

Abstract

Cancer cell cytotoxicity was used to guide the isolation of nine new swinholide-related compounds, named samholides A–I (1–9), from an American Samoan marine cyanobacterium cf. Phormidium sp. Their structures were determined by extensive analysis of 1D and 2D NMR spectroscopic data. The new compounds share an unusual 20-demethyl 44-membered lactone ring composed of two monomers, and they demonstrate structural diversity arising from geometric isomerization of double bonds, sugar units with unique glyceryl moieties and varied methylation patterns. All of the new samholides were potently active against the H-460 human lung cancer cell line with IC50 values ranging from 170 to 910 nM. The isolation of these new swinholide-related compounds from a marine cyanobacterium reinvigorates questions concerning the evolution and biosynthetic origin of these natural products.
Introduction
Cyanobacteria (blue-green alga) are a monophyletic bacterial phylum containing more than 60 genera with more than 400 species.1 As a group, they are extraordinarily rich in structurally diverse and biologically active natural products.2 Almost 500 new compounds have been isolated from cyanobacteria,3 with peptides, polyketides, and hybrids thereof being the major representatives. Several of these natural products have inspired the development of new pharmaceutical agents, and 17 cyanobacterial-derived or inspired agents are in phase I or II drug development (in 2018) and are part of the global marine pharmaceutical clinical pipeline.4
The swinholides represent a family of macrolide natural products having a unique dimeric 44-membered or larger lactone ring. The first example was swinholide A, originally isolated by Kashman and Carmeli from the marine sponge Theonella swinhoei in 1985.5 However, prior to this discovery, swinholide-type natural products had also been isolated from cultured cyanobacteria. Moore et al.6 reported in 1977 a novel monomeric macrolide, tolytoxin, from the terrestrial cyanobacterium Tolypothrix conglutinata var. colorata collected from Fanning Island. Tolytoxin showed potent cytotoxic and fungicidal properties and ultimately was found to possess a structure closely related to a monomeric half of swinholide A. In 1986, the same research group reported a series of tolytoxin-related macrolides, scytophycins A–E, from the cultured terrestrial cyanobacterium Scytonema pseudohofmanni.7 The scytophycins were also structurally related to the swinholides and possessed potent cytotoxicity as well as broad-spectrum antifungal activity. In 2005, our laboratory reported two new glycosylated swinholides, ankaraholides A and B, from a Madagascar cyanobacterium of the genus Geitlerinema sp., together with swinholide A from a Fijian cyanobacterium Symploca cf. sp.8
To date, nearly 60 closely related swinholide-type compounds have been isolated from various species of mollusks, sea hares, nudibranchs, and red and brown algae, causing the true biosynthetic origin of this class of natural products to be confusing and ambiguous.2,3,9−11 In some cases, it is relatively clear that these metabolites are produced by associated or preyed upon microorganisms (e.g., in the cases of macroalgae or mollusks, respectively). In the case of the sponge Theonella spp., because large numbers of cyanobacteria are present in the sponge tissue, it was hypothesized that the swinholides are cyanobacterial secondary metabolites.9 However, Bewley et al. found that swinholide A was associated with a heterotrophic eubacterial fraction rather than the separated sponge and cyanobacterial cells.10 Subsequently, Piel et al. sequenced the biosynthetic gene cluster for misakinolide A from a metagenomic sample of Theonella sp. along with its complement of symbionts.11 Isolation of single filaments of the proposed bacterial source of the misakinolide cluster, Entotheonella serta, followed by multiple displacement amplification (MDA) and sequencing, confirmed that it was the source organism. Matrix-assisted laser desorption ionization (MALDI) imaging as well as catalyzed reporter deposition-fluorescence in situ hybridization (CARD-FISH) analysis provided additional support for this deduction. However, the same work provided highly analogous biosynthetic gene clusters from two cultured cyanobacteria, Scytonema sp. PCC 10023 and Planktothrix paucivesiculata PCC 9631, which produce scytophycin/tolytoxin and luminaolide, respectively. The gene sequencing studies of Piel et al. clearly revealed that two distinct phyla of microorganisms have the capacity to make this class of metabolite and likely reflect a complex set of vertical and horizontal evolutionary events.11 Recently, a study of the antifungal constituents of two freshwater cyanobacteria, Nostoc sp. UHCC 0450 and Anabaena sp. strain UHCC 0451, revealed swinholide and scytophycin-type natural products, respectively.12 Draft genomes of these two organisms revealed the presence of trans-AT biosynthetic gene clusters responsible for production of these molecules, and they were remarkably similar in sequence and architecture to those described by Piel for misakinolide A, scytophycin, and luminaolide. A phylogenetic analysis supported their origination through Horizontal Gene Transfer events.
As a class, the swinholides possess a number of significant bioactivities. Tolytoxin was found to inhibit the growth of a wide array of fungi with MIC values of 0.25–8 nM. It also showed strong cytotoxicity toward a variety of mammalian cells at similar IC50 values.13 Scytophycins A and B displayed antiproliferative activity to the KB human nasopharyngeal carcinoma cell line with an IC50 of 1.2 nM. Other swinholide-related compounds have been found to have similar cytotoxicities with IC50 values in the low nanomolar to picomolar range.14 The molecular target of the swinholides in mammalian cells has been determined to be actin, and at least some of these molecules exhibit nanomolar-potency binding characteristics at the same site of F-actin and G-actin. These potent biological features have thus inspired in-depth investigations of structurally diversified swinholide analogues.15
In the current study, nine new swinholide-type compounds, termed samholides A–I (1–9, Figure 1), were obtained from a field collection in American Samoa of the cyanobacterium cf. Phormidium sp. using a bioassay-guided isolation approach in combination with the MS2-based molecular networking dereplication tool.16 Their structures were determined by extensive analysis of 1D and 2D NMR spectra, and they share a unique 20-demethyl (compared with swinholide A) 44-membered lactone ring that is composed of two monomers. Structural variations among these new compounds result from differences in the methylation and esterification patterns decorating the sugars, as well as the double-bond geometries. Cytotoxicity was evaluated using the H-460 human lung carcinoma cell line and showed that all nine were potently active with IC50 values ranging from 170 to 910 nM. The variations in structure and attendant biological activities provide some initial structure–activity relationships (SAR) and reveal that the sugar units are important for high potency. Finally, their isolation from a marine cyanobacterium provides further evidence for cyanobacteria being a frequent and plentiful source of these actin-binding dimeric metabolites of the swinholide family.
Figure 1.

Structures of compounds 1–9.
Results and Discussion
Tufts of a marine cyanobacterium, morphologically identified as cf.Phormidium sp., were collected at approximately 2 m water depth near American Samoa in July 2014. The preserved collection was repetitively extracted (2:1 CH2Cl2/MeOH) and fractionated using normal-phase vacuum liquid chromatography (VLC) to obtain nine fractions (Fr. A–I). Fractions and the crude extract were screened for cytotoxicity to H-460 human lung cancer cells at 1 and 10 μg/mL.17 While several fractions showed cytotoxicity at the higher concentration, only the two most polar fractions (H, I) were active at 1 μg/mL. By contrast, the extract and all fractions were inactive in a neuromodulatory assay that evaluated the ability of these materials to modulate intracellular calcium mobilization.18 LC–MS/MS molecular networking techniques revealed a distinct cluster of 19 nodes in fractions H and I (Figure 2 and p S104 in the Supporting Information), none of which matched any GNPS MS2 library standards.16 Reversed-phase HPLC of these two fractions yielded nine new compounds, given the names samholides A–I (1–9, Figure 1) to reflect their geographical origins, and these were collectively responsible for the potent cytotoxicity of these fractions.
Figure 2.

MS/MS-based molecular network of the samholides.16 The cosine level was adjusted to 0.7. Nodes display measured masses of the molecular ions. The yellow nodes are samholide analogues that were isolated and defined in the current study, whereas the green nodes are potential samholide analogues not yet described due to the small amounts that were present. The size of the node is reflective of the relative amount of the indicated compounds.
Samholide A (1) gave a sodiated molecular ion peak by HRESIMS spectrum at m/z 1880.0657 [M + Na]+ for a molecular formula of C96H160O34Na+ (calcd 1880.0683). However, the 13C NMR (Table 1) of compound 1 showed only 48 distinct carbon signals, suggesting that 1 possessed a homodimeric structure. The 1H NMR of samholide A displayed four doublet methyl resonances, each of which was composed of six protons for two symmetrical methyl groups at δH 0.83 (d, J = 7.02 Hz), 0.91 (d, J = 6.93 Hz), 0.98 (d, J = 6.74 Hz), and 1.20 (d, J = 6.18 Hz) as well as two overlapping symmetrical olefinic methyl signals at δH 1.80 (s). A cluster of oxygenated methines and methoxy groups was located between δH 3.10 and δH 5.00, and 10 olefinic protons appearing as 5 distinct signals were located between δH 5.67 to δH 7.61. Three olefinic signals, each composed of two isochronous protons, were deshielded to shifts of δH 5.78 (m), 6.34 (dd, J = 9.44, 3.17 Hz), and 7.61 (d, J = 15.54 Hz), indicating the presence of two trisubstituted, conjugated dienones. 13C NMR analysis of compound 1 showed four ester carbonyls with two each at δC 170.7 and 170.64; the latter two reinforced the above assignments in that they were conjugated to two symmetrical dienes with carbon resonances at δC 153.1, 142.9, 123.3, and 113.5. There were also two symmetrical isolated double bonds at δC 133.9 and 129.7.
Table 1. NMR Data for Compound 1 in CDCl3 at 600 MHz (1H)a and 125 MHz (13C)b, Respectively.
| 13C | 1H | COSY | HMBC | HSQC-TOCSY | ROESY | |
|---|---|---|---|---|---|---|
| 1/1′ | 170.6 | |||||
| 2,2′ | 113.5 | 5.78 (overlapped) | 3/3′ | 3/3′, 1/1′ | 2/2′, 3/3′ | 4/4′-Me |
| 3,3′ | 153.1 | 7.6 d (15.5) | 2/2′ | 2/2′, 4/4′, 4/4′-Me, 5/5′ | 2/2′, 3/3′ | 2/2′, 5/5′, 15/15′, 38/38′-OH, 39/39′-OH |
| 4/4′ | 133.9 | |||||
| 4,4′-Me | 12.1 | 1.74 s | 5/5′ | 3,4,5 | 4/4′-Me, 5/5′ | 2/2′, 6/6′, 8/8′ |
| 5,5′ | 142.9 | 6.34 dd (9.44, 3.17) | 6/6′, 4/4′-Me | 4/4′-Me, 3/3′, 6/6′, 7/7′ | 4/4′-Me, 5/5′, 6/6′, 7/7′, 8/8′, 9/9′ | 3/3′, 7/7′, 39/39′-OH |
| 6,6′ | 33.5 | 2.50 d (12.76) | 5/5′, 7/7′ | 4/4′, 5/5′, 7/7′ | 4/4′-Me, 5/5′, 6/6′, 7/7′, 8/8′, 9/9′ | 4/4′-Me, 8/8′, 9/9′, 10/10′ |
| 2.37 m | ||||||
| 7,7′ | 79.0 | 4.10 m | 6/6′, 8/8′ | 5/5′, 6/6′, 32/32′ | 5/5′, 6/6′, 7/7′, 8/8′, 9/9′ | 5/5′, 32/32′ |
| 8,8′ | 39.9 | 2.33 m | 7/7′, 9/9′ | 6/6′, 7/7′, 9/9′, 10/10′ | 5/5′, 6/6′, 7/7′, 8/8′, 9/9′ | 4/4′-Me, 13/13′ |
| 1.53 m | ||||||
| 9,9′ | 68.7 | 4.21 d (11.81) | 8/8′, 10/10′ | 8/8′, 10/10′, 11/11′, 13/13′ | 5/5′, 6/6′, 7/7′, 8/8′, 9/9′, 10/10′, 11/11′, 12/12′ | 6a/6a′, 14/14′ |
| 10,10′ | 129.7 | 5.68 d (10.26) | 9/9′, 11/11′, 12/12′ | 9/9′, 12/12′ | 8/8′, 9/9′, 10/10′, 11/11′, 12/12′, 13/13′, 14/14′ | 8/8′ |
| 11,11′ | 123.3 | 5.77 m | 10/10′, 12/12′ | 9/9′, 12/12′, 13/13′ | 8/8′, 9/9′, 10/10′, 11/11′, 12/12′, 13/13′, 14/14′ | |
| 12,12′ | 31.5 | 2.08 d (17.58) | 11/11′, 13/13′ | 10/10′, 11/11′, 13/13′, 14/14′ | 7/7′, 10/10′, 11/11′, 12/12′, 13/13′, 14/14′ | |
| 1.96 m | ||||||
| 13,13′ | 63.4 | 3.69 m | 12/12′, 14/14′a | 12/12′, 13/13′, 14/14′, 15/15′ | 8a/8a′ | |
| 14,14′ | 36.4 | 1.86 m | 13/13′, 15/15′ | 13/13′, 15/15′, 16/16′ | 12/12′, 13/13′, 14/14′, 15/15′ | |
| 1.64 m | ||||||
| 15,15′ | 75.0 | 4.07 m | 14/14′ | 14/14′, 15/15′-OMe, 16/16′-Me, 16/16′, 17/17′ | 12/12′, 13/13′, 14/14′, 15/15′ | 6a/6a′, 5/5′, 17/17′–OH |
| 16,16′ | 41.2 | 1.59 m | 16/16′-Me, 15/15′ | 16/16′-Me, 18/18′ | 13/13′, 14/14′, 15/15′, 16/16′, 16,16′-Me | |
| 16,16′-Me | 9.3 | 0.83 d (6.85) | 16/16′ | 15,16,17 | 16,16′-Me, 16/16′, 17/17′, 18/18′, 19/19′ | |
| 17,17′ | 73.5 | 3.84 t (9.5) | 18/18′ | 15/15′, 16/16′-Me, 16/16′, 18/18′, 19/19′ | 16/16′-Me, 18/18′, 17/17′, 19/19′ | 16/16′-Me |
| 18,18′ | 41.4 | 1.76 m | 17/17′, 19/19′ | 17/17′, 18/18′, 19/19′, 20/20′ | ||
| 19,19′ | 69.6 | 3.95 m | 18/18′, 19/19′, 20/20′, 21/21′ | 17/17′-OH | ||
| 20,20′ | 42.1 | 1.94 m | 21/21′, | 20/20′, 21/21′, 19/19′ | ||
| 1.52 m | ||||||
| 21,21′ | 70.3 | 5.84 d (11.37) | 20/20′ | 1′/1′, 19/19′, 23/23′, 20/20′, 22/22′, 22/22′-Me | 19/19′, 20/20′ | 23/23′, 23/23′-OH, 39/39′-OH, 38/38′-OH |
| 22,22′ | 40.5 | 1.65 m | 22/22′-Me, 23/23′ | 22,22′-Me, 22/22′, 23/23′, 24/24′,25/25′, 26/26′ | ||
| 22,22′-Me | 9.7 | 0.90 d (6.85) | 22/22′ | 21,22,23 | 22,22′-Me, 22/22′, 23/23′ | |
| 23,23′ | 75.8 | 3.18 d (9.01) | 22/22′, 24/24′ | 21/21′, 22/22′, 24/24′-Me, 25/25′ | 22/22′-Me, 22/22′, 23/23′ | 21/21′, 22/22′-Me, 24/24′-Me |
| 24,24′ | 33.4 | 1.65 m | 23/23′, 26/26′ | 22/22′, 23/23′, 24/24′-Me, 24/24′, 25/25′, 26/26′ | ||
| 24,24′-Me | 17.2 | 0.95 d (6.67) | 24/24′ | 23,24,25 | 24,24′-Me, 24/24′, 25/25′, 26/26′, 27/27′ | |
| 25,25′ | 23.2 | 1.22 m | 26/26′, | 24,24′-Me, 24/24′, 25/25′, 26/26′, 27/27′ | ||
| 26,26′ | 29.3 | 1.80 m | 25/25′, 27/27′, | 24/24′-Me, 25/25′, 26/26′, 27/27′ | ||
| 1.22 m |
| 27,27′ | 71.5 | 3.94 m | 29/29′, 31/31′ | 24/24′-Me, 25/25′, 26/26′, 28/28′, 29/29′, 30/30′ | ||
| 28,28′ | 35.1 | 1.77 m | 29/29′, 30/30′ | 27/27′, 28/28′, 29/29′, 31/31′, 30/30′ | ||
| 1.55 m | ||||||
| 29,29′ | 73.5 | 3.50 m | 28/28′, 31/31′ | 28/28′, 29/29′, 30/30′, 31/31′, 31/31′-Me, | ||
| 30,30′ | 38.9 | 1.94 m | 29/29′ | 28/28′, 29/29′, 30/30′, 31/31′, 31/31′-Me | ||
| 1.15 m | ||||||
| 31,31′ | 64.7 | 3.64 m | 31/31′-Me | 27/27′, 29/29′, 31/31′-Me | 28/28′, 29/29′, 30/30′, 31/31′, 31/31′-Me | |
| 31,31′-Me | 21.9 | 1.16 d (6.19) | 31/31′ | 30,31 | 28/28′, 29/29′, 30/30′, 31/31′, 31,31′-Me | |
| 32,32′ | 102.1 | 4.78 d (6.09) | 7/7′, 33/33′, 36/36′ | 32/32′, 33/33′, 34/34′, 35/35′, 36/36′ | 5/5′, 7/7′, 34/34′ | |
| 33,33′ | 73.5 | 4.76 dd (8.21, 6.29) | 34/34′ | 32/32′, 34/34′, 37/37′ | 32/32′, 33/33′, 34/34′, 35/35′, 36/36′ | 34/34′-OMe, 35/35′ |
| 34,34′ | 82.4 | 3.25 t (7.50) | 33/33′ | 33/33′, 34/34′-OMe, 35/35′ | 32/32′, 33/33′, 34/34′, 35/35′, 36/36′ | 32/32′ |
| 35,35′ | 79.3 | 3.33 m | 32/32′, 33/33′, 34/34′, 35/35′, 36/36′ | 33/33′ | ||
| 36,36′ | 62.3 | 4.00 dd (14.79, 7.83) | 35/35′ | 32/32′, 34/34′, 35/35′-OMe | 32/32′, 33/33′, 34/34′, 35/35′, 36/36′ | |
| 3.30 m | ||||||
| 37/37′ | 170.7 | |||||
| 38,38′ | 72.4 | 4.06 m | 38/38′–OH, 39/39′ | 37/37′, 39/39′ | 38/38′, 39/39′ | 3/3′ |
| 39,39′ | 64.7 | 3.70 m | 38/38′, 39/39′–OH | 38/38′ | 38/38′, 39/39′ | 24/24′-Me |
| 3.50 m | ||||||
| 15,15′-OMe | 57.5 | 3.37 s | 15/15′ | 15,15′-OMe | ||
| 29,29′-OMe | 55.4 | 3.32 s | 29/29′ | 29,29′-OMe | ||
| 34,34′-OMe | 60.1 | 3.45 s | 34/34′ | 34,34′-OMe | ||
| 35,35′-OMe | 58.3 | 3.42 s | 35/35′ | 35,35′-OMe | ||
| 17,17′–OH | 4.82 s | 17/17′ | 16/16′-Me, 17/17′, 18/18′ | |||
| 19/19′-OH | 5.77 m | 19/19′ | 19/19′, 18/18′ | |||
| 23,23′-OH | 4.64 br s | 23/23′ | 22/22′ | 22/22′-Me, 22/22′, 23/23′, 31/31′ | 21/21′, 24/24′-Me, 31/31′ | |
| 38,38′-OH | 5.03 d (3.00) | 38/38′ | 37/37′, 38/38′, 39/39′ | 38/38′, 39/39′ | 2/2′, 3/3′, 21/21′ | |
| 39,39′-OH | 4.81 br s | 39/39′ | 38/38′, 39/39′ | 3/3′, 5/5′, 21/21′ |
1H and 2D NMR spectra were run on a Bruker Advance III DRX-600 MHz NMR spectrometer.
13C NMR spectra were run on a Varian X-Sens 500 MHz NMR spectrometer (125 MHz).
HSQC–TOCSY (Table 1) further enabled assignment of the 1H and 13C NMR data to two symmetrical pentose units with anomeric carbons at δC 102.1 and four oxygenated carbons each at δC 82.4, 73.2, 79.3, and 62.3. These NMR data were generally indicative of a swinholide-related polyketide structure possessing a sugar unit, and thus was of a structure class related to that of ankaraholide A which had been previously isolated from another cyanobacterium, Geitlerinema sp. collected in Madagascar.8
The main differences in the NMR data between compound 1 and ankaraholide A were between C-16 and C-22, and C2 and C5, which nevertheless showed highly similar Δδ-values ranging from 0.8 to 1.4 ppm, relative to the average of 0.3 ppm for the remainder of the comparable carbon signals (Table S1, Supporting Information). Considering the variability of the methyl groups at C-16 and C-20 in the known swinholides,3 these differences between 1 and ankaraholide suggested that it might possess the uncommon 16-methyl-20-demethyl skeleton, a carbon framework only previously appearing in the partial monomer structure of swinholide G.18 This speculation was supported by HMBC analysis that revealed that the long-range correlations from the methyl groups at C-16 and C-22 formed two distinct and nonoverlapping spin networks. Confirmation of this framework was obtained by COSY, HMBC, and HSQC-TOCSY experiments (Table 1 and Figures S9–S13, Supporting Information) which outlined a linear spin system decorated with secondary methyl and oxymethine groups and yielded a homodimeric structure possessing a 20,20′-didemethylswinholide A skeleton (Figure 3). Three additional carbon signals were observed in 1 compared to ankaraholide A and included one ester carbonyl group at δC 170.7, one oxygenated methine at δC 72.4, and one oxygenated methylene at δC 64.7. HMBC and COSY data clarified that these latter resonances and their associated protons were connected and formed a glyceric acid moiety. A neutral fragment ion loss of m/z 88 was observed in the MS/MS spectrum of 1, supporting the nature of this substituent.
Figure 3.
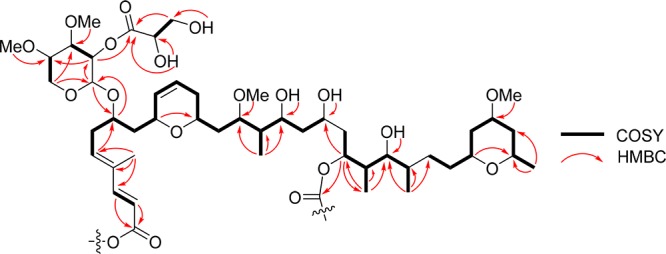
Key COSY and HMBC correlations of the monomeric structure present in samholide A (1).
While the spin system of H-32 to H-36 for a pentapyranose sugar was apparent in compound 1, H-32 and H-33 were overlapped and thus introduced a degree of uncertainty. However, this was resolved by consideration of the HMBC network between these proton and carbon atoms (Table 1 and Figure 3). Moreover, this spin connectivity between H-32/H-33/H-34/H-35/H2-36 was directly observed in the COSY spectrum of compound 3 (Table 2). The large vicinal diaxial coupling constants between H-32, H-33, H-34, H-35, and Ha-36 (all between 6 and 9 Hz, Table 1) in 1 were indicative of the β-xylopyranoside unit, and this was further confirmed by ROESY correlations between H-32 and H-34 and Ha-36 (Figure 4 and Figures S14 and S15). These xylose units were attached to C-7/7′ on the basis of reciprocal HMBC correlations between H-7/7′ and C-32/32′ and H32/32′ and C-7/7′. HMBC correlations between two OMe resonances at δH 3.45 and 3.42 with carbons at δC 82.4 and 79.3 were indicative of two di-O-methylxylopyranoside units, and by consideration of the 13C NMR assignments for this sugar (Table 1), these could be placed at C-34 and C-35. The glyceric acid moiety was connected through the oxygen atom at C-33 by HMBC correlation of H-33 with C-37.
Table 2. 1H NMR Data for Compounds 2–9 in CDCl3 at 600 MHz.
|
2 |
3 |
4 |
5 |
6 |
7 |
8a |
9 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δH | δH′ | δH/δH′ | δH | δH′ | δH | δH′ | δH | δH′ | δH | δH′ | δH | δH′ | δH/δH′ | |
| 2,2′ | 5.79 m | 5.67 m | 5.79 d (15.59) | 5.69 m | 5.82 m | 5.79 d (15.48) | 5.68 d (12.47) | 5.81 d (15.68) | 5.80 m | 5.66 d (12.72) | 5.80 d (15.6) | 5.80 d (15.62) | ||
| 3,3′ | 6.51 (d (12.6) | 7.50 d (15.6) | 7.53 (d 15.56) | 6.46 d (12.41) | 7.39 d (15.56) | 7.57 d (15.55) | 7.52 d (15.56) | 6.52 d (12.62) | 7.44 d (15.54) | 7.47 d (15.63) | 6.46 d (12.69) | 7.55 d (15.6) | 7.47 d (15.62) | |
| 4,4′-Me | 1.86 s | 1.77 s | 1.82 s | 1.84 s | 1.82 s | 1.75 s | 1.82 s | 1.86 s | 1.81 s | 1.78 s | 1.86 s | 1.83 s | 1.80 s | |
| 5,5′ | 5.84 dd (7.06, 6.27) | 6.1 dd, (6.53, 6.40) | 6.31 dd (7.55, 7.37) | 5.98 t (6.33) | 6.09 t (6.23) | 6.33 m | 6.32 m | 5.86 t (7.00) | 6.14 t (6.87) | 6.11 t (6.88) | 5.97 br s | 6.35 m | 6.19 t (7.03) | |
| 6,6′ | 2.55 dd (14.73, 5.62) | 2.62 br d (13.34) | 2.59 ddd (2.85, 6.14, 14.47) | 2.56 m | 2.64 m | 2.50 d (13.39) | 2.67 d (12.99) | 2.57 ddd (4.06, 6.48, 13.87) | 2.69 ddd (4.38, 6.03, 11.73) | 2.59 m | 2.57 m | 2.60 m | 2.47 ddd (3.24, 7.14, 14.94) | |
| 2.38 m | 2.28 m | 2.37 dt (8.86, 14.34) | 2.34 m | 2.36 m | 2.35 m | 2.35 m | 2.40 dt (7.02, 15.36) | 2.28 dt (7.45, 15.12) | 2.34 m | 2.41 dt (7.38, 14.56) | 2.38 m | 2.31 dt (7.56, 15.19) | ||
| 7,7′ | 4.06 m | 4.04 m | 4.01 m | 4.02 m | 3.99 m | 3.97 m | 4.00 m | 4.03 m | 3.97 m | 4.08 m | 4.04 ddd (4.25, 7.90, 11.76) | |||
| 8,8′ | 2.28 m | 2.25 ddd (5.02, 9.81, 15.0) | 2.10 m | 2.29 m | 2.10 m | 2.10 m | 2.19 m | 2.05 m | 2.26 m | 1.89 m | ||||
| 1.54 m | 1.63 m | 1.64 m | 1.67 m | 1.55 m | 1.67 m | 1.54 m | 1.64 m | 1.62 m | 1.52 m | |||||
| 9,9′ | 4.30 m | 4.30 m | 4.36 m | 4.21 d (11.37) | 4.31 brs | 4.29 m | 4.39 m | 4.28 m | 4.38 m | 4.29 m | 4.34 d (10.74) | |||
| 10,10′ | 5.70 m | 5.74 m | 5.74 m | 5.72 m | 5.78 m | 5.69 m | 5.78 m | 5.71 m | 5.74 m | 5.65 m | ||||
| 11,11′ | 5.80 m | 5.74 m | 5.80 m | 5.80 m | 5.71 m | 5.80 m | 5.69 m | 5.74 m | 5.77 m | 5.81 m | ||||
| 12,12′ | 2.01 m | 1.97 m | 1.96 m | 2.08 d (17.61) | 1.97 m | 1.95 m | 1.97 m | 1.98 m | ||||||
| 1.98 m | ||||||||||||||
| 13,13′ | 3.67 m | 3.63 m | 3.64 m | 3.69 m | 3.68 m | 3.79 m | 3.64 m | 3.68 m | ||||||
| 14,14′ | 1.87 m | 1.78 m | 1.87 m | 1.84 m | 1.88 m | 1.84 m | 1.77 m | 1.86 m | ||||||
| 1.62 m | 1.64 m | 1.55 m | 1.63 m | 1.56 m | 1.58 m | 1.64 m | 1.59 m | |||||||
| 15,15′ | 4.06 m | 4.01 m | 3.78 m | 4.05 m | 3.78 ma | 3.82 ma | 3.82 m | 4.08 m | 3.83 m | |||||
| 16,16′ | 1.54 m | 1.51 m | 1.55 m | 1.59 m | 1.65 m | 1.63 m | 1.66 m | 1.68 m | ||||||
| 16,16′-Me | 0.81 d (6.0) | 0.82 d (6.0) | 0.78 d (6.98) | 0.83 d (6.54) | 0.84 d (6.32) | 0.80 d (7.14) | 0.78 d (7.02) | 0.82 d (6.91) | 0.81 d (7.13) | 0.83 d (7.13) | 0.84 d (7.75) | 0.79 d (6.36) | 0.78 d (6.35) | 0.83 d (7.02) |
| 17,17′ | 3.85 m | 3.84 t (9.70) | 3.80 m | 3.88 m | 3.76 m | 3.86 m | 3.85 m | 3.84 m | ||||||
| 18,18′ | 1.76 m | 1.70 m | 1.79 m | 1.74 m | 1.70 m | 1.85 m | 1.72 m | 1.87 m | ||||||
| 1.63 m | 1.41 m | 1.41 m | 1.55 m | 1.55 m | 1.50 m | 1.41 m | 1.50 m | |||||||
| 19,19′ | 3.93 m | 3.88, m | 3.88 t, (10.55) | 3.85 m | 3.79 m | 3.99 m | 3.88 m | 3.88 m | 3.87 m | 3.85 m | 3.90 m | 3.84 m | ||
| 20,20′ | 1.94 m | 1.87 t (12.35) | 1.87 m | 1.90 m | 1.87 m | 1.87 m | 1.85 m | 1.89 m | ||||||
| 1.53 m | 1.52 m | 1.53 m | 1.55 m | 1.53 m | 1.57 m | 1.53 m | 1.55 m | |||||||
| 21,21′ | 5.63 d (9.82) | 5.71 m | 5.56 d (11.09) | 5.64 m | 5.66 m | 5.71 m | 5.60 d (10.39) | 5.63 d (10.00) | 5.65 m | 5.69 m | 5.55 d (11.17) | 5.65 m | ||
| 22,22′ | 1.64 m | 1.65 m | 1.64 m | 1.69 m | 1.76 m | 1.73 m | 1.76 m | 1.74 m | ||||||
| 22,22′-Me | 0.87 d (6.81) | 0.89 d (6.86) | 0.90 d (6.89) | 0.87 d (8.45) | 0.89 d (7.47) | 0.91 d (6.92) | 0.89 d (7.13) | 0.89 d (6.78) | 0.88 d (6.78) | 0.86 d (7.03) | 0.90 d (6.95) | 0.91 m | 0.91 d (6.93) | |
| 23,23′ | 3.17 d (9.69) | 3.12 d (9.59) | 3.19 m | 3.09 m | 3.20 m | 3.15 d (9.39) | 3.12 d (9.83) | 3.12 m | 3.20 m | 3.14 dd (1.52, 9.66) | ||||
| 24,24′ | 1.67 m | 1.68 m | 1.69 m | 1.69 m | 1.68 m | 1.66 m | 1.68 m | 1.66 m | ||||||
| 24,24′-Me | 0.97 d (7.15) | 0.95 d (7.07) | 0.93 d (6.73) | 0.97 d (6.10) | 0.96 d (7.17) | 0.95 d (7.23) | 0.98 d(6.80) | 0.96 d (6.74) | 0.97 d (6.18) | 0.96 d (6.3) | 0.93 m | 0.98 d (6.74) | ||
| 25,25″ | 1.25 m | 1.34 m | 1.37 m | 1.35 m | 1.39 m | 1.35 m | 1.36 m | 1.37 m | ||||||
| 1.17 m | 1.25 m | 1.20 m | 1.24 m | 1.24 m | 1.21 m | 1.23 m | ||||||||
| 26,26′ | 1.88 m | 1.83 m | 1.86 m | 1.86 m | 1.87 m | 1.86 m | 1.84 m | 1.87 m | ||||||
| 1.25 m | 1.26 m | 1.25 m | 1.27 m | 1.25 m | 1.22 m | 1.26 m | 1.24 m | |||||||
| 27,27′ | 3.99 m | 3.98 m | 3.98 m | 3.98 m | 3.99 m | 3.97 m | 3.97 m | 3.98 m | ||||||
| 28,28′ | 1.82 m | 1.79 m | 1.82 m | 1.81 m | 1.82 m | 1.80 m | 1.79 m | 1.81 m | ||||||
| 1.59 m | 1.59 | 1.56 m | 1.59 m | 1.58 m | 1.57 m | 1.58 m | 1.58 m | |||||||
| 29,29′ | 3.53 m | 3.52 ddd 4.4, (9.83, 14.17) | 3.52 m | 3.52 m | 3.52 m | 3.52 m | 3.52 m | 3.52 m | ||||||
| 30,30′ | 1.99 m | 1.95 m | 1.97 m | 1.98 m | 1.99 m | 1.94 m | 1.97 m | 1.98 m | ||||||
| 1.18 m | 1.18 m | 1.16 m | 1.19 m | 1.18 m | 1.17 m | 1.17 m | 1.16 m | |||||||
| 31,31′ | 3.67 m | 3.69 dddd (2.88, 6.13, 12.0, 15.0) | 3.68 m | 3.69 m | 3.69 m | 3.67 m | 3.70 m | 3.69 m | ||||||
| 31,31′-Me | 1.18 d (6.36) | 1.17 d (6.14) | 1.20 d (6.18) | 1.20 d (5.9) | 1.17 d (5.90) | 1.19 d (6.44) | 1.19d (6.25) | 1.18 d (6.24) | 1.20 d (6.3) | 1.21 d (6.6) | 1.20 d (6.18) | |||
| 32,32′ | 4.67 d (6.91) | 4.74 d (6.80) | 4.43 d (6.64) | 4.39 m | 4.46 d (6.6) | 4.81 d (4.98) | 4.46 d (5.45) | 4.65 d (6.77) | 4.39 d (6.72) | 4.71 m | 4.43 d (6.29) | 4.47 d (6.69) | 4.53 d (5.45) | |
| 33,33′ | 4.82 (dd, 8.33, 7.02) | 3.28 t 7.15 | 3.29 m | 4.74 t 6.11 | 3.27 m | 4.83 dd (6.85, 8.39) | 3.52 m | 4.83 dd (7.18, 8.13) | 3.37 m | 3.40 m | 3.36 m | |||
| 34,34′ | 3.34 m | 3.24 m | 3.20 m | 3.32 m | 3.25 m | 3.35 m | 3.21 m | 3.31 m | 3.21 m | 3.21 m | 3.26 m | |||
| 35,35′ | 3.34 m | 3.24 m | 3.25 m | 3.31 m | 3.30 m | 3.31 m | 3.26 m | 3.61 m | 3.36 m | |||||
| 36,36′ | 4.05 m | 3.98 m | 3.99 m | 4.01 m | 3.99 m | 3.99 m | 3.99 m | 3.96 m | ||||||
| 3.28 m | 3.16 m | 3.21 m | 3.25 m | 3.25 m | 3.23 m | 3.18 m | 3.31 m | |||||||
| 38,38′ | 4.28 m | 4.20 brs | 4.13 m | 4.27 m | 4.21 m | |||||||||
| 39,39′ | 3.88 m | 3.70 m | 3.80 m | 3.81 m | ||||||||||
| 3.66 m | ||||||||||||||
| 15,15′-OMe | 3.34 s | 3.35 s | 3.35 s | 3.35 s | 3.33 s | 3.34 s | 3.32 s | 3.34 s | 3.32 s | 3.35 s | 3.36 s | 3.35 s | ||
| 29,29′-OMe | 3.32 s | 3.32 s | 3.34 s | 3.32 s | 3.32 s | 3.34 s | 3.34 s | 3.34 s | ||||||
| 34,34′-OMe | 3.48 s | 3.56 s | 3.59 s | 3.47 s | 3.56 s | 3.48 s | 3.58 s | 3.48 s | 3.58 s | 3.55 s | ||||
| 35,35′-OMe | 3.44 s | 3.45 s | 3.45 s | 3.44 s | 3.43 s | 3.45 s | 3.44 s | 3.44 s | 3.45s | 3.45 | ||||
The data at the same atom position could be exchanged.
Figure 4.
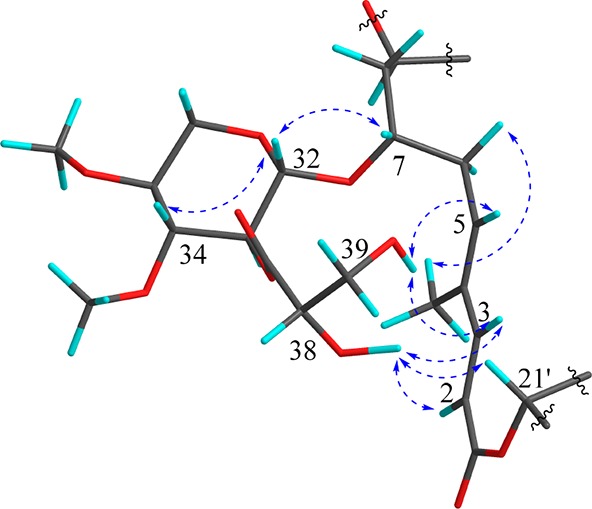
Key ROESY correlations of the 2,3-di-O-methyl-β-xylopyranoside and glyceryl moieties in 1 (energy minimized using standard settings for MM2 method in Chem3D 16.0).
All known swinholide-type compounds, irrespective of origin (sponge, cyanobacteria, algae, nudibranch), possess a highly analogous monomeric carbon skeleton as well as stereoconfigurations at comparable chiral centers (Figure 5); this latter aspect has been confirmed in two cases by X-ray crystallographic analysis7,9 as well as via total synthesis.19−21 As a result, it has been proposed that these swinholide-type metabolites are produced by highly similar polyketide synthase-type biosynthetic gene clusters.11 In the present case, similarities between the 1H and 13C NMR chemical shifts and coupling constants of the protons at the chiral centers of compound 1 and those of previously reported swinholides, as well as the ankaraholides, strongly suggest that they have the same configurations at comparable centers (Table 1 and Tables S1 and S2, Supporting Information).7,8 With the relative configuration of the sugar units defined as above from coupling constant analysis, these could be related to the aglycone stereoconfiguration by several ROESY correlations. ROESY correlations of H-32/H-7 (H-32′/H-7′), 39-OH/H-5 (39′-OH/H-5′), 39-OH/H-3 (39′-OH/H-3′), 38-OH/H-2 (38′-OH/H-2′), 38-OH/H-21′ (38′-OH/H-21), and 38-OH/H-3 (38′-OH/H-3′), in combination with molecular modeling, revealed that the sugar unit must be of l configuration (Figure 4). The configuration at C-38 (C-38′) was not discernible from these data, but given that all occurrences of glyceric acid in 26 cyanobacterial natural products are 2R, we predict samholide A to possess 38R (38′R) stereochemistry as well. Indeed, the glyceric acid unit configuration was rigorously established as d by chiral HPLC analysis of the acid hydrolysate of compound 5 in comparison with authentic standards. The geometry of the C-2-C-3 and C-4-C-5 double bonds were determined to both be E, the former by a characteristic 15.6 Hz J value between H-2 and H-3, and the latter from ROESY correlations between H-2 and the C-4 methyl group, H-3 and H-5, and H2-6 and the C-4 methyl group (Figure 4). Thus, both the constitutive and stereostructure of samholide A (1) was established.
Figure 5.

Monomeric structures and origins of the known swinholide-type compounds. The numbers in parentheses next to the structural diagram refer to the carbon position of dimerization.
The molecular formula of samholide B (2) was identical to that of compound 1 by HRESIMS, and it had a very similar profile of 1H and 13C NMR shifts as well (Table 2 and 3). The main difference in the 1H NMR of 2 was a different pattern of olefinic protons, including one that integrated for a single proton and was quite distinct from those in samholide A [δH 6.51 (d, J = 12.6 Hz)]. Moreover, the C-4 and C-4′ vinyl methyl groups were present as two distinct signals, and a number of other resonances appeared “twinned”. These data, along with a second proton ascribed to H-2′ that had the same large trans-type coupling as present in 1 (JH2′/H3′ = 15.6 Hz), along with ROESY correlations between H-2/H-3, H3/H-5, H-2′/4′-Me, and H-3′/H-5′, indicated that compound 2 was the C-2 double bond isomer of 1 with an overall heterodimeric structure.
Table 3. 13C NMR Data for Compounds 2–9 in CDCl3 at 125 MHz (δH/δH′ in ppm).
| 2 | 3 | 4 | 5 | 6 | 7 | 8c | 9 | |
|---|---|---|---|---|---|---|---|---|
| 1/1′ | 167.9/170.1 | 170.3 | 167.4/169.0 | 170.5/169.9 | 167.6/169.5 | 169.5/167.4 | 170.3 | 169.6 |
| 2/2′ | 116.3/114.3 | 114.2 | 116.7/115.8 | 113.5/114.4 | 116.5/115.3 | 114.5/116.2 | 114.3/113.9 | 114.5 |
| 3/3′ | 148.6/152.2 | 152.4 | 148.5/150.5 | 153.1/152.1 | 148.7/151.1 | 153.9/148.3 | 152.2/152.1 | 152 |
| 4/4′ | 134.1 | 134.6 | 133.6 | 133.7/134.5 | 134.7 | 134.1 | 134.4 | 134.2 |
| 4/4′-Me | 15.7/12.4 | 12.4 | 16.2/12.7 | 12.1/12.4 | 15.8/12.6 | 12.5/16.1 | 12.5/12.4 | 12.5 |
| 5/5′ | 135.0/140.6 | 140.8 | 134.8/137.9 | 142.7/140.3 | 134.1/138.5 | 140/134 | 141/134 | 140.3 |
| 6/6′ | 33.0/33.1 | 33.2 | 33.3/33.4 | 33.6/33.2 | 33 | 33.6/33.2 | 33.6/33.3 | 36.7 |
| 7/7′ | 77.4b/77.7b | 76.6b | 76.7b | 78.6/76.6 | 77.0/75.7 | 77.2b/76.7b | 76.0/76.3 | 69.8 |
| 8/8′ | 39.5/39.6 | 39.8 | 39.1 | 39.8/39.7 | 39.6/39.3 | 39.5/39.1 | 39.8/39.7 | 40.7a |
| 9/9′ | 69.5 | 69.6 | 69.2/68.6 | 68.9/69.2 | 69.0/69.6 | 69.4/69.7 | 69.6 | 72.6 |
| 10/10′ | 129.7a/129.6a | 129.8 | 129.6a/129.5a | 129.7/129.8 | 129.6 | 129.5/129.6 | 129.8 | 129.4 |
| 11/11′ | 123.8a/123.7a | 123.5 | 124.0/123.2 | 123.4/123.7 | 124.0/123.8 | 123.9/123.6 | 123.6 | 124.2 |
| 12/12′ | 31.5a/31.4a | 31.6 | 31.4/31.3 | 31.5 | 31.3/31.2 | 31.4 | 31.4/31.6 | 31.3 |
| 13/13′ | 64.1/63.8 | 63.6 | 64.4/64.3 | 63.5/64.1 | 64.4/64.3 | 64.5 | 63.7/63.5 | 64.5 |
| 14/14′ | 36.2/35.9 | 36.9 | 36.1/35.7 | 36.2/36.7 | 35.7/35.6 | 36.1/35.9 | 36.9/36.8 | 35.9 |
| 15/15′ | 77.2b | 76 | 78.0b | 75.7/75.4 | 77.0b | 77.0b | 75.8/76.0 | 77.6b |
| 15/15′-OMe | 57.1a/57.2a | 57.5 | 57.1 | 57.6/57.5 | 57.2/57.1 | 57.3/56.9 | 57.5 | 57.2 |
| 16/16′ | 41.5 | 41.5 | 41.0/41.2 | 41.6 | 40.9/40.5 | 40.8/40.6 | 41.5/41.4 | 40.5a |
| 16/16′-Me | 9.9a/9.6a | 9.3 | 10.0/10.2 | 9.2/9.0 | 10.2/9.9 | 9.9 | 9.4/9.3 | 9.8 |
| 17/17′ | 73.9/73.6 | 74 | 73.8/74.2 | 73.6/73.8 | 74.3/74.2 | 74.3/74.1 | 74.0/74.1 | 75 |
| 18/18′ | 40.9/41.2 | 40.8 | 41.7 | 40.9/40.6 | 41.7/41.4 | 41.7/41.5 | 40.8 | 40.8a |
| 19/19′ | 69.3/68.8 | 69.9 | 69.5 | 70.0/69.4 | 69 | 69.8/68.3 | 69.8 | 69.2 |
| 20/20′ | 41.1/41.5 | 42.2 | 41.9/42.1 | 42.4 | 41.8 | 42.1 | 42.1/42.0 | 41.7 |
| 21/21′ | 71.1/71.4 | 70.6 | 71.1/70.7 | 70.3/70.5 | 71.3/70.8 | 70.9/71.3 | 70.7/70.6 | 70.4 |
| 22/22′ | 41.1/40.9 | 40.8 | 40.5 | 41.2/40.9 | 41.1/40.9 | 41.3/40.9 | 40.8/40.7 | 40.9a |
| 22/22′-Me | 10.0/10.2 | 10.1 | 10.3/10.5 | 9.9/10.1 | 10.3 | 10.3/10.2 | 10.3/10.2 | 10.2 |
| 23/23′ | 76.6/76.1 | 76.3 | 76.5 | 76.1/76.5 | 76.7 | 76.3 | 76.8b | 76.6b |
| 24/24′ | 33.5 | 33.2 | 33.0/32.8 | 33.4/33.0 | 33.4 | 33 | 33.2/33.3 | 33.3 |
| 24/24′-Me | 17.6a /17.5a | 17.4 | 17.7/17.6 | 17.5 | 17.6 | 17.6 | 17.5/17.4 | 17.7 |
| 25/25′ | 24.0a /23.8a | 23.9 | 24.2/23.9 | 23.6/24.0 | 24 | 24.2/23.6 | 24 | 24.2 |
| 26/26′ | 29.4/29.3 | 29.4 | 29.8/29.3 | 29.8 | 29.3 | 29.3/29.2 | 29.4/29.5 | 29.4 |
| 27/27′ | 71.6/71.5 | 71 | 71.7 | 71.4/71.3 | 71.5 | 71.5/71.7 | 71.1/71.0 | 71.6 |
| 28/28′ | 35.1 | 35 | 35 | 35.1/35.0 | 35 | 35 | 35 | 35 |
| 29/29′ | 73.4 | 73.4 | 73.4 | 73.4 | 73.4 | 73.4 | 73.4 | 73.4 |
| 29/29′-OMe | 55.4 | 55.4 | 55.4 | 55.4 | 55.4 | 55.4 | 55.5 | 55.5 |
| 30/30′ | 38.9 | 38.7 | 38.9 | 38.8 | 38.9 | 38.9 | 38.7 | 38.8 |
| 31/31′ | 64.8 | 64.9 | 64.7 | 64.8/64.9 | 64.7 | 64.7 | 64.9 | 64.8 |
| 31/31′-Me | 22.0a/21.9a | 22 | 22 | 21.9/22.0 | 22 | 21.9 | 21.9 | 22 |
| 32/32′ | 100.7/101.2 | 103.3 | 101.9/102.6 | 101.4/102.9 | 100.4/102.1 | 100.8/102.5 | 103.2/102.9 | |
| 33/33′ | 73.4 | 73.4 | 73.4 | 73.0/73.3 | 73.4/72.9 | 73.4/73.0 | 73.1/72.1 | |
| 34/34′ | 82.1/82.4 | 83.5 | 83.4/83.5 | 82.1/83.3 | 82.0/83.5 | 82.4/83.6 | 83.2/83.3 | |
| 34/34′-OMe | 60.2a/60.1a | 60.2 | 60.3/60.2 | 60.1 | 60.2/60.3 | 60.2/60.1 | 60.1/59.8 | |
| 35/35′ | 79.0/79.3 | 79.4 | 79.0/79.3 | 79.2 | 79.1 | 79.3 | 79.3/68.9 | |
| 35/35′-OMe | 58.6a/58.7a | 58.6 | 58.5 | 58.5 | 58.7a/58.5a | 58.6a/58.5a | 58.5/– | |
| 36/36′ | 62.7 | 62.5 | 62.6 | 62.2/62.5 | 62.6 | 62.7 | 62.5/64.2 | |
| 37/37′ | 171.4/170.9 | 170.5/– | 171.5/– | 170.8/– | ||||
| 38/38′ | 72.5/72.4 | 72.6/– | 72.4/– | 72.4/– | ||||
| 39/39′ | 64.4/64.5 | 64.6/– | 64.3/– | 64.5/– |
Could be exchanged.
Overlapped with CDCl3, assigned using HMBC, HSQC, and HSQC-TOCSY.
The data at the same carbon atom position could be exchanged.
HRESIMS analysis of samholide C (3) indicated its molecular formula as C90H152O28, 176 mass units less than that of compound 1, and corresponded to the absence of both glyceric acid groups. This was consistent with the presence of a monomeric quasi-molecular ion peak at m/z 863.4 in the MS/MS spectrum of 3. The 1H and 13C NMR data of 3 were almost the same as those of 1, except that the glyceric acid signals were missing and the H-33/H33′ protons were shielded by 1.5 ppm.
Samholide D (4) had the same molecular formula as compound 3 from HRMS data. Similar to the relationship of compounds 1 and 2, the differences in its 1H and 13C NMR spectra were due to double-bond isomerization of the C-2-C-3 olefin in one-half of the dimer, as revealed by key chemical shift differences (e.g., δH 5.69 for H-2, δH 5.82 for H-2′) and twinning of many signals near this position of variance between the two monomeric halves. Thus, samholide D (4) was assigned as the C-2 double bond isomer of samholide C (3).
A molecular formula of C93H156O31 was established for samholide E (5) from HRESIMS data, indicating that it was 88 amu less than that of compound 1. This was consistent with the loss of one of the two glyceric acid residues from samholide A. The presence of ions at m/z 951.5 and 863.5, which result from cleavage of the two ester bonds, further indicated the loss of a glyceric acid residue from one of the two monomeric units. This change resulted again in a subtle twinning of many signals in the 1H and 13C NMR of 5; for example, the H-3 and H-3′ protons of the conjugated diene differed by 0.05 ppm in chemical shift (δH 7.57, d, J = 15.55 Hz, H-3; 7.52, d, J = 15.56 Hz, H-3′). Thus, samholide E (5) was assigned a structure identical to that of samholide A (1) but missing one of the two glyceric acid units.
Samholides F (6) and G (7) had the same molecular formula as that of samholide E (5), but differed in the Z and E geometry of the C-2–C-3 double bond in either the Western or Eastern halves of the dimer. For compound 6, there was evidence that the C-2–C-3 olefin was Z (δH 6.52, d, J = 12.62 Hz, H-3, and δH 7.44, d, J = 15.54 Hz, H-3′), whereas in compound 7 the C-2′-C-3′ olefin was Z (δH 6.46, d, J = 12.69 Hz, H-3′, and δH 7.47, d, J = 15.63 Hz, H-3), respectively. The sugar units, either with or without glyceric acid in the two monomeric halves of compound 6, were determined by key ROESY correlations between H-3 and H-5 (δH 5.86, t, J = 7.00 Hz), H-5 and H-7 (δH 4.00, m), and H-7 and H-32 (δH 4.65, J = 6.77 Hz) (Figures S69 and S70, Supporting Information). COSY correlations between H-5/H2-6 (δH 2.57, ddd, J = 4.06, 6.48, 13.87; 2.40, dt, J = 7.02, 15.36 Hz)/H-7 (Figures S63 and S64, Supporting Information), and HMBC correlations between the C-4 Me group (1.86, s) with C-3 (δC 148.7), C-4 (δC 134.7) and C-5 (δC 134.1), along with correlations between H-5 and C-7 (δC 77.0), H-7 and C-32 (δC 100.4), and H-32 and C-7 (Figures S65 and S66, Supporting Information), indicated that the glyceric acid residue was attached to the sugar unit located at C-7 of the 2Z,4E-monomeric structure. Further, lack of a glyceric acid residue in the 2E,4E-monomeric structure was shown by ROESY correlations between H-3′ and H-5′ (δH 6.14, t, J = 6.87 Hz), H-5′ and H-7′ (δH 3.97, m), and H-7′ and H-32′ (δH 4.39, J = 6.72 Hz). This was also supported by COSY correlations of H-5′/H2-6′ (δH 2.69, ddd, J = 4.38, 6.03, 11.73; 2.28, dt, J = 7.45, 15.12 Hz)/H-7′, together with HMBC correlations of the C-4′ Me group protons (1.81, s) with C-3′ (δC 151.1), C-4′ (δC 134.7) and C-5′ (δC 138.5), the H-5′ proton with C-7′ (δC 75.7), the H-7′ proton with C-32′ (δC 102.1), and the H-32′ proton with C-7′. The combination of these NMR data thus confirmed the overall structural assignment for compound 6 as the 2Z double bond isomer of samholide E (5). Accordingly, a similar analysis for compound 7 revealed it to be the 2′Z double bond isomer of samholide E (5).
Samholide H (8) had a molecular formula of C89H150O28 based on HRESIMS analysis. MS/MS data of 8 showed two different ions for the two monomeric halves of the molecule at m/z 863.4 and 849.4, indicating the lack of a glyceric acid residue in one-half and the lack of a methyl or methylene group in the other, relative to the monomeric structures present in compound 5. Further, an m/z 703.3 fragment ion derived from the m/z 849.4 fragment, indicating the lack of a methyl group on the sugar residue. HSQC–TOCSY revealed the signals of both sugar units (Tables 2 and 3); however, the unit lacking the glyceric acid residue also showed an upfield shift for C-35′ (δC 79.3 for C-35 vs δC 68.9 for C-35′). Combined with the absence of this midfield methyl group signal in its 1H and 13C NMR spectra, it was deduced that samholide H (8) possessed one-half of the samholide C structure combined with a second half in which the sugar lacked the C-35 methoxy methyl group. This was further supported by COSY, HMBC, and ROESY correlation networks for these two different sugar residues (Figures S86–S93, Supporting Information).
The molecular formula of samholide I (9) was determined as C76H128O20 from HRESIMS data. MS/MS fragmentation gave a prominent ion at m/z 703.4, suggesting that 9 was an analogue of compound 1 but lacked both the xylose sugar and glyceryl groups. Accordingly, the 1H NMR spectrum of compound 9 showed no sugar or glyceric acid signals, and the 2D NMR data set was fully consistent with the aglycone structure of samholide A (1) (see Supporting Information).
Because the samholides (1–9) are structurally similar to the highly cytotoxic swinholides, they were evaluated for activity against the H-460 human lung carcinoma cell line. All of the samholides showed significant activity with IC50 values less than 1 μM (Table 4), although it should be noted that these IC50 values are all considerably higher than those for the original swinholides.5 However, compound 9 (samholide I), which lacks both the glyceric acid and sugar units, showed the highest IC50 of 0.9 μM, indicating that sugar and glyceryl moieties enhance the cytotoxicity of the samholides. Compound 2 with the 2Z double bond, showed reduced activity compared to 1; however, compounds 3 and 4 as well as 5 and 6, two pairs which also differ only in the 2E versus 2Z geometry, showed equivalently high potency (0.17–0.21 μM). Compound 7 with the 2′Z configuration, was also a highly potent compound (0.21 μM). Compound 8, which had a less methylated xylose sugar, was of lower potency in this assay. In summary, it appears that highest potency among these analogs was obtained when the dimethoxylated xylose sugar was present, with or without glyceric acid residues attached.
Table 4. Cytotoxicity of Compounds 1–9 to H-460 Human Lung Carcinoma Cellsa.
| compd | IC50, mean ± standard errors (μM) |
|---|---|
| samholide A (1) | 0.17 ± 0.01 |
| samholide B (2) | 0.52 ± 0.02 |
| samholide C (3) | 0.21 ± 0.08 |
| samholide D (4) | 0.17 ± 0.06 |
| samholide E (5) | 0.17 ± 0.01 |
| samholide F (6) | 0.17 ± 0.00 |
| samholide G (7) | 0.21 ± 0.01 |
| samholide H (8) | 0.47 ± 0.04 |
| samholide I (9) | 0.91 ± 0.05 |
| doxorubicin | 0.30 ± 0.02 |
Cytotoxicity was assayed in triplicate, and doxorubicin was used as the positive control.
The samholides (1–9) possess several notable structural features, including the uncommon 20-demethyl 44-membered lactone ring, methoxylated xylose sugar, and unusual glyceric acid moiety. The previously described swinholide compounds isolated from sponges and cyanobacteria have been remarkably consistent in the structures of their fundamental carbon skeleton, and structural diversity has been observed in the side chains, position of lactonization, and overall ring size. Previous variations in methyl appendages have only been observed at positions C-16 and C-20. The glyceric acid group, generally a rare structural feature in natural products, is relatively common in cyanobacterium-derived peptides.23 However, the occurrence of such a residue in a polyketide such as the samholides, and as a substituent on a sugar unit, is quite unique.
In 1993, Carmeli et al.24 found that tolytoxin was a polyketide assembled from a glycine starter unit and 15 acetate repeater units, and that the one-carbon branches originated from the tetrahydrofolate Cl pool. Recently, Ueoka et al.11 showed that the architecture of the cyanobacterial PKS megasynthase which produces scytophycin and related compounds was highly similar to the trans-AT PKS cluster responsible for misakinolide production by “Entotheonella sp.”, a symbiont in the sponge Theonella swinhoei. Thus, the biosynthesis of the samholides most likely follows a similar PKS pathway for construction of the carbon chains but with extra sugar and glyceric acid units attached as novel substituents.25,26,27,28,29,30,31,32
Conclusions
The swinholide family of compounds has attracted much attention due to their intriguing structural, biosynthetic, and pharmacological features. In the present study, nine new swinholide-related compounds, samholides A–I (1–9), were obtained from an American Samoa field collection of the cyanobacterium cf. Phormidium sp. (Figure 6). A 20-demethyl 44-membered lactone ring distinguished these new compounds, with structural diversification occurring from geometrical isomerization of double bonds, presence of xylose sugar units with unique glyceric acid moieties, and varied O-methylations. The sugar and glyceric acid units appeared to enhance the relative potency of these agents in a cytotoxicity assay. Discovery of these additional representatives of the swinholide family from another cyanobacterium expands on the number of divergent species that contain this biosynthetic capacity and deepens questions concerning its evolutionary origins and history among the producing species of heterotrophic bacteria, cyanobacteria, and possibly additional sources.
Figure 6.

Photomicrograph of voucher sample of cf. Phormidium sp.
Experimental Methods
Optical rotation was measured on a Jasco P-2000 polarimeter. IR spectra were measured on a Thermo Electron Corp. Nicolet IR 100 FT-IR. UV/visual-light spectra were recorded on a Beckman Coulter DU 880 spectrophotometer.1H NMR and 2D NMR spectra were collected on a Bruker Avance III DRX-600 NMR with a 1.7 mm dual tune TCI cyroprobe (600 and 150 MHz for 1H and 13C NMR, respectively). 13C NMR spectra were run on a Varian X-Sens 500 MHz NMR (125 MHz) equipped with a 5 mm Xsens 13C{1H} cryoprobe. NMR spectra were referenced to residual solvent CDCl3 signals (δH 7.26 ppm and δC 77.16 ppm as internal standards). High-resolution mass spectra were carried out on an Agilent 6230 TOF-MS under positive ion ESI-TOF-MS conditions in the University of California, San Diego (UCSD) Small Molecule MS Facility. MS fragmentation experiments were run with a Biversa Nanomate electrospray source for a Finnigan LTQ-FTICR-MS instrument running Tune Plus software version 1.0. HPLC was performed using Chromeleon 7 software with Thermo Dionex UltiMate 3000 pump and a RS diode array detector. All solvents were HPLC grade except for water, which was produced by a Millipore Milli-Q system.
Cyanobacterial Collection and Taxonomy
The marine cyanobacterium cf. Phormidium sp. (voucher specimen available from W. H. G. as collection no. ASX22Jul14-1) was found growing in 1.0–2.0 m of water at Fagaalu Park in American Samoa, U.S. The sample was hand collected in July 2014, preserved in a 1:1 2-propanol–seawater solution, and stored in the laboratory at −20 °C until extraction. Microscopic examination indicated that this collection was morphologically consistent with the genus Phormidium sp.
Extraction and Isolation
The preserved cyanobacterium was filtered through cheesecloth, and the biomass (101.7 g dry wt) was extracted repeatedly by soaking in 500 mL of 2:1 CH2Cl2/MeOH with warming (<30 °C) for 30 min to afford 1.8 g of dried extract. A portion of the extract was fractionated by silica gel vacuum liquid chromatography (VLC) using a stepwise gradient solvent system of increasing polarity starting from 100% hexanes to 100% MeOH [nine fractions, 100% hexanes (Fr. A, 51.7 mg), 90% hexanes/10% EtOAc (Fr. B, 132.8 mg), 80% hexanes/20% EtOAc (Fr. C 597.7 mg), 60% hexanes/40% EtOAc (Fr. D, 358.5 mg), 40% hexanes/60% EtOAc (Fr. E, 68.2 mg), 20% hexanes/80% EtOAc (Fr. F, 78.2 mg), 100% EtOAc (Fr. G, 38.6 mg), 75% EtOAc/25% MeOH (Fr. H, 180.6 mg) and 100% MeOH (Fr. I, 248.1 mg)]. Fr. I was dissolved in 50% CH3CN/50% H2O, subjected to chromatography on C18 solid-phase extraction (SPE) with a Strata 6 mL column and 1 g of C18-E (55 μm, 70 Å), and eluted sequentially with 30 mL 50% CH3CN/50% H2O (Fr. I1, 15.2 mg), 65% CH3CN/35% H2O (Fr. I2, 10.4 mg), 80% CH3CN/20% H2O (Fr. I3, 73.2 mg), and 100% CH3CN (Fr. I4, 15.4 mg). Fr. I2 was further purified by HPLC using a Synergi 4 μm Hydro-RP 80 Å column (10.00 × 250 mm) and isocratic elution using 97% ACN/3% H2O at the flow rate of 3 mL/min over 45 min. This yielded six subfractions: Fr. I2A, Fr. I2B, Fr. I2C, Fr. I2D, Fr. I2E, and Fr. I2F at 13.0–16.0 min, 19.5–26.8 min, 27.2–31.0 min, 32.6–35.0 min, 36.2–38.8 min, and 41.0–44.0 min, respectively. Three fractions (Fr. I2C, Fr. I2E, and Fr. I2F) after reversed-phase HPLC were purified on the same type of column (10.00 × 250 mm Synergi 4 μm Hydro-RP 80 Å column) with different eluent solvents applied. Fr. I2C was purified using a gradient from 70% ACN/30% H2O to 90% ACN/10% H2O over 30 min, then 90% ACN/10% H2O for 10 min, finally ramping back to 70% ACN/30% H2O over 5 min, detection at 269 nm], giving compound 2 (1.4 mg) at 27.8–29.2 min. Fr. I2E was purified using 97% ACN/3% H2O, (detection at 269 nm), giving compound 5 (5.2 mg) at 32.0–34.5 min. Fr. I2F was purified using 97% ACN/3% H2O (detection at 269 nm) to yield 1 (5.5 mg) at 35.0–37.4 min and 3 (2.6 mg) at 39.5–42.2 min. In the same way, Fr. H was dissolved in 50% CH3CN/50% H2O, purified over C18 solid-phase extraction (SPE) with a Strata 6 mL column and 1 g of C18-E (55 μm, 70 Å), and eluted sequentially with 30 mL 50% CH3CN/50% H2O (Fr. H1, 15.2 mg), 65% CH3CN/35% H2O (Fr. H2, 10.4 mg), 80% CH3CN/20% H2O (Fr. H3, 73.2 mg), and 100% CH3CN (Fr. H4, 15.4 mg). Fr. H3 was separated further using RP HPLC (4 μ Phenomenex Kinetex column, isocratic 70% ACN/30% H2O for 30 min) to yield six subfractions (Fr. H3A, Fr. H3B, Fr. H3C, Fr. H3D, Fr. H3E, and Fr. H3F at 5.6–6.7 min, 8.0–10.0 min, 10.2–14.5 min, 14.5–16.8 min, 16.8–22.0 min, and 22.5–24.5 min, respectively. Three fractions (Fr. H3B, Fr. H3C, and Fr. H3D) were further purified by RP-HPLC using a Synergi Hydro-RP 80 Å column (10.00 × 250 mm, 4 μm) or Phenomenex Luna phenyl-hexyl column with different elution solvents at a flow rate of 3 mL/min. Fr. H3B was subjected to HPLC purification with Synergi Hydro-RP 80 Å using a gradient from 88% MeOH/12% H2O to 94% MeOH/6% H2O over 30 min, then 94% MeOH/6% H2O for 5 min, finally ramping back to 88% MeOH/12% H2O over 2 min, detection at 269 nm, giving compound 8 (0.5 mg) at 29.0–31.0 min. Fr. H3C was subjected to HPLC purification using a Synergi Hydro-RP 80 Å column [isocratic 80% ACN/20% H2O for 40 min, then a gradient from 80% ACN/20% H2O to 90% ACN/10% H2O over 5 min, isocratic 90% ACN/10% H2O for 10 min, finally ramping back to 80% ACN/20% H2O over 5 min, detection at 269 nm], giving compounds 9 (0.3 mg), 6 (0.8 mg) and 7 (0.8 mg). Fr. H3D was purified to yield compound 4 (0.3 mg) after reversed-phase HPLC using the same mobile condition.
LC–MS Analysis and Molecular Networking Generation
The crude extract and fractions A–I were dissolved in MeOH and passed through a Bond Elut-C18 OH cartridge (Agilent Technologies, USA) that was prewashed with 3 mL of CH3CN. Subsequently, the Bond Elut-C18 OH cartridge was washed with 3 mL of CH3CN, the solvent dried under N2, and the residue dissolved in MeOH. A 0.020 mL aliquot of each sample was injected and analyzed via LC–MS/MS on a ThermoFinnigan Surveyor Autosampler-Plus/LC-MS/MS/PDA-Plus system coupled to a Thermo Finnigan LCQ Advantage Max mass spectrometer with a gradient of 30–100% CH3CN in water with 0.1% formic acid. The MS/MS spectra of the crude extract and nine fractions were used to generate a molecular network following previously described methodology and visualized using Cytoscape (www.cytoscape.org).15 Algorithms assumed a cosine threshold set at 0.7 and nodes were color-coded according to the fractions from VLC isolation.
Cytotoxicity Assay
Cytotoxicity to H-460 human lung carcinoma cells was measured as cell viability using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction method.17 Cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium containing l-glutamine (Mediatech, Manassas, VA) and supplemented with 1 nM sodium pyruvate, 100 μg/mL streptomycin, 100 units penicillin, 0.15% sodium bicarbonate, and 10% fetal bovine serum (FBS) at 37 °C, 5% CO2. Cells were seeded in 96-well plates at 6660 cells/well in 180 μL. After 24 h, samholides A–I (1–9) were dissolved in DMSO at 1 mg/mL. The dose–response assays used 20 uL of this DMSO stock and were diluted with 180 μL of RPMI-1640 medium (without fetal bovine serum) to 100 μg/mL followed by nine serial 25 μL:54 μL (logarithmic scale) dilutions with RPMI-1640. Subsequently, 20 μL/well of all 10 mixtures were added to cells in duplicate, resulting in a final maximal DMSO concentration of 1%. Equal volumes of RPMI-1640 medium were added to 10 wells designated as negative controls for each plate. After 48 h, the medium was removed by aspiration and cell viability determined by MTT staining. All assays were validated using doxorubicin at 1.0 and 0.1 μg/mL as the positive control. OD values were measured on ThermoElectron Multiskan Ascent plate reader at 570 and 630 nm. Dose–response graphs were generated using GraphPad Prism (GraphPad Software Inc., San Diego, CA) for IC50 values determination.
Samholide A (1):
white amorphous solid; [α]27D −48 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 269 (4.72); 1H NMR (600 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) see Table 1; HRESIMS m/z 1880.0657 [M + Na]+ (calcd for C96H160O34Na+, 1880.0683).
Samholide B (2):
white amorphous solid; [α]27D −50 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 268 (4.68); 1H NMR (600 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Tables 2 and 3; HRESIMS m/z 1880.0654 [M + Na]+ (calcd for C96H160O34Na+, 1880.0683).
Samholide C (3):
white amorphous solid; [α]27D −52 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 269 (4.70); 1H NMR (600 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Tables 2 and 3; HRESIMS m/z 1704.0327 [M + Na]+ (calcd for C90H152O28Na+, 1704.0362).
Samholide D (4):
white amorphous solid; [α]27D −59 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 268 (4.66); 1H NMR (600 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Tables 2 and 3; HRESIMS m/z 1704.0359 [M + Na]+ (calcd for C90H152O28Na+, 1704.0362).
Samholide E (5):
white amorphous solid; [α]27D −66 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 269 (4.71); IR (neat, KBr) 3445, 3298, 2931, 1730, 1685, 1620, 1459, 1379, 1297, 1080, 1028 cm–1; 1H NMR (600 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Tables 2 and 3; HRESIMS m/z 1792.0508 [M + Na]+ (calcd for C93H156O31Na+, 1792.0523).
Samholide F (6):
white amorphous solid; [α]27D −25 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 269 (4.69); 1H NMR (600 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Tables 2 and 3; HRESIMS m/z 1792.0494 [M + Na]+ (calcd for C93H156O31Na+, 1792.0523).
Samholide G (7):
white amorphous solid; [α]27D −32 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 269 (4.70); 1H NMR (600 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Tables 2 and 3; HRESIMS m/z 1792.0503 [M + Na]+ (calcd for C93H156O31Na+, 1792.0523).
Samholide H (8):
white, amorphous solid; [α]27D −10 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 270 (4.70); 1H NMR (600 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Tables 2 and 3; HRESIMS m/z 1690.0211 [M + Na]+ (calcd for C89H150O28Na+, 1690.0206).
Samholide I (9):
white, amorphous solid; [α]27D −17 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 269 (4.66); 1H NMR (600 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Tables 2 and 3; HRESIMS m/z 1383.8881 [M + Na]+ (calcd for C76H128O20Na+, 1383.8891).
Absolute Configuration of Glyceric Acid Residue
An aliquot (0.2 mg) of compound 5 was hydrolyzed with 6 N HCl (0.3 mL) for 16 h at 90 °C. The hydrolysate was concentrated to dryness and subjected to chiral HPLC analysis (Phenomenex Chirex 3126 (D)-penicillamine (150 × 4.6 mm) HPLC column; flow rate 1 mL/min; UV detection at 254 nm; solvent 2 mM CuSO4:MeOH 85:15). The retention time of glyceric acid from the hydrolyzate was 17.4 min. This was compared to authentic standards whose retention times were 14.9 min for l-glyceric acid and 17.4 min for d-glyceric acid.
Acknowledgments
We thank Y. Su (UCSD Chemistry and Biochemistry Mass Spectrometry Facility) for some of the HRMS data, N. Moss for help in obtaining an IR spectrum, and B. Duggan and A. Mrse for assistance with NMR technical support. This work was supported by National Institutes of Health Grant No. CA100851 (W.H.G.), Guangdong Natural Science Foundation (2016A030313588), Special Fiscal Fund of Guangdong Provincial Oceanic and Fishery Administration in 2017 (A201701607), Fund of the Education Bureau of Guangzhou City (1201610155), National Natural Science Foundation of China (Grant No. 41522605), the project of “AoShan” excellent scholar for Qingdao National Laboratory for Marine Science and Technology, and the Chinese Scholarship Council (CSC) for financial support of this work. We thank the National Fisheries and Parks Service for permits for sample collection in American Samoa.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.8b00028.
Full NMR and MS data for samholides A–I (compounds 1–9), NMR data comparisons with known compounds, and MS2-based molecular network for cf. Phormidium sp. (PDF)
Author Contributions
⊗ Y.T. and P.L. contributed equally to this paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Komarek J.; Kastovsky J.; Mares J.; Johansen J. R. Preslia 2014, 86, 295–335. [Google Scholar]
- Nunnery J. K.; Mevers E.; Gerwick W. H. Curr. Opin. Biotechnol. 2010, 21, 787–793. 10.1016/j.copbio.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blunt J. W.; Copp B. R.; Keyzers R. A.; Munro M. H.; Prinsep M. R. Nat. Prod. Rep. 2015, 32, 116–211. 10.1039/C4NP00144C. [DOI] [PubMed] [Google Scholar]
- Mayer A.Marine Pharmaceuticals: The Clinical Pipeline. http://marinepharmacology.midwestern.edu/clinPipeline.htm.
- Carmely S.; Kashman Y. Tetrahedron Lett. 1985, 26, 511–514. 10.1016/S0040-4039(00)61925-1. [DOI] [Google Scholar]
- Moore R. E. In Marine Natural Products: Chemical and Biological Perspectives; Scheuer P. J., Ed.; Academic Press, 1981; Vol. 4, pp 1–52. [Google Scholar]
- a Ishibashi M.; Moore R. E.; Patterson G. M. L.; Xu C.; Clardy J. J. Org. Chem. 1986, 51, 5300–5306. 10.1021/jo00376a047. [DOI] [Google Scholar]; b Moore R. E.; Ishibashi M.; Patterson G. M. L.; Xu C.; Clardy J. Tennen Yuki Kagobutsu Toronkai Koen Yoshishu 1986, 28, 184–191. [Google Scholar]; c Moore R. E.; Patterson G. M.; Mynderse J. S.; Barchi J.; Norton T.; Furusawa E.; Furusawa S. Pure Appl. Chem. 1986, 58, 263–271. 10.1351/pac198658020263. [DOI] [Google Scholar]
- Andrianasolo E. H.; Gross H.; Goeger D.; Musafija-Girt M.; McPhail K.; Leal R. M.; Mooberry S. L.; Gerwick W. H. Org. Lett. 2005, 7, 1375–1378. 10.1021/ol050188x. [DOI] [PubMed] [Google Scholar]
- Kitagawa I.; Kobayashi M.; Katori T.; Yamashita M.; Tanaka J.; Doi M.; Ishida T. J. Am. Chem. Soc. 1990, 112, 3710–3712. 10.1021/ja00165a094. [DOI] [Google Scholar]
- Bewley C. A.; Holland N. D.; Faulkner D. J. Experientia 1996, 52, 716–722. 10.1007/BF01925581. [DOI] [PubMed] [Google Scholar]
- Ueoka R.; Uria A. R.; Reiter S.; Mori T.; Karbaum P.; Peters E. E.; Helfrich E. J. N.; Morinaka B. I.; Gugger M.; Takeyama H.; Matsunaga S.; Piel J. Nat. Chem. Biol. 2015, 11, 705–712. 10.1038/nchembio.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humisto A.; Jokela J.; Liu L.; Wahlsten M.; Wang H.; Permi P.; Machado J. P.; Antunes A.; Fewer D. P.; Sivonen K. Appl. Environ. Microbiol. 2018, 84, e02321. 10.1128/AEM.02321-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson G. M.; Carmeli S. Arch. Microbiol. 1992, 157, 406–410. 10.1007/BF00249096. [DOI] [PubMed] [Google Scholar]
- Smith C. D.; Carmeli S.; Moore R. E.; Patterson G. M. Cancer Res. 1993, 53, 1343–1347. [PubMed] [Google Scholar]
- a Yeung K.-S.; Paterson I. Angew. Chem., Int. Ed. 2002, 41, 4632–4653. 10.1002/anie.200290057. [DOI] [PubMed] [Google Scholar]; b Allingham J. S.; Klenchin V. A.; Rayment I. Cell. Mol. Life Sci. 2006, 63, 2119–2134. 10.1007/s00018-006-6157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Carver J. J.; Phelan V. V.; Sanchez L. M.; Garg N.; Peng Y.; Nguyen D. D.; Watrous J.; Kapono C. A.; Luzzatto-Knaan T.; Porto C.; Bouslimani A.; Melnik A. V.; Meehan Mi. J.; Liu W.-T.; Crusemann M.; Boudreau P. D.; Esquenazi E.; Sandoval-Calderon M.; Kersten R. D.; Pace L. A.; Quinn R. A.; Duncan K. R.; Hsu C.-C.; Floros D. J.; Gavilan R. G.; Kleigrewe K.; Northen T.; Dutton R. J.; Parrot D.; Carlson E. E.; Aigle B.; Michelsen C. F.; Jelsbak L.; Sohlenkamp C.; Pevzner P.; Edlund A.; McLean J.; Piel J.; Murphy B. T.; Gerwick L.; Liaw C.-C.; Yang Y.-L.; Humpf H.-U.; Maansson M.; Keyzers R. A.; Sims A. C.; Johnson A. R.; Sidebottom A. M.; Sedio B. E.; Klitgaard A.; Larson C. B.; Boya C. A.; Torres-Mendoza D.; Gonzalez D. J.; Silva D. B.; Marques L. M.; Demarque D. P.; Pociute E.; O’Neill E. C.; Briand E.; Helfrich E. J. N.; Granatosky E. A.; Glukhov E.; Ryffel F.; Houson H.; Mohimani H.; Kharbush J. J.; Zeng Y.; Vorholt J. A.; Kurita K. L.; Charusanti P.; McPhail K. L.; Nielsen K. F.; Vuong L.; Elfeki M.; Traxler M. F.; Engene N.; Koyama N.; Vining O. B.; Baric R.; Silva R. R.; Mascuch S. J.; Tomasi S.; Jenkins S.; Macherla V.; Hoffman T.; Agarwal V.; Williams P. G.; Dai J.; Neupane R.; Gurr J.; Rodriguez A. M. C.; Lamsa A.; Zhang C.; Dorrestein K.; Duggan B. M.; Almaliti J.; Allard P.-M.; Phapale P.; Nothias L.-F.; Alexandrov T.; Litaudon M.; Wolfender J.-L.; Kyle J. E.; Metz T. O.; Peryea T.; Nguyen D.-T.; Van Leer D.; Shinn P.; Jadhav A.; Muller R.; Waters K. M.; Shi W.; Liu X.; Zhang L.; Knight R.; Jensen P. R.; Palsson B. O.; Pogliano K.; Linington R. G.; Gutierrez M.; Lopes N. P.; Gerwick W. H.; Moore B. S.; Dorrestein P. C.; Bandeira N. Nat. Biotechnol. 2016, 34, 828–837. 10.1038/nbt.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alley M. C.; Scudiero D. A.; Monks A.; Hursey M. L.; Czerwinski M. J.; Fine D. L.; Abbott B. J.; Mayo J. G.; Shoemaker R. H.; Boyd M. R. Cancer Res. 1988, 48, 589–601. [PubMed] [Google Scholar]
- Dravid S. M.; Murray T. F. Brain Res. 2004, 1006, 8–17. 10.1016/j.brainres.2004.01.059. [DOI] [PubMed] [Google Scholar]
- Paterson I.; Yeung K.-S.; Ward R. A.; Smith J. D.; Cuimming J. G.; Lamboley S. Tetrahedron 1995, 51, 9467–9486. 10.1016/0040-4020(95)00549-N. [DOI] [Google Scholar]
- Nicolaou K. C.; Ajito K.; Patron A. P.; Khatuya H.; Richter P. K.; Bertinato P. J. Am. Chem. Soc. 1996, 118, 3059–3060. 10.1021/ja954211t. [DOI] [Google Scholar]
- Paterson I.; Watson C.; Yeung K.-S.; Ward R. A.; Wallace P. A. Tetrahedron 1998, 54, 11955–11970. 10.1016/S0040-4020(98)83051-2. [DOI] [Google Scholar]
- Tsukamoto S.; Ishibashi M.; Sasaki T.; Kobayashi J. J. Chem. Soc., Perkin Trans. 1 1991, 3185–3188. 10.1039/p19910003185. [DOI] [Google Scholar]
- Ishida K.; Matsuda H.; Murakami M.; Yamaguchi K. Tennen Yuki Kagobutsu Toronkai Koen Yoshishu 1997, 39, 667–672. [Google Scholar]
- Carmeli S.; Moore R. E.; Patterson G. M. L.; Yoshida W. Y. Tetrahedron Lett. 1993, 34, 5571–5574. 10.1016/S0040-4039(00)73884-6. [DOI] [Google Scholar]
- Todd J. S.; Alvi K. A.; Crews P. Tetrahedron Lett. 1992, 33, 441–442. 10.1016/S0040-4039(00)93962-5. [DOI] [Google Scholar]
- Kubanek J.; Jensen P. R.; Keifer P. A.; Sullards M. C.; Collins D. O.; Fenical W. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 6916–6921. 10.1073/pnas.1131855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M.; Tanaka J.; Katori T.; Kitagawa I. Chem. Pharm. Bull. 1990, 38, 2960–2966. 10.1248/cpb.38.2960. [DOI] [PubMed] [Google Scholar]
- Dumdei E. J.; Blunt J. W.; Munro M. H. G.; Pannell L. K. J. Org. Chem. 1997, 62, 2636–2639. 10.1021/jo961745j. [DOI] [PubMed] [Google Scholar]
- Youssef D. T. A.; Mooberry S. L. J. Nat. Prod. 2006, 69, 154–157. 10.1021/np050404a. [DOI] [PubMed] [Google Scholar]
- De Marino S.; Festa C.; D’Auria M. V.; Cresteil T.; Debitus C.; Zampella A. Mar. Drugs 2011, 9, 1133–1141. 10.3390/md9061133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinisi A.; Calcinai B.; Cerrano C.; Dien H. A.; Zampella A.; D’Amore C.; Renga B.; Fiorucci S.; Taglialatela-Scafati O. Bioorg. Med. Chem. 2013, 21, 5332–5338. 10.1016/j.bmc.2013.06.015. [DOI] [PubMed] [Google Scholar]
- Cui J.; Morita M.; Ohno O.; Kimura T.; Teruya T.; Watanabe T.; Suenaga K.; Shibasaki M. Chem. - Eur. J. 2017, 23, 8500–8509. 10.1002/chem.201701183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.