

Abstract
Base excision repair (BER) is a key genome maintenance pathway that removes endogenously damaged DNA bases that arise in cells at very high levels on a daily basis. Failure to remove these damaged DNA bases leads to increased levels of mutagenesis and chromosomal instability, which have the potential to drive carcinogenesis. Next Generation sequencing efforts of the germline and tumors genomes of thousands of individuals has uncovered many rare mutations in BER genes. Given that BER is critical for genome maintenance, it is important to determine whether BER genomic variants have functional phenotypes. In this chapter we present our in silico methods for the identification and prioritization of BER variants for further study. We also provide detailed instructions and commentary on the initial cellular assays we employ to dissect potentially important phenotypes of human BER variants and highlight the strengths and weaknesses of our approaches. BER variants possessing interesting functional phenotypes can then be studied in more detail to provide important mechanistic insights regarding the role of aberrant BER in carcinogenesis.
I. Introduction
DNA damage occurs as a result of normal metabolism resulting in oxidized, deaminated, and alkylated bases at a rate of at least 30,000 lesions per cell per day (Freidberg, Wood, Walker, & Siede, 2006). Base excision repair (BER) removes the majority of these lesions. The simplest and most common form of BER is short patch BER, which is initiated by one of several different DNA glycosylases, each having preferences for specific types of lesions [for a comprehensive review see (Wallace, Murphy, & Sweasy, 2012)]. Monofunctional DNA glycosylases recognize DNA lesions and catalyze the hydrolysis of the N-glycosyl bond that releases the damaged base and generates an abasic site. The abasic site is nicked at its 5′ side by the apurinic/apyrimidinic endonuclease 1 (APE1), leaving a 3′OH and a 5′deoxyribose phosphate (dRP). DNA polymerase β (Pol β) fills in the single nucleotide gap and catalyzes removal of the dRP group with its associated lyase activity. Bifunctional DNA glycosylases, which usually recognize and remove oxidative lesions, have associated lyase activity that cleaves the DNA backbone, leaving either a phosphate group or an α,β-unsaturated aldehyde attached to the 3′ end of the DNA. Phosphate groups at the 3′end of the DNA are removed by polynucleotide kinase (PNKP). The α,β-unsaturated aldehydes are processed APE1 endonuclease, which creates a 3′OH that is recognized by Pol β, which fills in the single nucleotide gap. The X-ray cross-complementing 1 (XRCC1)/Ligase IIIα complex catalyzes ligation of the DNA ends. Long patch BER is thought to be a minor DNA repair pathway in human cells and takes place if the 5′ sugar is modified or if the lyase activity of Pol β is defective [for reviews see (Balakrishnan & Bambara, 2013; Wallace et al., 2012)]. In this case, Pol β performs strand displacement synthesis to initiate long patch BER, and 2–12 nucleotides are added by DNA polymerases δ and/or ε, resulting in the generation of a 5′ DNA flap. The flap is removed by Flap endonuclease 1 (FEN1) and DNA ligase 1 or XRCC1/Lig3α seal the nick.
BER is critical for maintaining genomic stability. An inability to remove endogenous DNA damage can lead to the accumulation of point mutations that likely arise as a result of error prone lesion bypass by either replicative DNA polymerases, DNA polymerases λ or β, or translesion polymerases [for an excellent review see (Bacolla, Cooper, & Vasquez, 2014)]. Aberrant processing of base damage by mutant enzymes in the BER pathway may also lead to the accumulation of BER intermediates, including single- and double-strand breaks, which can result in chromosomal instability [for example see (Yamtich, Nemec, Keh, & Sweasy, 2012)].
Little is known about the relationship between single nucleotide polymorphisms (SNPs) and/or somatic variants in genes encoding for BER proteins and the etiology of human cancer. Recently, a linkage to colorectal and perhaps multiple cancers was identified for a truncation within the NTHL1 gene (Rivera, Castellsague, Bah, van Kempen, & Foulkes, 2015; Weren, Ligtenberg, Kets, de Voer, Verwiel, Spruijt et al., 2015). This DNA glycosylase recognizes and removes oxidized pyrimidines (Asagoshi, Yamada, Okada, Terato, Ohyama, Seki et al., 2000; Aspinwall, Rothwell, Roldan-Arjona, Anselmino, Ward, Cheadle et al., 1997; Eide, Luna, Gustad, Henderson, Essigmann, Demple et al., 2001; Ikeda, Biswas, Roy, Izumi, Boldogh, Kurosky et al., 1998). Previous to this it was shown that SNPs within the MUTYH gene predispose individuals to MUTYH-associated polyposis (MAP) and perhaps other types of cancer [for detailed reviews see (David, O’Shea, & Kundu, 2007; Wallace et al., 2012)]. MUTYH recognizes and removes adenine that has been inserted opposite 8-oxoguanine or FapyG (Au, Cabrera, Miller, & Modrich, 1988; Lu, Tsai-Wu, & Cillo, 1995; McGoldrick, Yeh, Solomon, Essigmann, & Lu, 1995; Pope & David, 2005; Slupska, Baikalov, Luther, Chiang, Wei, & Miller, 1996; Slupska, Luther, Chiang, Yang, & Miller, 1999). Mutations in MUTYH result in an inability of the protein to efficiently remove adenine opposite 8-oxoguanine, resulting in the accumulation of GC to TA transversions within the adenomatous polyposis coli (APC) gene and also within the KRAS gene (Al-Tassan, Chmiel, Maynard, Fleming, Livingston, Williams et al., 2002; Boparai, Dekker, Van Eeden, Polak, Bartelsman, Mathus-Vliegen et al., 2008).
There is general agreement among cancer geneticists that there is missing hereditability underlying the disease of cancer (Schork, Murray, Frazer, & Topol, 2009). This missing hereditability could result from multiple variants each having small effects that are additive or rare variants that have large phenotypic effects, referred to as “the rare variant hypothesis”. Because current methods of statistical analysis of the association of rare variants to the etiology of human cancer are underpowered, other methods must be considered. Our approach has been to characterize the function of coding SNPs and tumor-associated variants in specific human BER genes using a combined biological and biochemical approach. Using this approach, we have shown that expression of rare human germline or somatic BER genetic variants in human cells results in genomic instability and cellular transformation. These include human genetic variants in the NTHL1 (Galick, Kathe, Liu, Robey-Bond, Kidane, Wallace et al., 2013) and Thymine DNA glycosylase (TDG) genes (Sjolund, Nemec, Paquet, Prakash, Sung, Doublie et al., 2014), as well as in the POLB (Donigan, Hile, Eckert, & Sweasy, 2012; Donigan, Sun, Nemec, Murphy, Cong, Northrup et al., 2012; Lang, Dalal, Chikova, DiMaio, & Sweasy, 2007; Lang, Maitra, Starcevic, Li, & Sweasy, 2004; Murphy, Donigan, Jaeger, & Sweasy, 2012; Nemec, Donigan, Murphy, Jaeger, & Sweasy, 2012; Nemec, Murphy, Donigan, & Sweasy, 2014; Sweasy, Lang, Starcevic, Sun, Lai, Dimaio et al., 2005; Yamtich et al., 2012) and XRCC1 (Sizova, Keh, Taylor, & Sweasy, 2015) genes. Our work suggests that inherited or somatically acquired genetic alteration in the BER pathway has potential to drive carcinogenesis.
In this chapter, we describe our approach and methods for the characterization of human genetic BER variants. We initiate our studies with identification and prioritization of variants for further study. The variants are then expressed in human cells and assessed for their abilities to induce genomic instability and cellular transformation. Variants that induce these phenotypes undergo thorough characterization to uncover specific mechanisms underlying genomic instability and cellular transformation, the details of which can be found in our previously published work.
2. Identification and prioritization of base excision repair variants using in silico methods
We identified variants in silico based on public and private sequence data and linked them to features that reflect their significance for human health, for example predicted impact on the protein function and frequency in the general population, in order to prioritize variants for experimental studies (Figure 1).
Figure 1.
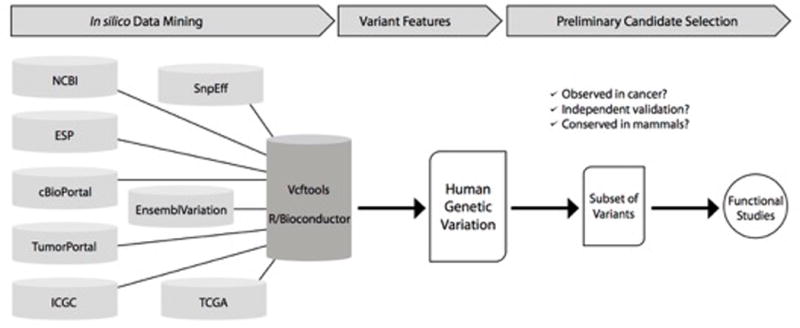
Structure and operation of the Base Excision Repair variant identification pipeline. See text for details.
2.1 In silico Data Analysis
For the in silico analysis, all known variants in BER genes are assembled from multiple databases. We use publicly available open source bioinformatics tools [SnpEff (Cingolani, Platts, Wang, Coon, Nguyen, Wang et al., 2012), Ensembl Variation (Chen, Cunningham, Rios, McLaren, Smith, Pritchard et al., 2010), vcftools (Danecek, Auton, Abecasis, Albers, Banks, DePristo et al., 2011) and R/Bioconductor packages (Gentleman RC, 2004)] to interface with a variety of databases including Ensembl (Chen et al., 2010; Yates, Akanni, Amode, Barrell, Billis, Carvalho-Silva et al., 2016), cBioPortal (Gao, Aksoy, Dogrusoz, Dresdner, Gross, Sumer et al., 2013) using the R/Bioconductor cgdsr package, over 200 restricted access TCGA germline whole exome sequence samples from tumor-normal pairs from patients with invasive breast cancer (dbGaP accession phs000178.v5.p5), the International Cancer Genome Consortium (ICGC), National Center for Biotechnology Information (NCBI), the NHLBI Exome Sequencing Project (ESP), and TumorPortal (Lawrence, Stojanov, Mermel, Robinson, Garraway, Golub et al., 2014). We combine the information from across these resources, and this comprehensive body of evidence about the variants is merged into a single matrix file that can be easily sorted and filtered with Microsoft Excel.
2.2 Variant Features
The Microsoft Excel worksheet generated from in silico analysis includes information on both germline and somatic variants. The germline variants have unique identifiers from NCBI’s dbSNP database and the somatic variants have unique identifiers from the Catalog of Somatic Mutations in Cancer (COSMIC). Amino acid alterations are annotated as is each type of mutation, namely, frameshift, stop-gained, missense, or nonsense mutations. These variant BER proteins are then assessed by PolyPhenII (Adzhubei, Schmidt, Peshkin, Ramensky, Gerasimova, Bork et al., 2010) to predict whether they are damaging, meaning that they have the potential to alter protein function. Ancestry corrected allele frequencies are also annotated in the Excel spreadsheet, as well as alternative transcript identifiers and associated PubMed identifiers for supporting literature. Variants are then filtered on any combination of features. We are generally interested in the BER variants that are predicted to be damaging to the protein product with a minor allele frequency <5%.
2.3 Preliminary evaluation of variants
A variant selected for experimental studies must be supported by evidence that it 1) reflects a correct variant call, 2) is infrequent in the general population, 3) is predicted to have a protein functional consequence, and 4) is either a germline or somatic mutation identified in a human tumor, or both. Evidence bearing on the confidence of the variant call is evaluated, including independent validation, sequencing quality metrics such as the quality score for the nucleotide call and of the mapping of sequence reads to the reference genome, as well as the depth of sequencing coverage of each variant. Interpretation of protein functional consequences is based on the collection of information and inspection by crystallographers and biochemists. Variants chosen for further study include those that are within important and conserved functional domains of proteins with amino acid alterations that would be predicted to affect protein function. For example, we identified the NTHL1 D239Y germline variant as altering a potentially important amino acid residue close to the active site of this DNA glycosylase. Subsequent studies demonstrated that the NTHL1 D239Y protein was catalytically inactive and that it induced genomic instability and cellular transformation upon expression in human epithelial cells (see below for additional details) (Galick et al., 2013).
3. Cellular characterization of BER variants
To accurately assess the potential for a BER variant to drive carcinogenesis, the cellular characterization of BER variants should be performed in a physiologically relevant system. The system described here enables the expression of BER variants of interest in human cells at levels roughly equivalent to those of the endogenous protein (as shown in Figure 2A) to generate a biologically relevant system for the study of BER variants. Using this method, the mechanisms by which BER variants induce cellular transformation and genomic instability are informative regarding directly to the etiology and progression of human disease. For cellular characterization of BER variants, the cDNA of the variant is stably expressed in immortal but non-transformed human cells. We have experience using the MCF10A or HME-1 mammary epithelial cells [for example see (Galick et al., 2013; Nemec, Bush, Towle-Weicksel, Taylor, Schulz, Weidhaas et al., 2016)]. Here we will describe our work with MCF10A, which is a non-transformed, immortalized mammary epithelial cell line that is near-diploid with a stable karyotype. Although the cell line does contain some karyotype abnormalities (including an extra chromosome), MCF10A cells are not able to form tumors in immunocompromised mice (Yoon, Wersto, Zhou, Chrest, Garrett, Kwon et al., 2002) and cannot form colonies in soft agar (Soule, Maloney, Wolman, Peterson, Brenz, McGrath et al., 1990). Therefore, the MCF10A cell line is extremely useful to study the effects of BER variants on premalignant progression.
Figure 2.
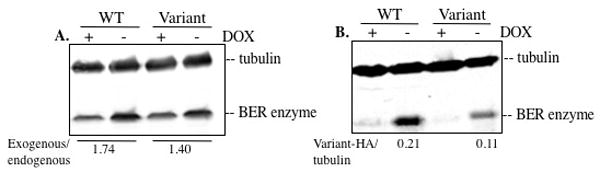
Western blot to quantify expression of exogenous WT and variant protein expression. A. The level of exogenous protein expression above endogenous protein expression is determined using a primary antibody against the BER enzyme of interest. The signal produced in lysates from cells not cultured with doxycycline (exogenous + endogenous protein) is divided by the signal produced in lysates from cells cultured with doxycycline (endogenous protein only) to calculate the fold increase of protein expression as a result of exogenous protein expression. To properly characterize the cellular effects of a human somatic or germline BER variant, it is important to maintain expression levels of the exogenous protein that are close to the levels of the endogenous protein. B. Expression of WT and variant is detected using a primary antibody against the HA epitope tag. Expression is normalized by dividing the HA signal by the tubulin signal.
3.1 Subcloning in pRVY-tet vector
Cellular expression of BER variants of interest is achieved using the pRVY-tet vector. The pRVY-tet vector is a retroviral vector encoding ampicillin resistance for selective bacterial expression and hygromycin resistance for selective expression in mammalian cells. The pRVY-tet vector conveniently provides tetracycline-controlled gene expression of the BER variants of interest. In the TET-off (or tTA-dependent) system, a tetracycline transactivator fusion protein (tTA), composed of a tetracycline repressor (TetR) and the C-terminal domain of VP16 (virion protein 16), is unable to bind to a tetracycline response element (TRE) located upstream of the promoter when tetracycline (or doxycycline) is added to the cell culture media thereby turning off gene expression (Gossen & Bujard, 1992). A major benefit of the TET-off system is the reversible and rapid induction of gene expression upon the removal of tetracycline (or doxycycline) from the media. However, “leakiness” can occur which results in detectable levels of gene expression even in the presence of tetracycline or doxycycline. Furthermore, tetracycline can cause toxicity in mammalian cells at higher concentrations so titration of tetracycline is necessary to achieve optimal inhibition of gene expression while minimizing toxicity. We generally use doxycycline (an analog of tetracycline) to regulate gene expression because it has lower toxicity in mammalian cells, a known half-life of 24 hours, and because it binds to tTA with high affinity, therefore reducing the likelihood of unintended gene expression (or “leakiness”). The TET-off feature of the pRVY-tet vector is particularly useful for expression of BER variants that may reduce cellular viability and/or cellular proliferation making study difficult with continuous expression.
The easiest and most time effective method for generating pRVY-tet constructs expressing BER variants of interest is to first subclone the WT sequence of the BER enzyme into the pRVY-tet vector then perform site-directed mutagenesis to generate the desired mutation. Since the pRVY-tet vector does not contain a multiple cloning site (MCS), the restriction enzymes available for sub-cloning are limited to the NotI and BamHI sites thereby requiring a multi-step approach to subcloning into the pRVY-tet vector. In contrast, site-directed mutagenesis is a relatively simple method involving PCR amplification with substitution of the desired nucleotide/s followed by digestion of the parental vector. In addition, if an epitope tag needs to be added to the BER enzyme of interest in order to distinguish the endogenous and exogenously expressed proteins then the sequence encoding the tag to be used is included in the primer design during subcloning. It is important to consider structural and catalytic constraints of an epitope tag of BER enzymes of interest. Furthermore, it is imperative to perform functional assays using the tagged BER enzyme to ensure the epitope tag does not interfere with its catalytic functions. BER enzymes studied in our laboratory have been successfully tagged with either the FLAG or human influenza hemagglutinin (HA) epitope tags without affecting catalytic activity (Galick et al., 2013).
3.1.1 Equipment
PCR Thermal cycler
37° water bath
PCR cleanup kit
Standard electrophoresis equipment
Gel extraction kit
Imaging equipment
Spectrophotometer
3.1.2 Buffers and Reagents
High fidelity DNA polymerase
PCR Buffer
dNTP stock solution
Primers
NotI
BamHI
Restriction enzyme buffer
DNA ligase
DNA ligase buffer
LB broth and LB agar
Ampicillin (100μg/mL final)
3.1.3 Procedure
Amplify sequence of BER enzyme of interest by PCR, using primers designed to add NotI and BamHI restriction sites to 5′ and 3′ ends respectively.
Isolate the amplified sequence using PCR cleanup kit.
Digest the PCR product from step 1 and the pRVY vector with NotI and BamHI for 1 hr at 37°C.
Run the digested vector on a 1% agarose gel. Cut out the appropriate band.
Gel purify the digested vector backbone.
Use a PCR cleanup kit to isolate the digested PCR product.
Determine the DNA concentration of the digested and purified pRVY vector and PCR product.
Perform a ligation reaction using 1:1, 1:2, and 1:4 digested pRVY vector (25ng): digested PCR product, DNA ligase, and DNA ligase buffer at 16°C overnight or at room temperature for 30 min-1hr.
Transform ligation reaction into competent E. coli (such as DH5α).
Plate onto pre-warmed LB/Ampicillin plates.
Isolate plasmid DNA from the colonies and sequence the insert to confirm its presence.
Upon successful subcloning of WT sequence of BER enzyme of interest into the pRVY-tet vector, perform site-directed mutagenesis to generate the BER variant following the manufacturer’s instructions (see note 4).
3.1.4 Notes
A two-step PCR reaction may be needed to successfully amplify the sequence of interest, particularly if your primers are long. If this is the case, use an appropriate annealing temperature (TA) for the sequence in the primers that overlaps with the sequence to be amplified (this would be the 8–12 base pairs that anneals with the target sequence only, not the TA for the entire primer sequence) for 5 cycles, followed by 30 cycles using a TA appropriate for the entire length of the primers.
Check for amplification by agarose gel electrophoresis before continuing.
We prefer to the New England Biolabs Monarch PCR purification/gel purification kit. We have found that the amount of DNA recovered is higher than other commercially available kits.
There are two commercially available site-directed mutagenesis kits that have been successful in our laboratory; the Q5® site-directed mutagenesis kit from New England Biolabs and the QuikChange II XL site-directed mutagenesis kit from Agilent. Primer design is quite different between the two kits and therefore it’s very important to design the primers using the appropriate primer design software for each kit. We most commonly use the Q5® site-directed mutagenesis kit from New England Biolabs.
3.2 Generation of stable MCF10A pools and clones expressing BER variant of interest
A major benefit of the pRVY expression system is the moderate level of expression of the exogenous protein of interest in the cells. As shown in Figure 2B, we measure the levels of exogenous protein expression by western blotting using a primary antibody against the epitope tag, which in this case is the HA tag. However, in order to compare the level of exogenous protein expression in relation to endogenous protein expression levels we also blot with an antibody against the BER enzyme of interest as shown in Figure 2A. It is also important to use pools and/or clones that exhibit similar expression levels of the exogenous WT enzyme and BER variant of interest so that any phenotypic changes detected cannot be attributed to an artifact as a result of higher expression levels of the variant enzyme than the WT enzyme. Some BER variants may not express well in cells, as seen in Figure 2B where expression of the variant is about half of WT expression levels. Lower expression of a variant compared to the WT enzyme may mask a possible cellular phenotype, which must be kept in mind during subsequent analyses. However, detection of a phenotype can be considered a positive result in these circumstances.
When performing downstream assays, there is a choice between using pools of cells or clones. Retroviral transduction results in random integration of the introduced sequence into the genome. Each cell will have a different number of integrations of the construct as well as different integration locations within the genome. Therefore, when selecting a pool of cells with stable integration, the resulting population will be heterogeneous and expression levels will reflect the heterogeneity of integration. In contrast, a clone will contain a population of cells that arose from a single cell and therefore all cells in that population will be homogeneous for the number and location of integrations. Although cellular transformation due to an integration event is not an issue when working with pools of cells, the heterogeneity of expression in pools may mask a cellular phenotype. In contrast, the homogeneous expression in clones makes it easier for cellular phenotypes to emerge, however the possibility of transformation due to an integration event needs to be addressed by screening at least 8–10 different clones with stable expression of the WT or variant enzyme. We typically select both pools and clones but perform our initial characterization assays using pools of cells. If the results using pools are inconsistent or hard to interpret we will then employ clones for our assays.
3.2.1 Equipment
Biosafety cabinet
CO2 incubator (5% CO2/37°C)
3.2.2 Buffers and Reagents
100 mm tissue culture plates
T25 and T75 tissue culture flasks
0.25M calcium chloride (3.68g into 100mls ddH2O), sterile filtered
2X HEBS (12mM Dextrose, 50mM HEPES, 10mM KCl, 280mM NaCl, 1.5mM Na2HPO4•2H2O), pH adjusted to 7.05 with 10N NaOH, sterile filtered
10μg of pRVY plasmid expressing WT and BER variant of interest
10μg of pVSV-G plasmid
0.45μm pore filter
10–30mL syringe (without a needle)
Dulbecco’s Modified Eagle’s Medium, high glucose
Fetal Bovine Serum (FBS)
Penicillin/streptomycin (P/S) (100X stock)
Polybrene (4mg/mL stock)
Hygromycin B, (50mg/mL stock)
Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F-12)
Horse Serum
Insulin, recombinant, 4mg/mL stock (Life Technologies)
Epidermal growth factor (EGF), 100μg/mL stock (Peprotech)
Hydrocortisone, 1mg/mL stock (Sigma)
Cholera Toxin from Vibrio cholera, 1mg/mL stock (Sigma)
MCF10A complete growth media (DMEM/F-12, 5% horse serum, 20ng/mL EGF, 0.5μg/mL hydrocortisone, 100ng/ml cholera toxin, 10μg/ml recombinant insulin, 1% P/S)
3.3.3 Procedure
Plate GP2–293 cells at 2.5×106 cells per 100mm plate in DMEM 10%FBS/1%P/S for each pRVY vector and one control plate (no pRVY vector included in transfection) 18–24 hrs prior to transfection.
Replace media with fresh DMEM 10% FBS/1%P/S about 1 hr prior to transfection.
Combine 500μL of 2X HEBS, 10μg of pRVY vector, and 10μg of pVSV-G vector in a sterile pre-labeled tube (one tube per pRVY expression vector).
Add dropwise 500μL of 0.25M CaCl2 with aeration to the 2XHEBS/DNA solution.
Add dropwise the CaCl2/2XHEBS/DNA solution to the GP2–293 cells.
Incubate at 37°C/5% CO2 for 6–8 hrs.
Replace the media with fresh DMEM 10%FBS/1%P/S and incubate overnight at 37°C/5% CO2.
The next day, change the media again. See note 4 below.
48 hrs post-transfection begin selection with 200μg/mL Hygromycin B. If the cells are >80–85% confluent, split at a ratio of 1:4 and begin selection 24 hrs later.
Select for stable integration for 5–8 days until all cells on the control plate are dead and visible colonies have formed on pRVY transfected plates.
Generation of high titer virus: Plate the selected GP2–293 cells at 2.5×106 cells per 100mm plate 18–24 hrs prior to transfection.
Replace the media with fresh DMEM 10%FBS/1%P/S about 1 hr prior to transfection.
Perform transfection as described in steps 3–8 using 10μg of pVSV-G vector only for each pRVY expressing GP2–293 cell line.
Plate 5×105 MCF10A cells in T25 flasks in MCF10A complete growth media 24 hrs prior to infection. Plate two-three flasks for each stable GP2–293 cell line generated, as well as one flask as a control for selection (no virus-containing media + polybrene).
Change media on MCF10A cells about 1 hr prior to infection.
For the infection: collect media 72 hrs after transfection (contains high titer virus) and filter through a 0.45μM pore filter.
Add polybrene to the filtered virus containing media at a final concentration of 8μg/mL.
Replace media on plated MCF10A cells in T25 flasks with 3mls of virus-containing media or non-virus containing media + polybrene (as a control for selection).
Incubate MCF10A cells with virus-containing media at 37°C/5% CO2, rocking the flasks every 30 min.
Add 2mls MCF10A complete media and incubate overnight at 37°C/5% CO2.
The next day, replace the media with MCF10A complete growth media.
For the generation of pools with stable expression, begin selection 48 hrs after infection with 200μg/mL hygromycin B. Add 2μg/mL of doxycycline to prevent expression of BER enzymes during selection.
For the generation of clones, serially dilute the infected cells and plate several dilutions in 100mm plates. Begin selection with 200μg/mL hygromycin B the next day.
Once selection is complete, change media to MCF10A complete media containing 15μg/mL hygromycin B to maintain selective pressure and 2μg/mL of doxycycline to prevent expression of the BER variant of interest until prepared to plate for downstream assays.
Expression of BER enzyme/variant of interest is then measured by Western blot. Plate each pool or clone with and without doxycycline. Collect cell lysates for Western blot.
3.2.4 Notes
Test for precipitate for every new stock of CaCl2 and 2X HEBS before transfection.
The pVSV-G construct expresses a viral envelope protein that is necessary for virus production and viral infection.
Add CaCl2 dropwise using a pipetman while adding aeration using a Pasteur pipet and pipet-aid.
Check for precipitate 20 min after adding CaCl2/2XHEBS/DNA solution to the GP2–293 cells.
At the point prior to selection of transfected GP2–293 cells, the cells are producing low titer virus. Low titer virus can be used to infect MCF10A cells with reasonable infectivity. However, we typically select GP2–293 cells with stable integration of pRVY construct and perform a second transfection with the pVSV-G vector to generate high titer virus.
A viral titer assay can be performed to calculate the colony forming units (CFU)/mL using 3T3 mouse fibroblasts if optimization is needed.
Do not attempt selection with the cells at >80–85% confluence. Split the cells 1:4 and begin selection 24 hrs later.
The virus-containing media can be diluted into MCF10A complete media if the CFU was determined to be high or if lower integration events are desired. In this case, 1:2 and 1:4 dilutions of MCF10A complete media: virus containing media is a good place to start.
Infection of MCF10A cells can be performed at 32°C/5% CO2 to increase virus viability.
If the infected MCF10A cells are confluent 48 hrs after infection, split each T25 flask into a T75 flask then begin selection the next day.
We have performed a dose titration with hygromycin B for MCF10A cells and found 200μg/mL hygromycin B to be the optimal concentration for selection. If selection is not efficient or the entire cell population dies then determine the optimal selection concentration for your own purposes.
Doxycycline should be added every 48 hrs to maintain inhibition of gene expression.
3.3 Cellular transformation in human cells
The MCF10A cell line is a non-transformed human epithelial cell line that is used to measure cellular transformation upon expression of a BER enzyme of interest (the Brugge laboratory website is an excellent source for general MCF10A protocols: http://brugge.med.harvard.edu/protocols). The potential of a BER variant of interest to transform human cells is initially assessed by measuring changes in proliferation and anchorage-independent growth. Other characteristic cellular properties not described here but that emerge as a result of transformation including cellular migration and invasive potential [for example see (Galick et al., 2013)] can also be assessed following these initial studies. MCF10A cells cannot grow in soft agar and therefore serve as an excellent cell line to assess the ability of a BER variant to induce anchorage independent growth. We have adapted more high-throughput “kit-based” assays to test both proliferation and anchorage-independent growth for the purpose of efficient characterization of several BER variants at the same time. However, if use of a kit is not preferred then refer to the notes section where alternative methods are described briefly. The general methodology is similar between the two approaches with the differences mainly including plate size, cell number and end-point measurement (as is the case for the proliferation assay). The CYQUANT® NF cell proliferation assay measures DNA content to quantify proliferation, therefore the end-point measurement is relative fluorescence units (RFU) as opposed to cell number which is the end-point measurement if you choose to count cells manually. Anchorage-independent growth is measured by counting the number of colonies formed whether you use the Cytoselect™ 96-well cell transformation assay or the alternative soft agar assay. The main difference between the soft agar assays described is the agar used. Proliferation and anchorage-independent growth are measured starting at passage 2 and then measured every other passage (i.e. passage 4, passage 6, passage 8 etc.). If using pools, you can begin with one WT pool and one variant pool. If you are using clones, you should begin with at 3–5 WT clones and 3–5 variant clones. We have detected differences in anchorage-independent growth between WT and BER variant expressing cells as early as passage 6 and as late as passage 14. Each time you plate cells for proliferation and anchorage-independent growth you should also plate cells for a western blot to monitor expression of the exogenous WT and variant proteins in the cells over passages. For BER variants, we have found that some of the hallmarks of cellular transformation, including increased cellular proliferation and anchorage independent growth, occur as a function of the numbers of passages of the cells and have suggested that BER is a tumor suppressor mechanism (Sweasy, Lang, & DiMaio, 2006). During the growth and passaging of the cells, aberrant BER will occur if the variant under study is deficient in its ability to remove and process DNA damage. Aberrant BER can occur as a result of deficient removal of damaged bases by DNA glycosylases, inefficient end remodeling or backbone incision by enzymes including PNKP and APE1, slow, defective, or error-prone gap filling by Pol ß, defective scaffolding of the BER enzyme complex by XRCC1, or inefficient ligation. Each of these events has the potential to result in point mutations in growth control genes [for an example see (Donigan, Hile, et al., 2012)] and/or genomic instability [for an example see (Nemec et al., 2016)], but the cells must replicate and divide in order for the phenotype to manifest itself.
3.3.1 Equipment
Plate reader (480/520nm)
Light microscope
Water bath (40°C)
Microwave
0.22μm pore filter, bottle top.
3.3.2 Buffers and Reagents
MCF10A complete media
CYQUANT® NF cell proliferation assay (Invitrogen)
2X DMEM/F12 (Sigma)
2X MCF10A complete media (10% horse serum, 40ng/mL EGF, 1.0μg/mL hydrocortisone, 200ng/ml cholera toxin, 20μg/ml recombinant insulin, 2% P/S)
Cytoselect™ 96-well cell transformation assay (Cell Biolabs)
3.3.3 Procedure
Proliferation Assay: Plate 103 cells expressing either the WT or variant enzyme per well in triplicate in a 96-well plate in MCF10A complete media without hygromycin B (see Figure 3A for example of plate setup).
Add media to the wells to adjust the total volume per well to 200μL.
Begin measuring proliferation 48 hrs after plating by following the manufacturers’ instructions.
Measure proliferation twice a day while cells are in log growth (we typically measure twice a day beginning on day 4 until day 6.5).
Anchorage independent growth: Turn on the water bath and set to 40°C.
Prepare 2X DMEM/F12 as per the manufacturers’ instructions. Sterile filter using a bottle top 0.22μm pore filter and store at 4°C.
Prepare 2X MCF10A complete media using the prepared 2X DMEM/F12 prepared in the previous step.
Prepare the 1.2% agar solution as described by the manufacturer.
Boil the 1.2% Agar solution in the microwave until completely dissolved.
Place in water bath until cools slightly.
Warm 2X MCF10A complete media in same water bath.
Calculate the volume of media: agar solution required for the bottom agar, considering that 50μL per well is needed and that each cell line will be plated in quadruplicate, including at least two wells without cells to serve as a negative control.
Combine 1.2% agar solution with 2X MCF10A complete media at a 1:1 ratio in a 15mL conical tube.
Plate 50μL of the bottom agar solution per well. Avoid creating bubbles in the agar when plating.
Tap the side of the plate gently to evenly distribute the agar: media solution.
Place at 4°C for 30 min.
Remove the plate from 4°C and place in the incubator for 15 min.
Melt the 1.2% agar in the microwave again and put in the water bath to cool. Place the 2X MCF10A complete media in the water bath as well.
Prepare your cells: dilute 2×105 cells in 1mL of 2X MCF10A complete media.
Combine 125μL of 2×105 cells, 125μL 2X MCF10A complete media, and 125μL of 1.2% agar. Add 75μL per well of the [cell: media: agar solution] to the wells with the solidified bottom agar.
Place the plate at 4°C for 15 min to allow the agar to solidify.
Add 100μL of MCF10A complete media (without hygromycin) to each well and place in incubator. (Be sure you add 1X growth media, not the 2X media you’ve been using throughout this protocol).
Colonies should start forming in about 3–4 days. Count colonies about 8–10 days after plating.
Count 4–5 randomly chosen fields per well using a light microscope. See Figure 3C-D for representative fields and colonies.
Figure 3.
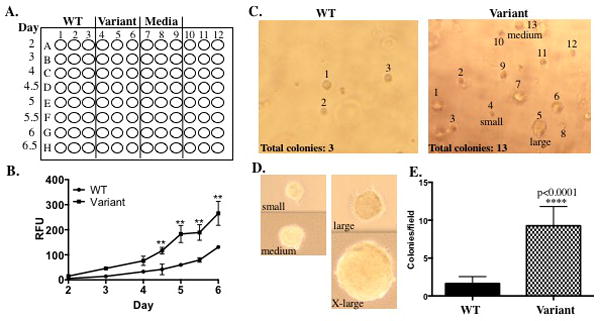
A. An example a 96-well plate setup for the proliferation assay. Plate WT expressing cells in the first three columns, variant-expressing cells in the next three columns and media only in the next three columns in rows A-H. Cells will be counted in triplicate (each column) over time (each row). The media only wells serve as a control for background. B. As seen in the graph, the CYQUANT® NF cell proliferation assay in a 96-well format measures proliferation during early log phase growth. Significant differences have been observed during early log phase growth in cells expressing a BER variant as shown in the graph. To generate a complete growth curve, the 6-well plate format with manual counting would be a better option. C-E. Representative fields of view of a soft agar assay using the Cytoselect™ 96-well cell transformation assay for a WT enzyme and BER variant. In each field of view, the number of colonies is provided and the colonies that were included in the count have been labeled. Colony size can vary, however colonies ranging from small to extra (X)-large are counted in the graph shown (E). Varying colony sizes are labeled in the variant field of view as well as in the panel below (D). If larger colonies are consistently formed by a BER variant, colonies of different sizes can be scored separately. To score the soft agar assay in the 96-well format, five fields of view are counted for each of four wells containing cells expressing either the WT enzyme or BER variant. As shown in (E), colonies per field of view are graphed on the Y-axis.
3.3.4 Notes
Do not pipet less than 1.5μL of cells when plating or pipet error will be high and results will be inconsistent.
As an alternative method to using the CYQUANT® NF cell proliferation assay, plate 104 cells expressing WT and variant enzyme in triplicate in 6-well plates, then count the cells 48 hrs after plating for a total of 8 days using a cell counter or hemacytometer.
The Cytoselect™ 96-well cell transformation assay is sold as a kit containing multiple components however we only use the Cytoselect™ Agar powder for this assay. We prepare our own 2X media because MCF10A cells grow in DMEM/F12 not DMEM. However, if you are using cells that grow in DMEM then prepare the 2X DMEM provided in the kit. The other reagents provided are for agar solubilization, cell lysis and cell quantification. We manually count the number of colonies that form in the agar.
You will need to move fast when plating the agar: media solutions so that the agar doesn’t solidify before you finish plating.
Be sure that the agar has cooled to about 40–45°C before combining with cells. The cells will lyse when combined with the agar: media solution if it is too hot.
Similar to the proliferation assay, anchorage independent growth can be measured without using the Cytoselect™ 96-well cell transformation assay. The alternative assay utilizes 1.0% Noble agar (Affymetrix) for the bottom agar and 0.7% Noble agar for the top agar, plated in a 6-well plate in triplicate for each cell line. For this type of assay, plate 104 cells per well using the same method described above, and simply adjust the volumes. The major difference is that colonies will be counted 30 days after plating instead of 8–10 days.
We recommend that cells from each passage be preserved in liquid nitrogen in order to perform the assays described below and also in case of contamination.
3.4 Genomic instability and mutagenesis
MCF10A cells expressing a BER variant that exhibit increased cellular proliferation and anchorage-independent growth, indicating cellular transformation, are then further interrogated for genomic instability and mutagenesis. While a few BER variants have exhibited increased proliferation and increased anchorage-independent growth, many BER variants exhibit increased anchorage-independent growth without increased proliferation. Increased proliferation may be counteracted by increased apoptosis or decreased cell viability and therefore is a more complex and less direct assessment of cellular transformation. Changes in anchorage-independent growth is a more direct reflection of cellular transformation and therefore is a key phenotype for continuing with characterization of a BER variant. To characterize the effects of a BER variant on genomic stability we measure the amount and types of chromosomal aberrations using metaphase spread preparations and we quantify the presence of micronucleus formation to assess chromosome damage. Increased chromosomal aberrations are an indication of structural changes in chromosomes and signify genomic instability. Micronuclei arise from lagging chromosomes (either whole chromosomes or fragments) during mitosis. Micronuclei are a reflection of chromosome damage, such as chromosome breakage or whole chromosome loss, and therefore increased levels of micronuclei is another biomarker for genomic instability. Scoring chromosomal aberrations and micronuclei can often be subjective based on the quality of the images or cellular staining and the investigator that is scoring. It is highly recommended that the slides be coded prior to imaging and/or scoring to avoid any bias. It is also imperative that chromosomal aberrations and micronuclei are scored in cells at passages prior to those in which significant levels of anchorage independent growth is observed, as described above. The purpose of these assays is to understand the mechanisms driving transformation. Genomic instability is an inherent feature of cells that are already transformed; therefore, no conclusions can be drawn as to whether or not the BER variant induced genomic instability or if increased genomic instability contributed to cellular transformation if transformed cells are utilized in these assays. In addition to DNA damage (i.e. breaks) and structural chromosomal aberrations, a BER variant may induce cellular transformation by increasing mutagenesis mainly in the form of point mutations. The ouabain assay is utilized to measure mutagenesis. Ouabain binds to and inhibits the Na+/K+-ATPase sodium potassium ion pump, leading to accumulation of intracellular sodium and eventual cell death. Mutations in the Na+/K+-ATPase sodium potassium ion pump can result in ouabain resistance, therefore increased ouabain resistance serves as a marker for increased mutagenesis. The purpose of the assays outlined in this section is to determine whether expression of a BER variant induces genomic instability or increased mutagenesis, providing key information that will guide preliminary mechanistic studies to determine the molecular mechanisms contributing to the cellular phenotypes observed.
3.4.1 Equipment
Centrifuge
37°C water bath
Clamp for pipetman
Glass slides and coverslips
Coplin staining jar
Microscope (500⁄526 filter for DNA visualization)
Two Hand Tally Counters
Counter pen (Thermo Scientific)
3.4.2 Buffers and Reagents
MCF10A complete media
KaryoMax® Colcemid™ (10μg/mL stock) (ThermoFisher)
Carnoy’s fixative (3:1 Methanol: Acetic Acid), pre-chilled to -20°C
1X PBS
0.05% trypsin
Potassium Chloride (Gibco, 0.075M), pre-warmed to 37°C
Mounting media with DAPI (we use Sigma Flouroshield™ with DAPI)
Cytochalasin-B in ddH2O
Methanol
Acridine Orange in PBS
Ouabain octahydrate (Sigma)
0.5% Crystal Violet (500mg diluted in 100mL 80% Methanol)
3.4.3 Procedure
Metaphase Spreads: Plate 5 × 105 cells of each MCF10A pool (or 3–5 clones of each line) in four-100mm plates.
When cells reach 50–65% confluency, add KaryoMax® Colcemid™ to each plate at a final concentration of 0.1μg/mL.
Incubate cells in KaryoMax® Colcemid™ for 3–4 hrs.
Label 50mL tubes for each MCF10A pool
After the KaryoMax® Colcemid™ incubation, collect the media from the plates into the 50mL tubes.
Wash plates with 5mL of 1XPBS and collect into the same 50mL tubes containing the collected media.
Add 4mL of 0.05% trypsin to each plate and incubate at 37°C/5% CO2 for 15–20 min, until the rest of the cells detach from the plate.
Collect the cells into the same 50mL tube.
Centrifuge the cells at 1000rpm for 5 min.
Aspirate supernatant leaving about 500μL left. Resuspend cells by flicking the tube. Avoid pipetting the cells from this point on.
Wash cells in 10mls of 1XPBS
Centrifuge at 1000rpm for 5 min.
Aspirate supernatant leaving about 500μL. Resuspend cells by flicking the tube.
Add 10mls of pre-warmed KCL (75mM) to the cells. Add the first 3mls dropwise slowly while gently mixing, add the last 7mls a little faster while still mixing.
Incubate the cells in KCl for 30 min in a 37°C water bath.
Add 10–15 drops of pre-chilled (-20°C) Carnoy’s fixative while gently mixing.
Centrifuge the cells at 1000rpm for 5 min.
Remove the supernatant by pipetting, taking care not to disturb the cell pellet, leaving about 500μL of liquid. Resuspend the cells by flicking the tube gently.
Slowly add 10mls of pre-chilled Carnoy’s fixative dropwise while gently mixing. It should take about 2 min to add the first 2mls of Carnoy’s fixative.
Incubate at room temperature for 10 min.
Centrifuge the cells at 1000rpm for 5 min.
Repeat fixation (steps 18–21) 3–4 times.
During the fixation steps, prepare the glass slides for later by rinsing with Carnoy’s fixative and allow to dry. Also, place a container (such as a tip box cover or plastic microcentrifuge box top) with wet Kimwipes at 37°C to prepare a pseudo-humidified chamber.
After centrifugation in the final fixation step, remove the supernatant as before leaving an appropriate amount of fixative based on the approximate number of cells presumed in the cell pellet. This volume is typically between 0.5–2mls of fixative. If the cells are overly dense or overly sparse after dropping the first slide, the volume can be adjusted.
Place your P20 pipetman into a clamp setup and place on a shelf above your bench such that the distance between the pipetman and the bench surface is about 3–4 feet. See Figure 4A for a picture of our setup.
Place the prepared glass slide into a humidified chamber, with the frosted side end of the slide on the edge of the chamber and the other end of the slide inside the chamber such that the slide angled.
Line up the slide under your pipetman, drop 10–15μL of cells onto the slide. Wait about a minute to allow the cells to spread out on the slide.
Check the spread under a light microscope (20X objective). In Figure 4B and C, cells in the metaphase stage with well spread chromosomes are annotated by (*). The spread in Figure 4B is a little sparse, however cells are fairly well dispersed. The spread in Figure 4C has several areas of clustered cells (indicated by the arrows), emphasizing the importance of step 16 in the procedure. If you don’t add several drops of fixative prior to centrifugation after the KCl incubation the cells can stick together. If the cells are sparse, add 5–6mls of Carnoy’s fixative, centrifuge, and resuspend the cells in a smaller volume. If the cells are closely-packed then increase the volume, drop another 10–15μL of cells, and check again under the microscope.
Allow slides to dry for 24–48 hrs.
Add 1–2 drops of mounting media containing DAPI and place coverslip on slide.
Image metaphase spreads 24 hrs after mounting.
Score metaphase spreads for aberrations. We typically score fragments, fusions and breaks. Figure 4D shows “normal” chromosomes in a metaphase spread from a pool of MCF10A cells expressing a WT BER enzyme. In contrast, the metaphase spread from cells expressing a BER variant in Figure 4E contains chromosomes with aberrations including fusions and breaks as annotated. Representative chromosomes with fusions or breaks are included below the metaphase spread to serve as a reference. Fragments, which are not represented in the images, are acentric pieces of DNA.
Micronucleus Assay: Plate 5 × 105 cells of each MCF10A pool (or 3–5 clones of each line) in three-100mm plates.
When cells reach 50–65% confluency, add 6μg/mL of Cytochalasin B (Cyt-B) per plate for 24 hrs at 37°C/5% CO2.
After 24 hrs, trypsinize cells and collect into a 50mL conical tube.
Centrifuge at 1000 rpm for 5 min.
Resuspend cells in 7mls of 0.075M KCl.
Incubate in KCl in a 37°C water bath for 10 min.
After 10 min, add 3mls of methanol to each tube and incubate for 1 hr at room temperature.
Centrifuge the cells at 1000 rpm for 5 min.
Fix cells twice in Carnoy’s fixative as described above.
Drop cells as described above for metaphase preparation.
Stain slides in 10μg/mL Acridine Orange for 10–20 min in a coplin jar wrapped in tin foil to protect from the light.
Briefly rinse slides with water. Keep the slides in a coplin jar in water until ready to score.
Slides should be scored immediately after staining using a microscope with a 500/520 excitation/emission filter. Take a slide out of the coplin jar in the dark and cover with a coverslip using the residual water as an aqueous buffer between the coverslip and the slide.
With hand tally counters in each hand score binucleated cells with one counter and binucleated cells with micronuclei with the other counter. Figure 5A shows a representative image of a binucleated cell with a distinct single micronucleus. For more detailed information on scoring micronuclei refer to The in vitro micronucleus technique (Fenech, 2000). Score 1000 binucleated cells per slide.
- Ouabain resistance assay: You will need to plate the following for each MCF10A pool in 100mm plates:
- Untreated cells
- 103 (cells)
- 102
- Ionizing radiation (IR) only
- 8 GY: 104, 103, 102
- Ouabain only
- 50nM ouabain: 104, 5×104
- 100nM ouabain: 5×104, 105
- Ouabain + IR
- 50nM ouabain + 8GY IR: 106, 7.5×105, 5×105
- 100nM ouabain + 8GY IR: 106, 7.5×105, 5×105
The following day, add 50nM or 100nM ouabain to the ouabain only plates. Also, irradiate the IR only and IR+ ouabain plates. IR serves as a positive control for this assay.
72 hrs after IR exposure, begin treatment with ouabain for IR+ ouabain plates.
Change the media on the ouabain and IR+ ouabain plates every 3–4 days.
Colonies that can be scored will form within about 8–10 days on the untreated plates and IR only plates. Colonies that can be scored will form within about 2–4 weeks on the ouabain and IR+ ouabain plates.
To accurately count colonies, use a counting pen. The plates will be marked with black marker after counting colonies, therefore take images of the plates prior to scoring.
To calculate the mutation frequency, the total number of colonies growing on ouabain is divided by the total number of colonies grown in the absence of ouabain.
Figure 4.
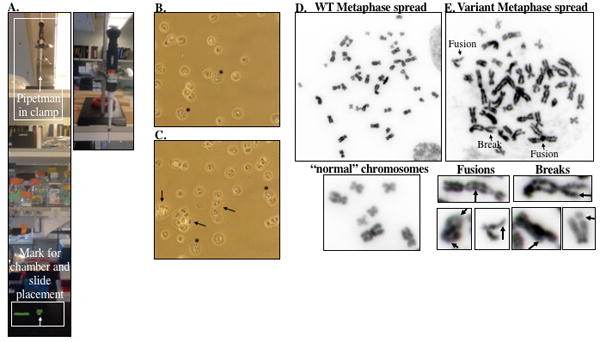
A. Picture of the pipetman clamp setup for dropping cells for metaphase preparation and micronucleus analysis. The pipetman is held in a clamp that is placed on a shelf about 3 ft above the bench surface. A mark is placed on the bench top for appropriate placement of the slides. B. Bright field images using a 20X objective of metaphase spreads prior to mounting. Cells in metaphase are indicated by (*). The arrows point to clumps of cells stuck together. Checking the slides after dropping cells serves both to adjust the volume if the cells are too sparse or confluent and to get an idea of how many cells are in metaphase prior to mounting. D-E. Representative image of metaphase spreads from a cell expressing WT or BER variant enzyme taken using a 100X objective on an Olympus BX50 research microscope with QImaging Retiga 2000R digital camera. The WT metaphase spread (D) contains “normal” chromosomes without aberrations. However, the BER variant metaphase spread (E) contains multiple chromosomes with aberrations, including breaks and fusions, as indicated in the image. Additional representative chromosomes with fusions or breaks are included below (E) to serve as a guide for scoring aberrations.
Figure 5.
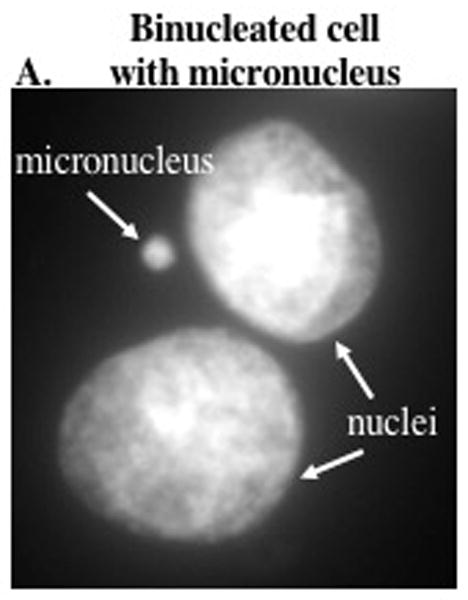
Representative image of a binucleated cell with a micronucleus.
3.4.4 Notes
If cells are more than 75% confluent you should re-plate and start again. KaryoMax® Colcemid™, also known as demecolcine, inactivates spindle fiber formation thus arresting cells in metaphase. If the cells are not in log phase growth, then incubation with KaryoMax® Colcemid™ will not have any effect on the number of cells arrested in metaphase.
Shorter incubations in KaryoMax® Colcemid™ will reduce the number of cells in metaphase at the time of collection, however longer incubations will condense the chromosomes which makes scoring aberrations more difficult. For MCF10A cells, we have found incubation with KaryoMax® Colcemid™ for 3–4 hrs is optimal, but this is likely cell type dependent.
Cells in mitosis become more rounded and therefore readily detach from the plate without trypsin. Therefore, it’s important to collect all washes to ensure optimal recovery of cells in metaphase.
Incubation in potassium chloride will swell the cells so that when dropped onto glass slides the chromosomes will spread nicely. The length of incubation is highly cell dependent. We have optimized 30 min in KCl for MCF10A cells, however if working with another cell line you will need to optimize the length of incubation.
It’s important to add 10–15 drops of Carnoy’s fixative prior to centrifugation to keep the cells from sticking together.
Incubation at 4°C for 30 min is also acceptable.
If you cannot setup a pipetman in a clamp, you can also hold the pipetman while dropping your cells onto the slides. However, it is more difficult to consistently get the drops onto the slide.
Different protocols recommend varying distances between the pipetman and the slide. For MCF10A cells, we found dropping the cells from heights of 3–4 feet produces excellent metaphase spreads that are easily scored for aberrations.
If exposing the cells to damaging agents, then add the Cyt-B 24 hrs after IR or H2O2 exposure or treat cells for 24 hrs with DNA damaging agent (i.e. cisplatin) then add Cyt-B upon removal of the agent.
Incubation in KCl is shorter for the micronucleus assay, as compared to preparation of metaphase spreads, because while it is desirable for the cells to swell it’s important that the nuclei and, if present, micronuclei remain in close proximity for scoring purposes. The optimal time in our hands is 10 min in KCl but this is a step that may need to optimized for each cell line and condition.
We have found that acridine orange produces high background if mounted in mounting media (such as Aquapolymount or antifade mounting media).
If the slide starts to dry out while imaging, place back in water and re-position the coverslip.
The dilutions listed are a starting point based on our results. The number of cells plated may need to be adjusted, particularly in the ouabain + IR (or any damaging agent) conditions.
We suggest trying 50nM and 100nM when beginning the assay. Once an optimal concentration of ouabain is determined then perform future assays using only that optimal concentration. The optimal concentration of ouabain will depend on the mutation frequency of the cells, the cytotoxicity of ouabain, and the combined cytotoxicity of ouabain and the chosen damaging agent.
The dose of irradiation may need to be adjusted. The dose recommended (8GY) generates mutations but is also cytotoxic. In combination with the cytotoxicity of ouabain, it may decrease the number of scoreable colonies. The dose can be decreased to 4GY, which will be less cytotoxic but will also generate fewer mutations and therefore may decrease the numbers of cells that acquire ouabain resistance (also leading to less colonies).
IR is just a suggested damaging agent to generate mutations. Other damaging agent can be used in place of IR, such as hydrogen peroxide or menadione.
The counting pen marker dries out quickly. If the black marker on the counter pen dries up and stops working, count colonies with the counter pen in one hand and a marker in the other in order to mark the colonies you have already counted.
3.5 Preliminary mechanistic analysis
The information gathered about a BER variant from the studies performed thus far should be used as a guide for the design of additional studies to determine the mechanisms contributing to the cellular outcomes. For instance, if expression of a BER variant increases the levels of chromosomal aberrations and micronuclei, as determined by the studies in the previous section, then one possibility is that the BER variant does not remove polymerase-blocking lesions from DNA thereby disrupting replication. Disruption of replication can be tested indirectly by measuring DNA double-strand breaks (DSBs) that arise during S-phase as a result of collapsed replication forks and directly by measuring the initiation and progression of sites of replication using the DNA fiber assay [for an example see (Nemec et al., 2016)]. A phosphorylated form of the histone variant H2A.X (γH2A.X) is used as a marker to measure DSBs and propidium iodide (PI) is used to measure DNA content to delineate G1, S and G2/M phases of the cell cycle by flow cytometry. Using this method, the percentage of cells positive for γH2A.X in S-phase can be compared between WT-expressing and BER variant expressing cells. The basic principles of flow cytometry and flow cytometric data analysis will not be described here but more information can be obtained from (Adan, Alizada, Kiraz, Baran, & Nalbant, 2016). The DNA fiber assay utilizes the incorporation of halogenated thymidine analogs into the DNA during replication in the cell allowing visualization of sites of DNA replication. The DNA fiber assay provides information about the efficiency of replication by permitting the measurement of mean replication tract length or replication fork speed as well as the measurement of replication structures including elongating replication forks and stalled replication forks. Described here are only two assays that can be used to tease out the mechanisms driving transformation. Depending on the specific function of your BER variant of interest and the results of the assays already performed, another avenue of investigation may be more appropriate.
3.5.1 Equipment
Microcentrifuge
Microcentrifuge tubes
Flow cytometer
Rocker
Software for flow cytometric analysis (we use FlowJo software)
Angled platform
Staining jar
Glass slides
3.5.2 Buffers and Reagents
PBS
MCF10A complete media
DMEM/F12 (no serum) supplemented with 1% penicillin/streptomycin
0.05% trypsin
Halt Protease/phosphatase inhibitor tablets (Roche)
EDTA (100mM stock)
1% paraformaldehyde with 5mM EDTA diluted in PBS
70% ethanol (pre-chilled -20°C)
Flow buffer: 1% bovine serum albumin (BSA)(w/v), 0.1% Triton X-100. 5mM EDTA diluted in PBS
Mouse monoclonal anti-phospho-Histone H2A.X (Ser139) (Millipore, clone JBW301), 1/300 dilution in flow buffer
AlexaFlour® 488 goat anti-mouse IgG (H+L) antibody (Invitrogen), 1/500 dilution in flow buffer
BD PI (Propidium iodide)/RNase Staining Buffer supplemented with 5mM EDTA
44μm pore filter mesh
12×75mm round-bottom polystyrene tubes, nonsterile without caps
Iododeoxyuridine (IdU), 5mM dissolved in DMSO (Sigma)
Chlorodeoxyuridine (CIdU), 2.5mM dissolved in ddH2O (Sigma)
Methanol: Acetic Acid fixative (3:1)
2.5M HCl
Fiber lysis solution: 50mM EDTA, 0.5% SDS, 200mM Tris-HCl pH 7.5 diluted in ddH2O.
5% BSA (w/v) in PBS
Rat monoclonal anti-BrdU primary antibody (Abcam, ab6326)
Mouse monoclonal anti-BrdU primary antibody (BD biosciences, 347580)
AlexaFlour® 647 goat anti-rat IgG (H+L) antibody (Invitrogen)
Mounting media (No DAPI)
3.5.3 Procedure
Plate 1×105 cells per well in triplicate for each cell line in 6-well plates in MCF10A complete growth media. You will need three wells for each line for 0, 2, 4, and 8 hr time-points as well as one well for each cell line for an unstained control, and PI only control).
The next day, remove the media and wash the cells 2X with PBS to remove any residual serum in the wells.
Add serum-free DMEM/F12 media (SFM) containing 1% pen-strep to each well. Incubate the cells in SFM for at least 24 hrs to halt the cells in G0/G1.
Replace SFM with MCF10A complete growth media 16–18 hrs prior to exposing cells to a damaging agent (i.e. IR).
Prior to exposing cells to damaging agent, prepare 0.05% trypsin with 5mM EDTA and one protease/phosphatase inhibitor tablet per 10mL of trypsin. Warm in a 37°C water bath.
Expose cells to DNA damaging agent 16 hrs post-release from serum deprivation.
Place 0 hr plates on ice until collection. Put the rest of the plates back into the incubator.
For each time point collection- wash the cells with PBS (unstained and PI only plates can be collected at any time point).
Trypsinize cells with 0.05% trypsin, 5mM EDTA, protease/phosphatase inhibitors prepared earlier until the cells are just detaching from the plate. Be careful to minimize the time the cells are in trypsin.
While the cells are trypsinizing, label microcentrifuge tubes for each sample containing 500μL of PBS with 5mM EDTA and place on ice.
Once the cells have detached, immediately pipet the cells into the labeled microcentrifuge tubes on ice.
Centrifuge cells in a microcentrifuge that is in a cold room or refrigerated (4°C) at 190xg for 5 min.
Using a vacuum aspirator, remove the supernatant carefully without disturbing the cell pellet, leaving about 50μL of supernatant to avoid losing cells.
Add 500μL of 1% paraformaldehyde with 5mM EDTA diluted in PBS and vortex gently to resuspend to cells.
Incubate cells in fixative for 20 min on ice.
Centrifuge samples at 4°C at 5200xg for 5 min. Aspirate supernatant leaving about 50μL of supernatant to avoid losing cells.
Add 1mL of cold (-20°C) 70% EtOH to tubes and vortex gently to resuspend cells. Incubate for at least 30 min at -20°C (see note 5 below).
Centrifuge samples at 5200xg for 5 min at 4°C. Aspirate supernatant leaving about 50μL of supernatant.
Add 1mL of flow buffer and vortex gently to resuspend cells. Incubate on ice for 15 min to rehydrate the cells.
Centrifuge samples at 5200xg for 5min. Aspirate the supernatant leaving about 50μL of supernatant.
Add 100μL of diluted (1/300) mouse monoclonal anti-phospho-Histone H2A.X primary antibody to each sample. Vortex gently to resuspend.
Incubate overnight on a slant on a rocker in a cold room (4°C).
The next day, add 1mL flow buffer to samples and vortex gently. Incubate on ice for 10 min.
Centrifuge samples at 5200xg for 5 min. Aspirate supernatant leaving about 50μL.
All steps starting from this point must be performed in the dark. Add 100μL of diluted (1/500) AlexaFlour® 488 goat anti-mouse secondary antibody to samples and vortex gently.
Incubate at room temperature for 1 hr with gentle shaking.
Add 1mL flow buffer and vortex gently. Incubate at room temperature for 10 min.
Centrifuge samples at 5200xg for 5 min. Aspirate the supernatant leaving about 50μL.
Add 450μL of PI/RNase buffer with 5mM EDTA to samples. Vortex gently to mix.
Incubate at room temperature for 15 min.
Pipet through 44μm pore filter mesh into 12×75mm round-bottom polystyrene tubes. Keep samples in the dark and at 4° until analysis.
Figure 6 provides a brief overview of cell cycle/γH2A.X gating analysis. However, for a comprehensive, detailed protocol describing gating and analysis of γH2A.X labeling refer to (Kataoka, Bindokas, Duggan, Murley, & Grdina, 2006) and (Tanaka, Huang, Halicka, Zhao, Traganos, Albino et al., 2007). If you have a flow cytometry facility at your institution, that would be the best resource for learning about flow cytometry.
DNA Fiber Assay: Plate 1×105 cells for each cell line in 35mm plates in MCF10A complete growth media.
Culture the cells until about 40–50% confluent.
Add IdU to the media at a final concentration of 25μM. Place plates back into the incubator for 20 min.
During the IdU incubation, prepare 250μM (final concentration) of CIdU in MCF10A complete growth media for the second pulse.
Aspirate the media and wash the cells three times with PBS.
Add MCF10A complete media containing 250μM CIdU. Place back into the incubator for 20 min.
After 20 min, trypsinize the cells and perform a cell count.
Resuspend the cells in PBS at 1×106–2.5×106 cells/mL (which will equal 2000–5000 cells in 2μL in the next step). Place cells on ice.
Prepare the slides and the setup to spread the DNA fibers. Label glass slides for each sample in pencil.
Place the labeled glass slides that will be spotted first into a container that will eventually be angled (see Figure 7A).
Setup a surface that will allow the container with the glass slides to be tilted to a 15° angle to allow the fibers to spread along the slide. See Figure 7A-E for an example setup. Once everything is setup, you are ready to spot your cells onto the slides.
Spot 2μL of cell suspension onto labeled glass slides.
Air dry the cell suspension for 5 min or until the volume is greatly reduced but not dry.
Pipet 7μL of fiber lysis solution into the cell suspension and gently mix with the pipet tip.
Incubate for 2 min to allow for the cells to lyse.
After 2 min of lysis, tilt the slides to a 15° angle. Watch carefully as the solution spreads down the slide. If the solution is traveling rapidly down the slide decrease the angle to 10°, however if the solution is not moving fast enough the angle can be increased to 25°-30°. It should take about 30 s-1 min for the solution to reach the bottom of the slide. The speed at which the solution spreads down the slide will significantly impact the dispersal of the DNA fibers along the slide.
Place the slides horizontally to allow to dry. Once dry a thin opaque line will be visible along the slide. Mark the beginning of the stretched fibers with a pencil to help with locating the fibers under the microscope. The line will not be visible after staining.
Immerse the slides in methanol: acetic acid (3:1) in a glass staining jar for 10 min.
Wash the slides with distilled H2O then immerse the slides in 2.5M HCl for 2.5 hrs.
Wash the slides three times in PBS for 5 min each.
Remove the slides from the staining jar and touch the bottom of the slide to a kimwipe to remove an excess PBS.
Place the slides horizontally in a humidified chamber and add 100μL of 5% BSA to each slide. Cover the slides carefully with a coverslip and incubate at room temperature for 20 min.
Dilute the primary antibodies in 5% BSA at the following concentrations: mouse anti-BrDU 1:25; rat anti-BrDU 1:200.
Gently slide the coverslip to the bottom of the slide to remove. If the coverslip sticks to the slide rehydrate the slide in PBS until the coverslip becomes loose and can be removed with ease.
Add 50μL of the diluted primary antibody solution to each slide. Cover with a coverslip carefully to avoid creating any bubbles or air pockets between the coverslip and slide.
Incubate at room temperature for 1.5 hrs.
During this incubation, dilute the secondary antibodies at 1/1000 in 5% BSA and keep in the dark at 4°C until ready to use.
Remove the coverslip as before. Wash the slides three times in PBS for 5 min each.
All steps from this point forward should be performed in the dark: Add 50μL of the diluted secondary antibodies and carefully cover with a coverslip as before.
Incubate in the dark for 2.5 hrs.
Remove the coverslips as before and wash the slides three times in PBS for 5 min each.
Add a drop of mounting media (such as Prolong Gold or Vectashield without DAPI) to the slides and apply a coverslip. Gently press the coverslip, pushing out any bubbles between the coverslip and the slide. Remove any excess mounting media along the sides of the slide with a kimwipe.
Image the DNA fibers using a 60x objective. Use the pencil mark made in step 49 as a guide to find the DNA fibers on the slide. Typically, there will be a main fiber bundle but the fibers in the main bundle are too entangled and therefore cannot be analyzed. Move away from the main fiber bundle down the slide to find areas with nicely separated fibers. Figure 7G is a great example of a field of view to image for analysis. The fibers are well separated and easily visible. Also, most of the fibers in Figure 7G have a single stretch of IdU incorporation and a single stretch of CIdU incorporation which is important for analysis. If you can find fields of view similar to Figure 7G, then 10 pictures of each slide will suffice. If you find all your slides are extremely crowded with DNA fibers and you cannot identify images that can be analyzed, in the next assay you will want to dilute your cells labeled with IdU and CIdU with unlabeled cells at a ratio that is appropriate to get images for analysis (for instance 200μL labeled cells: 800 μL unlabeled cells or 500 μL labeled cells: 500 μL unlabeled cells). For more information on the DNA fiber assay, as well as a step-by-step video of the procedure refer to the following link: http://www.jove.com/video/3255/visualization-dna-replication-vertebrate-model-system-dt40-using-dna.
Figure 6.
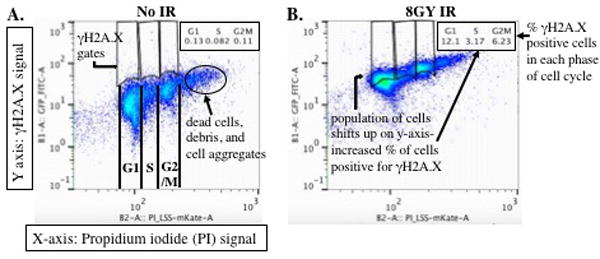
A-B. Sample 2-D dot plots of MCF10A cells synchronized using serum-deprivation then either untreated (No IR) or exposed to 8GY of IR. In these dot plots, the PI signal is plotted on the X-axis. PI binds to double stranded DNA by intercalating between base pairs and therefore provides a measurement of DNA content in the cells. DNA content is then used to identify cell populations in G1, S and G2/M phases of the cell cycle. The cell populations being analyzed in these dot plots were not previously filtered for live single cell populations using forward and side scatter. By gating for the live, single cell population using forward and side scatter, you will mostly eliminate the population clustered to the right of cells in G2/M (A) which includes cell doublets and aggregates, dead cells and debris. The γH2A.X signal is plotted on the Y-axis. To determine the percentage of γH2A.X positive cells in your samples, draw gates that delineate G1, S, and G2/M phases of the cell cycle. Use your untreated sample plots (A) to determine placement of the gates, serving as a baseline for γH2A.X expression. Copy and paste those gates from the untreated sample plots into the treated sample plots. The software will measure the percentage of cells within the entire cell population for each phase of the cell cycle (i.e. the number cells, referred to as events, that were measured during data collection on the flow cytometer) that is positive for γH2A.X expression based on the gates provided.
Figure 7.
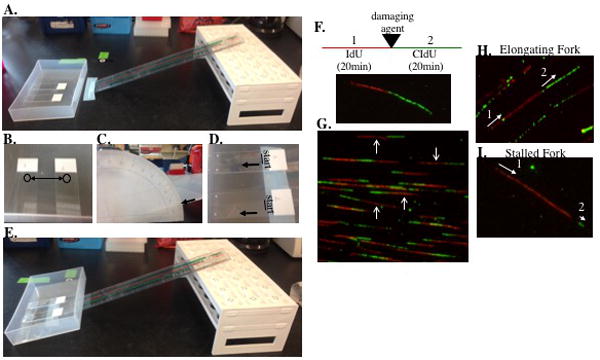
A. An image of our setup for the lysis step in the DNA fiber assay through to spreading the DNA fibers onto the slides. The slides are in a container that will sit flat and even on bench. The ruler and rack (turned upside down) serves as a platform for tilting the slides in the container to a 15° angle after lysis. B. An image of slides with 2μL of cells spotted at the top of the slide. C, E. After lysis, the container is moved up the ruler until the bottom edge of the container (corresponding to the bottom of the slides) reaches a 15° angle. D. The lines at the top of the slide mark the start of the DNA fibers and the arrows indicated the movement of the DNA fibers as they move down the slide. This is a critical step in the DNA fiber assay. How the fibers move down the slide will significantly impact the quality of images for analysis. Slide the container holding the lysed cells slowly up the ladder (or whatever you use to make an angled surface) watching the movement of the fiber solution carefully. The fibers should spread slowly down the slide. If the fiber solution is moving down the slide rapidly, lower the angle to a 10° angle or until the movement is slowed. F. A schematic of the experiment and an image of a representative DNA fiber with incorporation of IdU in the first pulse (which is red in the image) and then incorporation of CIdU in the second pulse (which is green in the image). To measure the effect of a damaging agent on fork progression or fork collapse, the cells are treated with a damaging agent after the first pulse with IdU. G. A representative image with well separated, elongating fibers. The arrows indicate a few of the elongating fibers in the image that can be used to measure mean tract length. H. A representative image of an elongating fiber indicating the progression of replication. 1-labels the first pulse with IdU then two-labels the second pulse with CIdU. Therefore, replication was moving from left to right in this fiber. I. A representative image of a stalled replication fork. IdU is incorporated during the first pulse, however CIdU is not incorporated or there is initial incorporation without continuation (as seen in this image). Elongating replication forks can be used to measure mean tract length. Replication structures, including elongating and stalled forks, can also be scored.
3.5.4 Notes
The easiest way to setup the experiment for collection is to plate three wells of WT-expressing cells and three wells of BER variant expressing cells in 1 6-well plate for each time point.
Residual serum in the wells will result poor synchronization.
MCF10A cells enter early S-phase about 16 hrs after release from serum-deprivation. Therefore, you want to time release from serum-deprivation with treatment with a damaging agent 16–18 hrs later. Cells will be collected at 0, 2, 4, and 8 hrs post-treatment, therefore we release the cells from serum deprivation in the late afternoon and treat with a damaging agent the next morning.
The addition of EDTA to reagents in this protocol is meant to prevent cells from aggregating or sticking together.
The antibody binds to a H2A.X that has been phosphorylated at serine 139 forming what is known as gamma H2A.X (γH2A.X), therefore it is imperative that the protease/phosphatase inhibitor tablet you choose to use contains phosphatase inhibitors, not just protease inhibitors.
Once the cells are in 70% EtOH, you can store your samples at -20°C for up to one month.
To filter the cells, you can also use the Falcon® 5mL Round Bottom Polystyrene Test Tube with Cell Strainer Snap Cap. The cell strainer is a 35μm mesh filter that can be detached from the round bottom tube once cells are filtered through by centrifugation. It’s a more convenient method however also more expensive and sometimes allows cell aggregates through probably because of the force applied during centrifugation.
It’s important to filter the cells prior to analyzing on the flow cytometer to remove large cell aggregates. If you do not perform this step, there is a good chance you will clog the sample injection port and the machine will need to be repaired prior to running any more samples.
DNA Fiber assay: Similar to metaphase preparation, the cells should be in log phase growth to maximize the number of cells in S phase when pulsing the cells with the thymidine analogs.
Washing the plates with PBS is a critical step to ensure complete removal of IdU from the cells. Incomplete washing of the cells will result in overlapping signals in the DNA fiber due to incorporation of both IdU and CIdU in the second pulse. The fiber will appear yellow instead of green and therefore cannot be scored.
Do not prepare more than 5 slides at a time. While adding lysis solution to the cells, you will not be able to move fast enough to avoid other slides drying out in the meantime. Once the cell solution dries out, lysis will be incomplete and the DNA will not spread down the slide as desired.
Once the slides have been fixed in methanol: acetic acid (3:1), they can be stored at 4°C until the next day.
We make a humidified chamber using a slide box and place wet paper towels in the center below the slides, however any container that allows the slides to lay flat horizontally with a source of humidity will work.
We have found that a 100x objective restricts to much light and makes the fibers difficult to image.
Microfluidics and other types of instruments can be purchased to spread DNA fibers on the slides.
References
- Adan A, Alizada G, Kiraz Y, Baran Y, Nalbant A. Flow cytometry: basic principles and applications. Crit Rev Biotechnol. 2016:1–14. doi: 10.3109/07388551.2015.1128876. [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, et al. Inherited variants of MYH associated with somatic G:C–>T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- Asagoshi K, Yamada T, Okada Y, Terato H, Ohyama Y, Seki S, et al. Recognition of formamidopyrimidine by Escherichia coli and mammalian thymine glycol glycosylases. Distinctive paired base effects and biological and mechanistic implications. J Biol Chem. 2000;275:24781–24786. doi: 10.1074/jbc.M000576200. [DOI] [PubMed] [Google Scholar]
- Aspinwall R, Rothwell DG, Roldan-Arjona T, Anselmino C, Ward CJ, Cheadle JP, et al. Cloning and characterization of a functional human homolog of Escherichia coli endonuclease III. Proc Natl Acad Sci U S A. 1997;94:109–114. doi: 10.1073/pnas.94.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au KG, Cabrera M, Miller JH, Modrich P. Escherichia coli mutY gene product is required for specific A-G----C.G mismatch correction. Proc Natl Acad Sci U S A. 1988;85:9163–9166. doi: 10.1073/pnas.85.23.9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Cooper DN, Vasquez KM. Mechanisms of base substitution mutagenesis in cancer genomes. Genes (Basel) 2014;5:108–146. doi: 10.3390/genes5010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan L, Bambara RA. Flap endonuclease 1. Annu Rev Biochem. 2013;82:119–138. doi: 10.1146/annurev-biochem-072511-122603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boparai KS, Dekker E, Van Eeden S, Polak MM, Bartelsman JF, Mathus-Vliegen EM, et al. Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology. 2008;135:2014–2018. doi: 10.1053/j.gastro.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Chen Y, Cunningham F, Rios D, McLaren WM, Smith J, Pritchard B, et al. Ensembl variation resources. BMC Genomics. 2010;11:293. doi: 10.1186/1471-2164-11-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donigan KA, Hile SE, Eckert KA, Sweasy JB. The human gastric cancer-associated DNA polymerase beta variant D160N is a mutator that induces cellular transformation. DNA Repair (Amst) 2012;11:381–390. doi: 10.1016/j.dnarep.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donigan KA, Sun KW, Nemec AA, Murphy DL, Cong X, Northrup V, et al. Human POLB gene is mutated in high percentage of colorectal tumors. J Biol Chem. 2012;287:23830–23839. doi: 10.1074/jbc.M111.324947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide L, Luna L, Gustad EC, Henderson PT, Essigmann JM, Demple B, et al. Human endonuclease III acts preferentially on DNA damage opposite guanine residues in DNA. Biochemistry. 2001;40:6653–6659. doi: 10.1021/bi0028901. [DOI] [PubMed] [Google Scholar]
- Fenech M. The in vitro micronucleus technique. Mutat Res. 2000;455:81–95. doi: 10.1016/s0027-5107(00)00065-8. [DOI] [PubMed] [Google Scholar]
- Freidberg EC, Wood RD, Walker GC, Siede W. DNA Repair and Mutagenesis. Second. Washington, DC: ASM Press; 2006. [Google Scholar]
- Galick HA, Kathe S, Liu M, Robey-Bond S, Kidane D, Wallace SS, et al. Germ-line variant of human NTH1 DNA glycosylase induces genomic instability and cellular transformation. Proc Natl Acad Sci U S A. 2013;110:14314–14319. doi: 10.1073/pnas.1306752110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Science Signaling. 2013;6:pl1–pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, C V, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biology. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S, Biswas T, Roy R, Izumi T, Boldogh I, Kurosky A, et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. J Biol Chem. 1998;273:21585–21593. doi: 10.1074/jbc.273.34.21585. [DOI] [PubMed] [Google Scholar]
- Kataoka Y, Bindokas VP, Duggan RC, Murley JS, Grdina DJ. Flow cytometric analysis of phosphorylated histone H2AX following exposure to ionizing radiation in human microvascular endothelial cells. J Radiat Res. 2006;47:245–257. doi: 10.1269/jrr.0628. [DOI] [PubMed] [Google Scholar]
- Lang T, Dalal S, Chikova A, DiMaio D, Sweasy JB. The E295K DNA polymerase beta gastric cancer-associated variant interferes with base excision repair and induces cellular transformation. Mol Cell Biol. 2007;27:5587–5596. doi: 10.1128/MCB.01883-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Maitra M, Starcevic D, Li SX, Sweasy JB. A DNA polymerase beta mutant from colon cancer cells induces mutations. Proc Natl Acad Sci U S A. 2004;101:6074–6079. doi: 10.1073/pnas.0308571101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu AL, Tsai-Wu JJ, Cillo J. DNA determinants and substrate specificities of Escherichia coli MutY. J Biol Chem. 1995;270:23582–23588. doi: 10.1074/jbc.270.40.23582. [DOI] [PubMed] [Google Scholar]
- McGoldrick JP, Yeh YC, Solomon M, Essigmann JM, Lu AL. Characterization of a mammalian homolog of the Escherichia coli MutY mismatch repair protein. Mol Cell Biol. 1995;15:989–996. doi: 10.1128/mcb.15.2.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DL, Donigan KA, Jaeger J, Sweasy JB. The E288K colon tumor variant of DNA polymerase beta is a sequence specific mutator. Biochemistry. 2012;51:5269–5275. doi: 10.1021/bi3003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemec AA, Bush KB, Towle-Weicksel JB, Taylor BF, Schulz V, Weidhaas JB, et al. Estrogen Drives Cellular Transformation and Mutagenesis in Cells Expressing the Breast Cancer-Associated R438W DNA Polymerase Lambda Protein. Mol Cancer Res. 2016;14:1068–1077. doi: 10.1158/1541-7786.MCR-16-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemec AA, Donigan KA, Murphy DL, Jaeger J, Sweasy JB. Colon cancer-associated DNA polymerase beta variant induces genomic instability and cellular transformation. J Biol Chem. 2012;287:23840–23849. doi: 10.1074/jbc.M112.362111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemec AA, Murphy DL, Donigan KA, Sweasy JB. The S229L colon tumor-associated variant of DNA polymerase beta induces cellular transformation as a result of decreased polymerization efficiency. J Biol Chem. 2014;289:13708–13716. doi: 10.1074/jbc.M114.550400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope MA, David SS. DNA damage recognition and repair by the murine MutY homologue. DNA Repair (Amst) 2005;4:91–102. doi: 10.1016/j.dnarep.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Rivera B, Castellsague E, Bah I, van Kempen LC, Foulkes WD. Biallelic NTHL1 Mutations in a Woman with Multiple Primary Tumors. N Engl J Med. 2015;373:1985–1986. doi: 10.1056/NEJMc1506878. [DOI] [PubMed] [Google Scholar]
- Schork NJ, Murray SS, Frazer KA, Topol EJ. Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev. 2009;19:212–219. doi: 10.1016/j.gde.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizova DV, Keh A, Taylor BF, Sweasy JB. The R280H X-ray cross-complementing 1 germline variant induces genomic instability and cellular transformation. DNA Repair (Amst) 2015;31:73–79. doi: 10.1016/j.dnarep.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjolund A, Nemec AA, Paquet N, Prakash A, Sung P, Doublie S, et al. A germline polymorphism of thymine DNA glycosylase induces genomic instability and cellular transformation. PLoS Genet. 2014;10:e1004753. doi: 10.1371/journal.pgen.1004753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slupska MM, Baikalov C, Luther WM, Chiang JH, Wei YF, Miller JH. Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J Bacteriol. 1996;178:3885–3892. doi: 10.1128/jb.178.13.3885-3892.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slupska MM, Luther WM, Chiang JH, Yang H, Miller JH. Functional expression of hMYH, a human homolog of the Escherichia coli MutY protein. J Bacteriol. 1999;181:6210–6213. doi: 10.1128/jb.181.19.6210-6213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- Sweasy JB, Lang T, DiMaio D. Is base excision repair a tumor suppressor mechanism? Cell Cycle. 2006;5:250–259. doi: 10.4161/cc.5.3.2414. [DOI] [PubMed] [Google Scholar]
- Sweasy JB, Lang T, Starcevic D, Sun KW, Lai CC, Dimaio D, et al. Expression of DNA polymerase {beta} cancer-associated variants in mouse cells results in cellular transformation. Proc Natl Acad Sci U S A. 2005;102:14350–14355. doi: 10.1073/pnas.0505166102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Huang X, Halicka HD, Zhao H, Traganos F, Albino AP, et al. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytometry A. 2007;71:648–661. doi: 10.1002/cyto.a.20426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace SS, Murphy DL, Sweasy JB. Base excision repair and cancer. Cancer Lett. 2012;327:73–89. doi: 10.1016/j.canlet.2011.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weren RD, Ligtenberg MJ, Kets CM, de Voer RM, Verwiel ET, Spruijt L, et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet. 2015;47:668–671. doi: 10.1038/ng.3287. [DOI] [PubMed] [Google Scholar]
- Yamtich J, Nemec AA, Keh A, Sweasy JB. A germline polymorphism of DNA polymerase beta induces genomic instability and cellular transformation. PLoS Genet. 2012;8:e1003052. doi: 10.1371/journal.pgen.1003052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, et al. Ensembl 2016. Nucleic Acids Research. 2016;44:D710–D716. doi: 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon DS, Wersto RP, Zhou W, Chrest FJ, Garrett ES, Kwon TK, et al. Variable levels of chromosomal instability and mitotic spindle checkpoint defects in breast cancer. Am J Pathol. 2002;161:391–397. doi: 10.1016/S0002-9440(10)64194-6. [DOI] [PMC free article] [PubMed] [Google Scholar]