

Abstract
Mammary stem cells (MSCs) expansion is associated with aggressive human breast cancer. The nuclear receptor peroxisome proliferator activated receptor γ (PPARγ) is a breast cancer tumor suppressor, but the mechanisms of this suppression are not completely characterized. To determine whether PPARγ regulates MSC expansion in mammary cancer, we deleted PPARγ expression in the mammary epithelium of an in vivo model of basal breast cancer. Loss of PPARγ expression reduced tumor latency, and expanded the CD24+/CD49fhi MSC population. PPARγ-null mammary tumors exhibited increased angiogenesis, which was detected in human breast cancer. In vivo inhibition of a PPARγ-regulated miR-15a/angiopoietin-1 pathway blocked increased angiogenesis and MSC expansion. PPARγ bound and activated a canonical response element in the miR-15a gene. PPARγ-null tumors were sensitive to the targeted anti-angiogenic drug sunitinib but resistant to cytotoxic chemotherapy. Normalization of tumor vasculature with sunitinib resulted in objective response to cytotoxic chemotherapy. Chemotherapy-treated PPARγ-null mammary tumors exhibited luminal phenotype and expansion of unipotent CD61+ luminal progenitor cells. Transplantation of chemotherapy-treated luminal progenitor cells recapitulated the luminal phenotype. These results have important implications for anti-angiogenic therapy in breast cancer patients.
INTRODUCTION
Mammary stem cells (MSCs) are the progenitor population for all breast epithelia.1–4 The MSC population is expanded in some mouse mammary cancer models,5,6 and tumorigenic progenitor populations have been isolated from human breast cancers.7,8 MSC expansion is associated with aggressive human breast cancer.9
Tumorigenic MSC expansion has been associated with increased angiogenesis and poor clinical prognosis in human breast cancer.10,11 However, the mechanisms of this expansion are unclear, and anti-angiogenic therapies have not resulted in significantly increased survival in breast cancer patients.12 The nuclear receptor peroxisome proliferator activated receptor γ (PPARγ13,14) has tumor suppressor effects in breast cancer.15–17 PPARγ has functional domains for ligand binding and interaction with recognition sequences in the promoter regions of its target genes to regulate transcription. Breast cancers express reduced levels of PPARγ compared with normal mammary tissue, consistent with its tumor suppressor function.18,19 However, mutant forms of PPARγ promoted tumor growth in animal models,20,21 and polymorphisms in PPAR genes were associated with increased breast cancer risk in humans.22 The mutant PPAR/PAX8 fusion protein was shown to regulate thyroid cancer phenotype via microRNA-122.23,24 MicroRNAs (miRNA) are single-stranded small non-coding RNA molecules.25 miRNAs regulate the stability of their target mRNAs by binding to their untranslated regions and inducing degradation or translational inhibition. Circulating tumor-associated miRNAs are found in breast cancer patients, and are associated with poor prognosis.26
To determine whether the PPARγ breast tumor suppressor regulated MSC expansion in vivo, we genetically deleted PPARγ expression in the mammary epithelium of a tumor prone model. We discovered a novel mechanism by which tumorigenic MSCs regulate the angiogenic niche in mammary cancer that is relevant to human breast cancer. Molecular inhibition of this mechanism disrupts the angiogenic niche, and suppresses MSC expansion associated with increased angiogenesis in this model. This model may inform the use of anti-angiogenic agents in human breast cancer, and how chemotherapy-mediated expansion of other tumorigenic cell fractions may result in poor clinical outcomes.
RESULTS
We created the MMTV-Cre;PPARγf/f;Wnt1 mouse in which exons 1 and 2 of the PPARγ gene are selectively deleted in mammary epithelium via Cre-mediated recombination. Wnt1 mammary tumors model human basal subtype breast cancer.5 PPARγ expression in MMTV-Cre;PPARγf/f;Wnt1 and MMTV-Cre;PPARγ+/+; Wnt1 control MSC is shown in Figure 1a. MMTV-Cre;PPARγf/f;Wnt1 mammary tumors developed with shorter mean latency of 140 days compared with 182 days for MMTV-Cre;PPARγ+/+;Wnt1 cancers (P < 0.04; Figure 1b). Tumor growth rate was significantly increased in MMTV-Cre;PPARγf/f;Wnt1 mammary cancers (P < 0.05; Figure 1c). Both genotypes developed poorly differentiated adenocarcinoma as determined by histopathologic analysis (Figures 1d and e). Mammary adenocarcinomas of both genotypes were composed of both basal and luminal epithelial cells. The CD24+/CD49f+ MSC population is one of two known tumorigenic populations in MMTV-Wnt1 cancers.3 MMTV-Cre; PPARγf/f;Wnt1 mammary tumors showed 36% relative expansion of the MSC population (P < 0.02, Figure 1f) as determined by FACS (Figures 1g and h). MSC was localized in MMTV-Cre;PPARγ+/+; Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 tumors by CD24/CD49f immunofluorescence microscopy (Figures 1i and j). MMTV-Cre; PPARγf/f;Wnt1 mammary tumors exhibited increased cell proliferation (36 vs 24%; P < 4 × 10−6; Figure 1k) as shown by proliferating cell nuclear antigen (PCNA) immunohistochemistry (Figures 1l and m). No significant differences in apoptotic cells were observed in mammary tumors of either genotype as shown by terminal transferase mediated dUTP nick end labeling (TUNEL) analysis (Figures 1n and o). These results indicate that loss of PPARγ expression decreases tumor latency, expands the MSC population and increases cell proliferation in Wnt1 mammary tumors.
Figure 1.
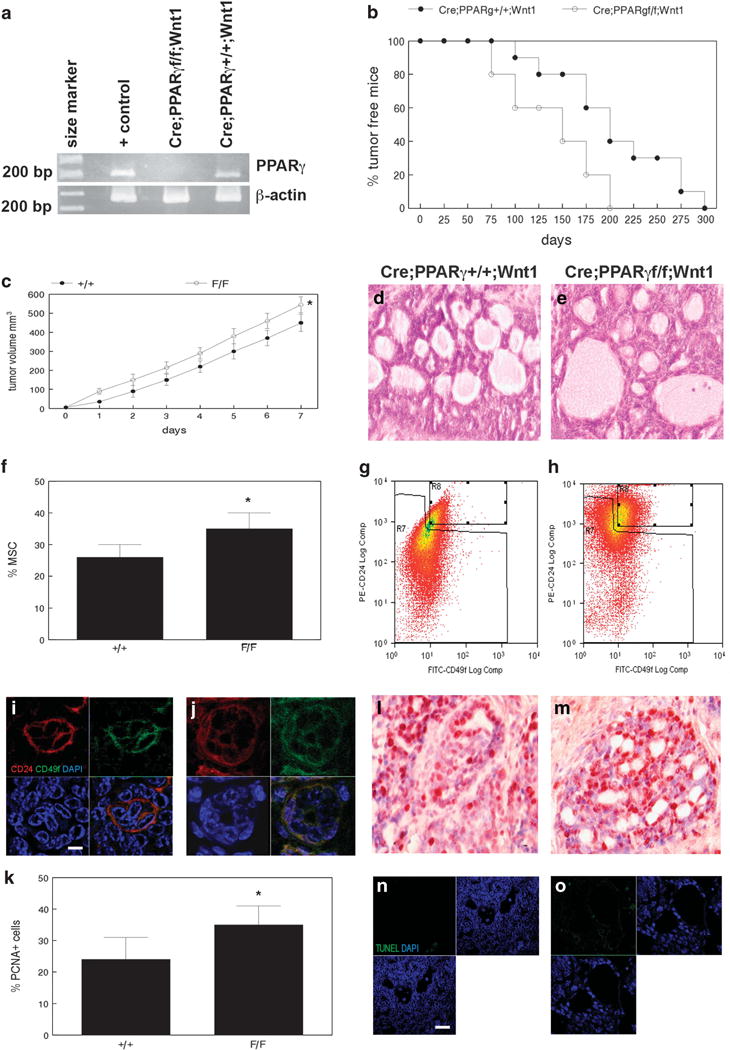
Loss of PPARγ expression reduces tumor latency, expands the tumorigenic CD24+/CD49fhi stem cell population and increases proliferation in Wnt1-induced mammary cancer. (a) PPARγ expression in adipocytes (+ control), MMTV-Cre;PPARγf/f;Wnt1 and MMTV-Cre; PPARγ+/+;Wnt1 mammary tumors is shown by RT–PCR. β-Actin amplification was used as the loading control. DNA size marker is shown at left. (b) Decreased tumor latency in MMTV-Cre;PPARγf/f;Wnt1 mammary cancer. Kaplan–Meier analysis of MMTV-Cre;PPARγf/f;Wnt1 and MMTV-Cre;PPARγ+/+;Wnt1 mammary tumor latency shows percent tumor-free mice over time (days). (c) MMTV-Cre;PPARγf/f;Wnt1 mammary tumors exhibit increased growth rate. Tumor volume in MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 cancers was measured daily. Error bars indicate s.e.m. (d, e) Histopathologic analysis of MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 mammary tumors indicates poorly differentiated adenocarcinomas as shown by H&E staining. Scale bar = 10 μm. (f) The tumorigenic CD24/CD49f stem cell population is expanded in MMTV-Cre;PPARγf/f;Wnt1 compared with MMTV-Cre;PPARγ+/+;Wnt1 mammary tumors. Quantitation of tumor stem cell fraction is shown for both genotypes. Representative flow-cytometric analysis of tumor stem cell fraction in MMTV-Cre;PPARγ+/+; Wnt1 (g) and MMTV-Cre;PPARγf/f;Wnt1 (h) is shown. Immunofluorescence microscopy shows CD24/CD49f stem cells in MMTV-Cre;PPARγ+/+; Wnt1 (i) and MMTV-Cre;PPARγf/f;Wnt1 (j) mammary tumors. CD24 and CD49f expression is shown by phycoerythrin and fluorescein-conjugated antibodies (red and green, respectively). DAPI nuclear counterstaining is shown (blue). Merged images are shown at lower right. Scale bar = 5 μm. (k) Loss of PPARγ expression increases cell proliferation compared with MMTV-Cre;PPARγ+/+;Wnt1 tumors as demonstrated by PCNA immunohistochemistry. Representative images of PCNA expression in MMTV-Cre;PPARγ+/+;Wnt1 (l) and MMTV-Cre;PPARγf/f;Wnt1 (m) mammary tumors. Sections were counterstained with hematoxylin. Scale bar = 10 μm. Representative TUNEL images of apoptotic cells in tumors from MMTV-Cre;PPARγ+/+;Wnt1 (n) and MMTV-Cre;PPARγf/f;Wnt1 (o) are shown. TUNEL-positive cells are shown by dUTP-fluorescein labeling (green) and DAPI nuclear counterstain (blue). Merged images are shown at lower left. Scale bar = 5 μm.
CD24+/CD49fhi MSC from MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 mammary tumors was grown as tumorspheres in MSC medium on low attachment plates. We observed no significant differences in MSC clonogenicity (Figure 2a) or tumorsphere proliferation (Figure 2b) between cells from MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Tumorspheres appeared morphologically similar by phase contrast microscopy (Figures 2c and d). These results indicate that loss of PPARγ expression does not affect in vitro clonogenicity and proliferation of tumor-derived MSC. We next performed gene expression analysis on in vivo-isolated MSC from both genotypes. Interestingly, the angiogenesis pathway was upregulated in MMTV-Cre;PPARγf/f;Wnt1 MSC (Angpt1, 2-fold; miR-15a, -2-fold; Figure 2e). There were no differences in VEGF, VEGFR or Tie2 expression between genotypes (data not shown). Searches of public databases identified a miR-15a/Angpt1 interaction in a previous genomic screening (Figure 2f).27 MMTV-Cre;PPARγf/f; Wnt1 mammary tumors expressed increased Angpt1 protein as shown by western blot (2- to 4-fold; Figure 2g). We then localized capillary endothelium by CD31 expression. We observed a 4-fold increase in capillary density in MMTV-Cre;PPARγf/f;Wnt1 mammary tumors (P < 0.007; Figures 2h–j). These results indicate that loss of PPARγ expression correlates with increased angiogenesis in MMTV-Cre;PPARγf/f;Wnt1 mammary tumors.
Figure 2.
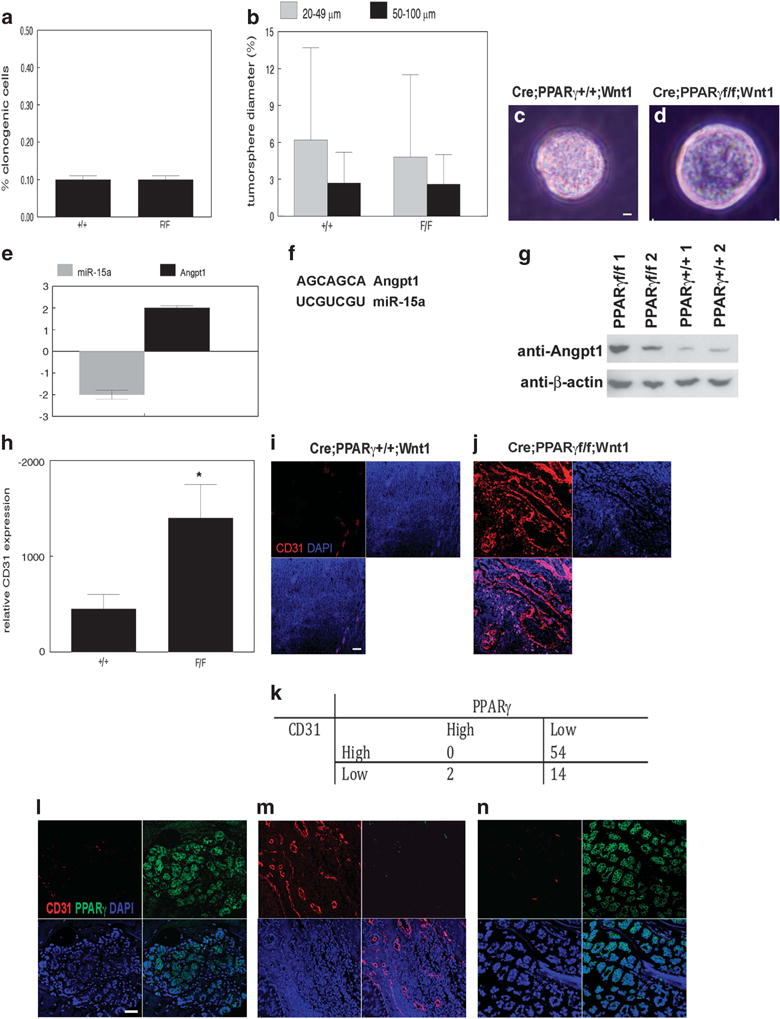
Loss of PPARγ correlates with stem cell expansion and increased angiogenesis in Wnt1 mammary tumors. (a) In vitro clonogenicity of MSC from MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. (b) In vitro proliferation of MSC from MMTV-Cre;PPARγ +/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Percent small (20–49 μm) and large (50–100 μm) tumorspheres from each genotype are shown. Representative phase contrast photomicrographs of tumorspheres from MMTV-Cre;PPARγ+/+;Wnt1 (c) and MMTV-Cre; PPARγf/f;Wnt1 (d) MSC. Scale bar = 10 μm. (e) miR-15a and angiopoietin-1 mRNA expression changes in MMTV-Cre;PPARγf/f;Wnt1 compared with MMTV-Cre;PPARγ+/+;Wnt1 mammary tumors. (f) Complementary sequences of miR-15a and Angpt1 3′-UTR. (g) Angpt1 protein expression is increased in MMTV-Cre;PPARγf/f;Wnt1 compared with MMTV-Cre;PPARγ+/+;Wnt1 tumors as shown by western blot. β-Actin expression is used as the loading control. Representative blots are shown. (h) Increased angiogenesis in MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Representative CD31 immunofluorescence images of capillary endothelium in MMTV-Cre;PPARγ+/+;Wnt1 (i) and MMTV-Cre;PPARγf/f; Wnt1 (j) mammary tumors. Nuclei were counterstained with DAPI (blue). Scale bar = 20 μm. (k) Decreased PPARγ expression correlates with increased angiogenesis in human breast cancer. PPARγ and CD31 expression in control human mammary gland (l) and human breast cancers (m, n). Representative immunofluorescence images of CD31 (red), PPARγ (green) and DAPI nuclear counterstain (blue). Merged images are shown at lower right. Scale bar = 20 μm.
We examined PPARγ and CD31 expression by immunofluorescence microscopy in 70 human breast cancers and normal breast tissue (Figure 2k). All normal human breast epithelium demonstrated diffuse PPARγ nuclear staining with minimal stromal capillaries (Figure 2l). In contrast, human breast cancer specimens were predominantly negative for PPARγ expression (68/70; 97%). These PPARγ-negative breast cancer specimens were highly angiogenic (54/70; 77%; P < 0.05; Figure 2m). The two breast cancer samples that expressed PPARγ did not exhibit the angiogenic phenotype (Figure 2n). These results indicate that loss of PPARγ expression correlates with increased angiogenesis in human breast cancer.
We treated MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f; Wnt1 tumors in vivo with the PPARγ agonist rosiglitazone. This PPARγ selective ligand significantly induced miR-15a expression and apoptosis, and inhibited Angpt1 expression, MSC fraction and tumor proliferation/growth in MMTV-Cre;PPARγ+/+;Wnt1 tumors (Supplementary Figure S1). These effects were not observed in MMTV-Cre;PPARγf/f;Wnt1 tumors. To determine whether miR-15a and Angpt1 were responsible for the angiogenic phenotype of MMTV-Cre;PPARγf/f;Wnt1 tumors, we stably transduced MMTV-Cre;PPARγf/f;Wnt1 MSC with Angpt1 shRNAs, miR-15a, or control lentiviruses followed by transplantation to mammary fat pads of immunodeficient mice. Angpt1 shRNAs reduced Angpt1 protein expression in MMTV-Cre;PPARγf/f;Wnt1 tumors by 90% as determined by western blot (Figure 3a). miR-15a lentivirus reduced Angpt1 protein expression in MMTV-Cre;PPARγf/f;Wnt1 tumors by 80%. Mammary tumors derived from Angpt1 shRNAs, miR-15a and control transduced MSC were classified as poorly differentiated adenocarcinomas by histopathologic analysis (Figures 3b–d). The angiogenic phenotype was dramatically suppressed in tumors derived from transplanted MSC transduced with Angpt1 shRNAs or miR-15a compared with control lentivirus as determined by CD31 immunofluorescence microscopy (75–80% reduction; P < 0.002; Figures 3e–h). Reduced angiogenesis correlated with threefold decreases in the CD24+/CD49fhi MSC fractions of tumors derived from Angpt1 shRNA and miR-15a transduced stem cells as determined by FACS (P < 0.02; Figures 3i–l). Angpt1 shRNAs and miR-15a inhibited growth of MMTV-Cre;PPARγf/f;Wnt1 tumors (P < 0.05; Figure 3m). Differences were not observed in PCNA+ (Figures 3n–p) or apoptotic (Figures 3q–s) cells in tumors derived from Angpt1 shRNA or miR-15a transduced MSC compared with control mammary cancers. These results indicate that Angpt1 and miR-15 regulate the angiogenic phenotype of MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Inhibition of the angiogenic phenotype dramatically suppressed MSC expansion in these cancers.
Figure 3.
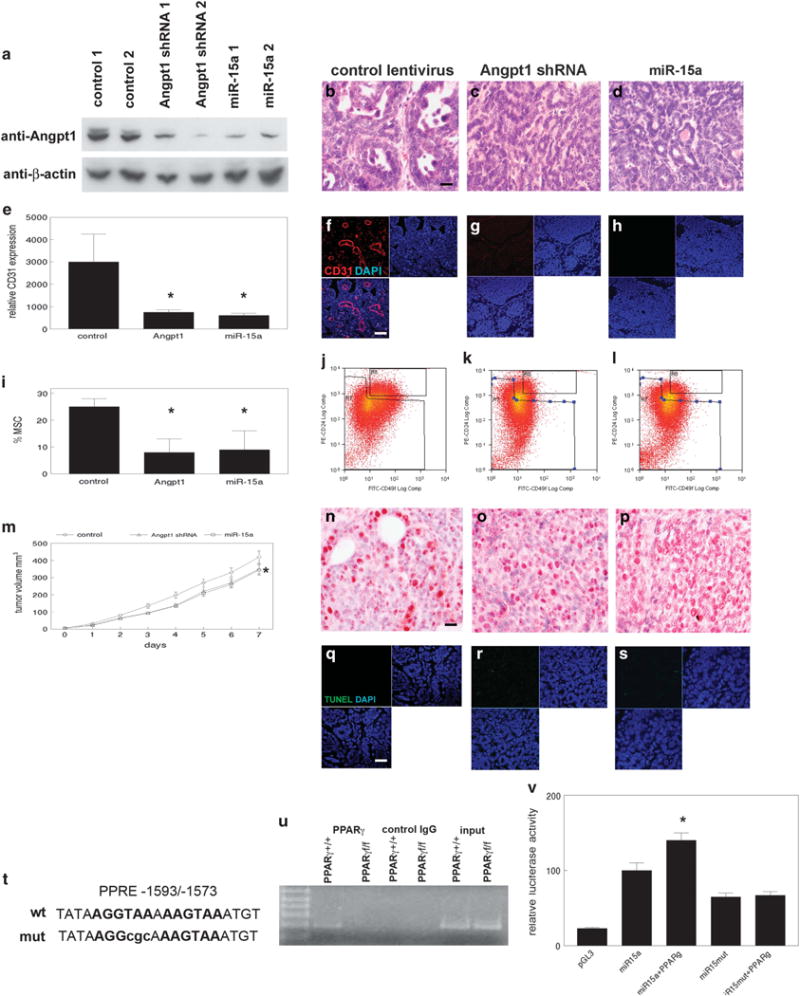
miR-15a and Angpt1 regulate angiogenesis in Wnt1 mammary tumors. (a) Angpt1 protein expression in MMTV-Cre;PPARγf/f;Wnt1 MSC transduced with control, Angpt1 shRNAs and miR-15a lentivirus is shown by western blot. β-Actin is expression used as the loading control. Histopathologic analysis of mammary tumors from control (b), Angpt1 shRNAs (c) or miR-15a lentivirus (d) transfected MMTV-Cre; PPARγf/f;Wnt1 MSC indicates poorly differentiated adenocarcinomas as shown by H&E staining. Scale bar = 10 μm. (e) Decreased angiogenesis in mammary tumors from Angpt1 shRNAs and miR-15a lentivirus transduced MMTV-Cre;PPARγf/f;Wnt1 MSC. Representative CD31 immunofluorescence images of capillary endothelium are shown in tumors from control (f), Angpt1 shRNAs (g) and miR-15a (h) lentiviral transduced MSC. Scale bar = 20 μm. (i) Decreased MSC fraction in tumors from MMTV-Cre;PPARγf/f;Wnt1 MSC transduced with Angpt1 shRNAs or miR-15a lentivirus. Representative FACS analyses of MSC fractions in tumors from control (j), Angpt1 shRNAs (k) and miR-15a (l) lentivirus transduced MSC. (m) miR-15a and Angpt1 shRNA inhibited growth of MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Tumor volume in miR-15a and Angpt1 shRNA transduced MMTV-Cre;PPARγf/f;Wnt1 cancers was measured daily. Error bars indicate s.e.m. Representative images of PCNA expression in tumors from MMTV-Cre;PPARγf/f;Wnt1 MSC transduced with control (n), Angpt1 shRNAs (o) and miR-15a lentivirus (p). Sections were counterstained with hematoxylin. Scale bar = 10 μm. Representative TUNEL images of apoptotic cells in tumors from control (q), Angpt1 shRNAs (r) and miR-15a (s) lentivirus transduced MSC are shown. TUNEL-positive cells are shown by dUTP-fluorescein labeling (green) and DAPI nuclear counterstain (blue). Merged images are shown at lower left. Scale bar = 5 μm. The 5′ flanking region of miR-15a gene has promoter activity and a DR1 element that is activated by PPARγ. (t) Sequence of the putative PPARγ response element (PPRE) located 1593 bp upstream of the transcriptional start (miR-15a wt). Site directed mutation in PPRE is shown in lowercase letters (miR-15a mut). (u) PPARγ binds to the miR-15a PPRE in genomic DNA. Chromatin immunoprecipitation of miR-15a PPRE from MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre; PPARγf/f;Wnt1 MSC was performed using anti-PPARγ antibody. Control IgG was used as the negative immunoprecipitation control. Non-immunoprecipitated genomic DNA (input) was used as the amplification control. DNA size marker is shown at left. (v) PPARγ induces miR-15a promoter activity via the DR1 element. Relative luciferase activities in miR-15a promoter and site directed DR1 mutant are shown.
Sequence analysis of the miR-15a gene revealed a potential PPARγ binding site in the 5′ flanking region at − 1593/− 1573 bp (Figure 3t). Chromatin immunoprecipitation using tumor-derived CD24/CD49f MSC revealed PPARγ binding to the DR1 region, but not in PPARγ-null cells (Figure 3u). We cloned 1.8 kb of the miR-15a 5′ flanking region into the pGL3 luciferase reporter vector. The miR-15a 5′ flanking region induced luciferase reporter activity by fourfold (Figure 3v). Transient transfection of a mouse PPARγ expression vector significantly induced transcription of the miR-15a reporter construct, but not when the putative PPRE was mutated (P < 0.008). These data indicate that PPARγ activates expression of the miR-15a gene by binding to a DR1 element in the 5′ flanking region.
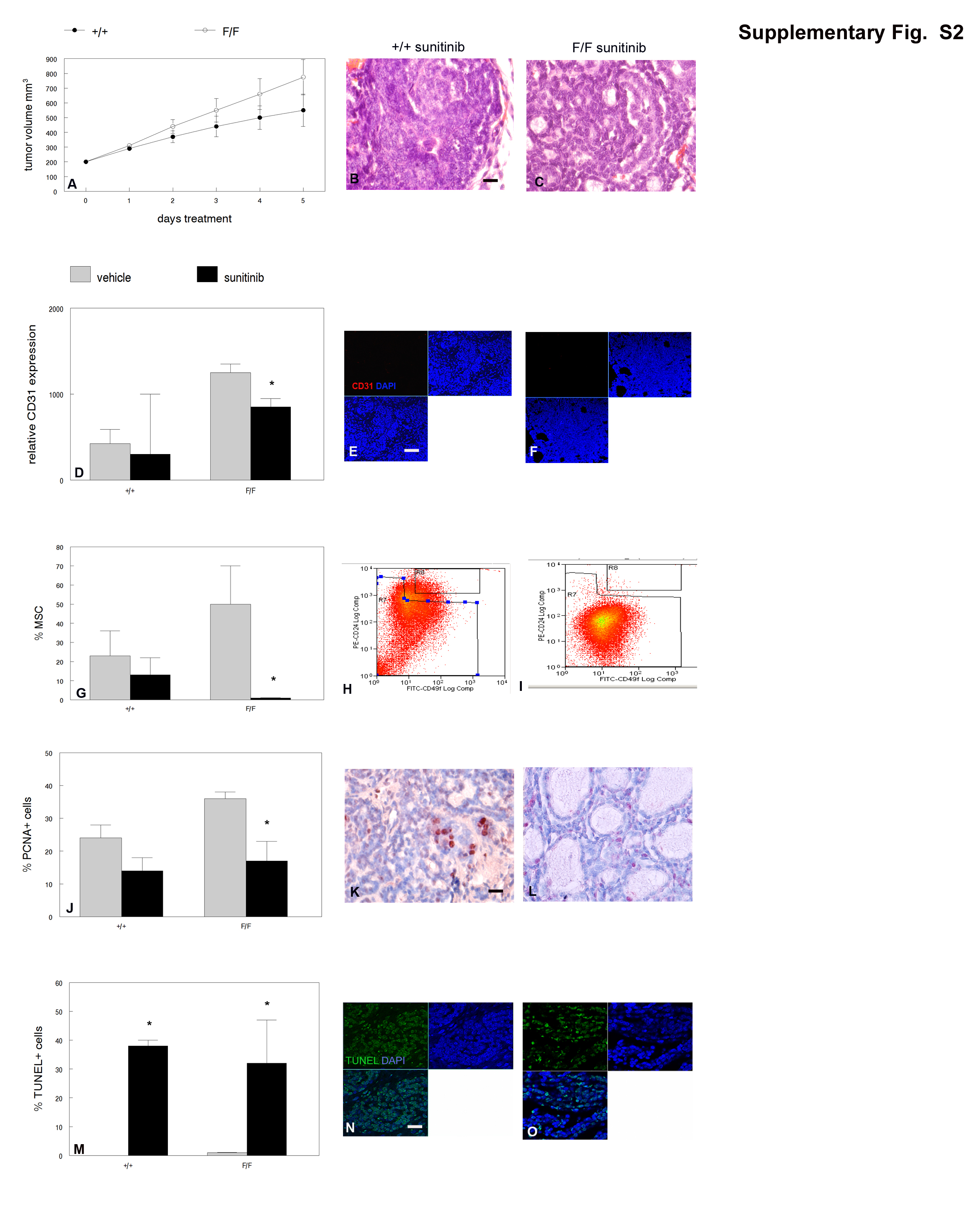
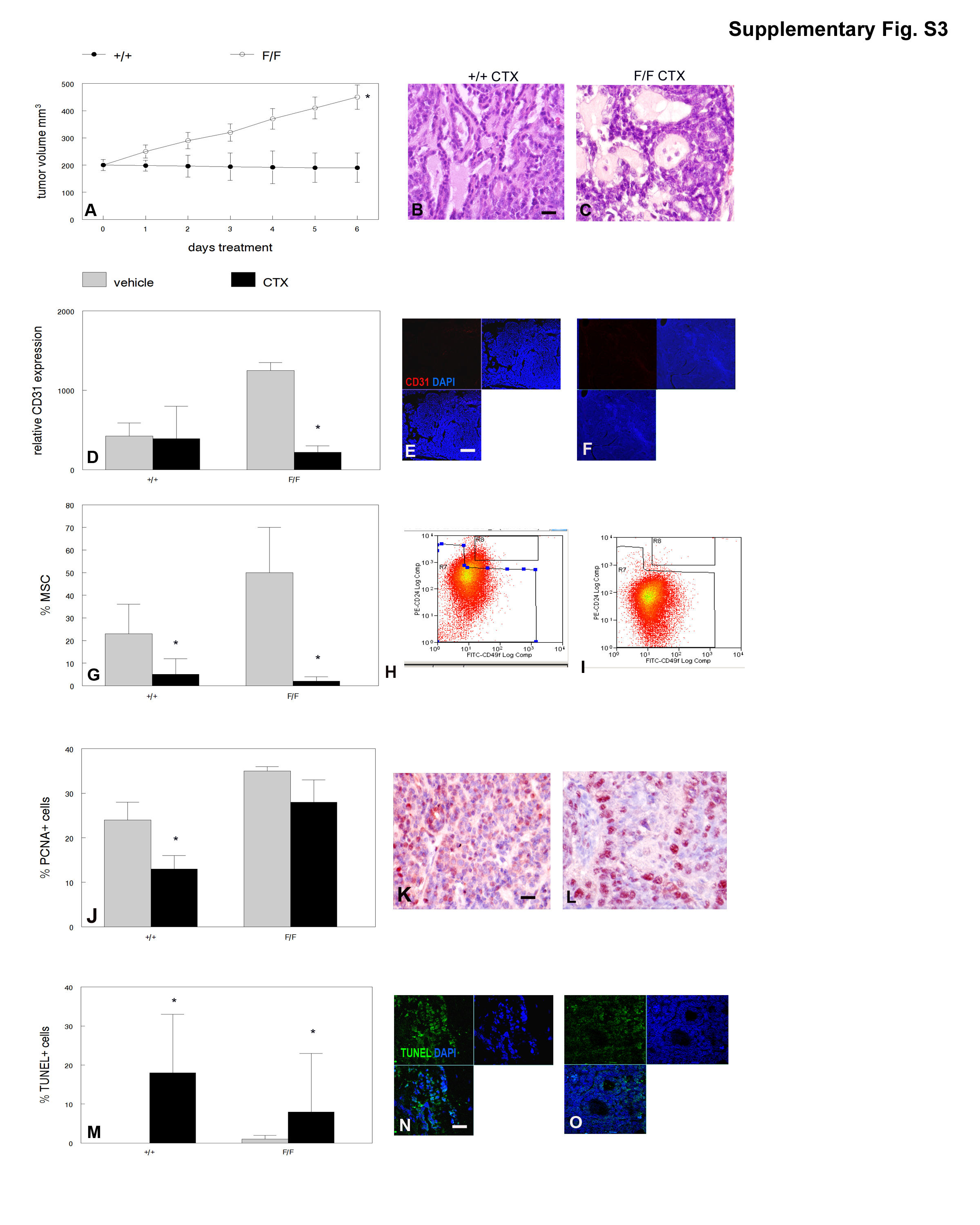
MMTV-Cre;PPARγf/f;Wnt1 mammary tumors were resistant to the anti-angiogenesis drug sunitinib and cyclophosphamide chemotherapy when used as single agents (Supplementary Figures S2 and S3). However, treatment with sunitinib followed by cyclophosphamide produced significantly greater tumor reduction in MMTV-Cre;PPARγf/f;Wnt1 than in MMTV-Cre;PPARγ +/+;Wnt1 cancers (P < 0.0004; Figure 4a). Tumors from both groups treated with sunitinib and cyclophosphamide resembled poorly differentiated luminal adenocarcinomas with reduced basal cells (Figures 4b and c). Sunitinib and cyclophosphamide treatment significantly inhibited the angiogenic phenotype in MMTV-Cre;PPARγf/f;Wnt1 (P < 0.002; Figures 4d–f) but not in MMTV-Cre;PPARγ+/+;Wnt1 mammary tumors. Sunitinib and cyclophosphamide treatment significantly decreased the MSC population in MMTV-Cre;PPARγf/f;Wnt1 (1 vs 49%; P < 0.05; Figures 4g–i) but not in MMTV-Cre;PPARγ+/+;Wnt1 mammary tumors. Sunitinib and cyclophosphamide treatment dramatically decreased cell proliferation in MMTV-Cre;PPARγ+/+;Wnt1 (mean 13 vs 24% in control-treated tumors; P < 0.04; Figures 4j and k) and MMTV-Cre;PPARγf/f;Wnt1 (mean 27 vs 34% in control-treated tumors; P < 0.03; Figures 4j and l) mammary tumors. Sunitinib and cyclophosphamide treatment significantly increased apoptotic basal and luminal cells in MMTV-Cre;PPARγ+/+;Wnt1 (42 vs 0.1%; P < 0.02; Figures 4m and n) and MMTV-Cre;PPARγf/f;Wnt1 (38 vs 0.2%; P < 10−5; Figures 4m and o) mammary tumors. These results indicate that MMTV-Cre;PPARγf/f;Wnt1 mammary tumors are sensitive to sunitinib and cyclophosphamide chemotherapy.
Figure 4.
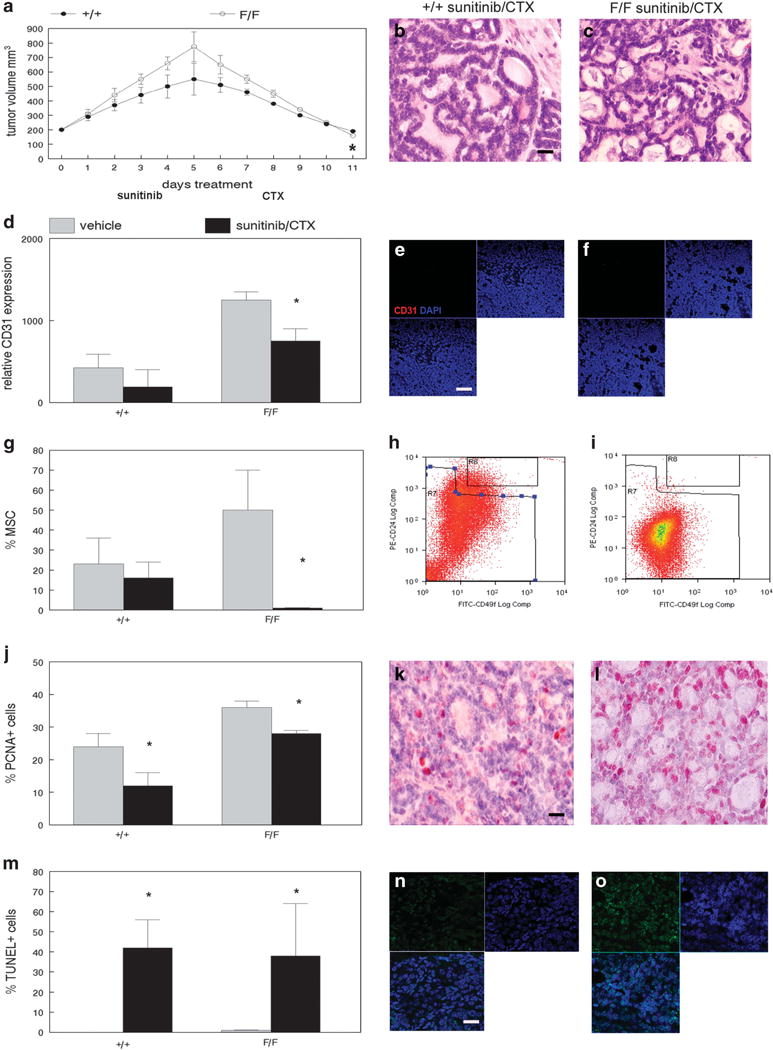
MSC from MMTV-Cre;PPARγf/f;Wnt1 mammary tumors is sensitive to sunitinib/cyclophosphamide chemotherapy. (a) Greater reduction in volume of sunitinib/cyclophosphamide-treated MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Tumor volume in sunitinib/cyclophosphamide-treated MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 cancers was measured daily. Error bars indicate s.e.m. Histopathologic analyses of sunitinib/cyclophosphamide-treated MMTV-Cre;PPARγ+/+;Wnt1 (b) tumors and MMTV-Cre;PPARγf/f;Wnt1 (c) mammary tumors indicate poorly differentiated luminal adenocarcinomas as shown by H&E staining. Scale bar = 10 μm. (d) Decreased angiogenesis in sunitinib/cyclophosphamide-treated MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Representative CD31 immunofluorescence images of capillary endothelium in sunitinib/cyclophosphamide-treated MMTV-Cre;PPARγ+/+;Wnt1 (e) and MMTV-Cre;PPARγf/f;Wnt1 (f) mammary tumors. Nuclei were counterstained with DAPI (blue). Merged images are shown at lower left. Scale bar = 20 μm. (g) Decreased MSC fraction in sunitinib/cyclophosphamide-treated MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Representative FACS analyses of MSC fractions in sunitinib/cyclophosphamide-treated MMTV-Cre;PPARγ+/+;Wnt1 (h) tumors and MMTV-Cre;PPARγf/f;Wnt1 (i) mammary tumors are shown. (j) Decreased cell proliferation in sunitinib/cyclophosphamide-treated MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 mammary tumors as demonstrated by PCNA immunohistochemistry. Representative PCNA immunohistochemical images of sunitinib/cyclophosphamide-treated MMTV-Cre;PPARγ+/+;Wnt1 (k) and MMTV-Cre;PPARγf/f;Wnt1 (l) mammary tumors. Sections were counterstained with hematoxylin. Scale bar = 10 μm. (m) Increased apoptotic cells in sunitinib/cyclophosphamide MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre; PPARγf/f;Wnt1 mammary tumors. Representative TUNEL images of apoptotic cells in tumors from MMTV-Cre;PPARγ+/+;Wnt1 (n) and MMTV-Cre;PPARγf/f;Wnt1 (o) mammary tumors are shown. TUNEL-positive cells are shown by dUTP-fluorescein labeling (green) and DAPI nuclear counterstain (blue). Merged images are shown at lower left. Scale bar = 5 μm.
Despite chemotherapy-mediated depletion of the MSC population, we observed increased tumor volume with sunitinib and cyclophosphamide monotherapies. CD61+ luminal progenitor cells were previously identified as tumorigenic and capable of regenerating both basal and luminal cells in the Wnt1 mammary tumor model.28 To determine whether CD61+ luminal progenitor cells contribute to mammary tumorigenesis following chemotherapy, we examined the CD61+ cell fraction in control and chemotherapy-treated MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 tumors using FACS. Control-treated MMTV-Cre; PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 tumors contained mean CD61+ cell fractions of 2 and 2.5%, respectively (Figures 5a and b). Sunitinib treatment resulted in dramatic expansion of the CD61+ cell population (63% in MMTV-Cre;PPARγ+/+;Wnt1 and 73% in MMTV-Cre;PPARγf/f;Wnt1 tumors; P < 10−7; Figures 5c and d). Cyclophosphamide treatment increased the CD61+ cell fraction in MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 tumors to 66 and 63%, respectively (Figures 5e and f). Sunitinib and cyclophosphamide chemotherapy increased the CD61+ cell fraction in MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f; Wnt1 tumors to 44 and 72%, respectively (Figures 5g and h). To confirm failure of basal cell regeneration in chemotherapy-treated tumors, we performed immunohistochemistry using the basal and luminal cell markers keratin 5 and keratin 18. We observed consistent reduction of keratin 5 expressing basal cells in chemotherapy-treated MMTV-Cre;PPARγ+/+;Wnt1 (Figures 5i and j) and MMTV-Cre;PPARγf/f;Wnt1 (Figures 5k and l) tumors. In contrast, no significant changes in keratin 18-positive cells were observed due to chemotherapy treatment in MMTV-Cre;PPARγ+/+; Wnt1 (Figures 5m and n) and MMTV-Cre;PPARγf/f;Wnt1 (Figures 5o and p) tumors. To determine whether loss of the angiogenic phenotype in MMTV-Cre;PPARγf/f;Wnt1 mammary tumors was due to basal cell-specific Angpt1 expression, we examined this protein by immunofluorescence microscopy in control and chemotherapy-treated tumors from both genotypes. Angpt1 protein was expressed in both basal and luminal cells in control-treated tumors from both genotypes (Figures 5r and t). Angpt1 expression was twofold higher in MMTV-Cre;PPARγf/f;Wnt1 tumors consistent with our western blot results (Figure 5q). Chemotherapy treatment significantly decreased Angpt1 expression in MMTV-Cre;PPARγf/f;Wnt1 mammary tumors (−3-fold; P < 0.01; Figures 5s and u), which correlated with reduced angiogenesis in these cancers. These results indicate that the CD61+ luminal progenitor population undergoes dramatic expansion in the context of chemotherapy induced MSC depletion.
Figure 5.
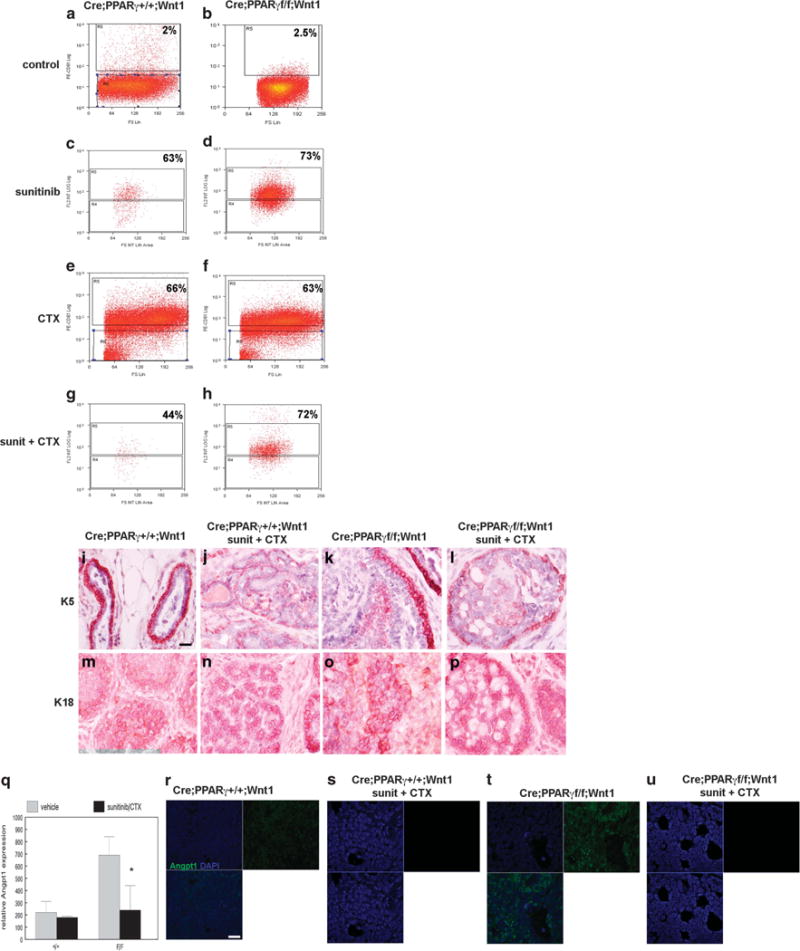
CD61+ luminal progenitor cell expansion in chemotherapy-treated MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Representative flow-cytometric analyses of CD61+ luminal progenitor cell fraction in control MMTV-Cre;PPARγ+/+;Wnt1 (a) and MMTV-Cre;PPARγf/f;Wnt1 (b), sunitinib-treated MMTV-Cre;PPARγ+/+;Wnt1 (c) and MMTV-Cre;PPARγf/f;Wnt1 (d), cyclophosphamide-treated MMTV-Cre;PPARγ+/+;Wnt1 (e) and MMTV-Cre;PPARγf/f;Wnt1 (f), sunitinib and cyclophosphamide-treated MMTV-Cre;PPARγ+/+;Wnt1 (g) and MMTV-Cre;PPARγf/f;Wnt1 (h) mammary tumors. Reduced basal cell marker keratin 5 expression in MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 tumors treated with sunitinib/cyclophosphamide chemotherapy. Representative immunohistochemical images of keratin 5 expression in control (i) and sunitinib/cyclophosphamide-treated (j) MMTV-Cre;PPARγ+/+;Wnt1 mammary tumors, and control (k) and sunitinib/cyclophosphamide-treated (l) MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Representative immunohistochemical images of keratin 18 luminal marker expression in control (m) and sunitinib/cyclophosphamide-treated (n) MMTV-Cre;PPARγ+/+;Wnt1 mammary tumors, and control (o) and sunitinib/cyclophosphamide-treated (p) MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Scale bar = 10 μm. (q) Decreased Angpt1 expression in MMTV-Cre;PPARγf/f;Wnt1 tumors treated with sunitinib/cyclophosphamide. Representative immunofluorescence images of Angpt1 expression (green) with DAPI nuclear counterstain (blue) in control (r) and sunitinib/cyclophosphamide-treated (s) MMTV-Cre;PPARγ+/+;Wnt1 mammary tumors, and control (t) and sunitinib/cyclophosphamide MMTV-Cre;PPARγf/f;Wnt1 (u) mammary tumors. Merged images are shown at lower left. Scale bar = 5 μm.
To determine whether chemotherapy-treated MSC and CD61+ luminal progenitor cells are tumorigenic, we transplanted these populations from MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre; PPARγf/f;Wnt1 tumors to mammary fat pads of immunodeficient mice. Chemotherapy-treated MSC transplants from both genotypes failed to form tumors (data not shown). However, transplanted chemotherapy-treated CD61+ luminal progenitor cells from both genotypes consistently formed tumors. There were no significant differences in growth rate between MMTV-Cre; PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 tumors derived from chemotherapy-treated CD61+ luminal progenitor cells (Figure 6a). These cancers were classified as poorly differentiated luminal adenocarcinomas (Figures 6b and c). Notably basal layer cells failed to regenerate in these transplanted tumors. Strikingly, the angiogenic phenotype was suppressed in tumors derived from transplanted CD61+ luminal progenitor cells sorted from chemotherapy-treated MMTV-Cre;PPARγf/f;Wnt1 cancers as determined by CD31 expression (Figures 6d–f). Angpt1 expression was significantly suppressed in tumors derived from transplanted CD61+ luminal progenitor cells sorted from chemotherapy-treated MMTV-Cre;PPARγf/f;Wnt1 tumors (P < 0.008; Figures 6g–i). We confirmed failure of basal cell regeneration using keratin 5 immunohistochemistry on tumors derived from transplanted CD61+ luminal progenitor cells sorted from chemotherapy-treated MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 tumors (Figures 6j and k). In contrast, luminal cells were regenerated in tumors derived from transplanted CD61+ luminal progenitor cells sorted from chemotherapy-treated MMTV-Cre; PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 cancers as confirmed by keratin 18 immunohistochemistry (Figures 6l and m). The PCNA-positive cell fraction was not significantly different in MMTV-Cre;PPARγ+/+;Wnt1 cancers (15 vs 23%; Figure 6n), but remained suppressed in MMTV-Cre;PPARγf/f;Wnt1 (20 vs 35%; P < 0.00005; Figures 6o and p) tumors derived from transplanted CD61+ luminal progenitor cells sorted from chemotherapy-treated mammary tumors. The TUNEL+ cell fraction was low in transplanted tumors, and no significant differences were observed between genotypes (Figures 6q–s). Consistent with failure of basal cells to regenerate, we observed suppression of the MSC fraction in transplanted tumors by FACS (0.5%; Figure 6t). Similarly consistent with observed luminal histopathology, the CD61+ luminal progenitor population remained expanded in transplanted tumors (72%; Figure 6u). These results indicate that the CD61+ luminal progenitor (but not MSC) population drives tumorigenesis in chemotherapy-treated mammary tumors. However, chemotherapy treatment results in persistent suppression of angiogenesis, cell proliferation and basal cell regeneration.
Figure 6.
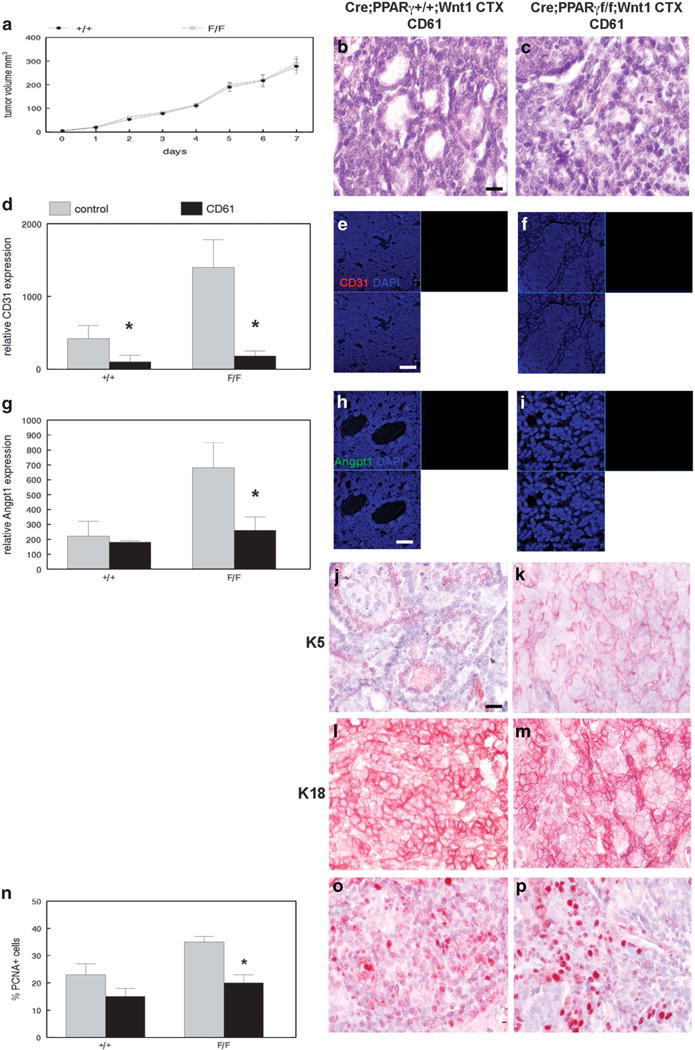
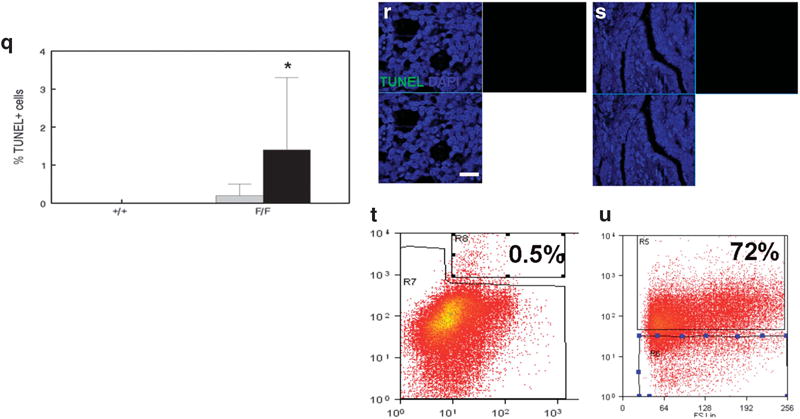
Chemotherapy-treated CD61+ luminal progenitor cells are tumorigenic but unipotent and not angiogenic. (a) No significant differences in growth rate between MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 tumors derived from chemotherapy-treated CD61+ luminal progenitor cells. Tumor volume was measured daily. Error bars indicate s.e.m. Histopathologic analysis of tumors from CD61+ luminal progenitor cells sorted from chemotherapy-treated MMTV-Cre;PPARγ+/+;Wnt1 (b) and MMTV-Cre;PPARγf/f;Wnt1 (c) indicates poorly differentiated luminal adenocarcinomas as shown by H&E staining. Scale bar = 10 μm. (d) Reduced CD31 expression in mammary tumors derived from transplanted CD61+ luminal progenitor cells from chemotherapy-treated cancers of each genotype. Representative CD31 (red) immunofluorescence images of capillary endothelium in mammary tumors from CD61+ luminal progenitor cells sorted from chemotherapy-treated MMTV-Cre;PPARγ+/+;Wnt1 (e) and MMTV-Cre;PPARγf/f;Wnt1 (f) mammary tumors. Nuclei were counterstained with DAPI (blue). Merged images are shown at lower left. Scale bar = 20 μm. (g) Reduced Angpt1 expression in transplanted mammary tumors derived from CD61+ luminal progenitor cells sorted from chemotherapy-treated MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Representative immunofluorescence images of Angpt1 expression (green) and DAPI nuclear counterstain (blue) in transplanted mammary tumors from CD61+ luminal progenitor cells sorted from chemotherapy-treated MMTV-Cre;PPARγ+/+;Wnt1 (h) and MMTV-Cre; PPARγf/f;Wnt1 (i) mammary tumors. Merged images are shown at lower left. Scale bar = 5 μm. Lack of basal cell regeneration in tumors derived from transplanted CD61+ luminal progenitor cells sorted from chemotherapy-treated MMTV-Cre;PPARγ+/+;Wnt1 (j) and MMTV-Cre; PPARγf/f;Wnt1 (k) mammary tumors is demonstrated by keratin 5 immunohistochemistry. Scale bar = 10 μm. Luminal cell regeneration in tumors derived from transplanted CD61+ luminal progenitor cells sorted from chemotherapy-treated MMTV-Cre;PPARγ+/+;Wnt1 (l) and MMTV-Cre;PPARγf/f;Wnt1 (m) mammary tumors is demonstrated by keratin 18 immunohistochemistry. Scale bar = 10 μm. (n) Quantitation of cell proliferation in tumors derived from transplanted CD61+ luminal progenitor cells sorted from chemotherapy-treated tumors of both genotypes is demonstrated by PCNA immunohistochemistry. Representative images of PCNA expression in tumors derived from transplanted CD61+ luminal progenitor cells sorted from chemotherapy-treated MMTV-Cre;PPARγ+/+;Wnt1 (o) and MMTV-Cre;PPARγf/f;Wnt1 (p) tumors are shown. Scale bar = 10 μm. (q) Quantitation of apoptotic cells in tumors derived from transplanted CD61+ luminal progenitor cells sorted from chemotherapy-treated cancers from both genotypes as shown by TUNEL analysis. Representative TUNEL images of apoptotic cells in tumors derived from transplanted CD61+ luminal progenitor cells sorted from chemotherapy-treated MMTV-Cre;PPARγ+/+;Wnt1 (r) and MMTV-Cre; PPARγf/f;Wnt1 (s) tumors are shown. TUNEL-positive cells are shown by dUTP-fluorescein labeling (green) and DAPI nuclear counterstain (blue). Merged images are shown at lower left. Scale bar = 5 μm. (t) Representative flow-cytometric analysis of the CD24+/CD49fhi tumor stem cell fraction in tumors derived from transplanted CD61+ luminal progenitor cells sorted from chemotherapy-treated mammary tumors. (u) Representative flow-cytometric analysis of the CD61+ luminal progenitor cell population in tumors derived from transplanted CD61+ luminal progenitor cells sorted from chemotherapy-treated mammary tumors.
DISCUSSION
Our study is the first to report a miRNA target gene for the tumor suppressor PPARγ. A genomic study of PPARγ target genes using chromatin immunoprecipitation followed by high throughput sequencing in adipocytes failed to identify the PPARγ binding site in the miR-15a gene.29 These results suggest that factors other than sequence similarity to the canonical PPARγ binding site are required for receptor binding in specific cell types.30 miR-15a expression was decreased in human breast cancers and cell lines,31,32 although target genes were not identified. Other miRNAs have been shown to regulate angiogenesis in human breast cancer. miR-126 expression targeted VEGF levels and is downregulated in human breast cancer.33 miR-148a expression is reduced in breast cancer and inhibits angiogenesis by targeting ERBB3.34 Loss of PTEN in breast cancer stromal fibroblasts downregulates miR-320, thereby upregulating its target ETS2 that promotes tumor angiogenesis.35 miR-126 inhibits angiogenesis by targeting signaling pathways such as insulin-like growth factor.36 miR-145 inhibits tumor angiogenesis by targeting N-RAS and VEGF in breast cancer.37 miR-98 inhibits tumor angiogenesis in breast cancer cells by targeting activin receptor like kinase 4 and matrix metalloproteinase 11.38 Expression of the transcription factor GATA3 is reduced in breast cancer; GATA3 inhibits breast cancer metastasis by inducing expression of miR-29b that regulates angiogenesis.39 miR-155 promotes tumor angiogenesis by targeting the von Hippel-Lindau tumor suppressor and is associated with poor prognosis and triple negative breast cancer.40 miR-542-3p inhibits angiogenesis in breast cancer xenografts by targeting angiopoietin-2 and is associated with poor prognosis in humans.41 These studies indicate that the breast cancer angiogenic niche is regulated by highly complex transcriptional and post-transcriptional mechanisms.
One of the novel findings of our study is the existence of an MSC angiogenic niche in mammary cancer. The angiogenic phenotype of MMTV-Cre;PPARγf/f;Wnt1 tumors is associated with MSC expansion and increased cellular proliferation. Inhibition of this phenotype by genetic and pharmacologic methods dramatically reduced the MSC fraction and cellular proliferation. This is in contrast to a previous study, which indicated that tumor hypoxia resulting from anti-angiogenic therapy results in stem cell expansion.42 An important difference in our study is the angiogenic phenotype of MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Our results suggest that anti-angiogenic therapy would be more effective if targeted at tumors expressing the angiogenic phenotype.
Our results indicate that Angpt1 expression is critical to angiogenesis and MSC expansion. Angpt1 is overexpressed in mouse mammary and human breast cancer specimens and cell lines.43,44 Angpt1 expression was higher in estrogen receptor-negative breast cancers and correlated with increased angiogenesis.45 BRCA1 mutant and triple negative breast cancers also expressed higher Angpt1 levels.46
Human breast cancer specimens were predominantly negative for PPARγ expression. These PPARγ-negative breast cancer specimens were highly angiogenic. Breast cancer samples that expressed PPARγ did not exhibit the angiogenic phenotype. These human breast cancer specimens that expressed PPARγ showed a different pattern of nuclear localization compared with normal breast epithelium. PPARγ expression in these cancers was confined to the nuclear periphery. Previous studies indicated that the nuclear periphery sequesters transcription factors from their chromatin binding sites.47 Transcription factor localization to the nuclear periphery restricts access to target genes, limits transactivation and is associated with transcriptional repression.48 In PPARγ expressing breast cancers, altered nuclear localization may result in transcriptional repression of genes regulating the angiogenic phenotype. Genomic analysis of short and long range PPARγ target genes in breast cancer will provide important insight into these findings.
Previous studies hypothesized that normalization of abnormal tumor vasculature leads to more efficient chemotherapy drug delivery (for review, see Jain49). Our study indicated that sunitinib or cyclophosphamide monotherapy did not decrease tumor volume in MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. Treatment with sunitinib followed by cyclophosphamide was required to reduce tumor volume in MMTV-Cre;PPARγf/f;Wnt1 mammary tumors. These results indicate that normalization of the tumor vasculature conferred chemotherapeutic sensitivity in angiogenic mammary tumors. Tumors lacking the angiogenic phenotype were more sensitive to first-line cytotoxic chemotherapy such as cyclophosphamide. These results suggest that evaluation of angiogenesis in human breast cancers may improve clinical response to targeted chemotherapy.
Our data showed that the tumorigenic CD61+ luminal progenitor population dramatically expanded following sunitinib or cyclophosphamide chemotherapy in conjunction with increased tumor volume. BRCA1 mutant human breast cancers exhibited expansion of luminal progenitor cells.50 However, our paper is the first report of luminal progenitor cell expansion in response to anti-proliferative therapy. In contrast, chemotherapy-treated MSC failed to generate tumors following transplantation. Currently, it is not clear whether tumor hypoxia resulting from anti-angiogenic therapy or drug resistance mechanisms result in luminal progenitor cell expansion. However, this expanded luminal progenitor population was not capable of regenerating basal layer cells, indicating that chemotherapy treatment induced unipotency in the CD61+ tumor fraction. Angpt1 expression and the angiogenic phenotype were suppressed in mammary tumors derived from chemotherapy-treated luminal progenitor cells, indicating that anti-proliferative therapy has long-term effects on tumor differentiation and the angiogenic phenotype. Future studies will determine the mechanisms responsible for these altered tumor phenotypes.
MATERIALS AND METHODS
Mouse breeding and procedures
We bred female MMTV-Cre;PPARγf/f;Wnt1 mice and MMTV-Cre;PPARγ+/+; Wnt1 littermate controls which were assigned to experimental and control groups based on genotype. Mouse background strain was identical in both groups. All mouse strains were obtained from The Jackson Laboratories (Bar Harbor, ME, USA) and approved by institutional animal care committee. PPARγ expression is deleted in mammary epithelium of these mice by Cre-mediated recombination of exons 1 and 2. Mammary tumorigenesis is driven by Wnt1 oncogene expression, which is a model of basal subtype breast cancer.5 All mice were genotyped using PCR amplification of extracted tail DNA according to Jackson Laboratories protocols. Twenty tumors were obtained from each group for analysis (α = 0.05, P = 0.8, s.d. = 0.5). The latency, number and volume of tumors were recorded for each mouse. Tumors from each group were analyzed at a uniform volume of 500 mm3. Complete necropsy was performed on each mouse. Portions of each tumor were fixed in 10% formalin, flash frozen for storage at − 80°C, and trypsin dissociated for cryopreservation in liquid nitrogen. For chemotherapy experiments, tumor bearing mice were treated with five daily doses of the PPARγ agonist rosiglitazone (25 mg/kg; Sigma, St Louis, MO, USA), five daily doses of the angiogenesis inhibitor sunitinib (60 mg/kg; Sigma), three doses of the cytotoxic chemotherapeutic drug cyclophosphamide (200 mg/kg on alternating days; Sigma), sunitinib followed by cyclophosphamide, or vehicle. A minimum of five tumors per group were harvested as described above for analysis. Data were analyzed by t-test.
qRT-PCR
RNA was extracted from sorted MSC from MMTV-Cre;PPARγf/f;Wnt1 and MMTV-Cre;PPARγ+/+;Wnt1 tumors and reverse transcribed according to manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). cDNA was amplified using mPPARγ primers 5′-AGCTGAATCACCCAGAGTCC-3′ and 5′-TGCAATCAATAGAAGGAACACG-3′. β-Actin was amplified using primers 5′-AAAAGCCACCCCCACTCCTAAG-3′ and 5′-TCAAGTCAGTGTACAGGCCAGC-3′. Angpt1 cDNA was amplified using primers 5′-GGGGGAGGTTGGACAGTAA-3′ and 5′-CATCAGCTCAATCCTCAGC-3′. PCR was performed using thermal cycling parameters of 94°C for 25 s, 55°C for 1 min and 72°C for 1 min (Stratagene, La Jolla, CA, USA). miScript Primer Assay (Qiagen, Valencia, CA, USA) was used to quantitate miR-15a expression. Data were analyzed by t-test.
Histopathology, immunohistochemistry and immunofluorescence microscopy
All experiments were performed at least three times on all tumors. Investigators were blinded to genotype for all experiments. Formalin-fixed tumor tissue was dehydrated in ethanol, cleared in xylene and embedded in paraffin. Sections were deparaffinized and stained with hematoxylin and eosin. For immunohistochemistry and immunofluorescence studies, sections were rehydrated in phosphate-buffered saline (PBS, pH 7.4) and blocked with 10% normal serum. For immunohistochemistry studies, sections were incubated with anti-PCNA, keratin 5 (K5) or keratin 18 (K18) primary antibodies overnight at room temperature. Following washing in PBS, sections were incubated with biotinylated secondary antibody and streptavidin-conjugated horseradish peroxidase. Antigen–antibody complexes were detected by incubation with peroxide substrate solution containing aminoethylcarbazole chromogen followed by hematoxylin counterstaining. The percentage of PCNA+ cells in 10 random high power fields was determined by counting. Data were analyzed by t-test. For immunofluorescence studies, sections were incubated with anti-CD24, -CD49f, -CD31, -PPARγ or -Angpt1 primary antibodies overnight at room temperature. Sections from 70 human breast cancer cases and matching normal breast tissue were incubated with anti-PPARγ and -CD31 antibodies overnight at room temperature. After washing in PBS, sections were incubated with secondary antibodies conjugated to AlexaFluor 488 or AlexaFluor 555 and visualized by fluorescence microscopy following coverslipping with anti-fade mounting medium containing DAPI (Vector, Burlingame, CA, USA). Data were analyzed by Fischer’s exact test or two-sided t-test.
Fluorescence-activated cell sorting
Dissociated tumor cells were incubated with phycoerythrin-conjugated anti-CD24 and AlexaFluor 488-conjugated anti-CD49f antibodies, washed in PBS, and the CD24+/CD49fhi MSC fraction sorted by flow cytometry (MoFlo Astrios, Becton Dickinson, Franklin Lakes, NJ, USA). The CD24 +/CD49flo/CD61+ luminal progenitor fractions were sorted in separate experiments. Data were analyzed by t-test.
Cell death analysis
Tumor tissue sections were incubated with terminal deoxynucleotidyl transferase and dUTP-fluorescein for 1 h at 37°C according to manufacturer’s recommendations (Roche Applied Sciences, Indianapolis, IN, USA). After washing, apoptotic cells were visualized by fluorescence microscopy following coverslipping with anti-fade mounting medium containing DAPI. The percentage of fluorescent cells in 10 random high power fields was determined by counting. Data were analyzed by t-test.
Cell culture, lentiviral transduction and transplantation
In all, 104 sorted MSCs from MMTV-Cre;PPARγf/f;Wnt1 and MMTV-Cre; PPARγ+/+;Wnt1 tumors were cultured in 3:1 Dulbecco’s modified Eagle medium:F12 medium containing 1 × B27 supplement, 10 ng/ml epidermal growth factor, 25 ng/ml basic fibroblast growth factor, 0.2% heparin, 40 μg/ml gentamicin, 2.5 μg/ml amphotericin B (MSC medium) at 37°C in a humidified atmosphere of 5% CO2. For tumorsphere analysis, clonogenicity and proliferation were determined by counting and tumorsphere diameter measurements every second day for 2 weeks. Tumorspheres were photographed using phase contrast microscopy.
miR-15a interaction with Angpt1 mRNA27 was identified in searches of TarBase using the DIANA software. In all, 104 sorted MSCs from MMTV-Cre; PPARγf/f;Wnt1 tumors were cultured in MSC medium and separately transduced with Angpt1 shRNAs, miR-15a or control lentiviruses with 5 μg/ml polybrene overnight according to manufacturer’s protocol (Thermo Scientific, Waltham, MA, USA) at 37°C in a humidified atmosphere of 5% CO2. MSC medium was replaced and cells cultured for 24 h. Two μg/ml puromycin was added, and cells were incubated for 48 h. MSC medium was replaced, and cells were injected into the fat pads of 2-month-old immunocompromised NU/J mice. Mice were examined weekly for tumor formation for up to 6 months. The latency and volume of tumors were recorded for each mouse. Complete necropsy was performed on each mouse. Tumors were processed for histopathology, immuno-histochemistry, immunofluorescence, FACS and cell death analysis. Data were analyzed by t-test.
Gene expression analysis
Total RNA was extracted from sorted MSC from MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 tumors. The integrity of the ribosomal RNA bands was confirmed by Northern gel electrophoresis. Total RNA (1 μg) was converted to labeled cRNA targets. The biotinylated cRNA targets were then purified, fragmented and hybridized to mouse genome 2.0 ST expression arrays (Affymetrix, Santa Clara, CA, USA) to interrogate transcript abundance in each sample. Affymetrix GCOS software was used to generate raw gene expression scores and normalized to the relative hybridization signal from each experiment. All gene expression scores were set to a minimum value of two times the background determined by GCOS software in order to minimize noise associated with less robust measurements of rare transcripts. Data were analyzed by t-test with a value of P < 0.005 followed by ratio analysis (minimum twofold change). Expression changes for selected genes were validated by qRT-PCR.
Western blot
Sorted MSC from MMTV-Cre;PPARγ+/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 tumors was lysed in 1 × Laemmli buffer. Sorted MSC from lentiviral transduced MMTV-Cre;PPARγf/f;Wnt1 mammary tumors was analyzed in separate experiments. Fifty μg total cellular proteins were separated by SDS–PAGE. Proteins were electroblotted onto PVDF membranes (Roche Applied Sciences, Indianapolis, IN, USA). Blots were incubated with blocking solution followed by anti-Angpt1 and anti-β-actin antibodies for 16 h at 4°C. After washing in Tris-buffered saline containing 0.1% Tween-20, blots were incubated for 30 min at room temperature with anti-IgG secondary antibody conjugated to horseradish peroxidase. Bands were visualized by the enhanced chemiluminescence method and quantitated by densitometry. Data were analyzed by t-test.
Chromatin immunoprecipitation
The putative PPAR response element in the miR-15a promoter was identified using the UCSC Genome Browser. MSC from MMTV-Cre;PPARγ +/+;Wnt1 and MMTV-Cre;PPARγf/f;Wnt1 mammary tumors was fixed in 1% formaldehyde for 10 min followed by native lysis and DNA fragmentation. Purified chromatin was immunoprecipitated using anti-PPARγ antibody and protein A/G agarose beads. Eluted DNA fragments were purified for PCR templates. The input fraction before immunoprecipitation was amplified as the positive control. Control IgG immunoprecipitates were used as the negative control. DNA templates were amplified with primers 5′-TTCAAAAAGTATCCCATTATTCTG-3′ and 5′-TGTCCTATTTCCCCCTTCAA-3′ flanking the putative PPAR response element in the miR-15a gene. The 197-bp fragment was separated by agarose gel electrophoresis and visualized by ethidium bromide staining.
Promoter cloning and reporter gene analysis
A 1.8-kb fragment of the promoter region of the mouse miR-15a gene was amplified by PCR from mouse genomic DNA using primers 5′-CGG GGTACCACTAGAATAACAGCCATGGGAGACAC-3′ and 5′-CCGCTCGAGAGTA TGGCCTGCACCTTTTCAACA-3′. This fragment was cloned into the KpnI and XhoI sites of the pGL3 luciferase vector. We generated a mutant miR-15a promoter construct with a 3-bp substitution in the putative PPARγ response element using the QuikChange site directed mutagenesis kit (Stratagene). Wild-type and mutant constructs were confirmed by sequencing. Promoter constructs were transfected with mouse PPARγ expression vector or control plasmid and Renilla luciferase reporter vector pRL-TK into the mouse mammary tumor cell line NF639 (American Type Culture Collection, Manassas, VA, USA) using Lipofectamine (Life Technologies, Grand Island, NY, USA). Promoter activity was determined using the dual luciferase assay kit (Promega, Madison, WI, USA) and luminometry (Berthold, Bad Wildbad, Germany). miR-15a promoter activity was normalized to TK promoter activity. Data were analyzed by ANOVA.
Antibodies
Antibodies used were CD31, AB5690; K5, AB24647; K18, AB668 (Abcam, Cambridge, MA, USA); β-actin, NB600-503SS (Novus Biologicals, Littleton, CO, USA); Angpt1, SC-6320; CD61, SC-19671PE; PCNA, SC-7907; PPARγ, SC-7273 (Santa Cruz Biotechnology, Santa Cruz, CA, USA); CD24-PE, 10814; CD49f-AF488, 60037 (Stem Cell Technologies, Vancouver, BC, Canada); and anti-rabbit IgG-AF488, A11008; anti-rabbit IgG-AF555, A21428 (Thermo Fisher Scientific, Waltham, MA, USA).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Dr Ke Ma and Jewell Graves (University of Illinois Research Resources Center) for assistance with microscopy and flow cytometry. KK was supported by NIH National Research Service award DE18381. This study was supported by Department of Defense Breast Cancer Research Program award W81XWH-10-1-0081.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Crowe DL, Parsa B, Sinha UK. Relationships between stem cells and cancer stem cells. Histol Histopathol. 2004;19:505–509. doi: 10.14670/HH-19.505. [DOI] [PubMed] [Google Scholar]
- 2.Sleeman KE, Kendrick H, Robertson D, Isacke CM, Ashworth A, Smalley MJ. Dissociation of estrogen receptor expression and in vivo stem cell activity in the mammary gland. J Cell Biol. 2007;176:19–26. doi: 10.1083/jcb.200604065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Visvader JE. Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev. 2009;23:2563–2577. doi: 10.1101/gad.1849509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Keymeulen A, Rocha AS, Ousset M, Beck B, Bouvencourt G, Rock J, et al. Distinct stem cells contribute to mammary gland development and maintenance. Nature. 2011;479:189–193. doi: 10.1038/nature10573. [DOI] [PubMed] [Google Scholar]
- 5.Li Y, Welm B, Podsypanina K, Huang S, Chamorro M, Zhang X, et al. Evidence that transgenes encoding components of the Wnt signaling pathway preferentially induce mammary cancers from progenitor cells. Proc Natl Acad Sci USA. 2003;100:15853–15858. doi: 10.1073/pnas.2136825100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu BY, McDermott SP, Khwaja SS, Alexander CM. The transforming activity of Wnt effectors correlates with their ability to induce the accumulation of mammary progenitor cells. Proc Natl Acad Sci USA. 2004;101:4158–4163. doi: 10.1073/pnas.0400699101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 9.Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 10.Nieto Y, Woods J, Nawaz F, Baron A, Jones RB, Shpall EJ, et al. Prognostic analysis of tumor angiogenesis, determined by microvessel density and expression of vascular endothelial growth factor, in high risk primary breast cancer patients treated with high dose chemotherapy. Br J Cancer. 2007;97:391–397. doi: 10.1038/sj.bjc.6603875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fantozzi A, Gruber DC, Pisarsky L, Heck C, Kunita A, Yilmaz M, et al. VEGF mediated angiogenesis links EMT induced cancer stemness to tumor initiation. Cancer Res. 2014;74:1566–1575. doi: 10.1158/0008-5472.CAN-13-1641. [DOI] [PubMed] [Google Scholar]
- 12.Chekhonin VP, Shein SA, Korchagina AA, Gurina OI. VEGF in tumor progression and targeted therapy. Curr Cancer Drug Targets. 2013;13:423–443. doi: 10.2174/15680096113139990074. [DOI] [PubMed] [Google Scholar]
- 13.Lehrke M, Lazar MA. The many faces of PPARγ. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 14.Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid activated nuclear receptors. Nature. 2008;454:470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- 15.Asano A, Irie Y, Saito M. Isoform specific regulation of vascular endothelial growth factor (VEGF) family mRNA expression in cultured mouse brown adipocytes. Mol Cell Endocrinol. 2001;174:71–76. doi: 10.1016/s0303-7207(00)00450-0. [DOI] [PubMed] [Google Scholar]
- 16.Fauconnet S, Lascombe I, Chabannes E, Adessi GL, Desvergne B, Wahli W, et al. Differential regulation of vascular endothelial growth factor expression by peroxisome proliferators activated receptors in bladder cancer cells. J Biol Chem. 2002;277:23534–23543. doi: 10.1074/jbc.M200172200. [DOI] [PubMed] [Google Scholar]
- 17.Panigraphy D, Singer S, Shen LQ, Butterfield CE, Freedman DA, Chen EJ, et al. PPARγ ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J Clin Invest. 2002;110:923–932. doi: 10.1172/JCI15634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang WG, Douglas-Jones A, Mansel RE. Expression of peroxisome proliferator activated receptor γ (PPARγ) and the PPARγ coactivator, PGC-1, in human breast cancer correlates with clinical outcomes. Int J Cancer. 2003;106:752–757. doi: 10.1002/ijc.11302. [DOI] [PubMed] [Google Scholar]
- 19.Watkins G, Douglas-Jones A, Mansel RE, Jiang WG. The localization and reduction of nuclear staining of PPARγ and PGC-1 in human breast cancer. Oncol Rep. 2004;12:483–488. [PubMed] [Google Scholar]
- 20.Saez E, Rosenfeld J, Livolsi A, Olson P, Lombardo E, Nelson M, et al. PPARγ signaling exacerbates mammary gland tumor development. Genes Dev. 2004;18:528–540. doi: 10.1101/gad.1167804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian L, Zhou J, Casimiro MC, Liang B, Ojeifo JO, Wang M, et al. Activating peroxisome proliferator activated receptor γ mutant promotes tumor growth in vivo by enhancing tumorigenesis. Cancer Res. 2009;69:9236–9244. doi: 10.1158/0008-5472.CAN-09-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golembesky AK, Gammon MD, North KE, Bensen JT, Schroeder JC, Teitelbaum SL, et al. Peroxisome proliferator activated receptor α (PPARA) genetic polymorphisms and breast cancer risk: a Long Island ancillary study. Carcinogenesis. 2008;29:1944–1949. doi: 10.1093/carcin/bgn154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lacroix L, Lazar V, Michiels S, Ripoche H, Dessen P, Talbot M, et al. Follicular thyroid tumors with the PAX8-PPARγ1 rearrangement display characteristic genetic alterations. Am J Pathol. 2005;167:223–231. doi: 10.1016/s0002-9440(10)62967-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reddi HV, Driscoll CB, Madde P, Milosevic D, Hurley RM, McDonough SJ, et al. Redifferentiation and induction of tumor suppressor miR-122 and miR-375 by the PAX8/PPAR fusion protein inhibits anaplastic thyroid cancer: a novel therapeutic strategy. Cancer Gene Ther. 2013;20:267–275. doi: 10.1038/cgt.2013.16. [DOI] [PubMed] [Google Scholar]
- 25.O’Day E, Lal A. MicroRNAs and their target networks in breast cancer. Breast Cancer Res. 2010;12:201. doi: 10.1186/bcr2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roth C, Rack B, Muller V, Janni W, Pantel K, Schwarzenbach H. Circulating microRNAs as blood based markers for patients with primary and metastatic breast cancer. Breast Cancer Res. 2010;12:R90. doi: 10.1186/bcr2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaillant F, Asselin-Labat ML, Shackleton M, Forrest NC, Lindeman GJ, Visvader JE. The mammary progenitor marker CD61/β3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Res. 2008;68:7711–7717. doi: 10.1158/0008-5472.CAN-08-1949. [DOI] [PubMed] [Google Scholar]
- 29.Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, et al. Genome wide profiling of PPARγ:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008;22:2953–2967. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt SF, Jorgensen M, Chen Y, Nielsen R, Sandelin A, Mandrup S. Cross species comparison of C/EBPα and PPARγ profiles in mouse and human adipocytes reveals interdependent retention of binding sites. BMC Genomics. 2011;12:152–167. doi: 10.1186/1471-2164-12-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu YH, Hong LQ, Yu WQ, Li XY, Zheng XY. Effect of miR-15a on induction of apoptosis in breast cancer MCF-7 cells. Chinese J Oncol. 2011;33:827–830. [PubMed] [Google Scholar]
- 32.Luo Q, Li X, Li J, Kong X, Zhang J, Chen L, et al. miR-15a is underexpressed and inhibits the cell cycle by targeting CCNE1 in breast cancer. Int J Oncol. 2013;43:1212–1218. doi: 10.3892/ijo.2013.2034. [DOI] [PubMed] [Google Scholar]
- 33.Zhu N, Zhang D, Xie H, Zhou Z, Chen H, Hu T, et al. Endothelial specific intron derived miR-126 is downregulated in human breast cancer and targets both VEGFA and PIK3R2. Mol Cell Biochem. 2011;351:157–164. doi: 10.1007/s11010-011-0723-7. [DOI] [PubMed] [Google Scholar]
- 34.Yu J, Li Q, Liu L, Jiang B. miR-148a inhibits angiogenesis by targeting ERBB3. J Biomed Res. 2011;25:170–177. doi: 10.1016/S1674-8301(11)60022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bronisz A, Godlewski J, Wallace JA, Merchant AS, Nowicki MO, Mathsyaraja H, et al. Reprogramming of the tumor microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol. 2012;14:159–167. doi: 10.1038/ncb2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Png KJ, Halberg N, Yoshida M, Tavazoie SF. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature. 2012;481:190–194. doi: 10.1038/nature10661. [DOI] [PubMed] [Google Scholar]
- 37.Zou C, Xu Q, Mao F, Li D, Bian C, Liu LZ, et al. miR-145 inhibits tumor angiogenesis and growth by N-RAS and VEGF. Cell Cycle. 2012;11:2137–2145. doi: 10.4161/cc.20598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siragam V, Rutnam ZJ, Yang W, Fang L, Luo L, Yang X, et al. MicroRNA miR-98 inhibits tumor angiogenesis and invasion by targeting activin receptor like kinase 4 and matrix metalloproteinase 11. Oncotarget. 2012;3:1370–1385. doi: 10.18632/oncotarget.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chou J, Lin JH, Brenot A, Kim JW, Provot S, Werb Z. GATA3 suppresses metastasis and modulates the tumor microenvironment by regulating microRNA-29b expression. Nat Cell Biol. 2013;15:201–213. doi: 10.1038/ncb2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kong W, He L, Richards EJ, Challa S, Xu CX, Permuth-Wey J, et al. Upregulation of miRNA-155 promotes tumor angiogenesis by targeting VHL and is associated with poor prognosis and triple negative breast cancer. Oncogene. 2014;33:679–689. doi: 10.1038/onc.2012.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He T, Qi F, Jia L, Wang S, Song N, Guo L, et al. MicroRNA-542-3p inhibits tumor angiogenesis by targeting angiopoietin 2. J Pathol. 2014;232:499–508. doi: 10.1002/path.4324. [DOI] [PubMed] [Google Scholar]
- 42.Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA. 2012;109:2784–2789. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caine GJ, Lip GY, Blann AD. Platelet derived VEGF, Flt-1, angiopoietin-1, and P-selectin in breast and prostate cancer: further evidence for a role of platelets in tumor angiogenesis. Ann Med. 2004;36:273–277. doi: 10.1080/07853890410026098. [DOI] [PubMed] [Google Scholar]
- 44.Reiss Y, Knedia A, Tal AO, Schmidt MH, Jugold M, Kiessling F, et al. Switching of vascular phenotypes within a murine breast cancer model induced by angiopoietin-2. J Pathol. 2009;217:571–580. doi: 10.1002/path.2484. [DOI] [PubMed] [Google Scholar]
- 45.Harfouche R, Echavarria R, Rabbani SA, Arakelian A, Hussein MA, Hussain SN. Estradiol dependent regulation of angiopoietin expression in breast cancer cells. J Steroid Biochem Mol Biol. 2011;123:17–24. doi: 10.1016/j.jsbmb.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 46.Danza K, Pilato B, Lacalamita R, Addati T, Giotta F, Bruno A, et al. Angiogenetic axis angiopoietins/Tie2 and VEGF in familial breast cancer. Eur J Hum Genet. 2013;21:824–830. doi: 10.1038/ejhg.2012.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heesen S, Fonorod M. The inner nuclear envelope as a transcription factor resting place. EMBO Rep. 2007;8:914–919. doi: 10.1038/sj.embor.7401075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steglich B, Sazer S, Ekwall K. Transcriptional regulation at the yeast nuclear envelope. Nucleus. 2013;4:379–389. doi: 10.4161/nucl.26394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jain RK. Normalization of tumor vasculature: an emerging concept in anti-angiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 50.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.