclized by TS into taxa-4(5), 11(12)-diene, that was catalyzed by T5αH into taxa-4(20), 11(12)-dien-5α-ol, where two branches arose (Jennewein and Croteau 2001). One recognized branch was that the taxa-4(20), 11(12)-dien-5α-ol was catalyzed by taxa-4(20),11(12)-dien-5α-ol-O-acetyltransferase (TAT) into taxa-4(20),11(12)-dien-5α-yl acetate and further catalyzed by taxane 10β-hydroxylase (T10βH) into taxa-4(20),11(12)-dien-5α-acetoxy-10β-ol. Subsequently, the taxa-4(20),11(12)-dien-5α-acetoxy-10β-ol was catalyzed by several undefined steps into 2-debenzoyltaxane, that was hydrolyzed by TBT to produce 10-DAB. However, none of unigenes homologous to the TAT gene and T10βH gene were detected in the C. cladosporioides MD2 transcriptome (Fig. 5), suggesting that C. cladosporioides MD2 might not have this same branch as the recognized branch of Taxus. Another indistinct branch arised from taxa-4(20), 11(12)-dien-5α-ol was that the taxa-4(20), 11(12)-dien-5α-ol was catalyzed by T13αH into taxa-4(20),11(12)-dien-5α,13α-diol, that was subsequently catalyzed by several indeterminate enzymatic steps to directly synthesize taxol (Jennewein and Croteau 2001); Or, the taxa-4(20), 11(12)-dien-5α-ol was catalyzed by T13αH into taxa-4(20),11(12)-dien-5α,13α-diol and subsequently catalyzed by several indeterminate steps into 2-debenzoyltaxane, that was hydrolyzed by TBT to produce 10-DAB (Liu et al. 2016). A total of 17 unigenes homologous to the T13αH gene and one unigene homologous to the TBT gene were detected in the C. cladosporioides MD2 transcriptome, and this branch participated by T13αH and TBT might be a novel and potential branch for the 10-deacetyl-2-debenzoylbaccatin III biosynthesis of C. cladosporioides MD2.
Additionally, an interesting and important finding was that unigenes homologous to those genes for enzymes functioning in the biosynthetic steps from 10-DAB to taxol, such as 10-deacetylbaccatin III-10-O-acetyltransferase (DBAT), C-13 phenylpropanoid side chain-CoA acyltransferase (BAPT) and 3′-N-debenzoyltaxol N-benzoyltransferase (DBTNBT), were not detected in the C. cladosporioides MD2 transcriptome. DBAT catalyzed the 10-DAB into Baccatin III, one immediate diterpenoid precursor of taxol (Walker and Croteau 2000b), BAPT catalyzed the selective 13-O-acylation of Baccatin III with β-phenylalanoyl-CoA as one acyl donor to form N-debenzoyl-2′-deoxytaxol (i.e. catalyzed the attachment of biologically important taxol side chain precursor) (Walker et al. 2002a), and DBTNBT catalyzed the final acylation reaction of taxol biosynthesis (Walker et al. 2002b). None of candidate unigenes homologous to those genes for DBAT, BAPT and DBTNBT might be a reason for the loss of taxol productivity in the subcultured C. cladosporioides MD2. This result clued us for future research that, (1) whether some genes homologous to the genes for DBAT, BAPT and DBTNBT exited in the genome but did not transcribed in the transcriptome of the subcultured C. cladosporioides MD2; (2) Or, some genes, that encoded products with similar function as DBAT, BAPT, DBTNBT but had low sequence identities to their genes, were not annotated and detected by sequence alignment in the transcriptome of C. cladosporioides MD2.
Collectively, this study not only provided an available transcriptome resource for further functional genomic research and comparative transcriptome analysis of C. cladosporioides MD2, but also revealed one potential and novel branch for the 10-deacetyl-2-debenzoylbaccatin III biosynthesis and valuable candidate genes for future investigation of the taxol biosynthesis mechanism in C. cladosporioides MD2.
Additional file
Additional file 1: Figure S1. Reverse-transcribed PCR validation of the C. cladosporioides MD2 unigenes. Ten of the C. cladosporioides MD2 unigenes were randomly selected for reverse-transcribed PCR (RT-PCR) validation. The results showed that these selected unigenes got right amplifications. M: DL2000 marker; lane 1-10 was the amplification products of unigenes of c3348_g1, c14778_g1, c2571_g1, c10254_g1, c11322_g1, c6738_g1, c13171_g1, c11554_g1, c13227_g1 and c13492_g1, respectively. Table S1. The primers of the ten unigenes for Reverse-Transcribed PCR (RT-PCR) validation. Table S2. GO annotation of the C. cladosporioides MD2 unigenes. Table S3. KEGG classification of the C. cladosporioides MD2 unigenes. Table S4. Enzymes and related unigenes in the potential fungal taxol biosynthesis pathway of C. cladosporioides MD2.
Authors’ contributions
ZP and JWW conceived and designed the research. MLY, MXC and XXY performed the experiments. ZL, DG, KYS, LP and SFJ performed analysis and interpretation of data. MLY, MXC and ZP wrote the manuscript. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank the Wuhan Bio-Broad Co., Ltd. (Wuhan, China) for sequencing of the C. cladosporioides MD2 transcriptome used in this study. And we would like to thank all the colleagues at National Demonstration Center for Experimental Ethnopharmacology Education, South-Central University for Nationalities, for their technical assistance throughout this study.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All datasets supporting the conclusions of this article were included in the main manuscript file and the additional file. The transcriptome data of C. cladosporioides MD2 used in this study were deposited in NCBI Sequence Read Archive (SRA) Sequence Database with accession number SRR6147052.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This study was funded by the National Natural Science Foundation of China (Grant Numbers: 31370118 and 31770134).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- 10-DAB
10-deacetylbaccatin III
- AACT
acetyl-CoA C-acetyltransferase
- BAPT
C-13 phenylpropanoid side chain-CoA acyltransferase
- DBAT
10-deacetylbaccatin III-10-O-acetyltransferase
- DBTNBT
3′-N-debenzoyltaxol N-benzoyltransferase
- FDPS
farnesyl diphosphate synthase
- GGPP
geranylgeranyl diphosphate
- GGPPs
geranylgeranyl diphosphate synthase
- HMGR
hydroxymethylglutaryl-CoA reductase
- HMGS
hydroxymethylglutaryl-CoA synthase
- IPI
isopentenyl-diphosphate delta-isomerase
- IPP
isopentenyl diphosphate
- MDD
diphosphomevalonate decarboxylase
- MK
mevalonate kinase
- T10βH
taxane 10β-hydroxylase
- T13αH
taxane 13-alpha-hydroxylase
- T5αH
taxadiene 5-alpha-hydroxylase
- TAT
taxa-4(20),11(12)-dien-5α-ol-O-acetyltransferase
- TBT
2-alpha-hydroxytaxane 2-O-benzoyltransferase
- TS
taxadiene synthase
Footnotes
Li-Yun Miao, Xin-Chun Mo and Xiao-Yuan Xi contributed equally to this work
Electronic supplementary material
The online version of this article (10.1186/s13568-018-0567-6) contains supplementary material, which is available to authorized users.
Contributor Information
Li-Yun Miao, Email: miaoliyun_928@163.com.
Xin-Chun Mo, Email: 26315125@qq.com.
Xiao-Yuan Xi, Email: 1043896809@qq.com.
Lan Zhou, Email: 2272045923@qq.com.
Ge De, Email: 2391002358@qq.com.
You-Sheng Ke, Email: keyuu.me@qq.com.
Pan Liu, Email: 1067419936@qq.com.
Fa-Jun Song, Email: 449822345@qq.com.
Wen-Wen Jin, Phone: +86 (27) 87792264, Email: jww@hust.edu.cn.
Peng Zhang, Phone: +86 (27) 67842689, Email: zhangpenghust@126.com.
References
- Choi J, Park JG, Ali MS, Choi SJ, Baek KH. Systematic analysis of the anticancer agent taxol-producing capacity in Colletotrichum species and use of the species for taxol production. Mycobiology. 2016;44:105–111. doi: 10.5941/MYCO.2016.44.2.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Croteau R, Ketchum REB, Long RM, Kaspera R, Wildung MR. Taxol biosynthesis and molecular genetics. Phytochem Rev. 2006;5:75–97. doi: 10.1007/s11101-005-3748-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Hao C, Ge G, Xiao P, Zhang Y, Yang L. The first insight into the tissue specific Taxus transcriptome via Illumina second generation sequencing. PLoS ONE. 2011;6:e21220. doi: 10.1371/journal.pone.0021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Bustamante ZR, Rivera-Orduña FN, Martínez-Cárdenas A, Flores-Cotera LB. Microbial paclitaxel: advances and perspectives. J Antibiot (Tokyo) 2010;63:460–467. doi: 10.1038/ja.2010.83. [DOI] [PubMed] [Google Scholar]
- Gond SK, Kharwar RN, White JF., Jr Will fungi be the new source of the blockbuster drug taxol? Fungal Biol Rev. 2014;28:77–84. doi: 10.1016/j.fbr.2014.10.001. [DOI] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng QD, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo BH, Kai GY, Jin HB, Tang KX. Taxol synthesis. Afr J Biotechnol. 2006;5:15–20. [Google Scholar]
- Iseli C, Jongeneel CV, Bucher P (1999) ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. In: Proceedings of the international conference on intelligent system for molecular biology. Menlo Park: AAAI Press, pp 138–148 [PubMed]
- Jennewein S, Croteau R. Taxol: biosynthesis, molecular genetics, and biotechnological applications. Appl Microbiol Biotechnol. 2001;57:13–19. doi: 10.1007/s002530100757. [DOI] [PubMed] [Google Scholar]
- Jennewein S, Rithner CD, Williams RM, Croteau RB. Taxol biosynthesis: taxane 13alpha-hydroxylase is a cytochrome P450-dependent monooxygenase. Proc Natl Acad Sci USA. 2001;98:13595–13600. doi: 10.1073/pnas.251539398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennewein S, Long RM, Williams RM, Croteau R. Cytochrome P450 taxadiene 5α-hydroxylase, a mechanistically unusual monooxygenase catalyzing the first oxygenation step of taxol biosynthesis. Chem Biol. 2004;11:379–387. doi: 10.1016/j.chembiol.2004.02.022. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusari S, Singh S, Jayabaskaran C. Rethinking production of taxol (paclitaxel) using endophyte biotechnology. Trends Biotechnol. 2014;32:304–311. doi: 10.1016/j.tibtech.2014.03.011. [DOI] [PubMed] [Google Scholar]
- Langmead B (2010) Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics, New York: Wiley, Chapter 11: Unit 11.7 [DOI] [PMC free article] [PubMed]
- Li ST, Zhang P, Zhang M, Fu CH, Zhao CF, Dong YS, Guo AY, Yu LJ. Transcriptional profile of Taxus chinensis cells in response to methyl jasmonate. BMC Genom. 2012;13:295. doi: 10.1186/1471-2164-13-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WC, Gong T, Zhu P. Advances in exploring alternative Taxol sources. RSC Adv. 2016;6:48800–48809. doi: 10.1039/C6RA06640B. [DOI] [Google Scholar]
- Miller HC, Biggs PJ, Voelckel C, Nelson NJ. De novo sequence assembly and characterisation of a partial transcriptome for an evolutionarily distinct reptile, the tuatara (Sphenodon punctatus) BMC Genom. 2012;13:439. doi: 10.1186/1471-2164-13-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miziorko HM. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch Biochem Biophys. 2011;505:131–143. doi: 10.1016/j.abb.2010.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BH, Karpinets TV, Syed MH, Leuze MR, Uberbacher EC. CAZymes Analysis Toolkit (CAT): web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology. 2010;20:1574–1584. doi: 10.1093/glycob/cwq106. [DOI] [PubMed] [Google Scholar]
- Qiao F, Cong H, Jiang X, Wang R, Yin J, Qian D, Wang Z, Nick P. De novo characterization of a Cephalotaxus hainanensis transcriptome and genes related to paclitaxel biosynthesis. PLoS ONE. 2014;9:e106900. doi: 10.1371/journal.pone.0106900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stierle A, Strobel G, Stierle D. Taxol and taxane production by Taxomyces andreanae, an endophytic fungus of Pacific yew. Science. 1993;260:214–216. doi: 10.1126/science.8097061. [DOI] [PubMed] [Google Scholar]
- Sun G, Yang Y, Xie F, Wen JF, Wu J, Wilson IW, Tang Q, Liu H, Qiu D. Deep sequencing reveals transcriptome re-programming of Taxus x media cells to the elicitation with methyl jasmonate. PLoS ONE. 2013;8:e62865. doi: 10.1371/journal.pone.0062865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopalan A, Srivastava S. Endophytes as in vitro production platforms of high value plant secondary metabolites. Biotechnol Adv. 2015;33:873–887. doi: 10.1016/j.biotechadv.2015.07.004. [DOI] [PubMed] [Google Scholar]
- Walker K, Croteau R. Taxol biosynthesis: molecular cloning of a benzoyl-CoA: taxane 2α-O-benzoyltransferase cDNA from Taxus and functional expression in Escherichia coli. Proc Natl Acad Sci USA. 2000;97:13591–13596. doi: 10.1073/pnas.250491997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker K, Croteau R. Molecular cloning of a 10-deacetylbaccatin III-10-O-acetyl transferase cDNA from Taxus and functional expression in Escherichia coli. Proc Natl Acad Sci USA. 2000;97:583–587. doi: 10.1073/pnas.97.2.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker K, Croteau R. Taxol biosynthetic genes. Phytochemistry. 2001;58:1–7. doi: 10.1016/S0031-9422(01)00160-1. [DOI] [PubMed] [Google Scholar]
- Walker K, Fujisaki S, Long R, Croteau R. Molecular cloning and heterologous expression of the C-13 phenylpropanoid side chain-CoA acyltransferase that functions in Taxol biosynthesis. Proc Natl Acad Sci USA. 2002;99:12715–12720. doi: 10.1073/pnas.192463699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker K, Long R, Croteau R. The final acylation step in taxol biosynthesis: cloning of the taxoid C13-side-chain N-benzoyltransferase from Taxus. Proc Natl Acad Sci USA. 2002;99:9166–9171. doi: 10.1073/pnas.082115799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc. 1971;93:2325–2327. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- Wildung MR, Croteau R. A cDNA clone for taxadiene synthase, the diterpene cyclase that catalyzes the committed step of taxol biosynthesis. J Biol Chem. 1996;271:9201–9204. doi: 10.1074/jbc.271.16.9201. [DOI] [PubMed] [Google Scholar]
- Yang Y, Zhao H, Barrero RA, Zhang B, Sun G, Wilson IW, Xie F, Walker KD, Parks JW, Bruce R, Guo G, Chen L, Zhang Y, Huang X, Tang Q, Liu H, Bellgard MI, Qiu D, Lai J, Hoffman A. Genome sequencing and analysis of the paclitaxel-producing endophytic fungus Penicillium aurantiogriseum NRRL 62431. BMC Genomics. 2014;15:69. doi: 10.1186/1471-2164-15-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Zhou PP, Yu LJ. An endophytic taxol-producing fungus from Taxus media, Cladosporium cladosporioides MD2. Curr Microbiol. 2009;59:227–232. doi: 10.1007/s00284-008-9270-1. [DOI] [PubMed] [Google Scholar]
- Zhang L, Jia H, Yin Y, Wu G, Xia H, Wang X, Fu C, Li M, Wu J. Transcriptome analysis of leaf tissue of Raphanus sativus by RNA sequencing. PLoS ONE. 2013;8:e80350. doi: 10.1371/journal.pone.0080350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Reverse-transcribed PCR validation of the C. cladosporioides MD2 unigenes. Ten of the C. cladosporioides MD2 unigenes were randomly selected for reverse-transcribed PCR (RT-PCR) validation. The results showed that these selected unigenes got right amplifications. M: DL2000 marker; lane 1-10 was the amplification products of unigenes of c3348_g1, c14778_g1, c2571_g1, c10254_g1, c11322_g1, c6738_g1, c13171_g1, c11554_g1, c13227_g1 and c13492_g1, respectively. Table S1. The primers of the ten unigenes for Reverse-Transcribed PCR (RT-PCR) validation. Table S2. GO annotation of the C. cladosporioides MD2 unigenes. Table S3. KEGG classification of the C. cladosporioides MD2 unigenes. Table S4. Enzymes and related unigenes in the potential fungal taxol biosynthesis pathway of C. cladosporioides MD2.
Data Availability Statement
All datasets supporting the conclusions of this article were included in the main manuscript file and the additional file. The transcriptome data of C. cladosporioides MD2 used in this study were deposited in NCBI Sequence Read Archive (SRA) Sequence Database with accession number SRR6147052.