

Abstract
Aim:
Until now, identification of drug targets for treatment of patients with specific stages of colorectal cancer (CRC) has remained a challenging field of research. Herein, we aimed to identify the key genes and regulatory networks involved in each stage of CRC.
Results:
The results of gene expression profiles were integrated with protein–protein interaction networks, and topologically analyzed. The most important regulatory genes (e.g., CDK1, UBC, ESR1 and ATXN1) and signaling pathways (e.g., Wnt, MAPK and JAK-STAT) in CRC initiation, progression and metastasis were identified. In vitro analysis confirmed some in silico findings.
Conclusion:
Our study introduces functional hub genes, subnetworks, prioritizes signaling pathways and novel biomarkers in CRC that may guide further development of targeted therapy programs.
Keywords: : biomarkers, CRC stages, hub genes, PPI networks, signaling pathways
Lay abstract
A comprehensive view of the molecular mechanisms involved in colorectal cancer (CRC) pathogenesis has been demonstrated. Cell cycle and signal transduction genes were contributors to CRC initiation and progression. In contrast to stage IV, the stages I–III of CRC shared a notable number of common genes suggesting their close gene signatures. The efficacy of these predictions has been validated using two colon cancer cell lines in vitro.
Graphical abstract
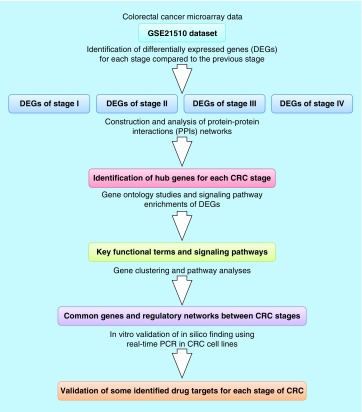
Colorectal cancer (CRC) is known as the third leading cause of cancer mortality worldwide, propagating by the acquisition of several genetic alternations influencing the expression and function of target genes. Adenomatous Polyposis Coli and TP53 are the most commonly mutated genes in familial/inherited and sporadic CRC [1]. CRC metastasis is a common phenomenon and the major lethal cause of CRC cases, as approximately 50% of CRC patients die within 5 years due to extensive metastatic stage [2]. So far, however, there has been little information about the molecular mechanisms underlying the ability of CRC cells to become invasive. Numerous studies have proved that the transition of CRC cells from early stages to late stages (metastasis) is associated with a significant change in the cellular gene expression profile [3]. These genetic alterations have led to the emergence of the novel strategies for personalized medicine programs [4]. As a result, Zhao et al. found an apparent difference between protein expression profile of nonmetastatic and metastatic CRC cell lines. They concluded that overexpression of HSP27 might play a key role in the acquisition of invasion and migratory properties by CRC cells [5]. Furthermore, Tan et al. introduced STMN1 gene as a crucial player in CRC cell migration and prognostic marker through a comparative proteome analysis of primary CRC cell lines HCT-116 and its metastatic derivative E1 [6]. Most recently, we have identified HOXB family as the most significant functional modules in metastatic CRC samples using a network biology approach. Consequently, we proposed a rationally designed anti-HOXB7 peptide for preventing CRC metastasis [7]. Despite, these valuable studies, the benign-to-malignant transition of CRC is a complicated multistep process and needs to be further elucidated in greater details.
Nowadays, computational systems biology has become a focus of many researchers to decipher complex biological systems through integrating the experimental and computational approaches [8]. Among the currently available methods, reconstruction of protein–protein interaction (PPI) networks are widely used for identification of key genes and functional modules involved in the cancer initiation and progression [9,10]. There are several network-based methodologies for modeling of biological systems that result in an identification of novel biomarkers in the human cancers and complex diseases [11]. To the best of our knowledge, no attempt has yet been made to monitor the dynamic changes of regulatory networks from normal to distant metastasis of the patients with CRC. Accordingly, no effective biomarkers have been introduced for detecting the early events of CRC metastasis. Hence, we assessed the gene expression changes of normal versus stage I (N-SI), stage I versus stage II (SI-SII), stage II versus stage III (SII-SIII) and stage III versus stage IV (SIII-SIV) of CRC cells to propose a biological model for CRC initiation, progression and invasion to other tissues. Additionally, the overlapping mechanisms between four studied CRC stages were elaborated in more details The observations uncovered that several common genes and subnetworks seem to be associated with CRC initiation and metastasis. These findings would help to gain insights into the major contributors involved in the regulation of CRC initiation, progression and invasion. Our study also provides novel potential biomarkers which can be considered to be therapeutic candidates for designing novel drugs with anti-CRC activity.
Materials & methods
Microarray gene expression data
A gene expression dataset (Gene Expression Omnibus [GEO] accession number: GSE21510) consisting of total 148 microarray samples were chosen from (GEO; www.ncbi.nlm.nih.gov/geo/). The samples had been accumulated from LCM and homogenized tissues of patients with colorectal cancer. Subsequently, the samples separately normalized using robust multiarray average method under R 2.6.2 statistical software with affy package from BioConductor. There was only one sample for stage 0, therefore, we excluded this stage from the study. Differentially expressed genes (DEGs) were determined by using the GEO2R tool [12]. Fold change > 1 or < −1 and p-value < 0.05 are the thresholds used in DEGs determination.
PPI networks
A comprehensive PPI was constructed for each stage using BisoGenet plugin of Cytoscape [13]. This plugin is a multitier application that builds the PPI based on the biomolecular relationships data curated from several PPI databases, including Database of Interacting Proteins, Biological General Repository for Interaction Datasets, Human Protein Reference Database, Biomolecular Interaction Network Database, Molecular INTeraction database (MINT) and IntAct [14–19]. In parallel, an intensive literature search was conducted to retrieve new PPIs from the data of most recent studies. The PPI networks were visualized and analyzed by Cytoscape and FunRich packages [20].
Identification of hub genes
Topological properties of the PPI networks were interpreted to detect the paramount functional hub genes. We used Network Analyzer, a network analysis plug-in of Cytoscape, to calculate the different topological parameters of resulted PPI networks. To this end, nine measures including Number of Undirected Edges, Degree, Betweenness Centrality, Clustering Coefficient, Closeness Centrality, Eccentricity, Neighborhood Connectivity, Topological Coefficient and Average Shortest Path Length were calculated for each PPI network.
Gene ontology (GO) & signaling pathway enrichment
Gene ontology (GO) studies were carried out utilizing Biological Networks Gene Ontology tool, a flexible and extendable Cytoscape's plugin [21]. This plugin finds GO terms over-represented in the given biological networks. Enrichr server was used as an alternative tool for validating the results of Biological Networks Gene Ontology tool [22]. A p-value less than 0.05 was chosen as the cut-off criterion for statistically over-represented GO terms. A signaling pathway enrichment was performed using SPEED web tool to identify the paramount signaling pathways underlying CRC initiation and invasion [23]. This server is a signaling pathway annotation tool that performs pathway enrichments based on the data obtained from previous pathway perturbation experiments.In parallel, the signaling pathways corresponding to the DEGs were also checked with the Kyoto Encyclopedia of Genes and Genomes [24]. This database consists of throughput information about genomic, cellular pathways and chemical compounds.
Construction of DEG-GO network for SIV of CRC
After determining DEGs of stage IV gene expression profile compared with the normal samples, a DEG-GO network was constructed to distinguish the major regulators of significantly over-represented GO terms. First, the up-regulated DEGs were separated and their biological roles were determined using DAVID server to understand the activated molecular mechanisms and signaling pathways in metastatic CRC cells compared with their adjacent normal tissue. The GO terms with p-value < 0.05 containing at least ten genes were considered to be significantly over-represented categories. Finally, a DEG-GO network was constructed and topological analysis of the network was performed using out-degree (the number of outgoing edges) and in-degree (the number of incoming edges) measures.
Identification of common regulatory networks between CRC stages
Stages 1–4 of CRC may share several common genes or modules and identification of these similarities can be helpful in CRC therapy programs. To this end, all DEGs obtained from the comparison of four stages with their adjacent normal tissues (with fold change >1 and <−1 and p-value < 0.05) were clustered using Venny 2.1 web tool [25]. The main subnetwork for both common up and down-regulated DEGs were separately extracted and interpreted. In parallel, GO analysis of common DEGs was conducted to understand the regulatory functions of these genes.
Cell culture, RNA extraction & real-time PCR
In order to validate the in silico findings, the expression level ATXN1 and CDK1, identified as hub genes in SIV of CRC, was quantified in human metastatic colon carcinoma cell line (SW620 cells) compared with NCM460, a normal human colon mucosal epithelial cell line. Briefly, SW620 and NCM460 cell lines were seeded in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Total RNA was extracted from SW620 and NCM460 cell lines (15–30 mg) using the miRNeasy Kit according to the manufacturer's instructions (Qiagen, USA). Subsequently, cDNA synthesis was performed utilizing a commercial cDNA synthesis kit (2-step RT-PCR kit, Vivantis, USA). The standard SYBR green-based quantitative real-time PCR (qRT–PCR) analyses were carried out in Real-time PCR Detection System ABI 7300 (Applied Biosystems, USA). Primer sequences of ATXN1, CDK1 and GAPDH, as internal control, are as follows: ATXN1-F (5′ CCCACCTTTCCCCTGCTGCG-3′), ATXN1-R (5′-GGTTCCTCCCCCGGGTCTCC-3′), CDK1-F (5′-GGAAACCAGGAAGCCTAGCA-3′), CDK1-R (5′-TGATTCAGTGCCATTTTGCC-3′), GAPDH-F (5′-TCCACCACCCTGTTGCTGTAG-3′) and GAPDH-R (5′-GGTTCCTCCCCCGGGTCTCC-3′). PCR cycling conditions were conducted in a total volume of 20 μl and containing 10 pmol of each primer and 2 μl of diluted cDNA template (800 ng cDNA). The thermal cycler conditions were composed of an initial step at 95°C for 5 min followed by 45 cycles of 95°C for 15 s, 60°C for 1 min and 72°C for 30 s and a step of 82°C for 5 s. To generate standard curves, the tenfold serial dilutions covering a range of the eluted cDNA was considered as a template in SYBR green qRT-PCR.
Statistical analysis
Statistical analysis of in vitro experiment was done using GraphPad Prism version 4 (GraphPad Software, CA, USA). P-values were calculated by two-sided Student's t-test and the values below 0.05 were considered to be significant.
Results
PPI networks properties
A PPI network was constructed for each CRC stage based on the experimentally validated PPI interactions. The PPI networks included 214 nodes and 169 edges (for PPI network of SI), 179 nodes and 219 edges (for PPI network of SII), 196 nodes and 152 edges (for PPI network of SIII), and 203 nodes and 146 edges (for PPI network of SIV). A decreasing tendency of the distribution of node degree implicated a biological scale-free pattern of four constructed PPI networks (data not shown). The main subnetworks were extracted from each PPI network which is illustrated in Supplementary Figure 1. CDK1, UBC, ESR1 and ATXN1 genes had the highest degree of connectivity in the main subnetworks of SI, SII, SIII and SIV, respectively.
Hub genes of each PPI network
Topological analysis of the PPI networks revealed several hub genes as major regulators of CRC initiation, progression and invasion. The results of all topological measures had a relatively high overlap. Interestingly, many of cell cycle-related genes were identified as hub genes in their corresponding PPI networks. The top ten hub genes derived from interpretation of each PPI network are listed in Table 1.
Table 1. . Top 10 hub genes identified from analysis of each protein–protein interaction network.
| NumUndE | D | BetCen | CluCoe | CloCen | E | NeiCon | TopCoe | Avg.ShoPat |
|---|---|---|---|---|---|---|---|---|
| Hub genes of SI of CRC | ||||||||
| CDK1 | CDK1 | PALB2 | CKS2 | NUP37 | PPAT | FBXO5 | NUP37 | CBX3 |
| NPM1 | NPM1 | CDK1 | NIFK | NUP160 | PAICS | TOP2A | NUP160 | EXOSC3 |
| CCNB1 | CCNB1 | CSE1L | SKP2 | NUP107 | DIMT1 | PTTG1 | NUP107 | EEF1E1 |
| SNRPD2 | SNRPD2 | GNL3 | CENPE | PALB2 | POLR1B | KIF20B | SKP2 | HELLS |
| BUB1 | BUB1 | BCCIP | NUP37 | CD44 | RPP40 | CEP55 | SPC25 | HSPE1 |
| CKS1B | CKS1B | NOP58 | NUP160 | MAD2L1 | HELLS | DDX21 | CENPE | RPP40 |
| DKC1 | DKC1 | NPM1 | DTL | PPARD | EEF1E1 | CPSF3 | DTL | POLR1B |
| RFC4 | GART | DKC1 | NUP107 | NCAPG2 | EXOSC3 | NUDCD1 | NIFK | DIMT1 |
| RFC5 | KIF11 | KPNA2 | SPC25 | SMC2 | CBX3 | CDC25C | NUF2 | EXOSC8 |
| DTL | EXOSC3 | CCNB1 | PPP40 | NUF2 | KIF20B | NUP107 | CDK1 | NPM1 |
| Hub genes of SII of CRC | ||||||||
| UBC | UBC | UBC | CEBPG | UBC | SORBS1 | DNAJC5 | SORBS1 | SLC35D1 |
| YWHAB | YWHAB | AURKA | ATXN3 | YWHAB | WIF1 | JAG1 | ZFP36L2 | ZNF713 |
| AURKA | AURKA | DNAJB12 | NCOA6 | AURKA | KLHL29 | SLC25A23 | PARD6B | MBTPS2 |
| SNRNP200 | SNRNP200 | YWHAB | CASK | SNRNP200 | YWHAB | PROCR | AAR2 | SYK |
| CEBPB | CEBPB | EIF6 | TPX2 | EIF4E2 | AURKA | G2E3 | TPX2 | PXMP4 |
| AMOTL2 | AMOTL2 | SNRNP200 | TLK2 | ARAF | SNRNP200 | TRERF1 | NUF2 | DYNLRB1 |
| TOP1 | COPS6 | EIF4E2 | PDHA1 | CDC25C | EIF4E2 | PIGU | TLK2 | TMEM189 |
| ARAF | ARAF | CEBPB | CTNNBL1 | TNFAIP3 | ARAF | ZNF687 | MAPRE1 | RALGAPB |
| COPS6 | TOP1 | AMOTL2 | HERC5 | CEBPB | CDC25C | DUSP1 | CEBPG | ARFGAP1 |
| EIF4EB | CDC25C | TPX2 | CEBPB | CASK | TOP1 | AURKA | PDHA1 | ZNF687 |
| Hub genes of SIII of CRC | ||||||||
| CABLES2 | ESR1 | TRIM23 | HIP1 | TRIM23 | HIP1 | RPS12 | HIP1 | CABLES2 |
| PCK1 | ARRB1 | ESR1 | ITSN1 | INTS9 | ITSN1 | AKT2 | ITSN1 | PCK1 |
| MGA | RYBP | ARRB1 | YY1 | TGFB1 | EPN1 | PAK1 | MED23 | MGA |
| CDK3 | YY1 | YY1 | E2F2 | INTS10 | PCK1 | IRS1 | FASN | CDK3 |
| EPN1 | CDK3 | AP2B1 | RYBP | ITGB8 | CABLES2 | EPN1 | RYBP | EPN1 |
| HIP1 | HIP1 | MED23 | AP2B1 | ESR1 | AP2B1 | PTAFR | CDK3 | HIP1 |
| ITSN1 | ITSN1 | E2F2 | TRIM23 | DMRT3 | FASN | YY1 | E2F2 | ITSN1 |
| PTAFR | FASN | CDK3 | ESR1 | CCDC25 | PTAFR | HIP1 | YY1 | PTAFR |
| FASN | AP2B1 | FASN | ARRB1 | MED23 | CDK3 | ITSN1 | AP2B1 | FASN |
| AP2B1 | E2F2 | HIP1 | EPN1 | HIP1 | ESR1 | AP2B1 | ESR1 | TRIM23 |
| Hub genes of SIV of CRC | ||||||||
| ATXN1 | ATXN1 | MAP4 | ATXN1 | UBXN1 | SNX2 | LDB1 | HIVEP1 | SCAMP1 |
| SMAD2 | SMAD2 | ATXN1 | SMAD2 | ARHGEF12 | PTPRK | WBSCR16 | FAM193B | SMG9 |
| TRIM23 | TRIM23 | SMAD2 | TRIM23 | SF3B6 | CREB1 | PUM1 | TRIP6 | FAM208B |
| INSR | INSR | FAM193B | INSR | AKT2 | SCAMP1 | LRSAM1 | FAM208B | SNX2 |
| YY1 | YY1 | TRIM23 | YY1 | ANKRD28 | INSR | HIVEP1 | YY1 | PTPRK |
| TRIP6 | TRIP6 | INSR | FAM208B | PHLDA3 | YY1 | FAM193B | INSR | CREB1 |
| FAM208B | FAM208B | YY1 | TRIP6 | MAP4 | RUNX1 | RUNX1 | TRIM23 | AFAP1L1 |
| SCAMP1 | SCAMP1 | TRIP6 | SCAMP1 | TAF9 | TNRC6B | TNRC6B | SMAD2 | TRIM23 |
| UBXN1 | UBXN1 | FAM208B | SMG9 | ARHGEF2 | AFAP1L1 | TRIP6 | ATXN1 | RUNX1 |
| SMG9 | SMG9 | UBXN1 | UBXN1 | YY1 | ATXN1 | SCAMP1 | MAP4 | PTPRK |
Avg.ShoPat: Average shortest path length; BetCen: Betweenness centrality; CloCen: Closeness centrality; CluCoe: Clustering coefficient; D: Degree; E: Eccentricity, NeiCon: Neighborhood connectivity; NumUndE: Number of undirected edge; TopCoe: Topological coefficient.
Cell cycle genes are remarkably involved in CRC initiation & progression
The results of GO demonstrated that, except for SII, the cell cycle pathway is remarkably involved in CRC initiation as well as its progression. The UBI–proteasome pathway, an intracellular process involved in protein degradation, was identified as the most significant enriched term for the PPI network which made from the DEGs of SII. Moreover, signal transduction seems to be critical in the regulation of gene expression in SII and SIII of CRC (Figure 1).
Figure 1. . Top five functional categories (categories with p-value < 0.05 containing at least 10 genes) that significantly enriched for each DEG list.

The right schematic representation of colon cancer progression is based on a model from Fearon and Vogelstein [26].
DEG: Differentially expressed gene; N: Normal stage; SI: Stage 1; SII: Stage 2; SIII: Stage 3; SIV: Stage 4.
Signaling pathway enrichment was carried out utilizing SPEED and Kyoto Encyclopedia of Genes and Genomes tools. In this step, only up-regulated DEGs were considered for pathway enrichment analyses to decipher activated signaling pathways during CRC progression. The results showed that the Wnt signaling pathway was the most significant signaling pathway in SI-SIII implying the indispensable role in initiation and progression of CRC from SI to SIII. However, this signaling pathway was not significantly enriched in SIV suggesting that the metastasis of CRC is not considerably dependent on Wnt signaling pathway activity. The MAPK signaling pathway was shown to be especially active in SI and SIII. In contrast, the JAK-STAT signaling pathway was substantially enriched in the DEGs of SII and SIV (Figure 2).
Figure 2. . Signaling pathway enrichment of the DEGs of four CRC stages.

MAPK signaling pathway (green line) and JAK-STAT signaling pathway (red line) had a negative correlation during colorectal cancer progression. Every significant hit in SPEED had an FDR <0.05.
DEG: Differentially expressed gene; CRC: Colorectal cancer; FDR: False discovery rate.
DEG-GO network construction for distant metastasis stage of CRC
The DEGs of SIV were obtained from analysis of gene expression profile of this stage compared with the adjacent normal cells. The up-regulated DEGs with fold change larger than 1 and p-value smaller than 0.05 were selected for GO studies. The results of GO revealed that the cell cycle related pathways are actively involved in CRC invasion. Furthermore, gene regulatory of these DEGs with their neighbors remarkably contributed to cell proliferation events. (Figure 3).
Figure 3. . Gene enrichment and network analysis of CRC's SIV gene signature.

(Left) GO of the DEGs obtained from gene expression analysis of CRC's SIV compared to adjacent normal tissue. (Right) Biological networks of these genes (green nodes) and their neighbors (gray nodes).
GO: Gene ontology; DEG: Differentially expressed gene; CRC: Colorectal cancer; SIV: Stage 4
In order to identify the most important up-regulated DEGs involved in the aforementioned functional categories of CRC's SIV, a DEG-GO network was also reconstructed. Detailed interpretation of this network based on out-degree measure resulted in the identification of several hub genes such as BUB1B, CENPE, CCNB1, FBXO5, MAD2L1, DLGP5, ANAPC10, CDK1, BRCA2, NUSAP1, CENPF, ANLN, NDC80 and KIF18A belong to the most significant enriched GO terms. The in-degree analysis of resultant DEG-GO network revealed several important intracellular molecular functions, particularly cell cycle associated pathways, as the most highly regulated pathways by the up-regulated DEGs of CRC's SIV (Figure 4).
Figure 4. . DEG-GO network constructed for up-regulated genes of CRC's SIV when compared to its adjacent normal tissue.

Nodes (DEGs) with higher and lower out-degree measures are shown in bigger and smaller circles, respectively. Nodes (GO terms) with higher in-degree measures are indicated in dark colors, while nodes with lower in-degree measures are shown in bright colors.
DEG: Differentially expressed gene; GO: Gene ontology; CRC: Colorectal cancer; SIV: Stage 4.
The SI-SIV of CRC share a high number of common genes
Clustering analysis of the DEGs obtained from each CRC stage compared to the respective normal tissues revealed that the four studied CRC stages (SI-SIV) share a notable number of overlapped genes involved in multiple cellular processes. Prior to clustering, up-regulated and down-regulated DEGs were determined and separated. The results of clustering identified 817 (24.2%) common up-regulated DEGs and 490 (19%) common down-regulated DEGs in all studied CRC stages. Nevertheless, the highest proportion of common genes was observed between SI, SII and SIII (1274 genes among up-regulated DEGs and 774 genes among down-regulated DEGs) (Figure 5).
Figure 5. . Clustering of CRC stages according to their gene signatures.

Clustering of (A) up-regulated and (B) down-regulated DEGs resulted from preprocessing of CRC's SI-SIV compared to their adjacent normal tissues.
CRC: Colorectal cancer; DEG: Differentially expressed gene.
Functional annotation of these common genes indicated that the majority of common up-regulated genes were actively involved in regulation of cell cycle (94 genes) and mitosis (57 genes). However, several other pathways such as regulation of RNA processing, DNA repair and protein metabolism were also significantly enriched. In contrast, the common down-regulated DEGs obviously participated in the regulation of apoptosis and intracellular signaling cascade (results not shown). Detailed analysis of these genes indicated that 21 genes (out of 94 genes) have a direct role in cell cycle pathway (Figure 6).
Figure 6. . Relationship between cell cycle and common up-regulated DEGs identified between SI-SIV of CRC (red color).

Activation, inhibition and undirected interactions are shown with solid green lines, solid red lines and dotted black lines, respectively. The cell cycle pathway was extracted from KEGG database.
DEG: Differentially expressed gene; SI-SIV: Stage 1 to Stage 4; CRC: Colorectal cancer; KEGG: Kyoto Encyclopedia of gene and genome.
The main subnetwork was extracted from the PPI network constructed for common up-regulated DEGs and is depicted in Supplementary Figure 2. Calculation of several centrality measures such as Betweenness centrality and degree showed that CAND1 gene is the most significant hub gene in the subnetwork. However, some other genes including CDK2, EED, BRCA1, CDK1, BARD1, SRPK1, NPM1, PCNA, SRSF2, EZH2, HNRNPA1, HIST1H4C and HNRNPU were also distinguished as important hub genes.
The enriched cellular functions for the SI-SIV common genes were examined. Our observations revealed that cell proliferation and apoptosis are the most significant functional categories enriched in common up-regulated and down-regulated genes, respectively. We further compared the expression levels of these genes during SI–SIV of CRC. The results indicated that cell proliferation genes had the highest expression levels in SI and SIV. However, apoptosis-related genes are down-regulated in SIII of CRC. ANLN and CDK1 genes had the highest overexpression level in all CRC stages compared with adjacent normal cells, whereas STAT1 was the most down-regulated gene in all CRC stages (Supplementary Figure 3).
ATXN1 &CDK1 have significantly deregulated in metastatic SW620 cells
The expression levels of ATXN1 and CDK1 were normalized to the level of the GAPDH housekeeping gene which showed the most homogenous expression in both SW620 and NCM460 cell lines. Consistent with our bioinformatics analyses, the results of real-time PCR indicated that the expression of ATXN1 (down-regulated, p-value < 0.001) and CDK1 (up-regulated, p-value < 0.05) have significantly altered in metastatic SW620 cells compared with the normal colon NCM460 cells (Figure 7). This in vitro validation suggests that the introduced novel biomarkers of CRC can be reliably considered to be promising candidates for therapeutic applications.
Figure 7. . Gene expression analysis using qRT-PCR.

Expression level of (A) ATXN1 and (B) CDK1 in SW620 and NCM460 cell lines. Changes in expression level were found to be significant (p < 0.05).
qRT-PCR: Quantitative real-time PCR.
Discussion
Our study was aimed at modeling CRC initiation and invasion using deep analyses of microarray data. The results of this in silico study propose several potential candidates for the treatment of CRC as well as early diagnosis of this cancer. Consistent with previous studies, we found numerous important regulatory genes and transcription factors associated with CRC progression. These findings provide support for the efficiency of systems biology approaches in the identification of novel CRC biomarkers.
We found out that CDK1, UBC, ESR1 and ATXN1 are the most important hub genes of SI, SII, SIII and SIV of CRC, respectively. Among these hub genes, CDK1 and UBC were up-regulated in CRC samples compared with adjacent normal cells, while ESR1 and ATXN1 were down-regulated. CDK1 is a member of Cyclin-dependent kinases (CDKs) family, which are serine/threonine kinases whose deregulation is associated with cancer progression. Hence, these cell cycle regulators have been targeted in a variety of human cancers [27]. Studies have shown that inhibition of CDK1 enhances the sensitivity of CRC cells to anticancer compounds through caspase-dependent signaling pathways [28,29]. The UBC, also called second ubiquitin-conjugating enzyme or E2, is a member of ubiquitin-proteasome degradation system which contributes to the regulation of cell proliferation by mediating the selective degradation of target proteins. This enzyme also catalyzes a particular type of polyubiquitination, ‘Lys-63’-linked polyubiquitination, which does not result in protein degradation and leads to an enhanced cell survival through regulating cell cycle and DNA repair pathways [30,31]. Therefore, up-regulation of UBC may enhance the survival of CRC cell through maintaining the cell cycle and DNA repair abilities of the cell. ESR1 is a nuclear hormone receptor that encodes estrogen receptor α involved in the regulation eukaryotic cell proliferation and differentiation. Although, ESR1 is frequently amplified in breast cancer cells [32], methylation-dependent silencing of this gene by CGI (CpG island) has been previously reported [33]. CGI regions are unmethylated short and dispersed sequences of DNA with a high frequency of CpG dinucleotides which are highly associated with the bulk genome. Furthermore, evaluation of the ESR1 methylation status uncovered that significant methylation of this gene occurred in patients with CRC and is associated with markedly diminished ESR1 expression in colorectal tumors examined [34]. This suggests the possibility of tumor suppressor activity for ESR1 in CRC. Accordingly, estrogens might reduce CRC development via preventing the DNA methylation of ESR1 [35]. ATXN1 is a chromatin-binding factor that functions as a corepressor involved in transcriptional repression by directly repressing CBF1, a transcription factor that is crucial for the Notch signaling pathway [36]. It is now widely accepted that Notch pathway is substantially required for initiation of CRC as well as maintenance and self-renewal of CRC initiating cells [37]. Furthermore, Sonoshita et al. found that activation of DAB1/Dab1, a Notch signaling target gene, can promote CRC invasion and metastasis [38]. Kim and colleagues have found a negative crosstalk between Notch and Wnt/β-catenin signaling pathways [39]. Interestingly, the signaling pathway enrichment revealed that Wnt signaling is actively involved in regulation of SI, SII and SIII of CRC, but the activity of this signaling pathway has significantly diminished during SIV.
Wnt signaling pathway was found to be a key player in the initiation and progression on CRC, but not in metastasis stage. It is now well understood that Wnt signaling is a critical event for initiating the genesis of most CRCs, especially through loss of its negative regulator, Adenomatous Polyposis Coli [40]. Moreover, both β-catenin-dependent and β-catenin-independent Wnt signaling enhance CRC progression via regulating epithelial–mesenchymal transition [41]. For decades, components of this signaling pathway served as interesting targets in human CRC. In addition to the importance of Wnt signaling pathway, we found a negative correlation between JAK-STAT and MAPK signaling pathways during CRC progression suggesting a dual regulatory role of JAK-STAT/MAPK cascades in CRC progression and invasion. Wu and colleagues reported that cooperation of PAR2/MAPKs/NF-κB signal transduction pathways promotes proliferation and migration of colon cancer cell line SW620 [42]. The crosstalk between these signaling pathways is complex and each pathway can regulate the activation of the other at multiple levels. Consistent with this, Krasilnikov and colleagues have found that PI3K and MAPK signaling down-regulate JAK-STAT signaling in human melanoma cells via inhibiting tyrosine phosphorylation of JAK/STATs [43]. However, STAT1 was identified as the most down-regulated gene in all CRC stages. In our previous study, this gene was also found to be down-regulated in metastatic CRC samples compared with the nonmetastatic samples. Modular analyses of CRC metastasis-derived PPI network corroborated that STAT1 functions in one of the most important protein complexes correlated with CRC metastasis [7]. Considering the role of MAPK and JAK-STAT signaling pathways in the regulation of cell proliferation, we suggest that down-regulation of each pathway is recouped by activation of the other pathway. This mechanism of regulation may be indispensable for providing the favorable microenvironment for CRC development [44,45].
Interestingly, the clustering analyses demonstrated a remarkable overlap between all studied CRC stages. Functional annotation of common DEGs revealed that majority of common up- and down-regulated genes are obviously contributed to the regulation of cell proliferation and apoptosis, respectively. CAND1, CDK2, EED, BRCA1 and BARD1 were identified as the most important hub genes up-regulated in all CRC stages. Several evidence have indicated the disruption of CAND1 in prostate cancer [46]. Nevertheless, the function of this gene in CRC has poorly been investigated. CDK2 is a serine/threonine kinase which its activity is dispensable for controlling cell cycle and mitosis [47]. Inhibition of CDK2 in CRC is substantially associated with enhanced cell death and suppressed cell proliferation implying its crucial function in CRC development [48,49]. Consistent with our results, EED and BARD1 have been proposed as promising prognostic markers in CRC and their expression correlate with CRC progression and aggressive clinical behavior [50,51]. BARD1 is an E3 ubiquitin-protein ligase which acts as a heterodimer with BRCA1 (BRCA1-BARD1). This protein heterodimer mediates the formation of ‘Lys-6’-linked polyubiquitin chains, and subsequently regulates a wide range of intracellular pathways such as DNA repair, regulation of protein degradation, transcription and genomic stability [52,53].
Conclusion
Our findings suggest a dynamic model of CRC initiation, progression and invasion. The results were highly compatible with previous studies. Moreover, several novel biomarkers were also proposed for each stage of CRC. The cell cycle and signal transduction genes were apparently involved in regulation of CRC stages. Moreover, Wnt, MAPK and JAK-STAT signaling pathways seemed to be crucial in the regulation of CRC stages. However, the results of this study need to be investigated in greater details by further experimental methods.
Future perspective
Despite decades of investigations, clinicians are still faced with numerous challenges in the diagnosis and treatment of patients with a specific stage of CRC. This might be due to deregulation of particular gene signatures in each CRC stage. Thus, identification of drug targets based on molecular mechanisms of each CRC stage can be very helpful in targeted therapy of CRC. In this study, we separately analyzed gene expression profiles of each CRC stage (i.e., SI-SIV) and integrated the results of gene expression analysis with PPI networks. Several genes and regulatory networks were found in each CRC stage. Cell cycle and signal transduction genes showed critical roles in early stages of CRC. Intriguingly, a crosstalk between MAPK and JAK-STAT signaling pathways during CRC progression was also predicted in silico. Although, a part of in silico findings was validated in vitro, there is a need to further validate this study's results. Moreover, parallel analysis of other datasets (e.g., microarray or next generation sequencing data) would increase the reliability of these predictions.
Summary points.
Background
Rapid and robust screening for biomarkers in each stage of colorectal cancer (CRC) may result in early diagnosis and reliable treatment programs.
Computational integration of gene expression profiles with protein–protein interaction networks have shown robustness in identifying key target genes and regulatory networks in human cancers.
Experiment
Microarray dataset GSE21510 was obtained from gene expression omnibus database and analyzed with GEO2R.
Cytoscape and FunRich packages were used for construction and analyses of protein–protein interaction networks.
BiNGO, SPEED and DAVID tools were used for functional enrichment of differentially expressed genes.
Results & discussion
CDK1, UBC, ESR1 and ATXN1 genes were detected as the most significant hub genes of SI, SII, SIII and SIV of CRC, respectively.
Cell cycle genes were highly involved in SI-SIII of CRC suggesting their crucial role in CRC initiation and progression.
Wnt signaling pathway was particularly enriched in SI-SIII, but not in SIV. Whereas, MAPK and JAK-STAT signaling pathways had a negative correlation during CRC stages.
A high number of common genes were identified between SI, SII and SIII implying their close biological properties.
Some in silico findings were validated in vitro implicating that the results of this study can be used in CRC diagnosis and prevention programs.
Supplementary Material
Footnotes
Authors’ contributions
M Poorebrahim studied design and concept, did main manuscript writing and bioinformatics analyses. M Asghari, MF Abazari, H Bokharaie, MN Aleagha, V Poortahmasebi, H Askari, S Torabinejad, A Ardalan and N Negaresh did manuscript writing, bioinformatics analyses and provided language editing. A Ataie and P Pazooki performed in vitro analysis.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Open access
This work is licensed under the Creative Commons Attribution 4.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Armaghany T, Wilson JD, Chu Q, Mills G. Genetic alterations in colorectal cancer. Gastrointest. Cancer Res. GCR. 2012;5(1):19. [PMC free article] [PubMed] [Google Scholar]; • Discusses the basic concepts of genetic integrity and the consequences of defects in the DNA repair as well as epigenetic alterations relevant to colorectal cancer.
- 2.Chirica M, Leconte M, Oberlin O, Dousset B. Surgical treatment of liver metastasis in patients with colorectal cancer. Presse. Medicale (Paris, France: 1983) 2012;41(1):58–67. doi: 10.1016/j.lpm.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 3.Riihimaki M, Hemminki A, Sundquist J, Hemminki K. Patterns of metastasis in colon and rectal cancer. Sci. Rep. 2016;6:29765. doi: 10.1038/srep29765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duffy MJ. Personalized treatment for patients with colorectal cancer: role of biomarkers. Biomarkers. 2015;9(4):337–347. doi: 10.2217/bmm.15.3. [DOI] [PubMed] [Google Scholar]
- 5.Zhao L, Liu L, Wang S, Zhang Y-F, Yu L, Ding Y-Q. Differential proteomic analysis of human colorectal carcinoma cell lines metastasis-associated proteins. J. Cancer Res. Clin. Oncol. 2007;133(10):771–782. doi: 10.1007/s00432-007-0222-0. [DOI] [PubMed] [Google Scholar]
- 6.Tan HT, Wu W, Ng YZ, et al. Proteomic analysis of colorectal cancer metastasis: stathmin-1 revealed as a player in cancer cell migration and prognostic marker. J. Proteome Res. 2012;11(2):1433–1445. doi: 10.1021/pr2010956. [DOI] [PubMed] [Google Scholar]
- 7.Maryam I, Seyede SM, Seyed SA, Sadegh Azimzadeh SA, Sadegh A, Mansour P. HOXB7 and Hsa-miR-222 as the potential therapeutic candidates for metastatic colorectal cancer. Recent. Pat. Anticancer Drug. Discov. 2016;11:1–10. doi: 10.2174/1574892811999160628114857. [DOI] [PubMed] [Google Scholar]
- 8.Kitano H. Computational systems biology. Nature. 2002;420(6912):206–210. doi: 10.1038/nature01254. [DOI] [PubMed] [Google Scholar]; •• An interesting overview of computational systems biology approaches.
- 9.Ahn J, Yoon Y, Park C, Shin E, Park S. Integrative gene network construction for predicting a set of complementary prostate cancer genes. Bioinformatics. 2011;27(13):1846–1853. doi: 10.1093/bioinformatics/btr283. [DOI] [PubMed] [Google Scholar]
- 10.Poortahmasebi V, Poorebrahim M, Najafi S, et al. How hepatitis C virus leads to hepatocellular carcinoma: a network-based study. Hepat. Mon. 2016;16(2):e36005. doi: 10.5812/hepatmon.36005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayashida M, Akutsu T. Complex network-based approaches to biomarker discovery. Biomarkers. 2016;10(6):621–632. doi: 10.2217/bmm-2015-0047. [DOI] [PubMed] [Google Scholar]
- 12.Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets – update. Nucleic Acids. Res. 2013;41(D1):D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin A, Ochagavia ME, Rabasa LC, Miranda J, Fernandez-De-Cossio J, Bringas R. BisoGenet: a new tool for gene network building, visualization and analysis. BMC Bioinformatics. 2010;11(1):1–9. doi: 10.1186/1471-2105-11-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xenarios I, Salwinski L, Duan XJ, Higney P, Kim SM, Eisenberg D. DIP, the database of interacting proteins: a research tool for studying cellular networks of protein interactions. Nucleic Acids. Res. 2002;30(1):303–305. doi: 10.1093/nar/30.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stark C, Breitkreutz B-J, Chatr-Aryamontri A, et al. The BioGRID interaction database: 2011 update. Nucleic Acids Res. 2011;39(Suppl. 1):D698–D704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bader GD, Betel D, Hogue CW. BIND: the biomolecular interaction network database. Nucleic Acids Res. 2003;31(1):248–250. doi: 10.1093/nar/gkg056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prasad TK, Goel R, Kandasamy K, et al. Human protein reference database – 2009 update. Nucleic Acids Res. 2009;37(Suppl. 1):D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zanzoni A, Montecchi-Palazzi L, Quondam M, Ausiello G, Helmer-Citterich M, Cesareni G. MINT: a molecular interaction database. FEBS. Lett. 2002;513(1):135–140. doi: 10.1016/s0014-5793(01)03293-8. [DOI] [PubMed] [Google Scholar]
- 19.Kerrien S, Aranda B, Breuza L, et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2011;40:D841–D846. doi: 10.1093/nar/gkr1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pathan M, Keerthikumar S, Ang CS, et al. FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics. 2015;15(15):2597–2601. doi: 10.1002/pmic.201400515. [DOI] [PubMed] [Google Scholar]
- 21.Maere S, Heymans K, Kuiper M. BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 22.Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parikh JR, Klinger B, Xia Y, Marto JA, Blüthgen N. Discovering causal signaling pathways through gene-expression patterns. Nucleic Acids Res. 2010;38(Suppl. 2):W109–W117. doi: 10.1093/nar/gkq424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oliveros JC VENNY. An interactive tool for comparing lists with Venn Diagrams. 2007. http://bioinfogp.cnb.csic.es/tools/venny_old/venny.php
- 26.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 27.Zhou Q. Targeting cyclin-dependent kinases in ovarian cancer. Cancer Invest. 2017;35(6):367–376. doi: 10.1080/07357907.2017.1283508. [DOI] [PubMed] [Google Scholar]
- 28.Huang W-W, Ko S-W, Tsai H-Y, et al. Cantharidin induces G2/M phase arrest and apoptosis in human colorectal cancer colo 205 cells through inhibition of CDK1 activity and caspase-dependent signaling pathways. Int. J. Oncol. 2011;38(4):1067. doi: 10.3892/ijo.2011.922. [DOI] [PubMed] [Google Scholar]
- 29.Abal M, Bras-Goncalves R, Judde J-G, et al. Enhanced sensitivity to irinotecan by Cdk1 inhibition in the p53-deficient HT29 human colon cancer cell line. Oncogene. 2004;23(9):1737–1744. doi: 10.1038/sj.onc.1207299. [DOI] [PubMed] [Google Scholar]
- 30.David Y, Ziv T, Admon A, Navon A. The E2 ubiquitin-conjugating enzymes direct polyubiquitination to preferred lysines. J. Biol. Chem. 2010;285(12):8595–8604. doi: 10.1074/jbc.M109.089003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bothos J, Summers MK, Venere M, Scolnick DM, Halazonetis TD. The Chfr mitotic checkpoint protein functions with Ubc13-Mms2 to form Lys63-linked polyubiquitin chains. Oncogene. 2003;22(46):7101–7107. doi: 10.1038/sj.onc.1206831. [DOI] [PubMed] [Google Scholar]
- 32.Holst F, Stahl PR, Ruiz C, et al. Estrogen receptor alpha (ESR1) gene amplification is frequent in breast cancer. Nat. Genet. 2007;39(5):655–660. doi: 10.1038/ng2006. [DOI] [PubMed] [Google Scholar]
- 33.Belshaw NJ, Elliott GO, Williams EA, et al. Methylation of the ESR1 CpG island in the colorectal mucosa is an ‘all or nothing’ process in healthy human colon, and is accelerated by dietary folate supplementation in the mouse. Biochem. Soc. Trans. 2005;33(Pt 4):709–711. doi: 10.1042/BST0330709. [DOI] [PubMed] [Google Scholar]
- 34.Issa J-PJ, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat. Genet. 1994;7(4):536–540. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 35.Kennelly R, Kavanagh DO, Hogan AM, Winter DC. Oestrogen and the colon: potential mechanisms for cancer prevention. Lancet Oncol. 2008;9(4):385–391. doi: 10.1016/S1470-2045(08)70100-1. [DOI] [PubMed] [Google Scholar]
- 36.Tong X, Gui H, Jin F, et al. Ataxin-1 and brother of ataxin-1 are components of the notch signalling pathway. EMBO Rep. 2011;12(5):428–435. doi: 10.1038/embor.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sikandar SS, Pate KT, Anderson S, et al. NOTCH signaling is required for colon cancer initiating cell tumor formation, self-renewal and repression of secretory cell lineage differentiation. Cancer Res. 2010;70(4):1469. doi: 10.1158/0008-5472.CAN-09-2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sonoshita M, Itatani Y, Kakizaki F, et al. Promotion of colorectal cancer invasion and metastasis through activation of Notch–Dab1–Abl–RhoGEF protein TRIO. Cancer Discov. 2015;5(2):198–211. doi: 10.1158/2159-8290.CD-14-0595. [DOI] [PubMed] [Google Scholar]
- 39.Kim H-A, Koo B-K, Cho J-H, et al. Notch1 counteracts WNT/β-catenin signaling through chromatin modification in colorectal cancer. J. Clin. Invest. 2012;122(9):3248–3259. doi: 10.1172/JCI61216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Novellasdemunt L, Antas P, Li VS. Targeting Wnt signaling in colorectal cancer. A review in the theme: cell signaling: proteins, pathways and mechanisms. Am. J. Physiol. Cell. Physiol. 2015;309(8):C511–C521. doi: 10.1152/ajpcell.00117.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vincan E, Barker N. The upstream components of the Wnt signalling pathway in the dynamic EMT and MET associated with colorectal cancer progression. Clin. Exp. Metastasis. 2008;25(6):657–663. doi: 10.1007/s10585-008-9156-4. [DOI] [PubMed] [Google Scholar]
- 42.Wu Y, Zhang X, Zhou H, et al. Factor VIIa regulates the expression of caspase-3, MMP-9, and CD44 in SW620 colon cancer cells involving PAR2/MAPKs/NF-kappaB signaling pathways. Cancer Invest. 2013;31(1):7–16. doi: 10.3109/07357907.2012.743556. [DOI] [PubMed] [Google Scholar]
- 43.Krasilnikov M, Ivanov VN, Dong J, Ronai ZE. ERK and PI3K negatively regulate STAT-transcriptional activities in human melanoma cells: implications towards sensitization to apoptosis. Oncogene. 2003;22(26):4092–4101. doi: 10.1038/sj.onc.1206598. [DOI] [PubMed] [Google Scholar]; • Describes a negative crosstalk between MAPK and JAK-STAT signaling pathways in human melanoma cells.
- 44.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007;7(1):41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 45.Wu M, Chen X, Lou J, et al. TGF-beta1 contributes to CD8+ Treg induction through p38 MAPK signaling in ovarian cancer microenvironment. Oncotarget. 2016;7(28):44534–44544. doi: 10.18632/oncotarget.10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Korzeniewski N, Hohenfellner M, Duensing S. CAND1 promotes PLK4-mediated centriole overduplication and is frequently disrupted in prostate cancer. Neoplasia. 2012;14(9):799–806. doi: 10.1593/neo.12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98(6):859–869. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 48.Lim T-G, Lee S-Y, Huang Z, et al. Curcumin suppresses proliferation of colon cancer cells by targeting CDK2. Cancer Prev. Res. 2014;7(4):466–474. doi: 10.1158/1940-6207.CAPR-13-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beale G, Haagensen EJ, Thomas HD, et al. Combined PI3K and CDK2 inhibition induces cell death and enhances in vivo antitumour activity in colorectal cancer. Br. J. Cancer. 2016;115(6):682–690. doi: 10.1038/bjc.2016.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y-L, Gao X, Jiang Y, et al. Expression and clinicopathological significance of EED, SUZ12 and EZH2 mRNA in colorectal cancer. J. Cancer Res. Clin. Oncol. 2015;141(4):661–669. doi: 10.1007/s00432-014-1854-5. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y, Pilyugin M, Kuester D, et al. Expression of oncogenic BARD1 isoforms affects colon cancer progression and correlates with clinical outcome. Br. J. Cancer. 2012;107(4):675–683. doi: 10.1038/bjc.2012.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu-Baer F, Lagrazon K, Yuan W, Baer R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J. Biol. Chem. 2003;278(37):34743–34746. doi: 10.1074/jbc.C300249200. [DOI] [PubMed] [Google Scholar]
- 53.Wu-Baer F, Ludwig T, Baer R. The UBXN1 protein associates with autoubiquitinated forms of the BRCA1 tumor suppressor and inhibits its enzymatic function. Mol. Cell. Biol. 2010;30(11):2787–2798. doi: 10.1128/MCB.01056-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.