

Abstract
SIRT6 and COX-2 are oncogenes target that promote the expression of proinflammatory and pro-survival proteins through a signaling pathway, which leads to increased survival and proliferation of tumor cells. However, COX-2 also suppresses skin tumorigenesis and their relationship with SIRT6, making it an interesting target for the discovery of drugs with anti-inflammatory and anti-cancer properties. Herein, we studied the interaction of thieno[3,2-c]pyran analogs and RONS species with SIRT6 and COX-2 through the use of molecular docking and molecular dynamic simulations. Molecular docking studies revealed the importance of hydrophobic and hydrophilic amino acid residues for the stability. The molecular dynamics study examined conformational changes in the enzymes caused by the binding of the substrates and how those changes affected the stability of the protein-drug complex. The average RMSD values of the backbone atoms in compounds 6 and 10 were calculated from 1000 ps to 10000 ps and were found to be 0.13 nm for both compounds. Similarly, the radius of gyration values for compounds 6 and 10 were found to be 1.87 ± 0.03 nm and 1.86 ± 0.02 nm, respectively. The work presented here, will be of great help in lead identification and optimization for early drug discovery.
Introduction
Skin cancer is the most common cancer in the United States1,2. The development of skin cancer requires both genomic and proteomic alterations. Specifically, significant oncogenic or tumor-suppressive gene modulation may augment cell survival and proliferation, and additionally induce inflammation in vivo3–5. SIRT6 is a well-known anti-aging protein, which is important for many aspects of cellular homeostasis, including longevity6–8. SIRT6 knockout mice have demonstrated genomic instability leading to accelerated premature aging9. SIRT6 overexpression in mice has been correlated to an increased lifespan in male mice but not in female mice10,11. Furthermore, SIRT6 promotes the reprogramming of induced pluripotent stem cells12 and adjusts collagen metabolism in dermal fibroblasts to control skin aging13. SIRT6 is predicted to act as a tumor suppressor principally because of its longevity potential and its regulation of genomic integrity. Certainly, it is reported to promote tumorigenesis in the liver and intestinal cancer in a mouse model14–16. SIRT6 has also been reported as an oncogene in skin cancer17 and prostate cancer18, and skin-cancer causing UVB rays reportedly induce SIRT6 expression by augmenting the AKT pathway. Furthermore, SIRT6 enhances the expression of the oncogenic and proinflammatory protein COX-2 by inhibiting AMPK signaling, leading to an increase in cell survival and proliferation, as was demonstrated in a study on the effects of UV radiation19,20. COX-2 is an inducible enzyme, which catalyzes the synthesis of prostaglandins, and its up-regulation has been reported in human skin cancer21. The genetic ablation of COX-2 was demonstrated to suppress skin tumorigenesis in chemical- and UV-carcinogenesis models22,23. Recent studies have revealed that COX-2 activity in intrinsic keratinocytes plays a major role in skin cancer24.
The up-regulation of COX-2 in malignant tumors or after acute UV exposure leads to increased PGE2 production followed by the activation of prostaglandin E (EP) receptor signaling, which leads to increased vascular permeability, epidermal proliferation, angiogenesis, and the induction of inflammation. COX enzyme activity is oxidative in nature and can lead to the production of reactive oxygen species (ROS) in activated inflammatory cells, which contributes to a pro-oxidant state25. All these lethal effects of chronic COX-2 up-regulation along with UV-induced p53 mutations combine to make a probable cause for the carcinogenic progression instigated and encouraged by chronic UV exposures. UV exposed skin cells generate a number of reactive oxygen and nitrogen species (RONS). However, the effect of generated RONS is regulated by host anti-oxidative defense mechanisms. High levels of H2O2 or severe oxidative stress were shown to cause increased proteasomal degradation of SIRT1, leading to cellular apoptosis. Similarly, exposure of human monocytes to high doses of H2O2, or human lung epithelial cells to H2O2, aldehyde-acrolein, and cigarette smoke extract significantly decreased SIRT1 activity and gene expression. H2O2 exposure (100 μM, 30 min) caused a drastic drop of 50% in SIRT1 activity, while low doses of H2O2 had no effect26. RONS has also been implicated in the modulation of several proteins linked to the SIRT gene family27–30.
The generation of various RONS in a controlled manner under various gases (O2, N2, He, etc.) is a new approach that is being used in cold-atmospheric thermal plasmas (CAPs). Thus using CAPs might serve as therapeutic areas for treating inflammation-induced cancer. The RONS includes H2O2, NO2, N2O, NO3, N2O3, and N2O5 species and their excessive production aggravates diverse physiological states, including oxidative stress-induced inflammation, which leads to the apoptosis of cancer cells31,32. Therefore, therapeutic drugs that inhibit COX-2 and that use CAPs to target SIRT may be an effective hybrid treatment strategy for non-melanoma skin cancers (NMSCs)33. Our continuing research is focused on the design and synthesis of a scaffold-based fused heterocyclic such as thieno[3,2-c]pyridin34, thia-and oxa-thia[5]helicenes35, thieno[3,2-c]pyran36, tetra substituted thiophenes37, benzo[h]quinolines38, and a new series of analogs based on thieno[3,2-c]pyran. The present study focuses on two objectives. The first is to explore the structure-activity relationship of SIRT6 with our in-house designed thieno[3,2-c]pyran analogs by considering the active-site ligand binding interaction. The second objective is to study the interaction and reactivity of various RONS with SIRT6 and COX-2 target proteins. Moreover, the relative stability and reactivity of designed thieno[3,2-c]pyran analogs and RONS were studied with the aid of unrestrained molecular dynamic simulation.
Results and Discussions
Pharmacokinetic parameters compliance
All of the designed thieno[3,2-c]pyran analogs possessed a good number of hydrogen bond donors and acceptors. Most of the designed compounds possess at least one hydrogen donor and 4 to 7 hydrogen acceptors. The analogs were designed so as to increase the binding affinity of the drug to the receptor mainly by hydrogen bonding39–44. The thieno[3,2-c]pyran moiety was added to increase the binding of the designed molecules with the target receptor. The pharmacokinetic parameters suggest that these analogs were found to follow Lipinski’s rule of 5 since it would increase drug-likeness (Table 1). The polar surface area was calculated to optimize the drug’s ability to permeate the cell membrane. Further, lipophilicity (ratio of octanol solubility to water solubility) was calculated as logP, which has been implicated in blood-brain barrier penetration and permeability prediction45,46. Metabolism and excretion of xenobiotic compounds from the human body depends on MW and logP46,47.
Table 1.
Compliance of designed thieno[3,2-c]pyran analogues and standard drugs to computational parameters of drug-likeness and ADME properties.
| Compounds | Pharmacokinetic property (ADME) dependent on chemical descriptors | Rule of 5 violation | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Oral bioavailability: TPSA (Å2) | MW | Log P | Amine group count | H-bond donor | H-bond acceptor | ||||
| Sec-amine group count | Hydroxyl group count | Nitrogen atom count | Oxygen atom count | ||||||
| 01 | 108.84 | 359.353 | 3.15 | 1 | 0 | 0 | 1 | 6 | 0 |
| 02 | 108.84 | 385.391 | 3.23 | 1 | 0 | 0 | 1 | 6 | 0 |
| 03 | 103.05 | 360.338 | 3.45 | 0 | 0 | 1 | 0 | 7 | 0 |
| 04 | 106.33 | 326.326 | 2.73 | 1 | 0 | 0 | 2 | 4 | 0 |
| 05 | 106.33 | 352.364 | 2.81 | 1 | 0 | 0 | 2 | 4 | 0 |
| 06 | 100.54 | 327.311 | 3.03 | 0 | 0 | 0 | 2 | 4 | 0 |
| 07 | 82.54 | 315.343 | 3.43 | 1 | 0 | 0 | 1 | 4 | 0 |
| 08 | 82.54 | 341.381 | 3.50 | 1 | 0 | 0 | 1 | 4 | 0 |
| 09 | 76.74 | 316.328 | 3.73 | 0 | 0 | 1 | 0 | 4 | 0 |
| 10 | 82.54 | 343.397 | 4.27 | 1 | 0 | 0 | 1 | 4 | 0 |
| 11 | 82.54 | 369.434 | 4.29 | 1 | 0 | 0 | 1 | 4 | 0 |
| 12 | 99.61 | 371.407 | 3.61 | 1 | 0 | 0 | 1 | 5 | 0 |
| Celecoxib | 77.99 | 381.372 | 3.61 | 1 | 1 | 0 | 3 | 0 | 0 |
| Diclofenac | 49.33 | 296.152 | 4.57 | 0 | 1 | 0 | 1 | 2 | 0 |
| Flurbiprofen | 37.30 | 244.265 | 4.05 | 0 | 0 | 0 | 0 | 2 | 0 |
| Indomethacin | 68.54 | 357.793 | 3.99 | 0 | 0 | 0 | 1 | 4 | 0 |
| Olaparib | 86.37 | 434.469 | 2.52 | 0 | 1 | 0 | 4 | 3 | 0 |
The calculated TPSA values of the designed compounds were within acceptable limits. Further, the distribution of compounds in the human body as indicated by blood-brain barrier coefficient (logBB), apparent Caco-2 permeability, log Kp for skin permeability, the volume of distribution, and plasma protein binding (log Khsa for serum protein binding) were determined to be within the standard range48,49. All compounds except 3 and 6 possessed good aqueous solubility (Table S1, Fig. 1), while compounds 1, 2, 4, and 5 showed medium apparent MDCK permeability. The calculated logS values of the analogs were within the acceptable interval, and all analogs showed compliance with a standard range of ADME (Table 2). The analogs were comparable to reference drugs, celecoxib, diclofenac, flurbiprofen, and indomethacin. All the designed compounds were within their permissible limits and hence are unlikely to have limited bioavailability.
Figure 1.
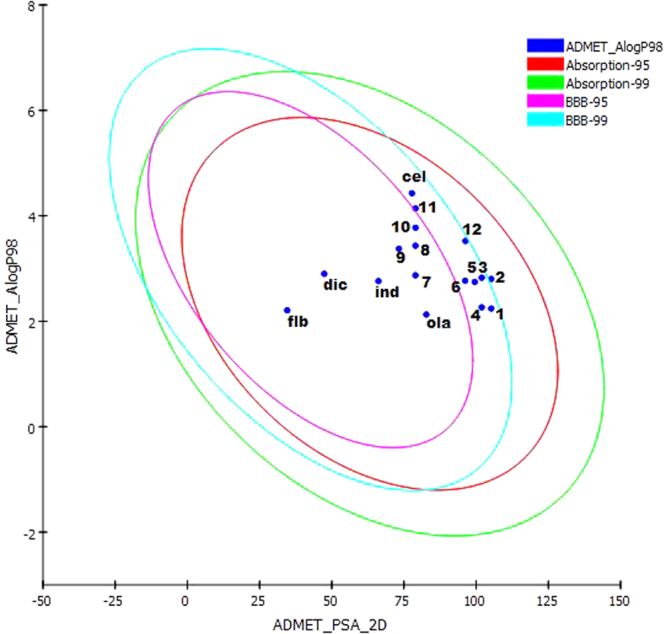
The plot of polar surface area (PSA) versus ALogP for designed thieno[3,2-c]pyran analogs showing the 95% and 99% confidence limit ellipses corresponding to the blood-brain barrier (BBB) and intestinal absorption.
Table 2.
Compliance of designed thieno[3,2-c]pyran analogues and standard drugs of computational parameters for pharmacokinetics (ADME).
| Compounds | log Khsa for Serum Protein Binding | log BB for brain/blood | No. of metabolic reactions | Predicted CNS Activity | log HERG for K + Channel Blockage | Apparent Caco-2 Permeability (nm/sec) | Apparent MDCK Permeability (nm/sec) | log Kp for skin permeability | % Human Oral Absorption in GI ( + −20%) | Qual. Model for Human Oral Absorption |
|---|---|---|---|---|---|---|---|---|---|---|
| 01 | 0.141 | −1.399 | 1 | −2 | −6.438 | 21.792 | 14.4 | −6.632 | 60.632 | Medium |
| 02 | 0.301 | −1.437 | 3 | −2 | −6.214 | 21.503 | 12.65 | −6.829 | 62.161 | Medium |
| 03 | −0.062 | −1.761 | 2 | −2 | −5.725 | 94.397 | 63.709 | −4.499 | 74.929 | HIGH |
| 04 | −0.012 | −1.424 | 1 | −2 | −6.246 | 16.134 | 10.399 | −6.783 | 55.551 | Medium |
| 05 | 0.141 | −1.447 | 3 | −2 | −5.979 | 16.036 | 9.216 | −6.981 | 57.051 | Medium |
| 06 | −0.22 | −1.785 | 2 | −2 | −5.532 | 70.311 | 46.344 | −4.644 | 70.243 | high |
| 07 | 0.31 | −0.627 | 2 | 1 | −6.073 | 77.715 | 56.875 | −5.626 | 74 | High |
| 08 | 0.465 | −0.649 | 4 | 1 | −5.792 | 77.204 | 50.364 | −5.828 | 75.486 | High |
| 09 | 0.208 | −0.985 | 3 | −1 | −5.343 | 338.668 | 253.487 | −3.49 | 88.938 | High |
| 10 | 0.548 | −0.747 | 3 | 0 | −5.954 | 76.556 | 49.386 | −5.719 | 77.164 | High |
| 11 | 0.707 | −0.756 | 4 | 0 | −5.881 | 77.473 | 50.566 | −5.813 | 79.28 | High |
| 12 | 0.197 | −1.463 | 4 | −2 | −5.355 | 168.045 | 101.097 | −4.158 | 80.447 | High |
| Celecoxib | 0.367 | −0.781 | 1 | −1 | −5.772 | 356.195 | 790.105 | −3.223 | 92.188 | High |
| Diclofenac | 0.034 | −0.163 | 4 | −1 | −2.892 | 384.134 | 809.104 | −1.785 | 100 | High |
| Flurbiprofen | 0.163 | −0.342 | 1 | −1 | −3.143 | 339.99 | 298.2 | −1.948 | 96.585 | High |
| Indomethacin | 0.062 | −0.608 | 3 | −1 | −3.243 | 187.398 | 253.764 | −2.565 | 92.548 | High |
| Olaparib | 0.104 | −1.188 | 1 | −2 | −4.182 | 196.841 | 207.7 | −3.287 | 84.563 | High |
| Stand. Range* | (−1.5/1.5) | (−3.0/1.2) | (1.0/8.0) | −2 (inactive) + 2 active) | (concern below −5) | (<25 poor, > 500 great) | (<25 poor, > 500 great) | (–8.0 to –1.0, Kp in cm/hr) | (<25% is poor) | (>80% is high) |
Compliance with toxicity risk assessment parameters
The toxicity risk assessment for all of the designed compounds and standard drugs were calculated using TOPKAT software by Discovery Studio 3.5 (Accelrys, USA). The toxicity risk parameter includes reproductive/developmental toxicity, TOPKAT Ames mutagenicity, TOPKAT skin irritant effects, TOPKAT ames score, TOPKAT rat oral LD50 (mg/kg), and TOPKAT rat carcinogenic potency TD50 (mg/kg) (Table 3). The TOPKAT computes probable toxicity values for a chemical structure by calculating a discriminant score based on its QSTR model. The discriminant score of all the compounds was found to be negative, indicating their non-carcinogenic property. The majority of the compounds were non-toxic for reproductive/developmental toxicity except for compounds 1, 3, 4, 6, 9, and 12. Likewise compounds 4 and 7 were mutagenic and compounds 9 and 12 showed skin irritability; these compounds were in compliance with drug-like parameters and comparable with the predicted results of the reference compounds celecoxib, diclofenac, flurbiprofen, indomethacin, and olaparib. Other designed compounds that showed toxicity still need to be further optimized.
Table 3.
Compliance of designed thieno[3,2-c]pyran analogues and standard drugs to computational toxicity risks parameters (i.e., mutagenicity, tumorigenicity, irritation, and reproduction).
| Compounds | Reproductive effects | TOPKAT Ames Mutagenicity | TOPKAT Skin Irritancy | TOPKAT Ames Score | TOPKAT Rat Oral LD50 (mg/kg) | TOPKAT Rat Carcinogenic potency TD50 (mg/kg) |
|---|---|---|---|---|---|---|
| 01 | Toxic | Non-Mutagen | Non-Irritant | −3.6508 | 2.6589 | 15.3886 |
| 02 | Non-Toxic | Non-Mutagen | Non-Irritant | −3.2304 | 3.9341 | 4.7794 |
| 03 | Toxic | Non-Mutagen | Non-Irritant | −4.8779 | 0.5851 | 32.7399 |
| 04 | Toxic | Mutagen | Non-Irritant | −0.5579 | 1.3455 | 6.7780 |
| 05 | Non-Toxic | Non-Mutagen | Non-Irritant | −1.1059 | 2.0047 | 2.1199 |
| 06 | Toxic | Non-Mutagen | Non-Irritant | −1.7774 | 0.1696 | 14.4274 |
| 07 | Non-Toxic | Mutagen | Non-Irritant | −0.3380 | 3.6710 | 10.3481 |
| 08 | Non-Toxic | Non-Mutagen | Non-Irritant | −1.4741 | 5.7619 | 3.2442 |
| 09 | Toxic | Non-Mutagen | Irritant | −1.5381 | 0.8082 | 22.0288 |
| 10 | Non-Toxic | Non-Mutagen | Non-Irritant | −2.0154 | 2.0326 | 9.6870 |
| 11 | Non-Toxic | Non-Mutagen | Non-Irritant | −3.3643 | 1.9045 | 3.0804 |
| 12 | Toxic | Non-Mutagen | Irritant | −3.9004 | 0.9968 | 7.8474 |
| Celecoxib | Non-Toxic | Non-Mutagen | Non-Irritant | −10.4112 | 4.0652 | 3.3486 |
| Diclofenac | Non-Toxic | Non-Mutagen | Non-Irritant | −10.8461 | 0.6185 | 61.8037 |
| Flurbiprofen | Non-Toxic | Non-Mutagen | Irritant | −4.9857 | 0.5641 | 44.0478 |
| Indomethacin | Non-Toxic | Non-Mutagen | Non-Irritant | −18.4359 | 0.2095 | 4.7492 |
| Olaparib | Non-Toxic | Non-Mutagen | Non-Irritant | −10.3633 | 0.8587 | 1.6315 |
Binding affinity by molecular docking
In the present study, we explored the interactions and binding affinities of designed thieno[3,2-c]pyran analogs, RONS, and the reference drugs olaparib50, celecoxib, and flurbiprofen with the protein targets SIRT6 and COX-2. The binding affinities were determined as the SYBYL-X ‘total’ docking score.
Molecular Docking Analysis of SIRT6
A docking simulation was performed in order to investigate the binding affinity of thieno[3,2-c]pyran analogs and RONS to SIRT6 (PDB ID: 3K35) protein. The docked poses of thieno[3,2-c]pyran analogs were ranked using the surflex-dock score and top score poses were selected for further analysis (Tables 4 and S2). The docking results of compound 6 and SIRT6 showed a high binding affinity represented by a high docking score (6.0956) relative to thieno[3,2-c]pyran analogs. Compound 6 forms three H-bonds (1.8, 1.9, 2.0 Å) to the polar, basic, positively charged nucleophilic residue made up of arginine (Arg)-63 and the backbone atom of alanine (Ala)-51 (Fig. 2a). In the docking pose, the binding site residues on SIRT6 within a radius of 3 Å from the bound compound were polar and uncharged. These residues consisted of the aliphatic, hydrophobic threonine (Thr)-213, glycine (Gly)-50, Ala-51, and isoleucine (Ile)-183, Ile-217; the polar and positively charged Arg-63, the aromatic, hydrophobic phenylalanine (Phe)-62 and trytophan (Trp)-186, the polar, uncharged, and nucleophilic serine (Ser)-214, the polar amide glutamine (Gln)-111, and the polar, acidic aspartic acid (Asp)-185.
Table 4.
Comparison of binding affinity of selected designed thieno[3,2-c] pyran analogues, standard drugs and RONS against SIRT6 (PDB ID: 3K35) enzyme.
| Compounds/RONS | Total Score | Amino acids in the binding site within 3.0 Å of ligand (H-bonding residues shown in bold) | Length of H-bond Å | No. of H-Bond |
|---|---|---|---|---|
| 6 | 6.0956 | Gly-50, Ala-51, Phe-62, Arg-63, Gln-111, Ile-183, Asp-185, Trp-186, Thr-213, Ser-214, Ile-217 | 1.85, 1.97, 2.01 | 3 |
| 10 | 6.0431 | Phe-62, Arg-63, Trp-69, Gln-111, Met-155, Lys-158, Gly-156, Leu-184, Asp-185, Trp-186, Ile-217 | 1.93, 2.09 | 2 |
| Olaparib | 8.3569 | Lys-13, Ala-51, Arg-63, Trp-69, Asn-112, Val-113, His-131, Ile-183, Leu-184, Asp-185, Trp-186 | 1.71, 2.09, 2.32 | 3 |
| NO3 | 5.2070 | Arg-63, Gln-111, His-131, Ile-217 | 2.37, 2.03, 2.11 | 3 |
| N2O3 | 4.6042 | Phe-62, Arg-63, Trp-69 | 2.35, 1.98, 1.98 | 3 |
| NO2 | 4.3973 | Arg-63, Gln-111, His-131, Ile-217 | 2.36, 2.07, 2.13 | 3 |
Note: Surflex-Dock scores (total scores) were expressed in - log10 (Kd) units to represent binding affinities.
Figure 2.

(a) Docked complex of compound 06, (b) compound 10, (c) Olaparib, (d) NO3 spe-cies, (e) N2O3 species and (f) NO2 species on SIRT6 (PDB ID: 3K35) revealing respective binding amino acid residues. Residues showing H-bond within 3 Å is displayed.
Similarly, docking results for compound 10 with SIRT6 showed a good binding affinity (6.0431) and H-bond lengths of 1.9 Å and 2.0 Å to the nucleophilic (polar, basic, positively charged) residue, Arg-63 (Fig. 2b). In the docking pose, the chemical nature of the binding site residues within a radius of 3 Å from the bound compound were polar amide (Gln-111), polar-acidic (Asp-185), aromatic-hydrophobic (Phe-62, Trp-69, Trp-186), hydrophobic-aliphatic(Gly-156, Leu-184, Ile-217, and methionie (Met)-155, polar, positively charged (Lys-158). The docking score for all the thieno[3,2-c]pyran analogs ranged from 4.8363–6.0956 and a careful analysis of all binding poses revealed that the stability of thieno[3,2-c]pyran analogs has been implicated by hydrophobic and hydrophilic amino acid residues, however, hydrophobic amino acid residues favor stability and activity.
Furthermore, the binding interaction and binding affinities of thieno[3,2-c]pyran analogs were compared with those of olaparib, an FDA approved targeted therapy for cancer. The docking results for olaparib onto SIRT6 showed a high binding affinity indicated by docking score of 8.3569, formed by three H-bonds of lengths 1.7, 2.0, and 2.3 Å to nucleophilic, polar, basic, positively charged residues (Arg-63 and Lys-13). In the docking pose, the chemical nature of binding site residues within a radius of 3 Å from bound olaparib was basic, polar, positively charged (Arg-63, His-131, Lys-13), aliphatic-hydrophobic (Ile-183, Leu-184, Val-113, aromatic-hydrophobic (Trp-69, Trp-186, Phe-62), polar amide (Asn-112, Gln-111), and polar acid (Asp-185) (Fig. 2c). Analyzing the binding amino acid residues for olaparib reveals that it binds to hydrophobic and hydrophilic residues, where the hydrophobic residues stabilize in the binding pocket and the H-bond network is established by hydrophilic residues. Moreover, the thieno[3,2-c]pyran analogs and olaparib share the same amino acid residues that bind with the SIRT6 protein. Thus these thieno[3,2-c]pyran analogs could be a potential starting point that could be optimized into a cancer therapy drug targets.
Likewise, SIRT6 has been implicated in numerous types of cancer and RONS produce oxidative stress modulates the function of target proteins. Thus, RONS targeting of SIRT6 may alter SIRT6 function to inhibit cancer progression. In order to understand the interaction and binding affinities of RONS towards SIRT6, all RONS were docked into the SIRT6 binding pocket and ranked using the surflex-dock score. The three RONS molecules with the top scores were selected for further discussion where NO3 emerged as the RONS species with the strongest binding affinity to SIRT6.
The docking results for NO3 showed good binding affinity to SIRT6, as indicated by a docking score of 5.2070. NO3 forms three H-bonds with SIRT6 with lengths of 2.3, 2.0, and 2.1 Å. These bonds bind to a basic, polar, positively charged residue (Arg-63) and a polar amide (Gln-111). In the docking pose, the chemical nature of binding site residues within a radius of 3 Å from bound NO3 was basic polar, positively charged (Arg-63), polar uncharged (Gln-111), nucleophilic basic, polar (His-131), and aliphatic-hydrophobic (Ile-217) (Fig. 2d). Similarly, N2O3 and NO2 showed comparable binding affinities, indicated by the docking scores of 4.6042 and 4.3973, respectively. N2O3 forms three H-bonds of lengths 2.3, 1.9 and 1.9 Å to a basic polar, positively charged residue (Arg-63) and an N-H moiety of an aromatic hydrophobic residue (Trp-69) (Fig. 2e). Moreover, NO2 forms three H-bonds of lengths 2.3, 2.0, and 2.1 Å to basic polar, positive charged residues (Arg-63) and polar uncharged residues (Gln-111) (Fig. 2f). In the docking pose, the chemical nature of the binding site residues was basic polar, positively charged (Arg-63, His-131), aromatic hydrophobic (Phe-62, Trp-69) and aliphatic hydrophobic (Ile-217), and polar uncharged (Gln-111). When comparing the amino acid residues for all RONS species that bound to SIRT6, majority of the binding site residues were hydrophilic and were able to modulate the function of the target protein.
Molecular Docking Analysis of Designed Analogies into COX-2
Similarly, all the designed thieno[3,2-c]pyran analogs were docked into the binding pocket of COX-2 to check the anti-inflammatory properties of the designed analogs. The molecular docking results exhibited good binding affinities with the target protein (PDB ID: 6COX) (Table S3). The majority of the compounds interact with active amino acids residues such as His-90, Gln-192, Tyr-348, Val-349, Leu-352, Ser-353, Tyr-385, Ala-516, Ile-517, Phe-518, and Ser-530 in the COX-2 receptor. Compounds with good binding affinities are shown in Tables 5 and S3 compared with standard drugs. Compound 6 possesses a high docking score of 6.8541 with an H-bond of length 1.7 Å to the polar uncharged residue (Ser-530) (Fig. 3a). Similarly, compound 10 possessed a docking score of 6.8841 with an H-bond length of 1.9 Å to the same polar uncharged residue (Ser-530) (Fig. 3b). The standard drugs celecoxib and flurbiprofen were also docked into the same binding pocket in order to compare their binding orientation and interaction with those of the thieno[3,2-c]pyran analogs. Celecoxib and flurbiprofin had total docking scores of 6.1641 and 6.3151, respectively. Flurbiprofen forms a H-bond length 1.8 Å to the backbone atom of Phe-518, while most of the designed compounds formed H-bonds with the polar uncharged residue (Ser-530) (Fig. 3c). In all the docking poses, the chemical nature of the residues at the binding sites within 3 Å from bound ligands were polar positively charged residue (His-90), polar uncharged residues (Gln-192, Ser-353, Ser-530); aromatic hydrophobic residues (Tyr-355, Tyr-385, Phe-381, Phe-518, Trp-387) and aliphatic hydrophobic residues (Val-349, Val-523, Leu-352, Leu-534, Ala-516, Ile-517. Comparing the binding affinities of compounds 6 and 10 with COX-2 effectively and bind to the same amino acid residues as standard drugs. The docking outcomes suggest that these analogs ought to act as a potential scaffold for the design of inhibitors for inflammation-induced COX-2.
Table 5.
Comparison of binding affinity of selected designed thieno[3,2-c] pyran analogues, standard drugs and RONS against the COX-2 (PDB ID: 6COX) enzyme.
| Compounds/RONS | Total Score | Amino acids in the binding site within 3.0 Å of ligand (H-bonding residues shown in bold) | Length of H-bond Å | No. of H-Bond |
|---|---|---|---|---|
| 6 | 6.8541 | His-90, Val-349, Leu-352, Ser-353, Tyr-355, Phe-381, Tyr-385, Trp-387, Ala-516, Ile-517, Phe-518, Val-523, Ser-530, Leu-534 | 1.7 | 1 |
| 10 | 6.8841 | His-90, Val-344, Val-349, Leu-352, Ser-353, Tyr-355, Phe-381, Leu-384, Tyr-385, Trp-387, Arg-513, Ala-516, Ile-517, Phe-518, Met-522, Val-523, Gly-526, Ser-530, Leu-534 | 1.9 | 1 |
| Flurbiprofen | 6.1641 | His-90, Gln-192, Val-349, Leu-352, Ser-353, Tyr-355, Arg-513, Ala-516, Ile-517, Phe-518, Val-523, Ala-527, Leu-531 | 1.8 | 1 |
| Celecoxib | 6.3151 | Met-113, Val-116, Trp-348, Val-349, Leu-352, Ser-353, Tyr-355, Leu-359, Tyr-385, Val-523, Gly-526, Ala-527, Ser-530, Leu-531 | — | — |
| NO3 | 3.2150 | Val-349, Leu-531, Ser-530 | 2.62, 2.60, 1.98 | 3 |
| H2O2 | 3.1340 | Tyr-355, Ser-353, His-90 | 2.02, 2.03 | 2 |
| N2O3 | 3.0906 | Arg-120, Ala-527, Val-523, Ser-353, Tyr-355 | 1.98, 2.30 | 2 |
Note: Surflex-Dock scores (total scores) were expressed in - log 10 (Kd) units to represent binding affinities.
Figure 3.

Docked complex of (A) Compound 06, (B) Compound 10, (C) Olaparib, (D) NO3, (E) N2O3 and (F) NO2 on anti-inflammatory target COX-2 (PDB: 6COX), residue showing H-bond within 3 Å is displayed.
Apart from the docking of the designed thieno[3,2-c]pyran analogs, we also studied the interaction of RONS with COX-2 and observed how RONS interact with amino acid residues at the binding site of COX-2. Thus, all RONS, such as H2O2, NO2, N2O, NO3, N2O3, and N2O5 were docked into the binding pocket of COX-2 and given a surflex-dock score. From the docking results, the binding interactions of the top three RONS species were discussed. Apparently, among the top-ranked docked RONS species, NO3 was found to possess good binding affinity as indicated by total docking score of 3.2150, along with a higher binding affinity for SIRT6 protein. In the docking pose, the chemical natures of the binding site residues within a radius of 3 Å from bound NO3 were nonpolar, aliphatic, and hydrophobic (Leu-531, Val-349). The docked pose of NO3 to COX-2 includes three H-bonds to nucleophilic polar, uncharged residues (Ser-530) (Fig. 3d). Similarly, docking results for H2O2 species showed a docking score of 3.1340 and the formation of two H-bonds of length 2.0 Å to the nucleophilic polar, uncharged residue (Ser-353), and basic (polar) residue (His-131). Tyr-355, an aromatic hydrophobic amino acid residue was also found within a radius of 3 Å (Fig. 3e). Furthermore, the docking results of N2O3 showed a strong binding affinity to COX-2, as shown in the docking score of 3.0906, and the formation of two H-bonds of lengths 1.9 and 2.3 Å to a nucleophilic polar, basic, positively charged residue (Arg-120) (Fig. 3f). In the docking pose, the chemical natures of the residues were hydrophobic (Ala-527, Val-523), nucleophilic, polar, uncharged (Ser-353), and aromatic hydrophobic (Tyr-355). The amino acid residues in COX-2 involved in binding of all docked RONS species were mainly hydrophilic in nature, thus it may be pointed out that all RONS species target hydrophilic amino acid residues in order to modulate the function of target proteins.
The docking results and analysis of all RONS poses when bound to SIRT6 or COX-2 revealed that hydrophilic residues play an important role in modulating the function of the target proteins. In addition, studies of the interaction of RONS with protein/lipid/membrane system, RONS also reveal skin cancer targets and hydrophilic/double bond-containing substituents on the protein/lipid membrane system to modulate the functions of the system51–53.
Molecular Dynamic Simulations for thieno[3,2-c]pyran analogs
The stability of the complex system in an aqueous solution was examined using the parameters RMSD (root mean square deviation), RMSFs (root means square fluctuations), and radius of gyration (Rg). For this simulation, a 10 ns unconstrained simulation was performed on the docked structure of SIRT6 (3K35) and COX-2 (6COX) bound to compounds 6 and 10 and to the reference drugs olaparib, flurbiprofen, and celecoxib.
Despite the initial structural arrangements of the docked complex, the average RMSD of the trajectories for bound protein backbone atoms showed relative stability. Figure 4a,b display the stable RMSD values of the atoms for docked compounds 3K35 and 6COX proteins respectively. The RMSD analysis for 3K35 and 6COX indicates that they reach equilibration and oscillate around an average value after 1000 ps. The average RMSDs from 1000 ps to 10000 ps for compounds 6 and 10 and olaparib bound to 3K35 protein were 0.13 nm, 0.13 nm, and 0.12 nm, while the values for compounds 6 and 10, celecoxib, and flurbiprofen bound to 6COX were 0.17 nm, 0.19 nm, 0.16 nm, and 0.18 nm, respectively.
Figure 4.

(a) Time dependence of root mean square deviations (RMSDs) for Compound06, Compound10, Olaparib in complex with 3K35 protein; (b) Time dependence of root mean square deviations (RMSDs) for Compound06, Compound10, Flurbiprofen, Celecoxib in com-plex with 6COX protein; (c) Root mean square fluctuations (RMSF) for Compound06, Com-pound10, Olaparib in complex with 3K35 protein; (d) Root mean square fluctuations (RMSF) for Compound06, Compound10, Flurbiprofen, Celecoxib in complex with 6COX protein; (e) Time evolution of the radius of gyration (Rg) for Compound06, Compound10, Olaparib in complex with 3K35 protein (f) Time evolution of the radius of gyration (Rg) for Compound06, Compound10, Flurbiprofen, Celecoxib in complex with 6COX protein during 10000 ps mo-lecular dynamics (MD) simulation.
These RMSD results show the relative stability of compounds 6 and 10 and olaparib bound to 3K35 throughout the simulation, while for 6COX, compound 10 and flurbiprofen had slightly higher RMSDs than compound 6 and celecoxib. This result suggests that compound 10 and flurbiprofen undergo a structural conformational change during the simulations.
Local protein mobility was analyzed by measuring the time-averaged RMSF values of selected compounds bound to 3K35 and 6COX proteins against residue numbers based on 10000 ps trajectory data. The average RMSFs measured for compounds bound to 3K35 were 0.074 nm (compound 6), 0.077 nm (compound 10), and 0.075 nm (olaparib), which reveals the relative stability of the complex upon binding (Fig. 4c). Further, the average atomic fluctuations of compounds bound to 6COX protein were also measured and found to be 0.088 nm (compound 6), 0.084 nm (compound 10), 0.088 nm (celecoxib), and 0.085 nm (flurbiprofen) (Fig. 4d). Moreover, comparing the RMSFs for both bound 3K35 and 6COX proteins near the binding site regions suggest that all the designed compounds show similar binding patterns profiles for their respective target proteins and stabilize them in their favorable conformations for inhibition.
We also determined the Rg value, which provides insight into the overall dimension and the shape of the protein. Figure 4e,f display Rg values for 3K35 and 6COX proteins in bound form, respectively. The average Rg values were 1.87 nm (compound 6), 1.86 nm (compound 10), 1.87 nm (olaparib) for 3K35 and 2.43 nm (compound 6), 2.43 nm (compound 10), 2.44 nm (celecoxib), and 2.43 nm (flurbiprofen) for 6COX protein. The results of the average Rg values indicate that overall shape of the protein was stable upon binding of the ligand.
Molecular Dynamic Simulations for RONS
We further investigated the 3K35 and 6COX proteins system in an aqueous environment supplemented with RONS. The properties studied, including the RMSD, RMSF, Rg, and mean square displacement (MSD), were acquired by performing a 10 ns unconstrained simulation of the docked complex structures of 3K35 and 6COX with top binding poses of NO3, N2O3, NO2, NO3, H2O2, and N2O3, respectively. Despite being made up of tiny molecules, RONS significantly affect the structural properties of proteins. The average RMSD of the trajectories for RONS bound to 3K35 protein backbone atoms (Fig. 5a) shows that the bound complex of NO3 and NO2 achieved equilibrium after 2000 ps. Thereafter, the complexes fluctuated with RMSD values of 0.24 nm and 0.25 nm, respectively. Equilibrium for N2O3 bound to SIRT6 occurred after 3000 ps, after which the complex fluctuated with an RMSD value of 0.40 nm, which is higher than that of NO3 or NO2. A similar RMSD pattern was observed for RONS bound to 6COX protein (Fig. 5b). Average RMSD values for the trajectories of H2O2 and N2O3 bound to 6COX protein backbone atoms became stable after 4000 ps, and oscillated afterwards with an average value of 0.26 nm for both complexes. In contrast, large conformational changes were observed when NO3 bound to 6COX; the system was stable after 5000 ps and oscillate afterwards with an average length of 0.42 nm. The average RMSD for the whole frame was recorded with values of 0.34 nm, 0.24 nm, 0.24 nm for NO3, H2O2, and N2O3, respectively. Furthermore, the RMSD results for backbone atoms in both complex systems suggest that NO3 and NO2 for 3K35 and H2O2 and N2O3 for 6COX formed the most stable systems.
Figure 5.

(a) Time dependence of root mean square deviations (RMSDs) for NO3, N2O3, NO2 in complex with 3K35 protein; (b) Time dependence of root mean square deviations (RMSDs) for NO3, H2O2, N2O3 in complex with 6COX protein; (c) Root mean square fluctuations (RMSF) for NO3, N2O3, NO2 in complex with 3K35 protein; (d) Root mean square fluc-tuations (RMSF) for NO3, H2O2, N2O3 in complex with 6COX protein; (e) Time evolution of the radius of gyration (Rg) for NO3, N2O3, NO2 in complex with 3K35 protein (f) Time evolution of the radius of gyration (Rg) for NO3, H2O2, N2O3 in complex with 6COX protein during 10000 ps molecular dynamics (MD) simulation.
Similarly, the local binding effects of RONS towards amino acid chains of 3K35 and 6COX proteins were analyzed by measuring the time-averaged RMSF values and plotted against residue numbers based on 10000 ps trajectory data. The average atomic fluctuations for NO3, N2O3, and NO2 bound to 3K35 were 0.12 nm, 0.14 nm, and 0.12 nm, respectively (Fig. 5c), while the corresponding fluctuations for NO3, H2O2, and N2O3 bound to 6COX were 0.16 nm, 0.13 nm, and 0.13 nm, respectively (Fig. 5d). These results were all similar and comparable to each other, however, larger fluctuations were observed for the binding of NO3 to 6COX. Furthermore, a comparison of the RMSF results for the binding site regions of both 3K35 and 6COX proteins reveals that the amino acids involved in binding the ligand were stable and within their average values throughout the simulation, and that RONS exhibited similar binding patterns profiles.
Moreover, the globularity indices for 3K35 and 6COX proteins bound to RONS were measured by calculating Rg. Figure 5f display the Rg values for RONS bound to 3K35 and 6COX respectively. The average Rg values calculated for RONS bound to 3K35 were 1.86 nm (NO3), 1.90 nm (N2O3), and 1.86 nm (NO2), while those for 6COX were 2.41 nm (NO3), 2.38 nm (H2O2), and 2.40 nm (N2O3). The Rg indices for N2O3 bound to 3K35 and NO3 bound to 6COX were higher than the Rg indices for other RONS. A similar pattern was also seen for the RMSD and RMSFs values, which indicates that the binding of NO3 to either 3K35 or 6COX significantly affects the conformational behavior of the proteins and that NO3 could be an influential species for both target proteins.
As mentioned above, since RONS are tiny in comparison to thieno[3,2-c]pyran analogs, we also investigated the diffusion coefficient of RONS in the binding pocket of 3K35 and 6COX by measuring the MSD values (Fig. 6a,b). The average MSD values for NO3, N2O3, and NO2 were calculated and found to be 0.1668 (±0.1232)cm2/s, 0.1059 (±0.0256)cm2/s, 0.0055 (±0.0471)cm2/s, respectively. The lower MSD values suggest the stability of RONS bound to 3K35 during the simulation. MSD values for NO3, H2O2, and N2O3 bound to 6COX were found to be 0.1047 (±0.0313)cm2/s, −0.0574 (±0.8425)cm2/s, and 0.1116 (±0.0646)cm2/s. Comparing the MSD values for RONS, a different pattern for H2O2 was observed, wherein the diffusion coefficient for H2O2 increases gradually, then decreases to the diffusibility level of NO3 and N2O3 species and then further increases. From these values, it appears that evidently H2O2, during their simulations, could have interacted with many atoms of protein residues, and consequently increased their diffusibility. However, the MSD values for NO3 and N2O3 species were normal and stable throughout their simulation.
Figure 6.

(a) Time dependence of mean square displacement (MSD) for NO3, N2O3, NO2 in complex with 3K35 protein; (b) mean square displacement (MSD) for NO3, H2O2, N2O3 in complex with 6COX protein during 10000 ps molecular dynamics (MD) simulation.
The analysis of the molecular dynamic simulation revealed that NO3 and N2O3 were found to be particularly influential amongst all species of target proteins. Similarly, earlier reports on the interaction of RONS with proteins/lipid membrane systems also revealed that concentration-dependent RONS were known to modulate the functions of the target system.
Metabolic Networks and Pathway Maps
UV light energy is absorbed by molecules within the cell, and this energy is then transferred to molecular oxygen which uses it to produce ROS. Many of these ROS then react to form cysteine amino acids present in the active site of tyrosine phosphatases (Fig. 7) and MAP kinases (i.e. JNK, ERK, and p38) (Fig. 8). Specifically, tyrosine type protein receptor is inhibited by ROS, which is responsible for the inactive and phosphorylated state of epidermal growth factor receptor (EGFR)54. Therefore, inactivation of tyrosine type protein receptor phosphatase-κ by ROS leads to ligand-independent activation of EGFR, which in turn activates numerous downstream signaling pathways including the Ras⁄Rac1⁄p38 mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase(PI3K)⁄Akt pathways55,56. Rac1 activation also induces additional ROS generation via the activation of NADPH oxidase57. p38 activation results in the phosphorylation of cyclic AMP response element (CRE) binding protein (CREB) and the activation of transcription factor-1, which then binds to the CRE site in the COX-2 gene promoter, activating transcription. PI3K activation by EGFR leads to phosphorylation (and thus activation) of Akt at both Thr-308 and Ser-473, which then in turn phosphorylates glycogen synthase kinase-3b (GSK-3β) at Ser-9, inactivating it. GSK-3β normally phosphorylates CREB at Ser-129, which is an inhibitory phosphorylation site, and thus inactivation of GSK-3β53–55 by Akt leads to dephosphorylation of CREB at Ser-129, relieving CREB inhibition57–59. This mechanism, in combination with the activating phosphorylation of CREB at Ser-133 by p38, is responsible for the binding of CRB at the CRE site on the COX-2 promoter. This binding, in turn, recruits CRB’s coactivator, CREB binding protein, which leads CRB to interact with the basal transcriptional machinery57. Several pathway inhibitors and overexpressing predominant negative specific kinases, both the PI3K/Akt and hep38 MAPK pathways have been shown to be necessary for the maximal induction of COX-2 expression by UVB57–59.
Figure 7.
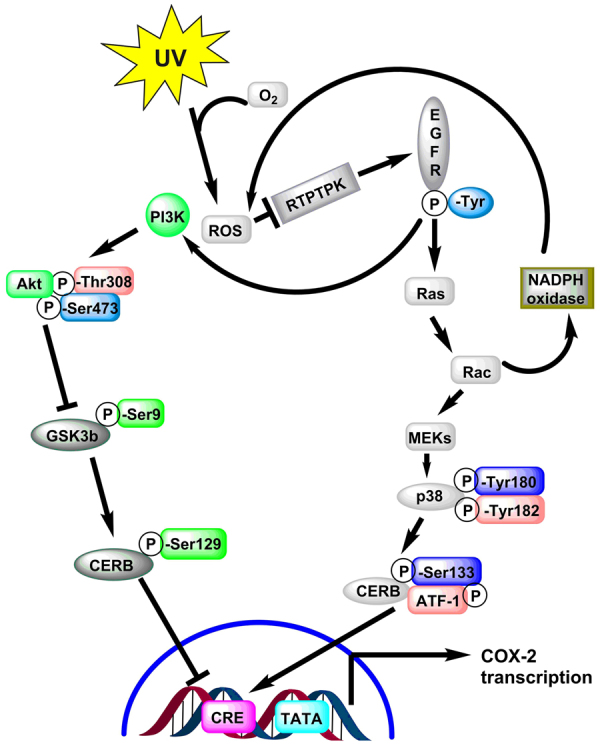
Molecular mechanisms by which UVB light induces cyclo-oxygenase-2 (COX-2) expression. Absorption of UV electromagnetic energy by molecules within a cell is transferred to molecular oxygen-generating ROS.
Figure 8.
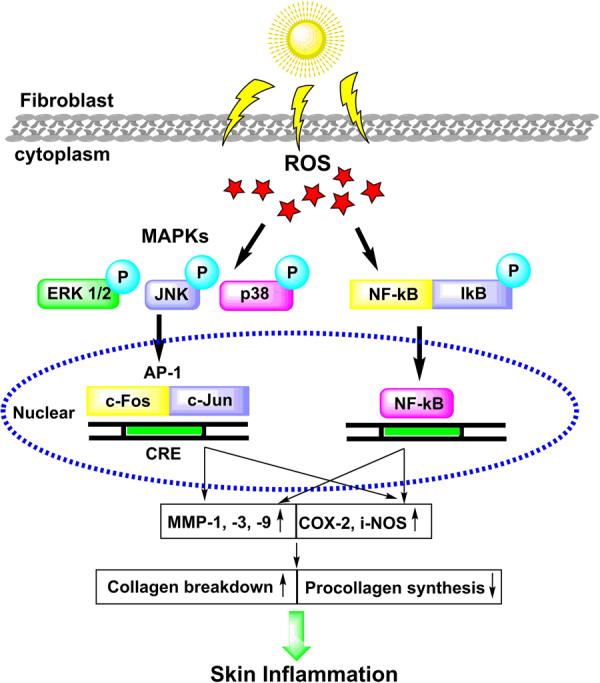
Schematic diagram showing effects of UVB induced in skin inflammatory and photo damage.
COX-2 and iNOS are enzymes that help mediate UV-induced inflammation60. UV-induced ROS drive the activation of MAP kinases (i.e., ERK, JNK, and p38), recruiting activator protein-1 (c-Fos and c-Jun) to the nucleus and, subsequently activating NF-κB and upregulating proinflammatory gene expression61,62. Another reported study revealed that activator protein-1 and MAP kinase are responsible for regulating COX-2 expression61. It was reported that UV radiation causes nuclear translocation of NF-κB and hence induces the release of MMP in order to degrade the collagen in human skin63,64. Additionally, in another study, NF-KB modulated COX-2 and iNOS gene transcription and protein expression, causing skin inflammation65,66. Our in-silico hypothesis also supported this experimental work. The details of this pathway presented in Figs 7–8.
Conclusion
UV-induced development of non-melanoma skin cancer (NMSCs) is the most prevalent existing cancer. The role of SIRT6 in the development of skin cancer is still debatable. However, previous experimental studies have reported that overexpressed SIRT6 results in increased inflammation, proliferation, and survival of sun-damaged skin cells. Likewise, COX-2 is also involved in the pathogenesis of UV-induced non-melanoma skin cancers, and its inhibition is presumed to prevent the development of NMSCs. The connection of SIRT6 and COX-2 has been widely determined; an increase in SIRT-6 expression leads to a greater abundance of COX-2 while inhibition of SIRT-6 expression leads to a decrease in SIRT-6 expression. The work presented here shows that chemical features of a set of designed compounds can be rationalized and that non-thermal cold atmospheric thermal plasmas (CAPs) can be utilized to modulate the functions of the enzyme. Herein, we report the design of thieno[3,2-c]pyran analogs, their binding to their targets, and the conformational changes they induce in the target proteins, using molecule docking and dynamics simulations. The molecular docking results of thieno[3,2-c]pyran analogs revealed that the active pocket of the protein target is hydrophobic in nature and can bind the molecules with hydrophobic groups. Hydrogen bonding features in the ligand also played an important role in their binding to the target protein. The pharmacokinetic compliance for the designed analogs was in agreement with the reference drugs. Moreover, they could be further optimized for lead/drug-like properties. The molecular docking and pharmacokinetic parameters suggested that compounds 6 and 10 possess good characteristics to be used as the lead-like molecule. The presence of a cyanide group and short alkyl group in the thieno[3,2-c]pyran scaffold could have caused the molecule to obtain the configuration necessary to bind to the target. The molecular dynamic studies revealed that compounds 6 and 10 bound to 3K35 and 6COX were stable and exhibited minimal conformational changes, compared to reference drugs. The effect of RONS (CAPs) was also studied within the binding pocket of the protein target and revealed the role of hydrophilic residues that modulating the structural properties of the target. Moreover, molecular dynamic studies revealed that NO3 and N2O3 influenced the conformational changes observed in the protein target. Thus, the present computational work reports the thieno[3,2-c]pyran scaffold for the development of analogs to targets SIRT6 and COX-2 enzymes, and highlights the role of CAPs in modulating the function of the enzyme.
Materials and Methods
Modeling Parameters and Geometry Optimization
The structures of all the designed thieno[3,2-c]pyran analogs were drawn using ChemBioDraw Ultra v12.0 software (Fig. 9). The 2D structures were converted into 3D using the Concord program, followed by geometry cleaning and energy minimization performed by SYBYL-X 2.1.1. The Tripos force field40,42,43,67 and Gasteiger-Hückel charge were assigned during the energy minimization. The maximum iterations for the minimization were set to 2000 steps. The molecular structures of reactive oxygen and nitrogen species (RONS), including H2O2, NO2, N2O, NO3, N2O3, and N2O5 were built using the builder features of the maestro. To ensure the correct geometry and charges, a hybrid density functional theory (DFT) with Becke’s three-parameter exchange potential and the Lee-Yang-Parr correlation functional (B3LYP) and 6–31 G** basis was employed for QM-based geometry optimization and charge calculation68. Finally, these optimized structures were used to compute the properties of the RONS.
Figure 9.

Structures of designed thieno[3,2-c]pyran analogues and reference drugs.
Molecular Docking Experiment Parameters
To find the potential bioactive conformation of the thieno[3,2-c]pyran analogs, the SYBYL-X 2.1.1 suite with the Geom-X mode of the Surflex-Dock engine was used to perform the molecular docking simulation. Thieno[3,2-c]pyran analogs were used as ligands, and SIRT6 (PDB ID: 3K35)69 and COX-2 (PDB ID: 6COX)70 were used as a potential drug targets. The standard protocol was adopted for protein preparation before docking; i.e. addition of H-atoms, assignment of charges, the addition of missing side chains, removal of co-crystallized water molecules, and energy minimization71,72. The prepared structure was further used for the docking of thieno[3,2-c]pyran analogs and RONS. The surflex-dock scoring function was used to score the docking interaction. The surflex-dock score considers several factors related to ligand-receptor interaction, hydrophobicity, polarity, repulsiveness, entropy, and solvation. The docking parameters included ligand flexibility and rigid protein structure, and all other parameters were set to their default values. Furthermore, for RONS docking, the same binding pockets of 3K35 and 6COX along with QM-based charges generated from hybrid density functional theory (DFT).
In Silico Pharmacokinetics Evaluation
Pharmacokinetic properties are described as the absorption, distribution, metabolism, excretion, and toxicity (ADMET) of a drug and provide key information about human therapeutic use of any compound. Pharmacokinetic data also includes information about drug solubility and the ability of the drug to be metabolized by cytochrome-P450 (CYP). This information may be used to determine the metabolism of possible future molecules. The metabolism of these predicted lead compounds was evaluated in detail by a plethora of diverse enzyme families that are involved in xenobiotic metabolisms, such as CYP450 enzymes, dehydrogenases, flavin-containing monooxygenases, hydrolases, peroxidases, UDP-glucuronosyl-transferases (UGTs), sulfotransferases, and glutathione S-transferases. The descriptors used herein correlate well with pharmacokinetic properties such as primary determinant of fractional absorption, and refer to the polar surface area (PSA) (cut-off ≤ 140 Å2), and low molecular weight (MW) for absorption67,73. For the secondary determinant of fractional absorption, we used the sum of H-bond donors and acceptors (cut-off ≤ 12); this descriptor was used to show passive membrane transport. The rotatable numbers of bonds number was used as a measure of flexibility (cut-off ≤ 10) and bioavailability. Drug distribution depends on several factors, including permeability (indicated by apparent Caco-2 and MDCK permeability, and log Kp for skin permeability), blood-brain barrier (log BBB), volume of distribution, and plasma protein binding (shown as logKhsa for serum protein binding)48,49,74. These ADME descriptors for the designed thieno[3,2-c]pyran analogs were calculated and checked for compliance with standard ranges. In addition, the octanol-water partition coefficient (log P) has been implicated in logBB penetration and permeability studies. The process of drug excretion from the body depends on log P and MW. Likewise, rapid renal clearance is associated with hydrophilicity and small molecules. In the liver, increased drug metabolism is associated with hydrophobicity and large molecules. Thus, higher lipophilicity leads to poor absorption and an increase in the metabolic process. The descriptors of 90% of orally active compounds follow Lipinski’s rule. Therefore, in order to ascertain the drug-like properties, all the parameters were calculated using QikProp v3.2 (Schrödinger, LLC, USA, 2015) and Discovery Studio 3.5 software75. Lastly, the toxicity parameter was evaluated by calculating different standard properties using TOPKAT software76–79.
Molecular Dynamics Simulations
The molecular dynamics simulation was carried out for 10000 ps using the GROMACS 4.6.5 package80. The topology parameters for SIRT6 (3K35) and COX-2 (6COX) were generated by Gromacs, whereas for thieno[3,2-c]pyran analogs and RONS, the ACPYE module of AmberTools1681 and the Automated Topology Builder server82 were used, respectively. Charges for RONS, were adopted from QM-based hybrid DFT. Prior to simulation, an energy minimization was performed to full system without constraints using steepest descent integrator for 2000 steps. The system was then equilibrated for 200 ps of NVT and NPT ensemble, applying the position restraints on protein, inhibitors, and counterions at 310.15 K with periodic boundary conditions. The temperature was kept constant by a Berendsen thermostat, while the pressure was maintained at 1 bar using a Parrinello-Rahman scheme. Electrostatic interactions were calculated using the particle mesh Ewald method and cut-off distances for the calculation of Coulomb and van der Waals interactions were 1.4 nm during the equilibration. Finally, the system was subjected to 10000 ps MD at a temperature of 310.15 K (V-rescale thermostat) and a pressure of 1 bar (Parrinello-Rahman barostat). A periodic boundary condition was imposed on the system and the motion equations were integrated by applying the leaf-frog algorithm with a time step of 2 fs.
Molecular Dynamic Simulation of SIRT6 and COX-2
The thieno[3,2-c]pyran analogs and RONS with the top docking scores were selected for a 10000 ps molecular dynamic simulation. The complex was placed in a cubic periodic box with a side length of 114.32 Å and the minimum distance between the complex and box walls was set to larger than 10 Å. The complex was comprised of 2736 atoms in the SIRT6 protein and 5835 atoms in the COX-2 protein. One inhibitor was solvated by single point charge (SPC) water molecules followed by the addition of counterions. The system was energy minimized and equilibrated prior to the molecular dynamic simulation run. The dynamic trajectories were recorded every 2 ps during the production stage for further analysis.
Electronic supplementary material
Acknowledgements
This study was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (No.: 2017R1C1B2003380) at Gachon University of Medicine and Science, Incheon city, Korea and Science & Engineering Research Board (SERB), New Delhi (www.serb.gov.in), for financial support as Young Scientist fellowship (No.: SB/YS/LS-130/2014) at the All India Institute of Medical Sciences, Jodhpur, India. SC acknowledges DST, New Delhi for DST-RFBR Indo-Russian Joint Research Project (INT/RUS/RFBR/P-169) and CSIR, New Delhi for EMR Grant [02(0189)/14/EMR-II]. Authors are thankful to The National Institute of Supercomputing and Network/Korea Institute of Science and Technology Information provided supercomputing resources including technical support (No. KSC-2017-C1-0013; KSC-2017-C2-0017) and NK acknowledges the Research Foundation-Flanders (FWO), grant numbers 12J5617N and Research group PLASMANT, Department of Chemistry, University of Antwerp, Antwerp, Belgium.
Author Contributions
D.K.Y. conceived and designed the project, collected data from literature and database. D.K.Y., S.K., Saloni performed computational studies and analysed the data and wrote the manuscript. M.T. and H.S.K. provided suggestions and helped with manuscript editing. S.K. created the Figures 7 to 8 and the Supplementary Material. L.Y., S.C., H.S.K., S.M., N.K., M.H.K., E.H.C. and P.S. provided the molecular modelling lab facility. All authors have read and approved the final version of the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Dharmendra K. Yadav and Surendra Kumar contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-22972-9.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dharmendra K. Yadav, Email: dharmendra30oct@gmail.com
Mi-hyun Kim, Email: kmh0515@gachon.ac.kr.
References
- 1.Rogers HW, et al. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Archives of dermatology. 2010;146:283–287. doi: 10.1001/archdermatol.2010.19. [DOI] [PubMed] [Google Scholar]
- 2.D’Orazio J, Jarrett S, Amaro-Ortiz A, Scott T. UV radiation and the skin. International journal of molecular sciences. 2013;14:12222–12248. doi: 10.3390/ijms140612222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bowden GT. Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling. Nature reviews. Cancer. 2004;4:23–35. doi: 10.1038/nrc1253. [DOI] [PubMed] [Google Scholar]
- 4.Cleaver JE. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nature reviews. Cancer. 2005;5:564–573. doi: 10.1038/nrc1652. [DOI] [PubMed] [Google Scholar]
- 5.Sundaresan NR, et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nature medicine. 2012;18:1643–1650. doi: 10.1038/nm.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lombard DB, Schwer B, Alt FW, Mostoslavsky R. SIRT6 in DNA repair, metabolism and ageing. Journal of internal medicine. 2008;263:128–141. doi: 10.1111/j.1365-2796.2007.01902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sebastian C, Satterstrom FK, Haigis MC, Mostoslavsky R. From sirtuin biology to human diseases: an update. The Journal of biological chemistry. 2012;287:42444–42452. doi: 10.1074/jbc.R112.402768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tennen RI, Chua KF. Chromatin regulation and genome maintenance by mammalian SIRT6. Trends in biochemical sciences. 2011;36:39–46. doi: 10.1016/j.tibs.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mostoslavsky R, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 10.Kanfi Y, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- 11.Schwer B, et al. Neural sirtuin 6 (Sirt6) ablation attenuates somatic growth and causes obesity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21790–21794. doi: 10.1073/pnas.1016306107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma A, et al. The role of SIRT6 protein in aging and reprogramming of human induced pluripotent stem cells. The Journal of biological chemistry. 2013;288:18439–18447. doi: 10.1074/jbc.M112.405928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baohua Y, Li L. Effects of SIRT6 silencing on collagen metabolism in human dermal fibroblasts. Cell biology international. 2012;36:105–108. doi: 10.1042/CBI20110268. [DOI] [PubMed] [Google Scholar]
- 14.Sebastian C, et al. The Histone Deacetylase SIRT6 Is a Tumor Suppressor that Controls Cancer Metabolism. Cell. 2012;151:1185–1199. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Min LH, et al. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin (vol 14, pg 1203, 2012) Nat Cell Biol. 2013;15:440–440. doi: 10.1038/ncb2726. [DOI] [PubMed] [Google Scholar]
- 16.Marquardt JU, et al. Sirtuin-6-Dependent Genetic and Epigenetic Alterations Are Associated With Poor Clinical Outcome in Hepatocellular Carcinoma Patients. Hepatology. 2013;58:1054–1064. doi: 10.1002/hep.26413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lefort K, et al. A miR-34a-SIRT6 axis in the squamous cell differentiation network. Embo J. 2013;32:2248–2263. doi: 10.1038/emboj.2013.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu YW, et al. Inhibition of SIRT6 in prostate cancer reduces cell viability and increases sensitivity to chemotherapeutics. Protein Cell. 2013;4:702–710. doi: 10.1007/s13238-013-3054-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischer SM, Pavone A, Mikulec C, Langenbach R, Rundhaug JE. Cyclooxygenase-2 expression is critical for chronic UV-induced murine skin carcinogenesis. Mol Carcinogen. 2007;46:363–371. doi: 10.1002/mc.20284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chun KS, Akunda JK, Langenbach R. Cyclooxygenase-2 inhibits UVB-induced apoptosis in mouse skin by activating the prostaglandin E2 receptors, EP2 and EP4. Cancer research. 2007;67:2015–2021. doi: 10.1158/0008-5472.CAN-06-3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buckman S, et al. COX-2 expression is induced by UVB exposure in human skin: implications for the development of skin cancer. Carcinogenesis. 1998;19:723–729. doi: 10.1093/carcin/19.5.723. [DOI] [PubMed] [Google Scholar]
- 22.Tiano HF, et al. Deficiency of either cyclooxygenase (COX)−1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer research. 2002;62:3395–3401. [PubMed] [Google Scholar]
- 23.Rundhaug JE, Mikulec C, Pavone A, Fischer SM. A role for cyclooxygenase-2 in ultraviolet light-induced skin carcinogenesis. Mol Carcinogen. 2007;46:692–698. doi: 10.1002/mc.20329. [DOI] [PubMed] [Google Scholar]
- 24.Jiao J, et al. Cell-type-specific roles for COX-2 in UVB-induced skin cancer. Carcinogenesis. 2014;35:1310–1319. doi: 10.1093/carcin/bgu020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilgus TA, et al. Inhibition of cutaneous ultraviolet light B-mediated inflammation and tumor formation with topical celecoxib treatment. Mol Carcinogen. 2003;38:49–58. doi: 10.1002/mc.10141. [DOI] [PubMed] [Google Scholar]
- 26.Caito S, et al. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. The FASEB Journal. 2010;24:3145–3159. doi: 10.1096/fj.09-151308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao C, et al. SIRT1 confers protection against UVB- and H2O2-induced cell death via modulation of p53 and JNK in cultured skin keratinocytes. Journal of cellular and molecular medicine. 2009;13:3632–3643. doi: 10.1111/j.1582-4934.2008.00453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang CY, Ting WJ, Huang CY, Yang JY, Lin WT. Resveratrol attenuated hydrogen peroxide-induced myocardial apoptosis by autophagic flux. Food & nutrition research. 2016;60:30511. doi: 10.3402/fnr.v60.30511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu W, et al. Effects of resveratrol on H2O2-induced apoptosis and expression of SIRTs in H9c2 cells. Journal of Cellular Biochemistry. 2009;107:741–747. doi: 10.1002/jcb.22169. [DOI] [PubMed] [Google Scholar]
- 30.Zhai M, et al. Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3-dependent regulation of oxidative stress and apoptosis. Journal of Pineal Research. 2017;63:e12419-n/a. doi: 10.1111/jpi.12419. [DOI] [PubMed] [Google Scholar]
- 31.Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Graves DB. Reactive species from cold atmospheric plasma: implications for cancer therapy. Plasma Processes and Polymers. 2014;11:1120–1127. doi: 10.1002/ppap.201400068. [DOI] [Google Scholar]
- 33.Elmets CA, Ledet JJ, Athar M. Cyclooxygenases: Mediators of UV-Induced Skin Cancer and Potential Targets for Prevention. Journal of Investigative Dermatology. 2014;134:2497–2502. doi: 10.1038/jid.2014.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sahu SN, Singh S, Shaw R, Ram VJ, Pratap R. One-pot and step-wise synthesis of thieno [3, 2-c] pyridin-4-ones. RSC Advances. 2016;6:85515–85520. doi: 10.1039/C6RA17315B. [DOI] [Google Scholar]
- 35.Pratap R, Kumar A, Pick R, Hüch V, Ram VJ. Metal-free synthesis of nitrile based partially reduced thia-and oxa-thia [5] helicenes: conformation and dynamics. RSC Advances. 2012;2:1557–1564. doi: 10.1039/C1RA00824B. [DOI] [Google Scholar]
- 36.Sahu SN, et al. Substituent dependent tunable fluorescence in thieno [3, 2-c] pyrans. RSC Advances. 2014;4:56779–56783. doi: 10.1039/C4RA11337C. [DOI] [Google Scholar]
- 37.Sahu SN, et al. One pot synthesis of tetrasubstituted thiophenes:[3 + 2] annulation strategy. RSC Advances. 2015;5:36979–36986. doi: 10.1039/C5RA01290B. [DOI] [Google Scholar]
- 38.Pratap R, Ram VJ. A non-catalytic approach to the synthesis of 5, 6-dihydrobenzo [h] quinolines. Tetrahedron letters. 2007;48:2755–2759. doi: 10.1016/j.tetlet.2007.02.049. [DOI] [Google Scholar]
- 39.Yadav DK, Kalani K, Khan F, Srivastava SK. QSAR and docking based semi-synthesis and in vitro evaluation of 18 beta-glycyrrhetinic acid derivatives against human lung cancer cell line A-549. Med Chem. 2013;9:1073–1084. doi: 10.2174/1573406411309080009. [DOI] [PubMed] [Google Scholar]
- 40.Yadav DK, Khan F. QSAR, Docking and ADMET studies of Camptothecin derivatives as inhibitors of DNA Topoisomerase-I. Journal of Chemometrics. 2013;27:21–33. doi: 10.1002/cem.2488. [DOI] [Google Scholar]
- 41.Yadav DK, et al. Molecular docking and ADME studies of natural compounds of Agarwood oil for topical anti-inflammatory activity. Current computer-aided drug design. 2013;9:360–370. doi: 10.2174/1573409911309030012. [DOI] [PubMed] [Google Scholar]
- 42.Yadav DK, et al. QSAR and docking studies on Chalcone derivatives for anti-tubercular activity against M. tuberculosis H37Rv. Journal of Chemometrics. 2014;28:499–507. doi: 10.1002/cem.2606. [DOI] [Google Scholar]
- 43.Yadav DK, et al. QSAR and Docking Based Semi-Synthesis and In Vivo Evaluation of Artemisinin Derivatives for Antimalarial Activity. Curr Drug Targets. 2014;15:753–761. doi: 10.2174/1389450115666140630102711. [DOI] [PubMed] [Google Scholar]
- 44.Yadav DK, et al. Design, synthesis and in vitro evaluation of 18beta-glycyrrhetinic acid derivatives for anticancer activity against human breast cancer cell line MCF-7. Current medicinal chemistry. 2014;21:1160–1170. doi: 10.2174/09298673113206660330. [DOI] [PubMed] [Google Scholar]
- 45.Dwivedi GR, et al. Drug Resistance Reversal Potential of Ursolic Acid Derivatives against Nalidixic Acid‐and Multidrug‐resistant Escherichia coli. Chemical biology & drug design. 2015;86:272–283. doi: 10.1111/cbdd.12491. [DOI] [PubMed] [Google Scholar]
- 46.Yadav DK, et al. New arylated benzo [h] quinolines induce anti-cancer activity by oxidative stress-mediated DNA damage. Scientific reports. 2016;6:38128. doi: 10.1038/srep38128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pratap, R. et al. In International Conference on Bioinformatics and Biomedical Engineering. 664–673 (Springer).
- 48.Gaur R, et al. Synthesis, antitubercular activity, and molecular modeling studies of analogues of isoliquiritigenin and liquiritigenin, bioactive components from Glycyrrhiza glabra (vol 24, pg 3494, 2015) Med Chem Res. 2015;24:3772–3774. doi: 10.1007/s00044-015-1420-y. [DOI] [Google Scholar]
- 49.Gaur R, et al. Molecular Modeling Based Synthesis and Evaluation of In vitro Anticancer Activity of Indolyl Chalcones. Curr Top Med Chem. 2015;15:1003–1012. doi: 10.2174/1568026615666150317222059. [DOI] [PubMed] [Google Scholar]
- 50.Sociali G, et al. Quinazolinedione SIRT6 inhibitors sensitize cancer cells to chemotherapeutics. Eur J Med Chem. 2015;102:530–539. doi: 10.1016/j.ejmech.2015.08.024. [DOI] [PubMed] [Google Scholar]
- 51.Gupta R, Sridhar D, Rai B. Molecular dynamics simulation study of permeation of molecules through skin lipid bilayer. The Journal of Physical Chemistry B. 2016;120:8987–8996. doi: 10.1021/acs.jpcb.6b05451. [DOI] [PubMed] [Google Scholar]
- 52.Van der Paal J, Neyts EC, Verlackt CC, Bogaerts A. Effect of lipid peroxidation on membrane permeability of cancer and normal cells subjected to oxidative stress. Chemical Science. 2016;7:489–498. doi: 10.1039/C5SC02311D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van der Paal J, Aernouts S, van Duin AC, Neyts EC, Bogaerts A. Interaction of O and OH radicals with a simple model system for lipids in the skin barrier: a reactive molecular dynamics investigation for plasma medicine. Journal of Physics D: Applied Physics. 2013;46:395201. doi: 10.1088/0022-3727/46/39/395201. [DOI] [Google Scholar]
- 54.Xu Y, Shao Y, Voorhees JJ, Fisher GJ. Oxidative inhibition of receptor-type protein-tyrosine phosphatase kappa by ultraviolet irradiation activates epidermal growth factor receptor in human keratinocytes. The Journal of biological chemistry. 2006;281:27389–27397. doi: 10.1074/jbc.M602355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu YR, Voorhees JJ, Fisher GJ. Epidermal growth factor receptor is a critical mediator of ultraviolet B irradiation-induced signal transduction in immortalized human keratinocyte HaCaT cells. Am J Pathol. 2006;169:823–830. doi: 10.2353/ajpath.2006.050449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wan YS, Wang ZQ, Shao Y, Voorhees JJ, Fisher GJ. Ultraviolet irradiation activates PI 3-kinase/AKT survival pathway via EGF receptors in human skin in vivo. Int J Oncol. 2001;18:461–466. doi: 10.3892/ijo.18.3.461. [DOI] [PubMed] [Google Scholar]
- 57.Bachelor MA, Cooper SJ, Sikorski ET, Bowden GT. Inhibition of p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase decreases UVB-induced activator protein-1 and cyclooxygenase-2 in a SKH-1 hairless mouse model. Mol Cancer Res. 2005;3:90–99. doi: 10.1158/1541-7786.MCR-04-0065. [DOI] [PubMed] [Google Scholar]
- 58.Tang Q, et al. Role of cyclic AMP responsive element in the UVB induction of cyclooxygenase-2 transcription in human keratinocytes. Oncogene. 2001;20:5164–5172. doi: 10.1038/sj.onc.1204667. [DOI] [PubMed] [Google Scholar]
- 59.Tang Q, Gonzales M, Inoue H, Bowden GT. Roles of Akt and glycogen synthase kinase 3beta in the ultraviolet B induction of cyclooxygenase-2 transcription in human keratinocytes. Cancer research. 2001;61:4329–4332. [PubMed] [Google Scholar]
- 60.Wen KC, Fan PC, Tsai SY, Shih IC, Chiang HM. Ixora parviflora Protects against UVB-Induced Photoaging by Inhibiting the Expression of MMPs, MAP Kinases, and COX-2 and by Promoting Type I Procollagen Synthesis. Evidence-based complementary and alternative medicine: eCAM. 2012;2012:417346. doi: 10.1155/2012/417346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bickers DR, Athar M. Oxidative stress in the pathogenesis of skin disease. The Journal of investigative dermatology. 2006;126:2565–2575. doi: 10.1038/sj.jid.5700340. [DOI] [PubMed] [Google Scholar]
- 62.Hong MJ, Ko EB, Park SK, Chang MS. Inhibitory effect of Astragalus membranaceus root on matrix metalloproteinase-1 collagenase expression and procollagen destruction in ultraviolet B-irradiated human dermal fibroblasts by suppressing nuclear factor kappa-B activity. The Journal of pharmacy and pharmacology. 2013;65:142–148. doi: 10.1111/j.2042-7158.2012.01570.x. [DOI] [PubMed] [Google Scholar]
- 63.Huang G-J, Huang S-S, Deng J-S. Anti-inflammatory activities of inotilone from Phellinus linteus through the inhibition of MMP-9, NF-κB, and MAPK activation in vitro and in vivo. PloS one. 2012;7:e35922. doi: 10.1371/journal.pone.0035922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Filip G, Postescu D, Tatomir C, Muresan A, Clichici S. Calluna vulgaris extract modulates NF-κB/ERK signaling pathway and matrix metalloproteinase expression in SKH-1 hairless mice skin exposed to ultraviolet B irradiation. J Physiol Pharmacol. 2012;63:423–432. [PubMed] [Google Scholar]
- 65.Lindström TM, Bennett PR. The role of nuclear factor kappa B in human labour. Reproduction. 2005;130:569–581. doi: 10.1530/rep.1.00197. [DOI] [PubMed] [Google Scholar]
- 66.Kuo Y-H, Chen C-W, Chu Y, Lin P, Chiang H-M. In vitro and in vivo studies on protective action of N-phenethyl caffeamide against photodamage of skin. PloS one. 2015;10:e0136777. doi: 10.1371/journal.pone.0136777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yadav, D. K. et al. Studies of the benzopyran class of selective COX-2 inhibitors using 3D-QSAR and molecular docking. Archives of pharmacal research, 10.1007/s12272-017-0945-7 (2017). [DOI] [PubMed]
- 68.Bochevarov AD, et al. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int J Quantum Chem. 2013;113:2110–2142. doi: 10.1002/qua.24481. [DOI] [Google Scholar]
- 69.Timucin AC, Basaga H. SIRT6 is a positive regulator of aldose reductase expression in U937 and HeLa cells under osmotic stress: in vitro and in silico insights. PloS one. 2016;11:e0161494. doi: 10.1371/journal.pone.0161494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Firke SD, Bari SB. Synthesis, biological evaluation and docking study of maleimide derivatives bearing benzenesulfonamide as selective COX-2 inhibitors and anti-inflammatory agents. Bioorgan Med Chem. 2015;23:5273–5281. doi: 10.1016/j.bmc.2015.07.070. [DOI] [PubMed] [Google Scholar]
- 71.Verma, S., Kumar, S. & Kumar, S. Design, synthesis, computational and biological evaluation of new benzodiazepines as CNS agents. Arabian Journal of Chemistry (2017).
- 72.Synthesis, b. e. a. Q. s. et al. Studies of the benzopyran class of selective COX-2 inhibitors using 3D-QSAR and molecular docking. Archives of pharmacal research, 1–12 (2017). [DOI] [PubMed]
- 73.Yadav DK, et al. Molecular docking, QSAR and ADMET studies of withanolide analogs against breast cancer. Drug Des Dev Ther. 2017;11:1859–1870. doi: 10.2147/DDDT.S130601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yadav DK, et al. Development of QSAR model for immunomodulatory activity of natural coumarinolignoids. Drug design, development and therapy. 2010;4:173–186. doi: 10.2147/dddt.s10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yadav DK, Khan F, Negi AS. Pharmacophore modeling, molecular docking, QSAR, and in silico ADMET studies of gallic acid derivatives for immunomodulatory activity. Journal of molecular modeling. 2012;18:2513–2525. doi: 10.1007/s00894-011-1265-3. [DOI] [PubMed] [Google Scholar]
- 76.Faudzi SM, et al. Synthesis, biological evaluation and QSAR studies of diarylpentanoid analogues as potential nitric oxide inhibitors. MedChemComm. 2015;6:1069–1080. doi: 10.1039/C4MD00541D. [DOI] [Google Scholar]
- 77.Singh S, et al. Design, synthesis, biological evaluation and toxicity studies of N, N-disubstituted biguanides as quorum sensing inhibitors. Medicinal Chemistry Research. 2015;24:1974–1987. doi: 10.1007/s00044-014-1255-y. [DOI] [Google Scholar]
- 78.Sachdeva, S., Bhatia, S., Mittal, A. & Sinha, M. Synthesis, Evaluation and in silico studies of 1, 8-Naphthyridine derivatives against antimicrobial activity (2015).
- 79.Dow G, et al. Utility of alkylaminoquinolinyl methanols as new antimalarial drugs. Antimicrobial agents and chemotherapy. 2006;50:4132–4143. doi: 10.1128/AAC.00631-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berendsen HJC, Vanderspoel D, Vandrunen R. Gromacs - a Message-Passing Parallel Molecular-Dynamics Implementation. Comput Phys Commun. 1995;91:43–56. doi: 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- 81.Sousa da Silva AW, Vranken WF. ACPYPE - AnteChamber PYthon Parser interfacE. BMC research notes. 2012;5:367. doi: 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Malde AK, et al. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. Journal of chemical theory and computation. 2011;7:4026–4037. doi: 10.1021/ct200196m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.