

Abstract
Introduction
Tumors in the large intestine have been postulated to arise via a stepwise accumulation of mutations, a process that takes up to 20 years. Recent advances in lineage tracing and DNA sequencing, however, are revealing new evolutionary models that better explain the vast amount of heterogeneity observed within and across colorectal tumors.
Areas Covered
A review of the literature supporting a novel model of colorectal tumor evolution was conducted. The following commentary examines the basic science and clinical evidence supporting a modified view of tumor initiation and progression in the colon.
Expert Commentary
The proposed “cancer punctuated equilibrium” model of tumor evolution better explains the variability seen within and across polyps of the colon and rectum. Small colorectal polyps (6–9mm) followed longitudinally by interval imaging with CT colonography have been reported to have multiple fates: some growing, some remaining static in size, and others regressing in size over time. This new model allows for this variability in growth behavior and supports the hypothesis that some tumors can be “born to be bad” as originally postulated by Sottoriva and colleagues, with very early molecular events impacting tumor fitness and growth behavior in the later stages of the disease process.
Keywords: Big Bang model, Colorectal cancer, CT colonography, Tumor heterogeneity, Tumorigenesis, Punctuated equilibrium
1.0 Introduction
As the technologies emerge that allow thousands of molecules including DNA, RNA, and proteins to be rapidly assayed (“multi-omics”) and big data evolve, new evolutionary theories of tumorigenesis have been proposed. These theories provide new insights into tumor initiation and progression to invasive disease. This commentary will briefly review longstanding historical models of tumorigenesis in the colon, and highlight emerging theories of tumor evolution.
2.0 Tumor Evolution
Historically, cancers in the large intestine were believed to arise via the gradual stepwise accumulation of mutations [1, 2]. The progression from benign adenomatous polyps, to more advanced pre-malignant polyps with foci of high-grade dysplasia, to locally invasive cancer, and eventually to metastatic disease was thought to be driven by the accumulation of mutations that perturb specific genetic pathways at each step in the tumorigenic process. Seminal papers in the field of cancer biology demonstrated that 80–90% of colorectal tumors are initiated following the loss of activity of the Adenomatous polyposis coli (APC) gene [3, 4]. This tumor suppressor gene is lost following two inactivating mutations or one mutation followed by a loss-of-heterozygosity event such as somatic recombination or chromosomal loss. The gene encodes a member of the β-catenin destruction complex. Loss of APC activity results in the stabilization of β-catenin, which then translocates from the cell membrane to the nucleus and consequently leads to aberrant WNT signaling. Dysregulated WNT signaling is often followed by mutations in KRAS/NRAS, the TGFβ pathway, PIK3CA, TP53 or any combination of several of these alterations. At each step, a new mutation was thought to generate a sub-clone with the most advantageous mutation profile and a strong selective advantage, outcompeting less fit clones in a Darwinian fashion and prompting progression to the next stage of the disease process.
Under a stepwise or linear evolution model, the resulting tumors should have three defining characteristics: (1) all tumors have the potential to progress from a benign to malignant state; (2) cancers take a long time to form, possibly even as long as 20 years; and (3) cancers can be relatively homogeneous, as natural selection would over time select for the most malignant tumor clone. All of these characteristics were supported by early studies. However, whether these three characteristics are required for colorectal tumor formation and progression or not is being questioned because of new evidence generated with emerging technologies.
2.1 Tumor Fate
The first defining characteristic of the linear evolution model is that all tumors have the potential to progress to cancer. This underlying assumption implies that the transformation from a benign to malignant merely requires the accumulation of the “necessary” driver mutations. Recent studies have demonstrated that colorectal polyps have different growth fates, and that these fates might be dictated in part by transcriptional and methylation changes. Pickhardt and colleagues reported that after a surveillance interval ranging from 1-7 years (mean interval, 2.3 years), only 22% of small (6-9 mm) polyps were significantly growing in volume (≥20%/year) (figure 1 A–B), whereas 28% of these benign colorectal tumors were shrinking in volume (≥-20%/year) (figure 1 E–F), with 10% showing complete regression [5]. While polyp growth alone is not indicative of progression, polyp size has been reported to be the major independent risk factor associated with high-grade dysplasia and cancer [6, 7, 8].
Figure 1.
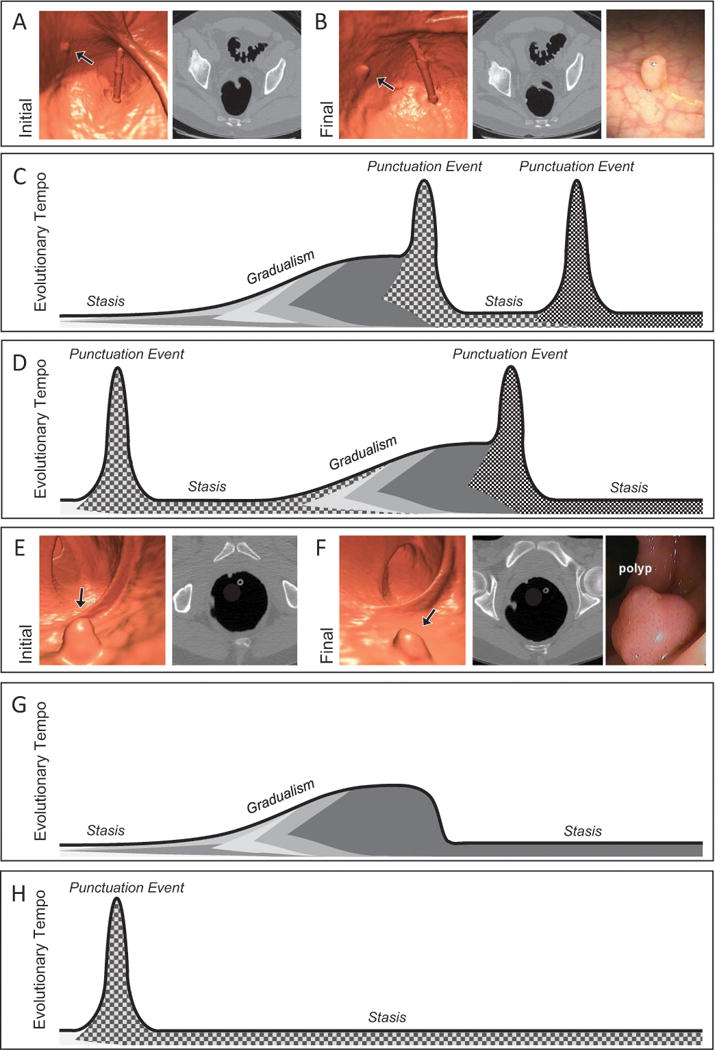
The cancer punctuated equilibrium model of colon tumor evolution better explains the variability of colorectal polyp growth. Computed tomography colonography (CTC) images from the initial (A) and final (B) scans from a patient with polyp that had an annual volumetric growth rate of 59% that was followed over 2.1 years prior to polypectomy. Black arrows point to the polyp that was followed longitudinally. (C) and (D) are possible evolutionary trajectories for a growing polyp. Shading under the line represent levels of intratumoral heterogeneity with punctuation events creating the greatest amount of heterogeneity. Tumorigenesis may begin with a punctuation event or periods of stasis and gradualism, a second punctuation event may provide enough molecular diversity allowing for malignant transformation. (E) and (D) are CTC images from the initial and final scans from a patient with polyp that had an annual volumetric growth rate of −33% that was followed over 0.9 years prior to polypectomy. (G) and (H) are possible evolutionary trajectories for a regressing polyp. Tumor regression may occur with the emergence of a negative or immunogenic phenotype acquired during a period of gradualism or via a punctuation event.
While well-controlled longitudinal progression studies in humans are not possible for obvious ethical reasons, multiple studies in mice have also reported differences in the growth and progression of adenomas. Tumors from mice with the simultaneous induction of pathogenic mutations in Apc, Kras and Pik3ca still progress through a histological adenoma-to-carcinoma sequence, but have an increased rate of progression, demonstrating that mutational profile of the tumor is not predictive of the stage of the disease [9, 10]. Furthermore, Paul Olson and colleagues reported that in a long-lived ApcMin/+ mouse model of colon cancer, adenomas fell into different fate categories, with some growing, some remaining static in size, and some regressing [11]. Interestingly, these authors also identified a microarray-based gene signature from biopsies taken at an early time point when the tumor first emerged that distinguished adenomas that remained as adenomas from adenomas that progressed to intramucosal carcinomas. They did not, however, identify differentially expressed genes between early and late biopsies of single tumors followed longitudinally, indicating that the transcriptional changes associated with progression were present from very early on in the lifetime of the tumor. These data taken from both human clinical observations as well as laboratory experiments in mouse models provide solid evidence that not all tumors will progress to invasive cancers via the accumulation of driver mutations.
2.2 Time
The second defining characteristic of the stepwise evolution model is that colorectal cancers (CRCs) form over many years. This conclusion arose primarily from the finding that colorectal polyp and cancer prevalence is associated with age as well as from observations from patients with colorectal polyps that were lost at follow-up in the clinic [2]. While the adenoma-to-carcinoma sequence still likely takes years, a stepwise, linear process is not supported. In 2008 Jones and colleagues concluded that the formation of adenomas takes on average 17 years, while the formation of liver metastases from a large adenocarcinoma took less than 2 years [12]. At the time, these authors still argued for a step-wise evolution model, even though their own data demonstrated a non-linear process. Additionally, the clinical observations of interval CRCs have demonstrated that an accelerated tumor formation rate can occur in some cases, assuming early cancers were not missed at the initial colonoscopy, compared to the 17–20 year prediction. Furthermore, the finding of clonally related cancer-synchronous polyps to adenocarcinomas by Yang and colleagues demonstrates unequal times to progression even in neoplastic tissue with a similar genomic changes [13]. Additionally, studies in the laboratory mouse reveal a subset of tumors with an accelerated time to progression. Leystra and colleagues reported a noncanonical model of CRC, in which mice with activated PI3K do not begin with a polypoid adenomatous precursor mass [14]. These mice rapidly develop mucinous invasive adenocarcinomas and share several histologic characteristics to human CRCs, including high-grade nuclear atypia, mucin lakes, and budding at the leading edge of the invasion fronts. This autochthonous model developing highly aggressive cancers again emphasizes that extended periods of time are not always necessary for the development of invasive colon cancer.
2.3 Tumor Fitness
The final underlying assumption of a stepwise evolution model is that mutations cause significant differences in tumor fitness so that natural selection can act upon the population of tumor cells driving clonal expansions. While cell-culture-based experiments have shown significant differences in proliferation, migratory ability, and colony formation owing to single mutations, these results are not necessarily duplicated in vivo. Interestingly, histologically normal-appearing epithelial cells from patients with FAP contain mutations in numerous genes, of which many (23%) were also present in colon polyps [15]. Some of the mutations found in non-tumor tissue from patients with sporadic colorectal cancer or even individuals who were disease-free altered a known driver gene, indicating that mutations in certain genes alone are insufficient to induce neoplastic transformation in vivo [15, 16, 17]. Along these lines, Bozic and colleagues calculated the selective advantage conferred by driver mutations and found that it was surprisingly small, 0.4% [19]. Thus, clones with mutations in key driver genes may not sweep through a tumor but clones with different mutation profiles may co-evolve.
2.4 Intratumoral Heterogeneity
Next-generation sequencing and multi-omic technologies are revealing vast amounts of intratumoral heterogeneity. Solid tumors can have sub-clonal passenger mutations and even some sub-clonal driver mutations [12, 20, 21, 22, 23, 24, 25]. The presence of sub-clonal driver mutations in frank cancers is inconsistent with a linear evolution model. Multiple research groups have demonstrated that while mutations in APC and KRAS are often clonal or founder events, oncogenic mutations in PIK3CA and the Notch and Hedgehog signaling pathways are often sub-clonal [26, 27].
2.5 “Born to be Bad”
In addition to the finding of intratumoral heterogeneity and sub-clonal driver mutations in cancers, classic cancer-associated events are now being found in premalignant precursors. Sottoriva and colleagues demonstrated the presence of both sub-clonal mutations and copy number events in colon adenomas [23, 28]. Similarly, Sievers and colleagues recently reported sub-clonal driver mutations, including the KRAS G12D variant, in small (6–9mm) colon polyps [29]. Utilizing a unique cohort of patients with small polyps for which volumetric growth rate was available, they were able to determine that detectable, sub-clonal mutations must have arisen before the polyp was of a detectable size. This finding supports the hypothesis that some precursor lesions might be “born to be bad” as originally postulated by Sottoriva and colleagues [28] and that early molecular events may dictate later tumor growth and progression.
Consistent with the finding of early genomic events, Luo and colleagues identified genome-wide DNA methylation alterations in non-tumor colon tissue and tubular adenomas have been recently identified [30]. Epigenetic alterations occur commonly in adenomas, sessile serrated polyps, and colorectal cancers and appear to cooperate with gene mutations to drive the adenoma-to-carcinoma sequence. The most prominent aberrations in DNA methylation related to the development of cancer include: 1) hypermethylation of CpG islands in gene promoters and other regulatory genomic domains (e.g. enhancer loci), which can silence tumor suppressor genes, and 2) hypomethylation of repetitive genetic elements, which may lead to genomic instability [31]. DNA methylation affects CpG rich regions, called ‘CpG islands’, in the 5′ region of genes and results in transcriptional silencing via effects on transcription factor binding and changes in chromatin structure [32]. The aberrant CpG island DNA methylation of genes is common in many cancers, including colorectal cancer [33, 34]. The process of aberrant gene methylation begins early in the adenoma-to-carcinoma sequence, is involved in both the canonical and alternative pathways to cancer, and likely affects the initiation and the progression of colorectal cancer [35]. The global patterns of aberrant DNA methylation are unique for different cancer types and may reflect the epigenome of the progenitor cell that gives rise to a cancer, which may impact on the malignant potential of different colon polyps [36]. Of interest, Luo, Grady, and colleagues [30] showed that molecular subtypes of adenomas exist (low-methylator vs. high-methylator subtypes). The low-methylator subtype has a methylation pattern similar to that of the normal colon, whereas the high-methylator subtype has a pattern similar to that of chromosomal unstable (CIN) colorectal cancer, which is the most common type of colorectal cancer.
Together, the results from all these studies indicate that new models are needed to more accurately describe tumorigenesis in the large intestine. Modern sequencing technologies and mathematical modeling techniques, in combination with annotated clinical surveillance information, have provided the data necessary for new insights.
3.0 New Models of Tumor Evolution
3.1 Big Bang Tumor Evolution Model
Recently, an alternative model of CRC evolution has emerged – the “Big Bang” theory of tumorigenesis [28]. In this model, the first few neoplastic cell divisions leading to the establishment of an adenoma generate many passenger mutations and copy number alterations. This increased evolutionary tempo may be due in part to the process of neoplastic transformation itself and chromosomal missegregation or some other form of genetic instability [37, 38, 39, 40]. Once all the necessary driver mutations are acquired, cancers grow from a single expansion of a diverse population of tumor cells, characterized by neutral evolution instead of Darwinian survival [27, 41, 42, 43]. This model accounts for the presence of intratumoral heterogeneity in CRCs not explained by the stepwise growth model. In this Big Bang growth model, intratumoral heterogeneity arises as a function of time, becoming detectable upon analysis when the population has expanded to a sufficient size, and not as a function of increased fitness. Therefore, the most recently acquired mutations will be at the lowest frequencies in the population, often undetectable by bulk genomic methodologies, and might explain why drug-resistant sub-clones are undetectable prior to treatment in the clinical setting.
Additionally, the Big Bang growth model allows for variation in tumor growth fates. Sets of mutations that are acquired early at the Big Bang event can contribute to the tumor fate, allowing for an accelerated growth rate if the right combination of alterations are present. This concept of some tumors being “born to be bad” attempts to explain those interval cancers and polyps that progress faster than the predicted time of 20 years. While the Big Bang growth model seeks to better explain some of the variation across tumor growth and progression, it is only one evolutionary phenomenon.
3.2 Punctuated Equilibrium Tumor Evolution Model
Recently, a more comprehensive model of tumor evolution has been described considering the process of Big Bang growth as well as updated theories of evolution [44, 45, 46, 47]. The cancer “punctuated equilibrium” model seeks to better explain how tumors evolve, a process that encompasses potentially long periods of stasis, punctuated by rapid periods of transformation and molecular changes (figure 1C) [48]. Within this model, three evolutionary processes are at work: (1) stasis—this phase is characterized by the neutral accrual of passenger mutations, which results in the stable phenotype; (2) gradualism—this phase is most similar to the stepwise growth model in which molecular changes are acquired in a sequential manner, under natural selection, which results in a quantifiable impact on the phenotype; and (3) punctuation events—this phase is derived from the Big Bang growth model where periods of genomic instability result in many molecular changes to occur simultaneously, which results in a dramatic phenotypic change. These punctuation events are not restricted to mutations, they are a mechanism of evolutionary change and thus can include epigenetic and transcriptional alternations that result in a change in the phenotype. Together, these three mechanisms change our understanding of how and when selective pressure is active and allows non-mutation events to contribute to the evolution of a tumor.
The inclusion of these three mechanisms of evolutionary change into this novel model of CRC development better explains the variability across adenoma growth patterns as well as the finding of intratumoral heterogeneity within adenomas and cancers. One major criticism of the classical stepwise acquisition model is the observation that adenomas can have different growth fates. The inclusion and timing of the three different mechanisms within the punctuated equilibrium model allow for variation in the evolutionary tempo, while functioning within the context of the adenoma-to-carcinoma sequence. For example, within the normal-appearing colonic epithelium during the static phase prior to the formation of a visible mass, eventual clonal passenger mutations are accumulating in a non-selective manner. From this point, gradualism or a punctuation event may occur, allowing for a phenotypic change resulting in the development of an adenomatous mass. This phenotypic change can be the result of mutations, copy number changes, and epigenetic or transcriptional changes. Following a period of gradualism or punctuation, another static phase could occur, allowing for the neutral expansion of multiple tumor clones. Polyps that progress likely undergo additional rounds of punctuation and stasis, allowing for the emergence of a clone that is better able to cope with hypoxia and metabolic stress, a clone that has a more mesenchymal phenotype, and in some cases a clone with an invasive phenotype (figure 1C–D). This would be in contrast to polyps that remain unchanged in their volumetric size. These polyps may remain in evolutionary stasis for extended periods of time (figure 1G–H). While the molecular basis of tumor regression is speculative, polyps that regress in size over time and those that resolve completely may become more immunogenic or even lose their ability for neoplastic growth through a punctuation event that impairs the fitness of the neoplastic cells.
A second criticism of the classical stepwise acquisition model is the finding of intratumoral heterogeneity within both adenomatous polyps and cancers. In the punctuated equilibrium model, multiple clones can co-exist due to the variation of when natural selection is active. Both punctuation events and stasis do not require natural selection to occur. In fact, the mechanism of stasis specifically lacks natural selection and allows for the neural expansion of all present tumor clones. This possibility allows a tumor as a whole to progress without necessitating that a clonal sweep of the most malignant clone had to occur. Therefore, tumors of any size can include various amounts of intratumoral heterogeneity. However, this model is not restricted to tumors with high levels of intratumoral heterogeneity. Relatively homogenous tumors can arise via the gradualism mechanism. Here, selection is active, creating a population bottleneck, and allowing only the most fit tumor clone to survive. While the punctuated equilibrium model is one way to explain the varying levels of intratumoral heterogeneity seen in CRCs, an alternate mechanism involving early short-range migratory ability and cell turnover [49] has been proposed.
Theoretically, the cancer punctuated equilibrium model does seem to better fit what has been observed both grossly and on a molecular level during the adenoma-to-carcinoma sequence. However, the precise timing of the three mechanisms during the evolution of a malignant cancer as well as the dynamics between multiple clones remains unknown.
4.0 Expert Commentary
Understanding tumor evolution from initiation through metastatic spread is critical for long-term CRC prevention, detection, and treatment. Resistance to targeted therapies has long been acknowledged as a major problem when treating and managing metastatic cancers. While precise mechanisms of pre-existing versus acquired drug resistance are under debate, likely both mechanisms can occur depending upon the specific biological context. In this sense, staying ahead of the process of evolution itself seems daunting. However, a deeper understanding of the mechanisms of tumor evolution may lead to novel drug targets and therapeutic approaches. In light of the cancer punctuated equilibrium model, targeting the mechanism behind punctuation events may be able to prevent additional drug resistance and may be useful in preventing cancerous tumors in high-risk populations. Furthermore, understanding how convergent evolution functions both within and among the hallmarks of cancer may lead to novel combination strategies to prevent further resistance to anti-tumor therapies. Moreover, having a biomarker or measure of when a punctuation or evolutionary event is occurring, such as CT-based radiomic metrics or changes in circulating tumor cells, may enable prevention of the selection of a more malignant tumor clone and allow for changes to a patient’s treatment regimen.
Immediate implications of this new model to CRC prevention are harder to pinpoint. However, long-term changes to screening guidelines and modalities could conceivably be implemented in the future. The cancer punctuated equilibrium model of tumor evolution postulates that early molecular events impact tumor growth behavior later on. With a greater understanding of early events on polyp growth and progression behavior, screening intervals may increase or decrease for patients with low- versus high-risk molecular characteristics and other factors. Additionally, a better understanding of colorectal tumor evolution and progression will allow for novel less-invasive screening mechanisms, increasing patient CRC screening compliance without the unnecessary burden on GI endoscopists.
In conclusion, theories of tumor evolution can themselves evolve as demonstrated by the shift from a stepwise accumulation of alterations model, to a Big Bang model, to a punctuated equilibrium model. This most recent model explains, via three evolutionary mechanisms, the observed confounding evidence against previous models. Under this cancer punctuated equilibrium model, increased intratumoral heterogeneity, rapid tumor progression, periods of tumor stasis, and neutral evolution can all be explained. While surely this model too will be subject to revision, at present it represents a major shift from the previous longstanding model of tumor evolution.
5.0 Five-year View
Over the next five years this new model of tumorigenesis is likely be subject to revisions as multi-omic and single cell resolution technologies improve. The previous five years have seen immense improvement in these areas and have given rise to this new model of tumor evolution. It is our hope that over the next five years researchers will be able to answer some of the lingering questions in the field of cancer biology and tumor evolution: can crypts of susceptible cells be identified prior to neoplastic transformation? Can adenomatous tumors that are “born to be bad” be distinguished from their more benign counterparts? Where and when does the malignant clone arise? Can tumor evolution be therapeutically targeted?
Reflecting on these questions in the context of the current model, we speculate that there are previously unidentified factors that make certain cells more susceptible to neoplastic transformation and that these factors, whether genetic, epigenetic, or signaling related, could be identified. As an example, patients with familial adenomatous polyposis, and the homologous ApcMin/+ mouse model, get many tumors throughout their GI tract. However, why doesn’t the entire intestinal epithelium become transformed? Others have demonstrated that a field of APC-deficient crypts may be a necessary intermediate in the establishment of adenomas [50]. Certainly there is more to neoplastic transformation than driver mutations alone. It is our hope that these additional factors could be identified and used as biomarkers in the future to determine personalized screening timelines and distinguish benign proliferative tumors from truly premalignant cancer precursors.
As our understanding of tumor biology and cancer evolution increases, we envision that the process of tumor evolution itself could be therapeutically exploited. While mechanisms of generating genetic diversity, such as the APOBEC family of DNA deaminases [51], are currently being investigated as novel targets, understanding the role of convergent evolution in the context of the hallmarks of cancer may also lead to promising therapeutic targets and combinations. Similar to the concept of synthetic lethality, understanding the pathways involved in tumor evolution, as well as the alternates when those fail, will be critical to halting the rise of drug-resistant clones. While many of the complex interactions between tumor cells, their environment, and the immune system remain unknown, we predict that a deeper understanding of these will lead to novel prevention and treatment options.
Key Issues.
Colorectal polyps have a variety of growth behaviors; some grow and progress, some grow for a period of time and then remain static in size, and some grow for a while and ultimately regress below limits of imaging-based detection.
Early molecular events including genetic, epigenetic, and transcriptional changes, contribute to the differential growth fates of colorectal polyps.
Some tumors appear to be “born to be bad”.
Improvements in multi-omic technologies are revealing vast amounts of intratumoral heterogeneity.
A stepwise model of tumor evolution is by itself insufficient to explain the molecular and phenotypic intra- and intertumoral heterogeneity that is observed in CRC tumorigenesis.
Two new models of CRC evolution have been described in the literature: Big Bang growth and Cancer Punctuated Equilibrium.
The cancer punctuated equilibrium model encompasses three evolutionary processes: stasis, gradualism, and punctuation.
Understanding tumor evolution will likely lead to novel prevention and treatment options.
Acknowledgments
The authors would additionally like to thank Linda Clipson, Rachel Van Doorn, and Thomas Guerin for their assistance in editing this manuscript.
Funding
This project was funded by the National Cancer Institute (P30 CA014520 to University of Wisconsin Carbone Cancer Center, T32 CA009135 supporting CKS); pilot funds from Department of Radiology, School of Medicine and Public Health at the University of Wisconsin; start-up funds from the Division of Gastroenterology and Hepatology, Department of Medicine, School of Medicine and Public Health at the University of Wisconsin; Wisconsin Dual Sport Riders; and Tomorrow’s Hope. This work was also supported by National Cancer Institute RO1CA194663, UO1CA152756, (WMG); the Lattner Foundation (WMG), R.A.C.E. Charities (WMG).
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1**.Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–32. doi: 10.1056/NEJM198809013190901. This seminal paper first described the tumor progression in the colon and rectum to occur via the stepwise accumulation of mutations in tumor suppressor genes and oncogenes. The diagram in which specific mutations were coupled to discrete stages became known as the “Vogelgram”. [DOI] [PubMed] [Google Scholar]
- 2.Muto T, Bussey HJ, Morson BC. The evolution of cancer of the colon and rectum. Cancer. 1975;36:2251–70. doi: 10.1002/cncr.2820360944. [DOI] [PubMed] [Google Scholar]
- 3.Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992;1:229–33. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- 4.Nagase H, Nakamura Y. Mutations of the APC (adenomatous polyposis coli) gene. Hum Mutat. 1993;2:425–34. doi: 10.1002/humu.1380020602. [DOI] [PubMed] [Google Scholar]
- 5*.Pickhardt PJ, Kim DH, Pooler BD, et al. Assessment of volumetric growth rates of small colorectal polyps with CT colonography: a longitudinal study of natural history. Lancet Oncol. 2013;14:711–20. doi: 10.1016/S1470-2045(13)70216-X. This paper was the first to demonstrate that colorectal polyps in humans could have different fates. Some grew, some remained static, some regressed, and some even completely resolved. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Brien MJ, Winawer SJ, Zauber AG, et al. The National Polyp Study. Patient and polyp characteristics associated with high-grade dysplasia in colorectal adenomas. Gastroenterology. 1990;98:371–9. [PubMed] [Google Scholar]
- 7.Pickhardt PJ, Kim DH. Colorectal cancer screening with CT colonography: key concepts regarding polyp prevalence, size, histology, morphology, and natural history. AJR Am J Roentgenol. 2009;193:40–6. doi: 10.2214/AJR.08.1709. [DOI] [PubMed] [Google Scholar]
- 8.Pickhardt PJ, Hain KS, Kim DH, et al. Low rates of cancer or high-grade dysplasia in colorectal polyps collected from computed tomography colonography screening. Clin Gastroenterol Hepatol. 2010;8:610–5. doi: 10.1016/j.cgh.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Hadac JN, Leystra AA, Paul Olson TJ, et al. Colon Tumors with the Simultaneous Induction of Driver Mutations in APC, KRAS, and PIK3CA Still Progress through the Adenoma-to-carcinoma Sequence. Cancer Prev Res (Phila) 2015;8:952–61. doi: 10.1158/1940-6207.CAPR-15-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hung KE, Maricevich MA, Richard LG, et al. Development of a mouse model for sporadic and metastatic colon tumors and its use in assessing drug treatment. Proc Natl Acad Sci U S A. 2010;107:1565–70. doi: 10.1073/pnas.0908682107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paul Olson TJ, Hadac JN, Sievers CK, et al. Dynamic tumor growth patterns in a novel murine model of colorectal cancer. Cancer Prev Res (Phila) 2014;7:105–13. doi: 10.1158/1940-6207.CAPR-13-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones S, Chen WD, Parmigiani G, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105:4283–8. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang CY, Tseng JY, Chen CF, et al. Genome-wide copy number changes and CD133 expression characterized distinct subset of colon polyps: differentiation between incidental polyps and cancer-associated polyps. Int J Colorectal Dis. 2015;30:1617–26. doi: 10.1007/s00384-015-2319-2. [DOI] [PubMed] [Google Scholar]
- 14.Leystra AA, Deming DA, Zahm CD, et al. Mice expressing activated PI3K rapidly develop advanced colon cancer. Cancer Res. 2012;72:2931–6. doi: 10.1158/0008-5472.CAN-11-4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borras E, San Lucas FA, Chang K, et al. Genomic Landscape of Colorectal Mucosa and Adenomas. Cancer Prevention Research (Phila) 2016;9:417–27. doi: 10.1158/1940-6207.CAPR-16-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamada S, Yashiro M, Maeda K, et al. A novel high-specificity approach for colorectal neoplasia: Detection of K-ras2 oncogene mutation in normal mucosa. Int J Cancer. 2005;113:1015–21. doi: 10.1002/ijc.20666. [DOI] [PubMed] [Google Scholar]
- 17.Tobi M, Luo FC, Ronai Z. Detection of K-ras mutation in colonic effluent samples from patients without evidence of colorectal carcinoma. J Natl Cancer Inst. 1994;86:1007–10. doi: 10.1093/jnci/86.13.1007. [DOI] [PubMed] [Google Scholar]
- 18.Zhu D, Keohavong P, Finkelstein SD, et al. K-ras gene mutations in normal colorectal tissues from K-ras mutation-positive colorectal cancer patients. Cancer Res. 1997;57:2485–92. [PubMed] [Google Scholar]
- 19.Bozic I, Antal T, Ohtsuki H, et al. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A. 2010;107:18545–50. doi: 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding L, Ellis MJ, Li S, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–10. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siegmund K, Shibata D. At least two well-spaced samples are needed to genotype a solid tumor. BMC Cancer. 2016;16:250. doi: 10.1186/s12885-016-2202-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang H, Salomon MP, Sottoriva A, et al. Many Private Mutations Originate From The First Few Divisions Of A Human Colorectal Adenoma. J Pathol. 2015 doi: 10.1002/path.4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–7. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardiman KM, Ulintz PJ, Kuick RD, et al. Intra-tumor genetic heterogeneity in rectal cancer. Lab Invest. 2016;96:4–15. doi: 10.1038/labinvest.2015.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McGranahan N, Favero F, de Bruin EC, et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7:283r. doi: 10.1126/scitranslmed.aaa1408. a54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uchi R, Takahashi Y, Niida A, et al. Integrated Multiregional Analysis Proposing a New Model of Colorectal Cancer Evolution. PLoS Genet. 2016;12:e1005778. doi: 10.1371/journal.pgen.1005778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28**.Sottoriva A, Kang H, Ma Z, et al. A Big Bang model of human colorectal tumor growth. Nat Genet. 2015;47:209–16. doi: 10.1038/ng.3214. This paper describes a new conceptual framework in which key mutations arise eartly during a burst instead of stepwise fashion over an extended period of time. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sievers CK, Zou LS, Pickhardt PJ, et al. Subclonal diversity arises early even in small colorectal tumours and contributes to differential growth fates. Gut. doi: 10.1136/gutjnl-2016-312232. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo Y, Wong CJ, Kaz AM, et al. Differences in DNA methylation signatures reveal multiple pathways of progression from adenoma to colorectal cancer. Gastroenterology. 2014;147:418–29. doi: 10.1053/j.gastro.2014.04.039. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Worthley DL, Whitehall VL, Buttenshaw RL, et al. DNA methylation within the normal colorectal mucosa is associated with pathway-specific predisposition to cancer. Oncogene. 2010;29:1653–62. doi: 10.1038/onc.2009.449. [DOI] [PubMed] [Google Scholar]
- 32.Bird A. The essentials of DNA methylation. Cell. 1992;70:5–8. doi: 10.1016/0092-8674(92)90526-i. [DOI] [PubMed] [Google Scholar]
- 33.Jubb AM, Bell SM, Quirke P. Methylation and colorectal cancer. J Pathol. 2001;195:111–34. doi: 10.1002/path.923. [DOI] [PubMed] [Google Scholar]
- 34.Lao VV, Grady WM. Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol. 2011;8:686–700. doi: 10.1038/nrgastro.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim YH, Petko Z, Dzieciatkowski S, et al. CpG island methylation of genes accumulates during the adenoma progression step of the multistep pathogenesis of colorectal cancer. Genes Chromosomes Cancer. 2006;45:781–9. doi: 10.1002/gcc.20341. [DOI] [PubMed] [Google Scholar]
- 36.Hernandez-Vargas H, Sincic N, Ouzounova M, et al. Epigenetic signatures in stem cells and cancer stem cells. Epigenomics. 2009;1:261–80. doi: 10.2217/epi.09.19. [DOI] [PubMed] [Google Scholar]
- 37.Rabinovitch PS, Reid BJ, Haggitt RC, et al. Progression to cancer in Barrett’s esophagus is associated with genomic instability. Lab Invest. 1989;60:65–71. [PubMed] [Google Scholar]
- 38.Stepanenko AA, Kavsan VM. Evolutionary karyotypic theory of cancer versus conventional cancer gene mutation theory. Biopolym Cell. 2012;28:267–80. [Google Scholar]
- 39.Maher CA, Wilson RK. Chromothripsis and human disease: piecing together the shattering process. Cell. 2012;148:29–32. doi: 10.1016/j.cell.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siegmund KD, Marjoram P, Tavare S, et al. Many colorectal cancers are “flat” clonal expansions. Cell Cycle. 2009;8:2187–93. doi: 10.4161/cc.8.14.9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ling S, Hu Z, Yang Z, et al. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc Natl Acad Sci U S A. 2015;112:E6496–505. doi: 10.1073/pnas.1519556112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams MJ, Werner B, Barnes CP, et al. Identification of neutral tumor evolution across cancer types. Nat Genet. 2016;48:238–44. doi: 10.1038/ng.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44**.Baca SC, Prandi D, Lawrence MS, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–77. doi: 10.1016/j.cell.2013.03.021. This paper extends the Big Bang theory of tumor evolution arguing that mutational burst or “puncuation events” can occur throughout tumorigenesis and be followed by periods of statsis or gradulaism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Navin N, Krasnitz A, Rodgers L, et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010;20:68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Humphries A, Cereser B, Gay LJ, et al. Lineage tracing reveals multipotent stem cells maintain human adenomas and the pattern of clonal expansion in tumor evolution. Proc Natl Acad Sci U S A. 2013;110:E2490–9. doi: 10.1073/pnas.1220353110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burrell RA, McGranahan N, Bartek J, et al. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–45. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 48.Cross W, Graham TA, Wright NA. New paradigms in clonal evolution: punctuated equilibrium in cancer. J Pathol. 2016;240:126–36. doi: 10.1002/path.4757. [DOI] [PubMed] [Google Scholar]
- 49.Waclaw B, Bozic I, Pittman ME, et al. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature. 2015;525:261–4. doi: 10.1038/nature14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50*.Fischer JM, Schepers AG, Clevers H, et al. Occult progression by Apc-deficient intestinal crypts as a target for chemoprevention. Carcinogenesis. 2014;35:237–46. doi: 10.1093/carcin/bgt296. This paper demonstrated that a field of Apc-deficient crypts might be necessary for a tumor to become established. Expansion of the field has broad implications with respect to understanding the origin of tumors and as potential target prevention strategies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swanton C, McGranahan N, Starrett GJ, et al. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015;5:704–12. doi: 10.1158/2159-8290.CD-15-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]