

Abstract
Although practiced clinically for more than 40 years, the use of hematopoietic stem cell (HSC) transplantation remains limited by the inability to expand functional HSCs ex vivo. To determine the role of phosphoinositide 3-kinase (PI3K)/AKT signaling in human hematopoietic stem and progenitor cell (HSPC) maintenance, we examined the effect of genetic and pharmacological inhibition of AKT on human umbilical cord blood (UCB) CD34+ cells. We found that knock-down of AKT1 in human UCB CD34+ cells using short interfering RNAs targeting AKT1 enhances their quiescence and colony formation potential in vitro. We treated human UCB CD34+ cells with an AKT-specific inhibitor (AKTi) and performed both in vitro and in vivo stem and progenitor cell assays. We found that ex vivo treatment of human HSPCs maintains CD34 expression and enhances colony formation in serial replating assays. Moreover, pharmacological inhibition of AKT enhances the short-term repopulating potential of human UCB CD34+ cells in immunodeficient mice. Mechanistically, genetic and pharmacological inhibition of AKT activity promotes human HSPC quiescence. These preclinical results suggest a positive role for AKTi during ex vivo culture of human UCB HSPCs.
Several decades of successful bone marrow transplantations have demonstrated the therapeutic importance of hematopoietic stem cells (HSCs) [1–4]. The use of noninvasively accessible umbilical cord blood (UCB)-derived HSCs provides many advantages over bone marrow cells, including enhanced long-term immune recovery and decreased graft versus host disease [1–4]. However, low cell numbers in single UCB units have limited the suitability of UCB transplantation for adult patients [1–4]. Methods to increase robustly the number of cells that give a rapid and sustained blood count recovery would enable the use of UCB in more patients [1–4].
Culture conditions optimized for HSC expansion (serum-free medium supplemented with cytokines, including thrombopoietin [TPO], stem cell factor [SCF], flt3 ligand, and interleukin [IL]-6) result in robust proliferation accompanied by differentiation, leading to loss of HSC activity [5]. Recent advances have allowed the ex vivo expansion of hematopoietic stem and progenitor cells (HSPCs) using cytokine mixtures combined with an array of factors, including aryl hydrocarbon receptor antagonists, Wnt activators, Notch ligands, angiopoietin-like proteins, prostaglandin E2, pleiotrophin, or glycogen synthase kinase 3 inhibitors, in combination with insulin [6–12]. These approaches are encouraging, but all require supplementation with a mixture of hematopoietic cytokines, which may promote lineage commitment at the cost of long-term HSC maintenance [9–12]. Therefore, the identification of molecules or factors that expand HSCs during ex vivo culture has remained an important goal [6–8].
Deciphering the molecular mechanisms controlling HSC self-renewal is essential for developing clinical strategies that can enhance ex vivo HSC expansion [13,14]. HSC self-renewal requires a complex cross-talk between extrinsic signals from the microenvironment and the cell-intrinsic regulators of HSCs to maintain an undifferentiated state [15,16]. The phosphoinositide 3-kinase (PI3K)/phosphatase and tensin homolog (PTEN)/AKT signaling pathway has been implicated in regulating mouse HSC self-renewal [17–19]. Although Pten deletion, which results in AKT activation, initially leads to a transient expansion of HSCs, the HSC pool is depleted over time [18]. In addition, overexpression of constitutively active AKT also exhausts HSCs [19]. The polycomb group protein Bmi1 plays an important role in regulating HSC self-renewal [20] and we found that AKT-mediated phosphorylation of Bmi1 inhibits HSC self-renewal [21], suggesting that activation of PI3K/AKT signaling impairs mouse HSC maintenance.
The role of the PI3K/PTEN/AKT signaling pathway in human HSCs is controversial [22,23]. Although transient silencing of PTEN in human CD34+ cells enhances their proliferation potential and short-term repopulation capability [22], ex vivo rapamycin treatment of human UCB CD34+ cells, which inhibits mTOR activity, enhances their engraftment of immunodeficient mice in serial transplantation assays [23]. Given that rapamycin can induce feedback activation of AKT signaling through an insulin-like growth factor-1 receptor-dependent mechanism [24], there is a critical need to use specific inhibitors to modulate PI3K/AKT signaling in human HSPCs.
There are three AKT isoforms in mammalian cells: AKT1, AKT2, and AKT3. AKT1 and AKT2 are expressed ubiquitously and in greater abundance in hematopoietic cells, whereas AKT3 expression is most pronounced in the testes and brain, but also can be expressed in lesser amounts in the hematopoietic system [17,25]. In this study, we found that knock-down of AKT1 in human UCB CD34+ cells using small interfering RNAs (siRNAs) targeting AKT1 enhances their quiescence and colony formation potential in vitro. Importantly, we discovered that pharmacological inhibition of AKT activity with an AKT-specific inhibitor (AKTi) in human UCB CD34+ cells promotes their quiescence and enhances their engraftment in immunodeficient mice. Our studies may facilitate the development of innovative clinical strategies that can enhance the engraftment of human UCB HSPCs.
Methods
Mice
For the repopulation assay, 6- to 8-week-old NSG (NOD.Cg-Prkdscid IL2rgtm1Wj1/Sz) mice were obtained from an on-site core breeding colony. The institutional animal care and use committee of the Indiana University School of Medicine approved all experimental procedures.
Western blot analysis
Cells were washed twice with ice-cold phosphate-buffered saline and lysed on ice in radioimmunoprecipitation assay buffer (Sigma-Aldrich) supplemented with protease inhibitor (Roche) and phosphatase inhibitor (Roche). Cell lysates were sonicated and centrifuged at 13,200 rpm for 10 min; boiled with lithium dodecyl sulfate sample buffer (ThermoFisher Scientific); separated by NuPAGE gel (ThermoFisher Scientific); transferred electrophoretically to a polyvinylidene fluoride membrane (EMD Millipore); and immunoblotted with phospho-AKT (Ser473), phospho-AKT (Thr308), AKT, and GAPDH (Cell Signaling Technology), followed by incubation with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology). The blots were developed using the enhanced chemiluminescence technique with SuperSignal West Femto Maximum Sensitivity Chemiluminescent Substrate (Fisher Scientific).
Flow cytometry analysis
Human HSPCs were identified and evaluated by flow cytometry using a single-cell suspension stained with antibodies against human CD34-fluorescein isothiocyanate (FITC), human CD34-phycoerythrin (PE), human CD3-FITC (BD Biosciences), GlyA (human CD235a)-PE (eBioscience), human CD71-allophycocyanin (APC), human CD34-PE/CY7, human CD3-APC/CY7, human CD45-APC, human CD19-PerCP-Cy5.5, human CD14-PE/CY7, and human CD33-FITC (BioLegend). Experiments were performed on Accuri C6 and FACSLSR II cytometers (BD Biosciences) and analyzed with FlowJo version 9.3.3 software (TreeStar).
Purification of CD34+ human UCB cells
Normal human UCB samples were collected with institutional approval. Mononuclear cells (MNCs) were isolated by Ficoll-Paque Plus density gradient centrifugation (1.077 g/mL, GE Healthcare Life Sciences). CD34+ cells were purified from MNCs by positive selection using the human CD34 MicroBead Kit and LS separation columns (Miltenyi Biotec) according to the manufacturer’s instructions.
Cell culture and expansion
A total of 40,000 purified CD34+ cells were expanded in Stem-Span Serum-Free Expansion Medium (STEMCELL Technologies) supplemented with recombinant human (rh) SCF (100 ng/mL), rhTPO (100 ng/mL), and rh-Flt-3L (100 ng/mL). To differentiate human HSPCs, purified CD34+ cells were cultured in myeloid-promoting conditions (SCF, 100 ng/mL; FLT-3 ligands, 10 ng/mL; IL-3, 20 ng/mL; IL-6, 20 ng/mL; granulocyte-macrophage colony-stimulating factor, 20 ng/mL; and granulocyte colony-stimulating factor, 20 ng/mL) and in erythroid-promoting conditions (EPO, 6 IU/mL; SCF, 100 ng/mL). Cytokines were from PeproTech. AKTi was from Merck [26].
Hoechst 33342/pyronin Y staining
Purified CD34+ cells were stained with 2.5 μmol/L Hoechst 33342 (Molecular Probes) for 45 minutes at 37°C. Pyronin Y was added at a final concentration of 1 μmol/L and cells were incubated for a further 45 minutes at 37°C. Thereafter, cells were kept on ice for 30 minutes. Cells were washed twice and stained with APC-conjugated antihuman CD34 antibody [27].
Colony-forming cell (CFC) assay
Hematopoietic clonogenic progenitors were determined in methyl-cellulose medium (MethoCult H4435, StemCell Technologies) using 1,000 cultured or 250 sorted CD34+ cells per well (six-well plate). Colonies were scored 14 days after plating. For secondary CFC assays, cells were harvested and washed with phosphate-buffered saline on day 14 after the first plating and 1 × 104 cells were replated as described above. Colonies were scored at day 14.
NSG mouse transplantation assay
NSG mice were from the Indiana University In Vivo Therapeutic Core Facility. At the age of 8 weeks, mice were irradiated with a single dose of 2.5 Gy and then 200,000 human CD34+ cells were injected into irradiated NSG mice via their tail vein. Peripheral blood of NSG mice was obtained every 4 weeks after transplantation, stained with anti-human CD45 FITC and anti-mouse CD45 PE antibodies, and then analyzed by flow cytometry. After 12 weeks, bone marrow cells were harvested and analyzed for human CD45 expression to define the engraftment efficiency.
Transfection of siRNA into human CD34+ cells
Accell nontargeting pool control siRNA (control siRNAs) and Accell human AKT1 smart pool siRNA (AKT1 siRNAs) were obtained from GE Dharmacon. A total of 20,000 human UCB CD34+ cells were suspended in Accell Delivery Medium (Dharmacon) with cytokines and seeded in 96-well plate. siRNAs were added in a final concentration of 1 μmol/L according to the manufacturer’s instructions (Dharmacon). Cells were transferred to CD34+ basic medium 48 hours after transfection [28].
Senescence analysis of human CD34+ cells
Sorted CD34+ cells were plated in eight-well chamber slides. Senescent cells were detected with the Senescence β-Galactosidase Staining Kit (Cell Signaling Technology) according to the manufacturer’s instructions [29].
Statistical analysis
Data were analyzed statistically using either the Student t test or one-way ANOVA (*p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant).
Results
Transient knock-down of AKT1 increases human HSPC quiescence
To determine whether knock-down of AKT1 expression alters the behavior of HSPCs, we transiently transfected either control or AKT1 siRNAs into purified CD34+ cells. AKT1 mRNA expression was decreased by 50% in AKT1 siRNA-transfected cells at 48 hours compared with control siRNA-transfected cells (Fig. 1A). To determine how transient knock-down of AKT1 affects the primitiveness of human HSPCs, we examined the proliferative capacity of CD34+ cells in early-acting cytokine (rhSCF, rhTPO, and rh-Flt-3L)-containing liquid cultures after transfection of control and AKT1 siRNAs. We quantified CD34 expression by fluorescence-activated cell sorting (FACS) over time and found that AKT1 deficiency altered neither the frequency nor the absolute number of CD34+ cells (Fig. 1B and 1C). We then examined the effect of AKT1 deficiency on HSPC quiescence using pyronin Y and Hoechst 33342 staining and found that transient knock-down of AKT1 was sufficient to increase human HSPC quiescence (Fig. 1D and 1E). To determine whether genetic inhibition of AKT affects CD34+ cell survival and senescence, we assessed the apoptosis and senescence of CD34+ cells 7 days after transfection. We observed a comparable level of apoptotic [Annexin V+/7-amino-actinomycin D-negative (7-AAD−)] cells in both control siRNA- and AKT1 siRNA-transfected CD34+ cells (Fig. 1F). We found that knock-down of AKT1 did not induce senescence in CD34+ cells (data not shown). These findings suggest that transient knock-down of AKT1 induces neither apoptosis nor senescence in HSPCs. To examine the effect of knocking down AKT1 on HSPC function, we performed CFC assays. When we used 250 sorted CD34+ cells 7 days after transfection, we observed that AKT1 siRNA-transfected CD34+ cells formed more colonies compared with control siRNA-transfected CD34+ cells (Fig. 1G). Therefore, transient knock-down of AKT1 increases human HSPC quiescence and enhances their colony formation potential.
Figure 1.

Transient knock-down of AKT1 is sufficient to increase human HSPC quiescence. (A) AKT1 expression levels were measured by quantitative real-time polymerase chain reaction analysis 48 hours after siRNA transfection and normalized to GAPDH (***p < 0.001, n = 3). (B) Percentage of human CD34+ cells in expanding medium measured by flow cytometric analysis 7 days after siRNA transfection (p > 0.05, nonsignificant [N.S.], n = 4). (C) Absolute number of human CD34+ cells in expanding medium measured 7 days after siRNA transfection (N.S., n = 4). (D) Flow cytometry analysis of G0 cells using cellular RNA staining (with pyronin Y) and DNA staining (with Hoechst 33342). Percentage of CD34+ cells in the G0 phase of the cell cycle 7 days after siRNA transfection is shown. (E) Quiescence analysis of CD34+ cells 7 days after siRNA transfection (*p < 0.05, n = 4). (F) Frequency of apoptotic CD34+ cells (Annexin V+/7-AAD−) measured 7 days after siRNA transfection (p > 0.05, n = 4). (G) Transient knock-down of AKT1 enhances colony formation of human CD34+ cells. CFC assays were performed using 250 sorted CD34+ cells 7 days after siRNA transfection (***p < 0.001, n = 3).
Pharmacological inhibition of AKT enhances the colony-forming capability of human HSPCs
We have shown previously that AKTi can inhibit AKT activation efficiently in both mouse and human HSPCs [21,30]. We found that treatment of CD34+ cells with AKTi (1 μmol/L) decreased AKT phosphorylation significantly at both serine 473 and threonine 308 (Fig. 2A), demonstrating that AKT activity was inhibited by AKTi treatment. To determine how AKTi affects the primitiveness of human HSPCs, we examined the proliferative capacity of CD34+ cells in early-acting cytokine (rhSCF, rhTPO, and rh-Flt-3L)-containing liquid cultures in the presence of AKTi (1 μmol/L). We quantified CD34 expression by FACS over time and found that a significantly greater proportion of AKTi-treated CD34+ cells retained CD34 expression (Fig. 2B and 2C). However, AKTi treatment did not increase the absolute number of CD34+ cells over time (Fig. 2D). We also examined the effect of AKT inhibition on primitive human HSCs (CD34+CD38−CD45RA−CD90+) and found that AKTi treatment did not increase the number of primitive HSCs (data not shown). Therefore, AKTi promotes the retention of CD34 expression in hematopoietic progenitor cells after ex vivo culture. We then performed CFC assays using 1,000 cells from dimethylsulfoxide (DMSO)- or AKTi-treated cultures at day 7 and found that AKTi-treated cells growing in liquid culture generated more colony-forming units (CFUs) than DMSO-treated cells during both primary and secondary CFC assays (Fig. 2E and 2F). When we used 250 sorted CD34+ cells 7 days after DMSO or AKTi (0.2 and 0.5 μmol/L, respectively) treatment in the CFC assays, we observed that AKTi-treated CD34+ cells formed more colonies compared with DMSO-treated CD34+ cells (Fig. 2G). Therefore, pharmacologic inhibition of AKT retains human HSPC primitiveness, leading to a greater capacity to generate CFUs.
Figure 2.

Pharmacological inhibition of AKT maintains human HSPCs and enhances their colony-forming capability. (A) Inhibiting AKT activity in CD34+ cells using an AKTi decreased AKT phosphorylation at both serine 473 and threonine 308. Western blot analysis of AKT phosphorylation in CD34+ cells 24 hours after DMSO or AKTi (1 μmol/L) treatment is shown. (B) Flow cytometric analysis of human CD34 expression 7 days after AKTi (1 μmol/L) or DMSO treatment. (C) Inhibition of AKT maintains the immature surface marker of human HSPCs in vitro. The percentage of human CD34+ cells in expanding medium supplemented with vehicle (DMSO) or AKTi (1 μmol/L) is shown (***p < 0.001, n = 3). (D) Absolute number of human CD34+ cells in expanding medium was measured 7 days after AKTi (1 μmol/L) or DMSO treatment (p > 0.05, n = 3). (E) Inhibition of AKT activity enhances colony formation of human CD34+ cells. CFC assays were performed using equal numbers of cultured cells treated with DMSO (0.01%) or AKTi (0.1 and 1 μmol/L) for 7 days (*p < 0.05, **p < 0.01, ***p < 0.001, n = 3). (F) The 10,000 cells collected from primary colonies used in the secondary CFC assays (*p < 0.05, ***p < 0.001, n = 3). (G) CFC assays performed using 250 sorted CD34+ cells 7 days after DMSO (0.01%) or AKTi (0.2 and 0.5 μmol/L) treatment (**p < 0.01, ***p < 0.001, n = 3).
Inhibition of AKT activity enhances human HSPC quiescence
Quiescence is an important feature of human HSPCs [31–34]. Given that knocking down AKT1 expression in human CD34+ cells enhances their quiescence (Fig. 1D and 1E), we speculated that pharmacological inhibition of AKT activity would promote human HSPC quiescence. To test this, we cultured UCB CD34+ cells in the presence or the absence of AKTi (0.1 or 1 μmol/L) for 7 days and then examined the frequency of quiescent CD34+ cells using pyronin Y and Hoechst 33342 staining. We found that both low (0.1 μmol/L) and high (1 μmol/L) concentrations of AKTi increased the frequency of quiescent CD34+ cells significantly compared with DMSO treatment (Fig. 3A and 3B), indicating that pharmacological inhibition of AKT activity promotes human HSPC quiescence. To determine whether inhibiting AKT activity affects CD34+ cell survival, we assessed the apoptosis of CD34+ cells 2 days after AKTi treatment and observed comparable level of apoptotic cells (Annexin V+/7-AAD−) in both DMSO- (0.01%) and AKTi (1 μmol/L)-treated CD34+ cells (Fig. 3C). We observed no increase in apoptotic CD34+ cells (Annexin V+/7-AAD−) 7 days after AKTi (0.1 or 1 μmol/L) treatment (Fig. 3D), suggesting that pharmacological inhibition of AKT activity does not induce apoptosis in HSPCs. To determine the effect of AKTi treatment on HSPC senescence, we performed senescence analysis using SA-β-gal staining [29] and found that neither DMSO nor AKTi treatment induced senescence in human CD34+ cells (Fig. 3E). We used late-passage Bmi1−/− mouse embryonic fibroblasts, which undergo senescence, as positive controls [35]. Therefore, AKT inhibition retains human HSPC primitiveness, at least in part through promoting quiescence.
Figure 3.

Inhibition of AKT activity increases human CD34+ cell quiescence. (A) Inhibiting AKT activity enhanced HSPC quiescence as measured by pyronin Y and Hoechst staining. Flow cytometric analysis of the G0 population of CD34+ cells 7 days after AKTi (0.1 and 1 μmol/L) or DMSO treatment using cellular RNA staining (with pyronin Y) and DNA staining (with Hoechst 33342). Percentage of CD34+ cells in the G0 phase of the cell cycle after AKTi treatment is shown. (B) Quiescence analysis of CD34+ cells 7 days after AKTi (0.1 and 1 μmol/L) or DMSO treatment (*p < 0.05, **p < 0.01, n = 3). (C) Flow cytometric analysis of apoptotic CD34+ cells 2 days after AKTi (1 μmol/L) or DMSO treatment using Annexin V and 7-AAD staining. Left lower gate, right lower gate, and right upper gate represent live cells, early apoptotic cells, and late apoptotic cells, respectively. (D) Frequency of apoptotic CD34+ cells (Annexin V+/7-AAD−) measured 7 days after DMSO or AKTi (0.1 or 1 μmol/L) treatment (p > 0.05, n = 3). (E) Senescence analysis of CD34+ cells using SA-β-gal staining after DMSO or AKTi treatment. Senescent Bmi1−/− mouse embryonic fibroblasts (MEFs) were used as positive controls.
Inhibition of AKT activity restricts myeloid and erythroid differentiation of human HSPCs
To investigate whether AKT activity is important for the differentiation of human HSPCs, we first placed purified CD34+ cells in myeloid-promoting medium along with DMSO (0.01%) or AKTi (1 μmol/L). The expression of the human myeloid marker CD11b was assessed by flow cytometry at day 9. The percentage of mature myeloid cells (CD11b+ cells) was significantly lower in the AKTi-treated group compared with DMSO-treated cells (Fig. 4A), revealing that myeloid differentiation of human CD34+ cells may be delayed upon AKT inhibition. We then analyzed the effect of AKT inhibition on erythroid differentiation by culturing purified CD34+ cells in erythroid-promoting medium along with DMSO (0.01%) or AKTi (1 μmol/L). The expression of human CD71 and GlyA was assessed by flow cytometry at day 5. The generation of mature erythroid precursor cells (CD71+/GlyA+ cells) was decreased significantly after AKTi treatment compared with DMSO-treated cells (Fig. 4B and 4C). These findings demonstrated that inhibition of AKT activity delays human HSPC differentiation toward the myeloid and erythroid lineages.
Figure 4.
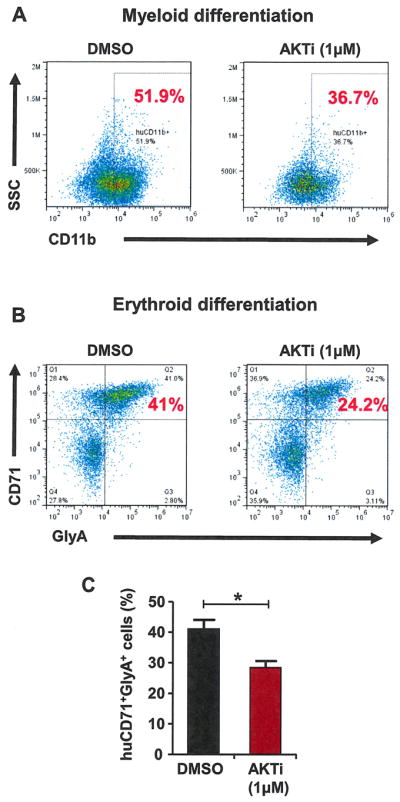
Inhibition of AKT activity decreases myeloid and erythroid differentiation of human HSPCs. (A) Inhibition of AKT activity delays myeloid differentiation of CD34+ cells. Purified CD34+ cells were cultured in myeloid-promoting medium containing DMSO (0.01%) or AKTi (1 μmol/L). The percentage of myeloid cells (CD11b+) was determined at day 9. (B) Purified CD34+ cells cultured in erythroid-promoting medium containing DMSO (0.01%) or AKTi (1 μmol/L). The percentage of mature erythroid cells (CD71+/GlyA+) was determined at day 5. (C) Inhibition of AKT activity in CD34+ cells deceases the generation of mature erythroid cells (*p < 0.05, n = 3).
Pharmacological inhibition of AKT enhances the repopulating capability of human UCB CD34+ cells
Quiescent HSCs exhibit a significantly higher engraftment potential than proliferating HSCs in transplantation assays [32–34]. Therefore, we sought to determine whether the enhanced quiescence seen in AKTi-treated CD34+ cells would improve their engraftability in immunodeficient mice. Whereas 1 μM AKTi treatment enhanced the colony formation potential of CD34+ cells (Fig. 2E–2G), inhibiting AKT activity decreased myeloid and erythroid differentiation (Fig. 4). Further, we found that CD34− cells showed increased apoptosis after 1 μmol/L AKTi treatment (data not shown). To avoid the potential negative impact of high concentrations of AKTi on transplantation outcome, we decided to use a low concentration of AKTi (0.1 μmol/L), which enhanced the colony formation potential as well as the quiescence of CD34+ cells (Figs. 2F and 3B), in transplantation experiments. An equal number of human CD34+ cells were treated with DMSO or AKTi (0.1 μmol/L) in vitro. Seven days later, we harvested cells and transplanted 200,000 cultured cells into sublethally irradiated NOD/SCID IL-2Rγ null mice. The engraftment of human cells in mice was analyzed every 4 weeks by monitoring human CD45 expression in peripheral blood harvested from the recipient mice. As shown in Figure 5A, we observed significantly increased engraftment in the AKTi-treated group compared with the control group at both 4 and 8 weeks after transplantation. Twelve weeks after transplantation, we examined human CD45 expression in the cells isolated from mouse bone marrow and observed a significant enhancement of AKTi-treated cell engraftment compared with DMSO-treated CD34+ cells (Fig. 5B). Next, we analyzed the multilineage reconstitution potential of AKTi-treated CD34+ cells in the bone marrow of recipient mice. Both the control group and the AKTi-treated group were capable of undergoing myeloid and lymphoid repopulation (Fig. 5C–5E). Whereas AKTi-treated CD34+ cells showed enhanced B-lineage reconstitution compared with the control group, inhibition of AKT activity decreased T-cell and myeloid differentiation of CD34+ cells (Fig. 5C–5E), which is consistent with the in vitro liquid culture experiment shown in Figure 4A and 4B. Therefore, AKT appears to restrict the self-renewal capacity of human HSPCs by promoting their cell-cycle entry and terminal differentiation and pharmacological inhibition of AKT activity leads to enhanced quiescence and better engraftability of human CD34+ cells.
Figure 5.

Pharmacological inhibition of AKT enhances the repopulating capability of human UCB CD34+ cells. (A) Percentage of human CD45+ cells in the peripheral blood of recipient mice 4 and 8 weeks after transplantation (*p < 0.05, **p < 0.01, n = 5). (B) Frequency of human CD45+ cells in the bone marrow of recipient mice 12 weeks after transplantation (**p < 0.01, n = 4). (C) Flow cytometric analysis of human myeloid cells (CD33+/CD14+) in the bone marrow of recipient mice 12 weeks after transplantation. (D) Flow cytometric analysis of human lymphoid cells (CD3+ or CD19+) in the bone marrow of recipient mice 12 weeks after transplantation. (E) Frequency of donor-derived myeloid cells, T cells, and B cells in the bone marrow of recipient mice 12 weeks after transplantation (**p < 0.01, n = 5).
Discussion
Clinical hematopoietic transplantation outcomes are strongly correlated with the numbers of cells infused, so low cell numbers in single UCB units have limited the suitability of UCB transplantation for adult patients [1–3]. The ability to increase the number of functional human HSCs in vitro or in vivo could provide improved treatment options for clinical transplantation and gene therapy protocols; however, clinically relevant expansion of HSCs has been difficult to achieve [1–4]. In this study, we report that pharmacological inhibition of AKT activity in human UCB CD34+ cells enhances their ability to engraft immunodeficient mice through promoting HSPC quiescence. In addition, our work uncovered an important role of PI3K/AKT signaling in regulating human HSPC maintenance and lineage commitments.
PI3K/PTEN/AKT signaling regulates murine HSC function because the constitutive activation of AKT depletes the HSC pool [17–19]. In addition, we have demonstrated that AKT-mediated phosphorylation of Bmi1 inhibits HSC self-renewal [21]. Based upon these observations, we hypothesized that suppressing PI3K/AKT signaling in human HSPCs promotes their quiescence and enhances their self-renewal capability. Using a serum-free culture system containing early-acting cytokines, we showed that pharmacological inhibition of AKT activity in UCB-derived CD34+ cells ex vivo improves human HSPC engraftment in NSG mice. Furthermore, inhibition of AKT activity promotes human HSPC quiescence. Transient knock-down of PTEN in human CD34+ cells enhances their proliferation, promoting quiescent HSPCs to enter the cell cycle [22]. Therefore, activation of PI3K/AKT signaling promotes human HSPC proliferation. Given that HSC quiescence is correlated with its self-renewal capability [32–34], the enhanced engraftability of human HSPCs treated with AKT inhibitor is at least in part due to their enhanced quiescence.
In addition to HSC maintenance, PTEN also regulates terminal differentiation [18]. Loss of PTEN promotes both myeloid and T-cell differentiation, but impairs B-cell production, in mice [18]. We found that pharmacological inhibition of AKT activity in human CD34+ cells results in decreased myeloid and T-cell differentiation in vivo. AKTi treatment also decreases erythroid differentiation of human HSPCs. Therefore, the PI3K/AKT signaling pathway promotes differentiation of human HSPCs.
Cancer patients that receive chemotherapy normally develop neutropenia, increasing the risk of infections and resulting in treatment delay [36,37]. Neutrophils are essential for the innate immune response and can protect patients from bacterial infections in the initial phase of transplantation, especially for patients undergoing myelosuppressive chemotherapy [38,39]. Therefore, the capability to generate matured blood cells is also critical for stem cell transplantation [11,40]. Although we observed increased engraftment of CD34+ cells treated with AKTi, the myeloid differentiation capacity of human HSPCs was decreased (Fig. 6). To compensate for the limitation of AKTi in human HSPC expansion, other factors, including Wnt activator [38,41,42], should be used in combination with AKTi. We expect that the combination of AKTi and the activator of canonical Wnt signaling will promote the ex vivo expansion of functional HSPCs without affecting their differentiation capability (Fig. 6).
Figure 6.

Working model. Pharmacological inhibition of AKT activity in human UCB CD34+ cells enhances their quiescence in vitro and promotes their engraftment in immunodeficient mice in vivo; however, the myeloid differentiation of human HSPCs was delayed. We predict that the combination of AKTi and other factors, including the activator of canonical Wnt signaling, will enhance HSPC quiescence and promote the ex vivo expansion of functional HSPCs without affecting their differentiation capability.
In summary, our work has identified an important role of PI3K/AKT signaling in regulating human HSPC maintenance. Our data suggest that modulating AKT activity in human HSPCs may be useful in enhancing their engraftability. Our findings may have a positive impact on the field of stem cell transplantation and provide novel clinical strategies that can enhance human HSPC transplantation outcome.
Acknowledgments
The authors thank the In Vivo Therapeutics Core of the Indiana University Simon Cancer Center, the nursing staff and Dr. Arthur Baluyut at the St. Vincent Hospital (Indianapolis, IN) for providing UCB samples for this study, Dr. Hal Broxmeyer at Indiana University for providing human UCB CD34+ cells and helpful advice, and Mrs. Marilyn Wales for helping in the preparation of the manuscript.
This work was supported in part by the Department of Defense (grant W81XWH-13-1-0187 to Y.L.), the St. Baldrick’s Foundation (scholar award to Y.L.), the Elsa Pardee Foundation (Y.L.), the Leukemia Research Foundation (Y.L.), the Children’s Leukemia Research Association (Y.L.), the Showalter Trust Fund (Y.L.), Alex’s Lemonade Stand Foundation (research grant to Y.L.), the American Cancer Society (institutional research grant to Y.L.), and the Indiana University Simon Cancer Center (pilot project grant to Y.L.).
Footnotes
Conflict of interest disclosure
The authors declare no competing financial interests.
References
- 1.Walasek MA, van Os R, de Haan G. Hematopoietic stem cell expansion: challenges and opportunities. Ann N Y Acad Sci. 2012;1266:138–150. doi: 10.1111/j.1749-6632.2012.06549.x. [DOI] [PubMed] [Google Scholar]
- 2.Gluckman E. History of cord blood transplantation. Bone Marrow Transplant. 2009;44:621–626. doi: 10.1038/bmt.2009.280. [DOI] [PubMed] [Google Scholar]
- 3.Wagner JE, Gluckman E. Umbilical cord blood transplantation: the first 20 years. Semin Hematol. 2010;247:3–12. doi: 10.1053/j.seminhematol.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Hofmeister CC, Zhang J, Knight KL, Le P, Stiff PJ. Ex vivo expansion of umbilical cord blood stem cells for transplantation: growing knowledge from the hematopoietic niche. Bone Marrow Transplant. 2007;39:11–23. doi: 10.1038/sj.bmt.1705538. [DOI] [PubMed] [Google Scholar]
- 5.Csaszar E, Kirouac DC, Yu M, et al. Rapid expansion of human hematopoietic stem cells by automated control of inhibitory feedback signaling. Cell Stem Cell. 2012;10:218–229. doi: 10.1016/j.stem.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Dahlberg A, Delaney C, Bernstein ID. Ex vivo expansion of human hematopoietic stem and progenitor cells. Blood. 2011;117:6083–6090. doi: 10.1182/blood-2011-01-283606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broxmeyer HE. Enhancing engraftment of cord blood cells via insight into the biology of stem/progenitor cell function. Ann N Y Acad Sci. 2012;1266:151–160. doi: 10.1111/j.1749-6632.2012.06509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norkin M, Lazarus HM, Wingard JR. Umbilical cord blood graft enhancement strategies: has the time come to move these into the clinic? Bone Marrow Transplant. 2013;48:884–889. doi: 10.1038/bmt.2012.163. [DOI] [PubMed] [Google Scholar]
- 9.Boitano AE, Wang J, Romeo R, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329:1345–1348. doi: 10.1126/science.1191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Himburg HA, Muramoto GG, Daher P, et al. Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med. 2010;16:475–482. doi: 10.1038/nm.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delaney C, Heimfeld S, Brashem-Stein C, Voorhies H, Manger RL, Bernstein ID. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat Med. 2010;16:232–236. doi: 10.1038/nm.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lechman ER, Gentner B, van Galen P, et al. Attenuation of miR-126 activity expands HSC in vivo without exhaustion. Cell Stem Cell. 2012;11:799–811. doi: 10.1016/j.stem.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Attar EC, Scadden DT. Regulation of hematopoietic stem cell growth. Leukemia. 2004;18:1760–1768. doi: 10.1038/sj.leu.2403515. [DOI] [PubMed] [Google Scholar]
- 14.Akala OO, Clarke MF. Hematopoietic stem cell self-renewal. Curr Opin Genet Dev. 2006;16:496–501. doi: 10.1016/j.gde.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 15.Zon LI. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature. 2008;453:306–313. doi: 10.1038/nature07038. [DOI] [PubMed] [Google Scholar]
- 16.Wang LD, Wagers AJ. Dynamic niches in the origination and differentiation of haematopoietic stem cells. Nat Rev Mol Cell Biol. 2011;12:643–655. doi: 10.1038/nrm3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Grindley JC, Yin T, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 19.Kharas MG, Okabe R, Ganis JJ, et al. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood. 2010;115:1406–1415. doi: 10.1182/blood-2009-06-229443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park IK, Qian D, Kiel M, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Liu F, Yu H, et al. Akt phosphorylates the transcriptional repressor Bmi1 to block its association with tumor suppressing Ink4a-Arf locus. Sci Signal. 2012;5:ra77. doi: 10.1126/scisignal.2003199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim I, Kim YJ, Métais JY, Dunbar CE, Larochelle A. Transient silencing of PTEN in human CD34(+) cells enhances their proliferative potential and ability to engraft immunodeficient mice. Exp Hematol. 2012;40:84–91. doi: 10.1016/j.exphem.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rohrabaugh SL, Campbell TB, Hangoc G, Broxmeyer HE. Ex vivo rapamycin treatment of human cord blood CD34+ cells enhances their engraftment of NSG mice. Blood Cells Mol Dis. 2011;46:318–320. doi: 10.1016/j.bcmd.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juntilla MM, Patil VD, Calamito M, Joshi RP, Birnbaum MJ, Koretzky GA. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood. 2010;115:4030–4038. doi: 10.1182/blood-2009-09-241000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.She QB, Chandarlapaty S, Ye Q, et al. Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS One. 2008;3:e3065. doi: 10.1371/journal.pone.0003065. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Miyata Y, Liu Y, Jankovic V, et al. Cyclin C regulates human hematopoietic stem/progenitor cell quiescence. Stem Cells. 2010;28:308–317. doi: 10.1002/stem.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonifazi P, D’Angelo C, Zagarella S, et al. Intranasally delivered siRNA targeting PI3K/Akt/mTOR inflammatory pathways protects from aspergillosis. Mucosal Immunol. 2010;3:193–205. doi: 10.1038/mi.2009.130. [DOI] [PubMed] [Google Scholar]
- 29.Smith LL, Yeung J, Zeisig BB, et al. Functional crosstalk between Bmi1 and MLL/Hoxa9 axis in establishment of normal hematopoietic and leukemic stem cells. Cell Stem Cell. 2011;8:649–662. doi: 10.1016/j.stem.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 30.Vu LP, Perna F, Wang L, et al. PRMT4 blocks myeloid differentiation by assembling a methyl-RUNX1-dependent repressor complex. Cell Rep. 2013;5:1625–1638. doi: 10.1016/j.celrep.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10:120–136. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 32.Jetmore A, Plett PA, Tong X, et al. Homing efficiency, cell cycle kinetics, and survival of quiescent and cycling human CD34(+) cells transplanted into conditioned NOD/SCID recipients. Blood. 2002;99:1585–1593. doi: 10.1182/blood.v99.5.1585. [DOI] [PubMed] [Google Scholar]
- 33.Srour EF, Tong X, Sung KW, et al. Modulation of in vitro proliferation kinetics and primitive hematopoietic potential of individual human CD34+CD38−/lo cells in G0. Blood. 2005;105:3109–3116. doi: 10.1182/blood-2004-05-1773. [DOI] [PubMed] [Google Scholar]
- 34.Broxmeyer HE, Orschell CM, Clapp DW, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307–1318. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 36.Yang BB, Savin MA, Green M. Prevention of chemotherapy-induced neutropenia with pegfilgrastim: pharmacokinetics and patient outcomes. Chemotherapy. 2012;58:387–398. doi: 10.1159/000345626. [DOI] [PubMed] [Google Scholar]
- 37.Mehdi I, Al Bahrani B. Chemotherapy-induced neutropenic necrotizing enterocolitis: a review. J Pak Med Assoc. 2012;62:718–723. [PubMed] [Google Scholar]
- 38.Chotinantakul K, Prasajak P, Leeanansaksiri W. Wnt1 accelerates an ex vivo expansion of human cord blood CD34+CD38− cells. Stem Cells Int. 2013;2013:909812. doi: 10.1155/2013/909812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McNiece I, Jones R, Bearman SI, et al. Ex vivo expanded peripheral blood progenitor cells provide rapid neutrophil recovery after high-dose chemotherapy in patients with breast cancer. Blood. 2000;96:3001–3007. [PubMed] [Google Scholar]
- 40.Shpall EJ, Quinones R, Giller R, et al. Transplantation of ex vivo expanded cord blood. Biol Blood Marrow Transplant. 2002;8:368–376. doi: 10.1053/bbmt.2002.v8.pm12171483. [DOI] [PubMed] [Google Scholar]
- 41.Willert K, Brown JD, Danenberg E, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 42.Perry JM, He XC, Sugimura R, et al. Cooperation between both Wnt/{beta}-catenin and PTEN/PI3K/Akt signaling promotes primitive hematopoietic stem cell self-renewal and expansion. Genes Dev. 2011;25:1928–1942. doi: 10.1101/gad.17421911. [DOI] [PMC free article] [PubMed] [Google Scholar]