

Abstract
An astute modification of the plectin-1-targeting peptide KTLLPTP by introducing a C-terminal cysteine preceded by a tyrosine residue imparted a reducing property to the peptide. This novel property is then exploited to fabricate gold nanoparticles (GNP) via an in situ reduction of gold (III) chloride in a one-pot, green synthesis. The modified peptide KTLLPTPYC also acts as a template to generate highly monodispersed, spherical GNPs with a narrow size distribution and improved stability. Plectin-1 is known to be aberrantly expressed in the surface of pancreatic ductal adenocarcinoma (PDAC) cells while showing cytoplasmic expression in normal cells. The synthesized GNPs are thus in situ surface modified with the peptides via the cysteine residue leaving the N-terminal KTLLPTP sequence free for targeting plectin 1. The visual molecular dynamics based simulations support the experimental observations like particle size, gemcitabine conjugation and architecture of the peptide-grafted nanoassembly. Additionally, GNPs conjugated to gemcitabine demonstrate significantly higher cytotoxicity in vitro in two established PDAC cell lines (AsPC-1 and PANC-1) and an admirable in vivo antitumor efficacy in a PANC-1 orthotopic xenograft model through selective uptake in PDAC tumor tissues. Altogether, this strategy represents a unique method for the fabrication of a GNP based targeted drug delivery platform using a multifaceted peptide that acts as reducing agent, template for GNP synthesis and targeting agent to display remarkable selectivity towards PDAC.
Keywords: Gold Nanoparticle, Plectin-1, Peptide, Gemcitabine, Drug delivery, Pancreatic cancer, Xenograft
Graphical abstract
Tumor-selective uptake of Plectin-1 targeting peptide-modified gold nanoparticles ameliorates targeted delivery of gemcitabine in pancreatic ductal adenocarcinoma
1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest human cancers with a five-year survival rate of 8% only.1 It is estimated to have 53,670 new cases and cause 43,090 deaths in 2017 in the United States and the numbers have been steadily increasing over the years.1 The high mortality rate of PDAC patients is mainly attributed to an aggressive malignant phenotype, wide-spread metastasis and late-stage diagnosis.2 Current frontline treatments for PDAC such as FOLFIRINOX (a combination of leucovorin, 5-fluorouracil, irinotecan, and oxaliplatin) or Gemcitabine (Gem) with Abraxane provide modest survival benefits only.3, 4 Moreover, the majority of these drugs are not tumor-selective which ultimately results in systemic toxicity.5, 6 Development of receptor-targeted nano drug-delivery systems with the ability to specifically bind to receptors that are overexpressed in malignant cells is emerging as an attractive therapeutic alternative.7 Among them gold nanoparticles (GNPs) have attracted massive interest due to their ease of synthesis, chemical stability, excellent biocompatibility and unique optical properties.8, 9
One of the key advantages of GNPs is their synthetic versatility.10 By modulating the synthesis conditions, the size, shape and surface properties of the GNPs can be exquisitely tuned to obtain GNPs with diverse physicochemical, electrical and optical properties.11 The surface of the GNPs can also be easily tailored to regulate particle stability, solubility and their interaction with the biological environment.12 Most importantly, the ability of the GNP surface to bind thiols and amines provides a convenient way to introduce various molecules including dyes, peptides, antibodies and drugs that can be exploited in a multitude of biomedical applications, including imaging, diagnosis, delivery and therapy.13–16 Specifically, surface modification of GNPs with ligands that bind to the extracellular domain of the receptors with significantly higher expression in cancer versus normal cells has allowed selective delivery of therapeutic payload to tumor cells.17 For example, cetuximab-conjugated GNPs had been utilized to deliver gemcitabine to PDAC cells in an epidermal growth factor receptor (EGFR)-targeted manner.18, 19 Similarly, GNPs conjugated to folic acid or tamoxifen-derivative demonstrated selective uptake in ovarian and breast cancer cells respectively.20, 21
Plectin-1, a high molecular weight protein (~500 kDa) that plays a vital role in cytoskeleton network organization by linking intermediate filaments to microtubules and microfilaments, has been shown to be implicated in pancreatic cancer pathogenesis.22, 23 Previous studies demonstrated that plectin-1 is expressed at a basal level in normal pancreatic ductal cells but overexpressed in PanINs and PDAC.24, 25 However, the most intriguing observation is the aberrant cell membrane expression of Plectin-1 in PDAC cells while being mostly localized in the cytoplasm and nucleus in normal fibroblasts.24 Not surprisingly, it has been suggested as a useful biomarker for PDAC.24, 25 A plectin-1-targeting peptide (KTLLPTP) has been successfully used by several groups to specifically target PDAC cells of both mouse and human origin for imaging.26–29 In addition, a slightly modified sequence of the above peptide (KTLLPTPC) has been used to decorate the surface of polylactic-co-glycolic acid-Polyethylene glycol (PLGA-PEG) nanoparticles to deliver therapeutic cargo to PDAC cells in a targeted manner.30 However, other than a patent which mentions to explore the conjugation of plectin-1-targeting peptides on the surface of pre-synthesized GNP, there is no report in our knowledge that illustrates the use of plectin-1-targeting peptide-GNP nanocomposites.31
A variety of ligands including DNA, proteins, antibodies, peptides, polymers and surfactants have been exploited to synthesize and stabilize GNPs.32–38 However, the interaction between thiols and GNPs is the most extensively studied.39, 40 Natural thiols including cysteine (Cys) and reduced glutathione (GSH) as well as synthetic peptides with a cysteine residue have been widely used for stabilizing GNPs.36, 41–43 The gold core of the peptide-stabilized GNPs is typically attached to the peptide either through a single terminal thiolate or via multiple Cys within the same peptide sequence.36, 43 The standard synthesis of peptide-capped GNPs is usually achieved by the addition of peptides to pre-formed gold colloids (as described in the patent mentioned earlier) or via in situ reduction of gold salt by a reducing agent in the presence of peptides.31, 43, 44
In the present study, we have developed a one-pot green synthetic procedure to fabricate GNPs by exploiting multiple functionalities of a modified plectin-1-targeting peptide (KTLLPTPYC) that reduces Gold (III) Chloride Trihydrate and simultaneously acts as a template for controlling the growth of nascent gold nanoparticles. This allowed us to generate highly stable monodispersed spherical GNPs with a narrow size distribution that are in situ surface-modified with plectin-1-targeting peptides (PTP). Additionally we have employed two separate control peptides to understand the mechanism behind the GNP synthesis: one having the same sequence without the cysteine (KTLLPTPY) and a different sequence but having a C-terminal YC (KGHSGLMYC) for synthesizing gold nanoparticles using same method. We have performed physical characterization and analyzed the stability and gemcitabine conjugation efficiency of the GNPs. In addition, we have evaluated the gemcitabine release kinetics from the gemcitabine-conjugated GNPs (GNP-Gem). We also evaluated the in vitro and in vivo therapeutic efficacy of as prepared targeted drug delivery platform GNP-Gem in two established pancreatic cancer cell lines and a PANC-1 xenograft model respectively.
2. EXPERIMENTAL
2.1. Reagents
Gold (III) chloride trihydrate, sodium chloride and sodium hydroxide were purchased from Sigma. The peptides KTLLPTPYC, KTLLPTPY and KGHSGLMYC (>90% purity) were obtained from Mayo Clinic Proteomics Core Facility. Luciferin sodium salt was purchased from Gold Biotechnology. Antibody against Ki67 was purchased from Santa Cruz Biotechnology. Celltiter 96 Aqueous One Solution Cell Proliferation Assay was purchased from Promega. Gemcitabine was obtained as a premade formulation Gemzar from Mayo Clinic Pharmacy. RPMI-1640 medium and Dulbecco’s Modified Eagle’s Medium (DMEM) were purchased from Life Technologies. Fetal Bovine Serum was obtained from Atlanta Biologicals. Immunohistochemistry was performed using the IHC Select HRP/DAB kit from Millipore.
2.2. Cell lines used in the current study
Two pancreatic cancer cell lines AsPC-1 and PANC-1 were used in the present study. AsPC-1 (CRL-1682; purchased on January, 2006) and PANC-1 (CRL-1469; purchased on November, 2005) cells were from American Type Culture Collection (ATCC). No authentication of the cell lines was done by the authors. AsPC-1 cell line was maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. PANC-1 was maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10% FBS and 1% penicillin-streptomycin.
2.3. Synthesis and characterization of peptide-templated GNPs
A simple one-pot green synthetic route was utilized to synthesize peptide-templated GNPs. Briefly, Gold (III) Chloride Trihydrate and KTLLPTPYC (or KTLLPTPY or KGHSGLMYC) peptide were dissolved in Milli-Q water in 10:1 molar ratio and the mixture was stirred at 37 °C for 2 minutes. Then, 1M NaOH was added dropwise to raise the pH of the solution to 12. The color of the reaction mixture instantly changed from yellow to dark brown. The reaction kinetics was monitored by taking out 100 μl of the reaction mixture at different time intervals, diluting it to 1 ml with milli-Q water and measuring the surface plasmon resonance peak (SPR) (typically ~520 nm) using a Shimadzu Spectrometer. Dilution was necessary as the high concentration of GNPs was not practical for measurements. Stirring was continued for 16 hours and the color of the reaction mixture turned to a dark red color. The GNPs were isolated by ultracentifugation at 38000 RPM in a Beckmann Optima L-80 XP ultracentrifuge. The supernatant was discarded carefully without disturbing the loose pellet of GNPs at the bottom of the ultracentrifuge tubes. Finally the volume was made up to the original reaction volume with Milli-Q water to get GNP stock solution. However, this stock solution was quite concentrated for physical characterizations, so a working stock was prepared by 10× dilution of the initial stock solution in Milli-Q water. The absorption spectra and hydrodynamic size of the synthesized GNPs were analyzed using Shimadzu Spectrometer and Malvern zetasizer respectively. Particle size was also analyzed by Transmission Electron Microscopy (TEM) analysis. TEM studies were carried out using a JEOL-JEM 2100 instrument operating at 200kV. For a given sample, 200 microliters of a given well-dispersed solution were placed on a carbon-coated 200-mesh copper grid, followed by drying the sample at ambient conditions.
2.4. Gemcitabine conjugation to GNPs
For Gem conjugation the GNP stock solution was tenfold diluted in Milli-Q water and mixed with increasing concentrations of Gem. The resulting mixtures were stirred for 16 hours followed by spectroscopic analysis and ultracentrifugation at 38000 rpm as described previously. The Gem content of the supernatant was determined by measuring the absorbance at 268 nm using Shimadzu Spectrometer and comparing with a standard curve prepared with known concentrations of Gem. The amount of Gem conjugated to GNPs was then calculated by subtracting the Gem content in the supernatant from the initial added amount.
2.5. Stability of GNPs and Gemcitabine-conjugated GNPs
The synthesized GNPs were either incubated with increasing concentrations of NaCl for one hour, or kept in two different storage temperatures (room temperature and 4 °C) and at two different pH values (pH 7.4 and pH 5).The stability was analyzed by measuring any change in hydrodynamic size distribution. For GNP-Gem, stability was tested at room temperature and in presence of 10% FBS at 37°C.
2.6. Gemcitabine release profile
2 mL of GNP-Gem were placed in a dialysis tube (MWCO 3.5 kDa) and the dialysis tube was incubated in 40 mL of PBS in a 50 mL Centrifuge tube in a 37 °C rotary incubator. At different time intervals 1 mL of the dialysate was withdrawn and replenished with 1 mL PBS. The amount of Gem released in the dialysate was determined by measuring the absorbance at 268 nm. The cumulative release was calculated and plotted against time to get a release curve for Gem. Similar experiment was performed in pH 5.
2.7. In vitro cell viability assay
Approximately, 5×103 cells were seeded in 96-well plates and treated with increasing doses of GNP, Gem and GNP-Gem diluted in respective media. After 1 hour incubation, cells were washed thrice with PBS, overlaid with 100 μL media containing serum and antibiotics and incubated for another 72 hours. At the end of the incubation, cell viability was measured using Celltiter 96 Aqueous One Solution Cell Proliferation Assay (Promega) as per the manufacturer’s protocol. Briefly, the media containing the treatments were aspirated from the plate and washed with PBS. Then 100 μL media containing 20 μL One Solution reagent was added to each well. The plate was incubated at 37 °C for 30 minutes and absorbance at 492 nm was measures using Spectramax i3x. Percentage viability is calculated as follows: Viability (%) = 100* (ATreated − ABlank) / (AUntreated − ABlank). Fresh batches of GNP-Gem were prepared for in vitro studies and they were analyzed for gemcitabine content as described before.
2.8. In vivo tumor growth inhibition
6–8 weeks old male and female NOD-SCID-Gamma (NSG) mice were obtained from NCI and housed in the institutional animal facilities. All animal work was performed under protocols approved by Mayo Clinic Institutional Animal Care and Use Committee. Approximately, 1×106 luciferase-transfected PANC-1 cells suspended in 50 μl PBS were injected orthotopically into the pancreas of the mice. Tumors were allowed to grow for three weeks. Then mice were randomized into four groups of ten (5 male and 5 female mice in each group) and 50 μg/mouse Gem, GNP-Gem with an equivalent Gem amount or unconjugated GNPs were injected intraperitoneally three times a week for three weeks. Fresh batches of GNP-Gem were prepared for each treatment and they were analyzed for gemcitabine content as described before. Tumor growth was measured weekly using an IVIS bio-imager after injecting Luciferin. After three weeks of treatment, mice were sacrificed and tumors were harvested, measured with slide calipers and weighed. Tumor volumes were calculated using the formula: V=0.5 × a × b2, where ‘a’ is the longest tumor axis and ‘b’ is the shortest tumor axis.
2.9. Histological study
Tumors were harvested and fixed in neutral buffered 10% formalin at room temperature for 24 hours prior to embedding in paraffin and sectioning. Sections were deparaffinized and then subjected Hematoxylin and Eosin (H&E) and Ki67 staining according to manufacturer’s instructions (DAB 150, Millipore). Stable diaminobenzidine was used as a chromogen substrate and the sections were counterstained with a hematoxylin solution. Slides were digitized with Aperio AT2 slide scanner (Leica) and analyzed with imagescope software (Leica).
2.10. Statistical analysis
Either OriginPro 2016 from Origin Lab Corporation or Microsoft Excel 2010 is used for data analysis. The independent-samples t-test was used to test the probability of significant differences between groups. Statistical significance was defined as p<0.05 (*) and statistical high significance was defined as p<0.01 (**). Error bars were given on the basis of calculated SD values where applicable.
3. RESULTS AND DISCUSSION
3.1. Synthesis of peptide-templated GNPs
A number of studies have mimicked the biomineralization process observed in microorganisms for fabrication of inorganic nanostructures where proteins obtained from biological sources are used as catalysts or templates for nucleation and growth of those nanostructures.45–48 Similarly, small peptides have also been utilized as templates for the fabrication of nanomaterials including noble metal nanoparticles.49–51 However, majority of peptide-templated synthesis of metal nanoparticles involve the use of an additional reducing agent to reduce metal salts to metal atoms, the peptides merely acting as a template for nucleation and growth control.14, 36, 43, 44 Only a few studies reported the usage of peptides as in situ reducing agents, however, the resulting nanoparticles are not uniform in size indicating lack of a controlled reaction environment.49
The current study employed a different approach to produce peptide-capped GNPs in a simple one-pot green synthetic route. This unique synthesis strategy is commonly well-acclaimed for commercialization purpose as it eliminates variability from multiple steps of complex reactions. Here, we utilized KTLLPTPYC peptide both as an in situ reducing agent and template to prepare PTP-grafted GNPs from gold (III) chloride trihydrate. We used this peptide since previous reports demonstrated the efficacy of the peptide sequence KTLLPTP in targeting pancreatic cancer via plectin-1.26–29 However, to suit our needs, we added two additional C-terminal residues without touching the N-terminal sequence that is essential for targeting. The tyrosine residue (Y) is included for its excellent reducing property at high pH and the cysteine residue (C) is included for its efficient binding to gold nanoparticle surface to confer stability.48 A similar approach was previously described for the synthesis of gold quantum clusters using peptides with an N-terminal CCY- sequence or proteins.52, 53 A recent study also utilized an in situ reduction method for the synthesis of protein-directed GNPs where hydrogen peroxide (H2O2) and the superoxide anion (O2. −) were used to activate the reducing potential of the proteins.34 However, the preparation of GNPs in that study essentially consisted of two steps. Gold (III) was first reduced to Gold (I) in presence collagen and sodium hydroxide (NaOH). Gold (I) was then reacted with luminol and H2O2 in presence of proteins to generate spherical monodispersed GNPs. In the present article, however, we used a peptide with C-terminal -YC sequence and utilized only NaOH to increase the reducing potential of the tyrosine moiety to prepare GNPs in a single step. We tested different molar ratios of the reagents and various reaction conditions, and the best was found to be a 1:0.1:10 molar ratio of gold (III) chloride: peptide: NaOH and a reaction temperature of 37 °C. The reaction kinetics showed that the reaction is almost complete within 16 hours as evident from the characteristic surface plasmon resonance (SPR) peak of synthesized gold nanoparticles at ~520 nm (Figure 1A and inset). Further reaction did not significantly increase the SPR peak and the monodispersity of the particles started to decline with time. Therefore we chose 16 hours as the standard reaction time for all of our experiments. The synthesized GNPs were purified from the reaction mixture by ultracentrifugation at 38000 RPM at 4 °C for one hour. The pellet was resuspended in Milli-Q water up to the original reaction volume to obtain a GNP stock solution. However, the stock solution was quite concentrated for practical measurements, so all the experiments were performed using a working stock prepared by 10× dilution of the initial stock solution in Milli-Q water.
Figure 1. Synthesis, characterization and stability of peptide-templated GNPs.
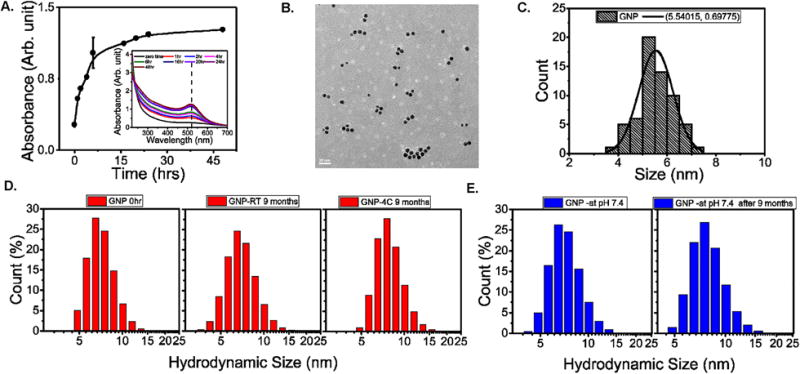
A. Absorption intensity of the reaction solution at SPR peak as a function of time. (inset) UV-vis spectra of the reaction solution obtained at different time intervals. B. TEM image of the synthesized GNPs, scale bar = 20 nm. C. GNP particle size distribution histogram obtained from TEM picture analysis. D. Long-term stability analysis of the synthesized GNPs after 9 months at room temperature (RT), 4 °C and E. at pH 7.4 by measuring respective hydrodynamic size distributions.
To understand the influence of YC at C terminals and the length of the peptide, we have designed two more peptides to synthesize GNPs. One was KTLLPTPY which was missing cysteine at C terminal and another nona- peptide which had nine amino acids and ends with YC at C terminal. All other reaction parameters like temperature and pH were maintained same. We observed that even if KTLLPTPY was able to make GNPs (GNP- Delta PTP), the sizes of the GNP- Delta PTP were almost doubled compared to those synthesized with KTLLPTYC. Also, the SPR peak intensity of these particles (~0.2346) was much lower compared to the particles synthesized with KTLLPTPYC (~1.27). This indicates that while KTLLPTPY preserves the reduction capability of the peptide, it is not as effective in providing a template for restricting the growth of GNPs as KTLLPTPYC. In addition, cysteine helps to attach the peptide on the GNP surface at proper orientation that is essential for targeting. On the other hand GNPs (GNP-PTPC) synthesized with the peptide KGHSGLMYC showed more or less comparable characteristics such as (average hydrodynamic diameter and SPR peak intensity), with GNPs synthesized using KTLLPTPYC. The results are depicted in Supplementary Figure 3.
3.2. Characterization and stability of the GNPs
The transmission electron microscopy (TEM) images of the synthesized GNPs showed uniform distribution of monodispersed spherical nanoparticles (Figure 1B; scale bar 20 nm). The particle size distribution obtained from figure 1B showed a narrow distribution of particles with diameter 5.5 ± 0.6 nm (Figure 1C). The hydrodynamic size distribution of the particles was also analyzed by dynamic light scattering (DLS) in a Malvern zetasizer. The histogram analysis showed a uniform distribution of particles centered around 6.5 nm for GNP (Figure 1D, left panel), and GNP-PTPC and that of 11.7 nm for GNP- Delta PTP (Supplementary Figure 3). The hydrodynamic sizes of these nanoparticles in aqueous suspension are likely to be larger than the core size of the same particles obtained from TEM analysis; these findings were consistent with the literature54, 55
Before we introduce pancreatic cancer specific targeting peptide assisted GNPs in cell culture and preclinical models, we evaluated their stability at various relevant conditions. The stability of the GNPs at two different storage conditions (room temperature and 4 °C) was tested by measuring their hydrodynamic size distributions after 9 months of storage. The GNPs showed excellent long-term stability without significant aggregation in both storage temperatures for 9 months as evident from their hydrodynamic size distributions (Figure 1D, middle and right panels). At physiological pH of 7.4, these GNPs show similar outstanding stability with no substantial change in hydrodynamic size distributions over a period of 9 months (Figure 1E). At pH 5, however, GNPs tend to aggregate more rapidly (data not shown). At this acidic conditions there will be significant influence on GNP’s surface charge which can result in aggregation.56
Furthermore, we incubated the synthesized GNPs with increasing concentrations of NaCl for 1 hour and measured any sign of aggregation by analyzing the hydrodynamic diameter. The results are depicted in Supplementary Figure 4. The GNPs demonstrated minimal increase in hydrodynamic diameter up to 200 mM NaCl concentration which is above physiological concentration. This indeed demonstrates the exceptional stability of the synthesized GNPs.
3.3. Conjugation of gemcitabine to the GNPs
Gemcitabine is a pyrimidine analog that contains a free –NH2 group. A few studies18 suggested that gem can bind to GNP surface either through electrostatic interaction via the pyrimidine ring or via an Au-N covalent bond with the free NH2 group. Since the primary objective of our study was to evaluate the efficacy of these GNPs as delivery vehicles, we analyzed the conjugation efficiency of Gem to the GNPs. For this purpose, we added increasing concentrations of Gem to 1 ml working stock of GNPs prepared by 10× dilution of the initial stock solution in Milli-Q water and stirred for 16 hours. Conjugation of Gem was confirmed by a slight red shift of the SPR peak (Figure 2A and inset) in agreement with previous reports.18 Ultracentrifugation at 38000 rpm for 1 hour allowed us to separate the Gem-conjugated GNPs (GNP-Gem) as a loose pellet from the reaction mixture and the Gem content of the pellet was calculated by subtracting the Gem content of the supernatant from the amount of Gem initially added (as shown in Supplementary Figure 5) The Gem content in the supernatants was determined by measuring the absorbance at 268 nm and comparing with a standard curve prepared against known concentrations of Gem as described previously.57 Finally the Gem content of the pelleted GNPs was plotted against the initial added amount to get a saturation curve (Figure 2B). The GNPs appear to be saturated at a Gem concentration of 25 μg/ml and approximately 4.25 μg/ml of Gem is found to be conjugated in the pellet. Therefore the conjugation efficiency is ~17%. To study the binding stability of Gem on GNPs surface, we further washed the gem conjugated GNPs with water and centrifuged at 38000 rpm for 1 hour. The collected soup was missing UV peak of Gem (Supplementary Figure 5) confirming that gem was strongly attached to GNP surface.
Figure 2. Fabrication, characterization and stability of GNP-Gem.
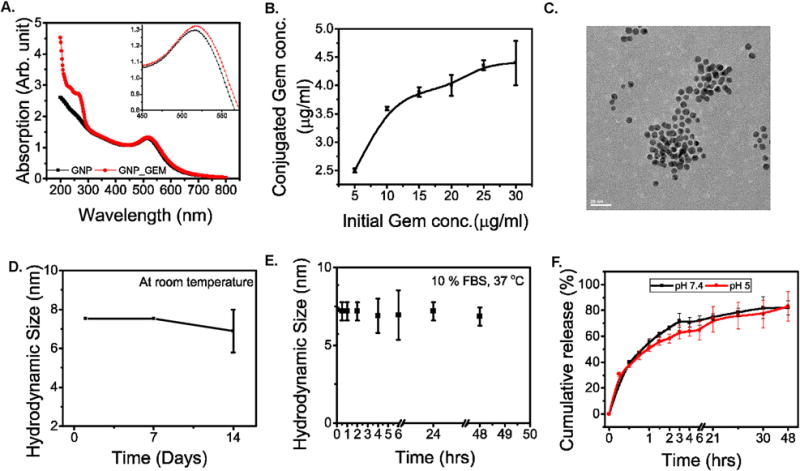
A. UV-vis spectra of GNP (Black) and GNP-Gem (Red). (inset) magnified portion of the spectra to highlight the red shift of the SPR peak upon gemcitabine conjugation. B. Saturation curve for gemcitabine conjugation to the GNPs. C. TEM image of the GNP-Gem, scale bar = 20 nm. D. Stability analysis of the GNP-Gem at room temperature by monitoring hydrodynamic diameter for 14 days and E. at 37 °C in presence of 10% FBS for 48 hours. F. In vitro cumulative gemcitabine release curve for GNP-Gem at 37 °C in two different pH environments. Black and red lines represent pH 7.4 and pH 5 respectively.
3.4. Characterization and stability of the gemcitabine conjugated GNPs
GNP-Gem was also analyzed for its size distribution and stability. The TEM image showed a uniform population of the particles (Figure 2C). The histogram analysis showed a narrow distribution of particles with a diameter of 5.4 ± 1nm (Supplementary Figure S1). The hydrodynamic size distribution showed a uniform distribution of particles as well centered around 7.5 nm (Supplementary Figure S2). Addition of Gem to GNP surface probably attracted more water molecules resulting in a significant increase in hydrodynamic size the nanoparticles. All these data pertaining to the increase in particle size suggest conjugation with Gem. The stability of the GNP-Gem was also analyzed by monitoring the hydrodynamic size at regular intervals at room temperature. No significant changes were observed from initial measurements up to 7 days indicating that the stable dispersion of GNPs was still maintained after conjugation of Gem (Figure 2D). However, the particles did show a decrease in size at day 14, presumably due to slow release of Gem from GNP-Gem. We also analyzed the stability of GNP-Gem in presence of 10% FBS at 37 °C and they fare very well with no significant change in hydrodynamic diameter up to 48 hours (Figure 2E).
3.5. In vitro gemcitabine release profile from gemcitabine-conjugated GNPs
Stability of the drug-loaded nanoparticles is important for reaching the target. However, once it reached the target it is also equally important to release the drug efficiently. So, the release kinetics at physiological conditions should be evaluated before moving forward with efficacy studies. To this end, we analyzed the Gem release profile from the Gem-conjugated GNPs at 37 °C in two different pH environments (pH 7.4 and pH 5). We did not find any significant difference in release kinetics in different pH environments suggesting that the release of Gem is not pH dependent. In both cases there was an initial burst release followed by a sustained release (Figure 2F). Approximately 60–70% of the drug was released within the initial 4-hour period followed by a sustained release of 15–20% over a period of 48 hours. These results demonstrated efficient drug release from the Gem-conjugated GNPs at physiological conditions.
3.6. Molecular modeling for gold nanoparticle identifies candidate structures amenable for Gemcitabine binding
Modeling Gem using statistical mechanics approaches and molecular mechanics with simulations gives improved results over a docking only approach.58 Small simulation was completed to study Gem affinity for the gold nanoparticle (GNP) with the 53 bound nonapeptides with sequence KTLLPTPYC (PTP) (Figure 3A–B). The water box size consists of over 2 million cubic Å (or 1728 nm3); giving the system over 205,000 atoms within the box and the box has dimensions of approximately 128 Å per edge (X, Y, Z). The 53-peptide found to be attached to the gold nanoparticle consists of 12,696 atoms by its C terminal cysteine leaving N-terminal of PTP available to act as targeting agents. It is to be noted here that the peptide by itself does not saturate the surface area of the gold nanoparticle leaving adequate space for gemcitabine to bind on the GNP’s surface. The gold nanoparticle has an inner diameter of 55.96 Å or approximately 5.6 nm (Figure 3C) which is a close match with our TEM observation. The particle is free to tumble and migrate during simulation; however periodic boundary conditions with Particle-Mesh Ewald are utilized to ensure accuracy (described in details in the supplementary methods).59, 60 When examining the particle up close (Figure 3B) the bulk of the captured drug has interaction via the peptides and some Van der Waals (VdW) contacts with the gold nanoparticle, but without the peptides bound to the gold nanoparticle, the drugs would not aggregate as strongly. We found that the Gem concentration increased from under 6% to nearly 25% over the course of the initial NPT simulations (Figure 3D) which is strongly in agreement with our experimental observations. This corresponds to having over 42 of the 200 drug molecules in the water box becoming captured by the nanoparticle. Simulations for the entire complex are provided in supplemental materials (Supplementary Movie S1–S2).
Figure 3. Computational modeling of nonapeptide bound gold nanoparticle with drug capture.
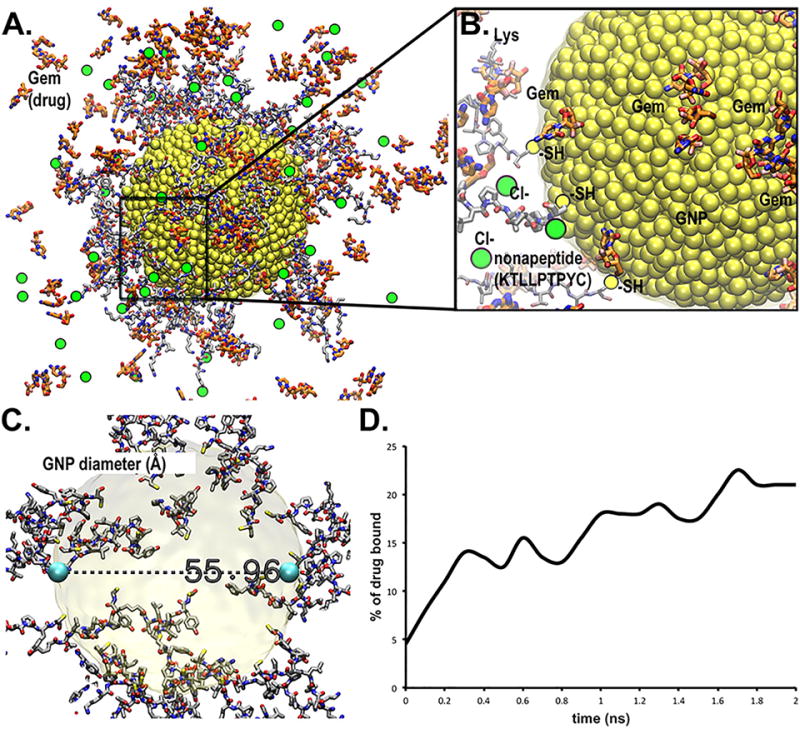
A model for a nonapeptide (KTLLPTPYC) bound to 5.6 nm wide GNP via thiol-gold covalent linkages was generated with capture of Gemcitabine (Gem) drug for study of drug delivery transport mechanism. A. Entire nanoparticle consists of 4,896 Au atoms at the central core shown in yellow VdW spheres. Licorice stick peptides are displayed using conventional color for standard elements (carbon-gray, oxygen-red, nitrogen-blue, sulfur-yellow and hydrogen-white). Thicker stick rendering is shown for the Gemcitabine drug (highlighted ligand carbons colored red and fluorine in pink). Thiol (-SH)-to-gold connected cysteine atoms are shown in olive-yellow. Chlorine counter ions are shown in green spheres. B. Magnification of a representative area on GNP that reveals interaction of peptides and Gem with the GNP surface. N-terminal sequence of the peptides pointing away into solution is available for interacting with target proteins (plectin-1 in this case). C. Physical dimensions of the inner gold nanoparticle (~56 Å or 5.6 nm). D. Binding efficiency of GNP indicated by “% of drug bound”, is increasing over time.
Free energy (ΔGassociation) of binding were implemented within the software algorithm considering factors such as: lipophilicity, displacement of water, hydrogen bonding and electrostatic interactions, and metal ion/ligand interactions as favorable interactions; while the desolvation of polar or charged groups, restriction of motion, and the entropic cost of binding adversely affect free energy.58, 61–63 These energetic measurements will help us determine optimal gold nanoparticle complexes for future drug delivery systems by measuring the total bound drug number and its combined free energy associated over time. This can then be correlated to the experimental value for conjugated Gem found in concentration directly from experiment, but more importantly act as a useful filter prior to experiment to guide the testing in a cost saving hypothesis generation.
3.7. In vitro therapeutic efficacy of gemcitabine-conjugated GNPs
Before analyzing the in vivo efficacy of GNP-Gem, it was important to test whether they were indeed demonstrating a superior inhibitory effect in pancreatic cancer cell viability. To this end, we selected two commercially available pancreatic cancer cell lines (AsPC-1 and PANC-1, obtained from ATCC) and treated the cells with increasing doses of GNP, Gem and GNP-Gem for 1 hour. After 1 hour, cells were washed three times with PBS to remove the treatments completely in order to reduce any non-specific effect and fresh media was added to the cells. The cell viability measurement was performed by MTS assay after 72 hours (Figure 4A–B). Among the treatment groups, GNP-Gem showed the maximum inhibitory effect in both cell lines. Interestingly, GNPs also showed a significant effect in PANC-1 cells especially at 5 μM but no dose dependent effect can be seen in AsPC-1 cells (Figure 4A). Similarly, Gem showed a nice dose dependent effect in AsPC-1 cells but not in PANC-1 cells (Figure 4B). This is not surprising, considering that previous studies demonstrated PANC-1 cells to be more resistant to Gem compared to AsPC-1.64, 65 On the other hand, the effects shown by GNPs may be partly due to the binding of the peptide on the GNP surface to plectin-1 since plectin-1 has been shown to have important functions in pancreatic cancer pathogenesis.23 Nonetheless, the results obtained from these experiments demonstrate that GNP-Gem exerts a superior inhibitory effect, especially at lower concentrations, on pancreatic cancer cell viability compared to GNP or Gem alone.
Figure 4. In vitro therapeutic efficacy of GNP-Gem.
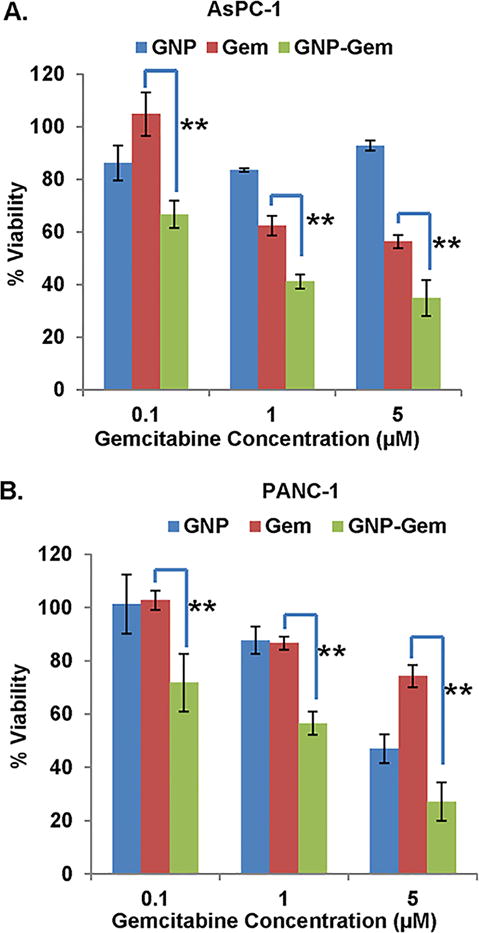
In vitro cell viability as measured by MTS assay in A. AsPC-1 and B. PANC-1 cells after treating with increasing concentrations of Gem (blue), GNP (red) and GNP-Gem (green) respectively. ** denotes p<0.01.
3.8. In vivo therapeutic efficacy of GNP-Gem
Since gemcitabine-conjugated GNPs demonstrated enhanced anti-proliferative activity in PDAC cells in vitro, we sought to evaluate the in vivo therapeutic potential of the same in a PANC-1 xenograft model. We selected PANC-1 for an in vivo experiment because it showed higher Gem resistance in vitro compared to AsPC-1 and we wanted to test whether conjugation of Gem to GNPs can increase the therapeutic efficacy. Approximately, 1 × 106 luciferase transfected PANC-1 cells were orthotopically implanted in the pancreas of 6–8 weeks old NSG mice. After three weeks, mice were imaged noninvasively using the Xenogen instrument to confirm tumor growth and then randomized into four groups before the initiation of treatment. We included both male and female mice in the experiment to delineate any differential sex-dependent therapeutic effect of GNP-Gem. The experiment was terminated after three weeks of treatment. Weekly measurement of luciferase bioluminescence showed very little tumor growth in mice receiving GNP-Gem (Figure 5A–C). Figures 5A and 5B demonstrate representative bioluminescent images from each treatment group (Day 0 and Day 22 in upper and lower panels respectively) in female and male mice respectively. Figure 5C depicts the tumor growth curve as measured by luciferase bioluminescence for female and male mice separately or in combination.
Figure 5. In vivo therapeutic efficacy of GNP-Gem.
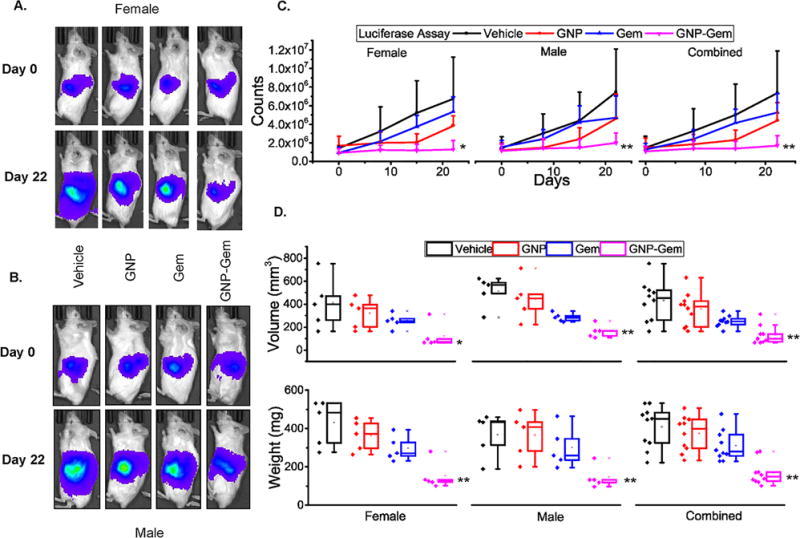
Representative in vivo bioluminescence images captured before start and after completion of treatment for each treatment group in A. female and B. male mice. C. In vivo bioluminescence plot against time for female and male separately or in combination. Only (+) error bar was provided to improve clarity. * and ** denote p <0.05 and p<0.01 compared to vehicle-treated group respectively. D. Box plot diagram depicting tumor volume and tumor weight for each treatment group in female and male separately or in combination after completion of the experiment. For comparison, individual data points from each treatment group are also provided side by side. Black = vehicle, Red = GNP, Blue = Gem and Purple = GNP-Gem. * and ** denote p <0.05 and p<0.01 compared to vehicle-treated group respectively.
These results were further validated by measuring the tumor volume (Figure 5D, upper panel) and weight (Figure 5D, lower panel) after sacrificing the mice at the end of the experiment. The tumor volumes in GNP-Gem treated female mice (mean 123.7 mm3; median 72 mm3; 25th–75th percentile 67.2, 98.6) were significantly smaller compared to vehicle-treated (mean 410.5 mm3; median 400.7 mm3; 25th–75th percentile 262.4, 471.6; p=0.0336), GNP-treated (mean 321.2 mm3; median 364.8 mm3; 25th–75th percentile 202.5, 395.3; p=0.0321) or Gem-treated female mice (mean 254.8 mm3; median 254.6 mm3; 25th–75th percentile 241.9, 272.9; p=0.047). Similarly, GNP-Gem treated male mice showed significantly smaller tumor volumes (mean 137.7 mm3; median 140.1 mm3; 25th–75th percentile 102.2, 140.9) compared to vehicle-treated (mean 450.5 mm3; median 500.2 mm3; 25th–75th percentile 434.6, 520.2; p=0.0007), GNP-treated (mean 392.04 mm3; median 396.6 mm3; 25th–75th percentile 314.6, 425.5; p=0.0099) or Gem-treated male mice (mean 247.17 mm3; median 245.3 mm3; 25th–75th percentile 223.6, 259.3; p=0.0038). Combined, GNP-Gem treatment caused a significant regression in tumor volume (mean 130.75 mm3; median 100.4 mm3; 25th–75th percentile 72, 140) compared to vehicle (mean 430.5 mm3; median 453.14 mm3; 25th–75th percentile 262.4, 520.2; p=0.000094), GNP (mean 356.6 mm3; median 380.09 mm3; 25th–75th percentile 202.5, 425.5; p=0.00039) or Gem (mean 251 mm3; median 250 mm3; 25th–75th percentile 223.6, 272.9; p=0.00069). Likewise, GNP-Gem treated female mice exhibited significantly lower tumor weights (mean 152.4 mg; median 126.1 mg; 25th–75th percentile 120.1, 133.5) compared to vehicle-treated (mean 429.9 mg; median 483.8 mg; 25th–75th percentile 326.4, 531.7; p=0.0023), GNP-treated (mean 363.24 mg; median 373.1 mg; 25th–75th percentile 297.6, 426.5; p=0.0025) or Gem-treated female mice (mean 295.9 mg; median 272.4 mg; 25th–75th percentile 256.5, 326.9; p=0.0038). The tumor weights in GNP-Gem treated male mice (mean 183 mg; median 170.1 mg; 25th–75th percentile 156.4, 171.8) were significantly smaller compared to vehicle-treated (mean 385.8 mg; median 446.4 mg; 25th–75th percentile 336.8, 451.6; p=0.0049), GNP-treated (mean 384.9 mg; median 424.5 mg; 25th–75th percentile 310.7, 448.2; p=0.0061) or Gem-treated male mice (mean 325.8 mg; median 288.3 mg; 25th–75th percentile 267.5, 367.7; p=0.0208). Combined, GNP-Gem treatment caused a significant regression in tumor weight (mean 167.7 mg; median 148.35 mg; 25th–75th percentile 126.1, 171.9) compared to vehicle (mean 407.86 mg; median 449.1 mg; 25th–75th percentile 326.4, 483.6; p=0.00001), GNP (mean 374.09 mg; median 396.8 mg; 25th–75th percentile 297.6, 448.2; p=0.000014) or Gem (mean 310.86 mg; median 280.35 mg; 25th–75th percentile 126.1, 171.9; p=0.00029). However, we did not find any significant difference between GNP-Gem treated female and male mice suggesting that the therapeutic efficacy of GNP-Gem is not sex-dependent.
Interestingly, accumulation of GNPs could be seen in the tumor tissues but not in the adjacent normal pancreas tissues (Figure 6A–B). This result is in agreement with a previous study where crosslinked iron oxides [CLIO]-Cy5.5 nanoparticles conjugated to the peptide KTLLPTP through a spacer selectively accumulate in PDAC tissues compared to normal pancreas.28 This validates excellent tumor-targeting efficacy of the synthesized GNPs for efficient delivery of Gem into the tumors. The hematoxylin and eosin staining of the formalin-fixed paraffin embedded tumor tissue sections showed a significant loss of cellularity in Gem-conjugated GNP treatment group (Figure 6C–D) which highlighted the in vivo antiproliferative efficacy of GNP-Gem compared to Gem or GNP alone. This was further confirmed by ki67 staining of the said tumors tissues (Figure 6E–G). The tissues from gemcitabine-conjugated GNP-treated group showed the least amount of Ki67-positive nuclei. Figure 6G depicts the quantitation of Ki67-positve nuclei in female and male mice separately or in combination. The percentage of Ki67-positive nuclei in GNP-Gem treated female mice (mean 22.2%; median 21.3%; 25th–75th percentile 17.9, 26.7) was significantly lower compared to vehicle-treated (mean 37.7%; median 37.1%; 25th–75th percentile 34.8, 41.8; p=9.81 × 10−14), GNP-treated (mean 32.5%; median 33.9%; 25th–75th percentile 28.8, 36.6; p=1.48 × 10−6) or Gem-treated female mice (mean 31.3%; median 32.3%; 25th–75th percentile 29.3, 39.5; p=0.0005). Similarly, GNP-Gem treated male mice showed significantly lower Ki67-positive nuclei (mean 23.3%; median 20.9%; 25th–75th percentile 14.3, 31.35) compared to vehicle-treated (mean 35.09%; median 34.3%; 25th–75th percentile 29.09, 41.35; p=1.95 × 10−5), GNP-treated (mean 32.3%; median 32.6%; 25th–75th percentile 28.36, 36.05; p=0.00037) or Gem-treated male mice (mean 31.5%; median 31.4%; 25th–75th percentile 24.6, 39.4; p=0.01). Combined, GNP-Gem treatment caused a significant regression in Ki67-positive nuclei (mean 22.7%; median 21.09%; 25th–75th percentile 16.6, 28) compared to vehicle (mean 36.4%; median 36.06%; 25th–75th percentile 32.5, 41.5; p=2.52 × 10−15), GNP (mean 32.4%; median 33.4%; 25th–75th percentile 28.7, 36.4; p=4.11 × 10−9) or Gem (mean 31.4%; median 32.07%; 25th–75th percentile 25.7, 39.5; p=2.41 × 10−5). Taken together, these data demonstrated excellent in vivo therapeutic efficacy of GNP-Gem in a PDAC xenograft model.
Figure 6. Immunohistochemical analysis.
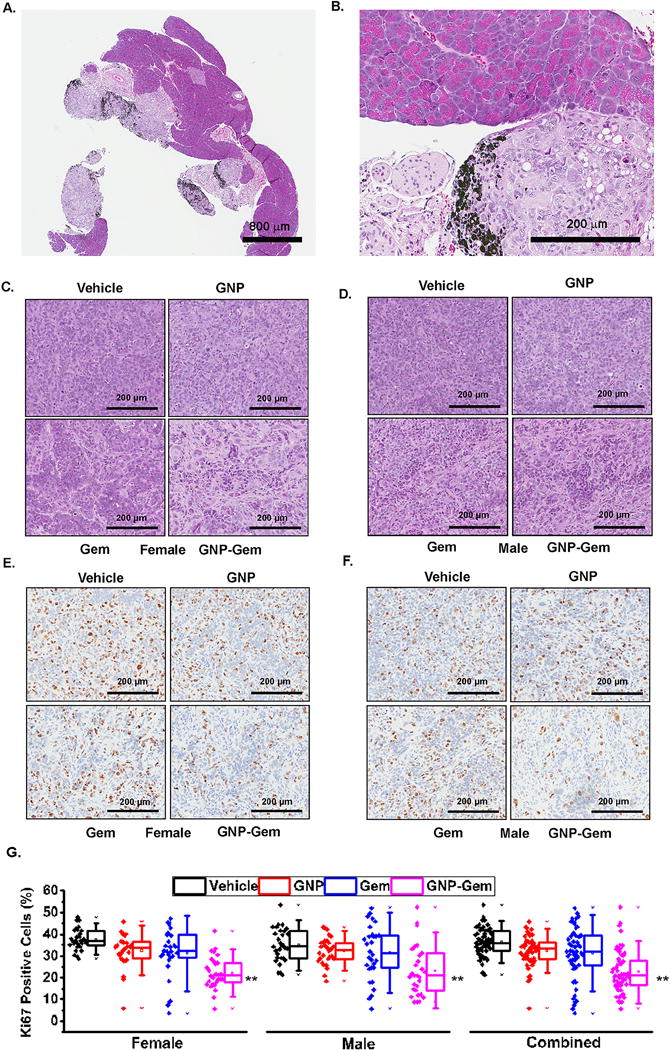
A. H&E staining of a tissue section from GNP-Gem treated group showing tumor-specific uptake of GNP-Gem without touching adjacent normal pancreas tissue. Scale bar =800 μm. B. Higher magnification showing accumulation of GNPs in tumor tissue. Scale Bar = 200 μm. Accumulated GNPs are shown using black arrows. C. & D. H&E staining of tissue sections obtained from each treatment group showing decrease in cellularity in GNP-Gem treated group in female and male mice respectively. Scale Bar = 200 μm. E. & F. Ki67 staining of tissue sections obtained from each treatment group showing reduction in number of Ki67 positive nuclei in GNP-Gem treated group in female and male mice respectively. Scale bar = 200 μm. G. Box plot diagram depicting percentage of Ki67 positive nuclei for each treatment group in female and male separately or in combination. Three slides were stained for Ki67 per treatment group and ten random areas of equal dimensions from each tissue sections were analyzed in Aperio Imagescope. For comparison, individual data points from each treatment group are also provided side by side. Black = vehicle, Red = GNP, Blue = Gem and Purple = GNP-Gem. * and ** denote p <0.05 and p<0.01 compared to vehicle-treated group respectively.
4. CONCLUSION
In summary, the present work reports an in situ reduction-based approach to synthesize highly monodispersed, spherical GNPs with a uniform size-distribution and extended stability that are surface modified with plectin-1-targeted multifunctional peptides. These GNPs demonstrated a remarkable efficiency in accumulating into the tumor tissue where plectin-1 is known to have an aberrant cell-surface expression, without evading into adjacent normal pancreas tissue. In addition, these GNPs displayed excellent anti-proliferative effect in established PDAC cell lines and antitumor effect in a PDAC xenograft by delivering gemcitabine selectively into the cancer cells. To our knowledge, this is the first report of utilizing plectin-1-targeted GNPs for targeted delivery of gemcitabine in PDAC. This simple but elegant method may be generalized for fabricating GNPs with suitable peptides that target cell-surface receptors or proteins overexpressed in any specific type of cancer.
Supplementary Material
Acknowledgments
The authors thank Dr. Debabrata Mukhopadhyay for his valuable suggestions. This work was partly supported by Mayo-UCF seed grant, Florida Department of Health (Cancer Research Chair Fund, Florida #3J to DM), NIH (CA78383, CA 150190 to DM) and McKenzie 16 – PAW funds. Financial support by the Robert A. Welch Foundation (Grant P-1212 to JLC) is also gratefully acknowledged.
Footnotes
Conflicts of interest: The authors declare that they have no competing financial interests.
References
- 1.Siegel RL, Miller KD, Jemal A. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Soo RA, Yong WP, Innocenti F. Curr Drug Targets. 2012;13:811–828. doi: 10.2174/138945012800564068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldstein D, El-Maraghi RH, Hammel P, Heinemann V, Kunzmann V, Sastre J, Scheithauer W, Siena S, Tabernero J, Teixeira L, Tortora G, Van Laethem JL, Young R, Penenberg DN, Lu B, Romano A, Von Hoff DD. J Natl Cancer Inst. 2015:107. doi: 10.1093/jnci/dju413. [DOI] [PubMed] [Google Scholar]
- 4.Papadatos-Pastos D, Thillai K, Rabbie R, Ross P, Sarker D. Expert Rev Anticancer Ther. 2014;14:1115–1125. doi: 10.1586/14737140.2014.957188. [DOI] [PubMed] [Google Scholar]
- 5.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, Fouchardiere C de la, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M, U. Groupe Tumeurs Digestives of and P. Intergroup N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 6.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu B, Tai HC, Xue W, Lee LJ, Lee RJ. Mol Membr Biol. 2010;27:286–298. doi: 10.3109/09687688.2010.521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sykes EA, Dai Q, Sarsons CD, Chen J, Rocheleau JV, Hwang DM, Zheng G, Cramb DT, Rinker KD, Chan WC. Proc Natl Acad Sci U S A. 2016;113:E1142–1151. doi: 10.1073/pnas.1521265113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X. Cell Biochem Biophys. 2015;72:771–775. doi: 10.1007/s12013-015-0529-4. [DOI] [PubMed] [Google Scholar]
- 10.Shah M, Badwaik V, Kherde Y, Waghwani HK, Modi T, Aguilar ZP, Rodgers H, Hamilton W, Marutharaj T, Webb C, Lawrenz MB, Dakshinamurthy R. Front Biosci (Landmark Ed) 2014;19:1320–1344. doi: 10.2741/4284. [DOI] [PubMed] [Google Scholar]
- 11.Daniel MC, Astruc D. Chem Rev. 2004;104:293–346. doi: 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- 12.Levy R, Thanh NT, Doty RC, Hussain I, Nichols RJ, Schiffrin DJ, Brust M, Fernig DG. J Am Chem Soc. 2004;126:10076–10084. doi: 10.1021/ja0487269. [DOI] [PubMed] [Google Scholar]
- 13.Swierczewska M, Lee S, Chen X. Phys Chem Chem Phys. 2011;13:9929–9941. doi: 10.1039/c0cp02967j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar A, Ma H, Zhang X, Huang K, Jin S, Liu J, Wei T, Cao W, Zou G, Liang XJ. Biomaterials. 2012;33:1180–1189. doi: 10.1016/j.biomaterials.2011.10.058. [DOI] [PubMed] [Google Scholar]
- 15.Pantic I, Markovic L. Rev Adv Mater Sci. 2011;29:126–129. [Google Scholar]
- 16.Vigderman L, Zubarev ER. Adv Drug Deliv Rev. 2013;65:663–676. doi: 10.1016/j.addr.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Arvizo R, Bhattacharya R, Mukherjee P. Expert Opin Drug Deliv. 2010;7:753–763. doi: 10.1517/17425241003777010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patra CR, Bhattacharya R, Wang E, Katarya A, Lau JS, Dutta S, Muders M, Wang S, Buhrow SA, Safgren SL, Yaszemski MJ, Reid JM, Ames MM, Mukherjee P, Mukhopadhyay D. Cancer Res. 2008;68:1970–1978. doi: 10.1158/0008-5472.CAN-07-6102. [DOI] [PubMed] [Google Scholar]
- 19.Patra CR, Bhattacharya R, Mukhopadhyay D, Mukherjee P. Adv Drug Deliv Rev. 2010;62:346–361. doi: 10.1016/j.addr.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patra CR, Bhattacharya R, Mukherjee P. J Mater Chem. 2010;20:547–554. doi: 10.1039/b913224d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dreaden EC, Mwakwari SC, Sodji QH, Oyelere AK, El-Sayed MA. Bioconjug Chem. 2009;20:2247–2253. doi: 10.1021/bc9002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinbock FA, Wiche G. Biol Chem. 1999;380:151–158. doi: 10.1515/BC.1999.023. [DOI] [PubMed] [Google Scholar]
- 23.Shin SJ, Smith JA, Rezniczek GA, Pan S, Chen R, Brentnall TA, Wiche G, Kelly KA. Proc Natl Acad Sci U S A. 2013;110:19414–19419. doi: 10.1073/pnas.1309720110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bausch D, Thomas S, Mino-Kenudson M, Fernandez-del CC, Bauer TW, Williams M, Warshaw AL, Thayer SP, Kelly KA. Clin Cancer Res. 2011;17:302–309. doi: 10.1158/1078-0432.CCR-10-0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moris M, Dawson DW, Jiang J, Lewis J, Nassar A, Takeuchi KK, Lay AR, Zhai Q, Donahue TR, Kelly KA, Crawford HC, Wallace M. Pancreas. 2016;45:1353–1358. doi: 10.1097/MPA.0000000000000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leung K. Molecular Imaging and Contrast Agent Database (MICAD) Bethesda (MD): 2004. [PubMed] [Google Scholar]
- 27.Leung K. Molecular Imaging and Contrast Agent Database (MICAD) Bethesda (MD): 2004. [PubMed] [Google Scholar]
- 28.Kelly KA, Bardeesy N, Anbazhagan R, Gurumurthy S, Berger J, Alencar H, Depinho RA, Mahmood U, Weissleder R. PLoS Med. 2008;5:e85. doi: 10.1371/journal.pmed.0050085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konkalmatt PR, Deng D, Thomas S, Wu MT, Logsdon CD, French BA, Kelly KA. Front Oncol. 2013;3:84. doi: 10.3389/fonc.2013.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanna V, Nurra S, Pala N, Marceddu S, Pathania D, Neamati N, Sechi M. J Med Chem. 2016;59:5209–5220. doi: 10.1021/acs.jmedchem.5b01571. [DOI] [PubMed] [Google Scholar]
- 31.US20160258940 A1. US Pat. 2016
- 32.Ohta S, Glancy D, Chan WC. Science. 2016;351:841–845. doi: 10.1126/science.aad4925. [DOI] [PubMed] [Google Scholar]
- 33.Sohn JS, Kwon YW, Jin JI, Jo BW. Molecules. 2011;16:8143–8151. doi: 10.3390/molecules16108143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leng Y, Fu L, Ye L, Li B, Xu X, Xing X, He J, Song Y, Leng C, Guo Y, Ji X, Lu Z. Sci Rep. 2016;6:28900. doi: 10.1038/srep28900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jazayeri MH, Amani H, Pourfatollah AA, Pazoki-Toroudi H, Sedighimoghaddam B. Sensing and Bio-Sensing Research. 2016;9:17–22. [Google Scholar]
- 36.Levy R. Chembiochem. 2006;7:1141–1145. doi: 10.1002/cbic.200600129. [DOI] [PubMed] [Google Scholar]
- 37.Corbierre MK, Cameron NS, Sutton M, Mochrie SG, Lurio LB, Ruhm A, Lennox RB. J Am Chem Soc. 2001;123:10411–10412. doi: 10.1021/ja0166287. [DOI] [PubMed] [Google Scholar]
- 38.Rogowski JL, Verma MS, Gu FX. Langmuir. 2016;32:7621–7629. doi: 10.1021/acs.langmuir.6b01339. [DOI] [PubMed] [Google Scholar]
- 39.Gao J, Huang X, Liu H, Zan F, Ren J. Langmuir. 2012;28:4464–4471. doi: 10.1021/la204289k. [DOI] [PubMed] [Google Scholar]
- 40.Xue Y, Li X, Li H, Zhang W. Nat Commun. 2014;5:4348. doi: 10.1038/ncomms5348. [DOI] [PubMed] [Google Scholar]
- 41.Chai F, Wang C, Wang T, Ma Z, Su Z. Nanotechnology. 2010;21:025501. doi: 10.1088/0957-4484/21/2/025501. [DOI] [PubMed] [Google Scholar]
- 42.Sousa AA, Morgan JT, Brown PH, Adams A, Jayasekara MP, Zhang G, Ackerson CJ, Kruhlak MJ, Leapman RD. Small. 2012;8:2277–2286. doi: 10.1002/smll.201200071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Porta F, Speranza G, Krpetić Ž, Dal Santo V, Francescato P, Scarì G. Materials Science and Engineering: B. 2007;140:187–194. [Google Scholar]
- 44.Serizawa T, Hirai Y, Aizawa M. Langmuir. 2009;25:12229–12234. doi: 10.1021/la9021799. [DOI] [PubMed] [Google Scholar]
- 45.Kroger N, Deutzmann R, Sumper M. Science. 1999;286:1129–1132. doi: 10.1126/science.286.5442.1129. [DOI] [PubMed] [Google Scholar]
- 46.Cha JN, Shimizu K, Zhou Y, Christiansen SC, Chmelka BF, Stucky GD, Morse DE. Proc Natl Acad Sci U S A. 1999;96:361–365. doi: 10.1073/pnas.96.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Douglas T, Strable E, Willits D, Aitouchen A, Libera M, Young M. Adv Mater. 2002;14:415–+. [Google Scholar]
- 48.Xie J, Zheng Y, Ying JY. J Am Chem Soc. 2009;131:888–889. doi: 10.1021/ja806804u. [DOI] [PubMed] [Google Scholar]
- 49.Naik RR, Jones SE, Murray CJ, McAuliffe JC, Vaia RA, Stone MO. Adv Funct Mater. 2004;14:25–30. [Google Scholar]
- 50.Kasotakis E, Mitraki A. Methods Mol Biol. 2013;996:195–202. doi: 10.1007/978-1-62703-354-1_11. [DOI] [PubMed] [Google Scholar]
- 51.Raha S, Paunesku T, Woloschak G. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2011;3:269–281. doi: 10.1002/wnan.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y, Cui Y, Zhao Y, Liu R, Sun Z, Li W, Gao X. Chem Commun (Camb) 2012;48:871–873. doi: 10.1039/c1cc15926g. [DOI] [PubMed] [Google Scholar]
- 53.Soptei B, Nagy LN, Baranyai P, Szabo I, Mezo G, Hudecz F, Bota A. Gold Bull. 2013;46:195–203. [Google Scholar]
- 54.Domingos RF, Baalousha MA, Ju-Nam Y, Reid MM, Tufenkji N, Lead JR, Leppard GG, Wilkinson KJ. Environ Sci Technol. 2009;43:7277–7284. doi: 10.1021/es900249m. [DOI] [PubMed] [Google Scholar]
- 55.De Palma R, Peeters S, Van Bael MJ, Van den Rul H, Bonroy K, Laureyn W, Mullens J, Borghs G, Maes G. Chem Mater. 2007;19:1821–1831. [Google Scholar]
- 56.Mihailescu G, Olenic L, Pruneanu S, Bratu I, Kacso I. J Optoelectron Adv M. 2007;9:756–759. [Google Scholar]
- 57.Celano M, Calvagno MG, Bulotta S, Paolino D, Arturi F, Rotiroti D, Filetti S, Fresta M, Russo D. BMC Cancer. 2004;4:63. doi: 10.1186/1471-2407-4-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lorber DM, Shoichet BK. Protein Sci. 1998;7:938–950. doi: 10.1002/pro.5560070411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Caulfield T, Devkota B. Proteins. 2012;80:2489–2500. doi: 10.1002/prot.24131. [DOI] [PubMed] [Google Scholar]
- 60.Caulfield TR, Devkota B, Rollins GC. J Biophys. 2011;2011:219515. doi: 10.1155/2011/219515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 62.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 63.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 64.Fryer RA, Barlett B, Galustian C, Dalgleish AG. Anticancer Res. 2011;31:3747–3756. [PubMed] [Google Scholar]
- 65.Huanwen W, Zhiyong L, Xiaohua S, Xinyu R, Kai W, Tonghua L. Mol Cancer. 2009;8:125. doi: 10.1186/1476-4598-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.