

Abstract
Bipolar Disorder (BD) is a prevalent and disabling condition, determined by gene-environment interactions, possibly mediated by epigenetic mechanisms. The present study aimed at investigating the transcriptional regulation of BD selected target genes by DNA methylation in peripheral blood mononuclear cells of patients with a DSM-5 diagnosis of type I (BD-I) and type II (BD-II) Bipolar Disorders (n = 99), as well as of healthy controls (CT, n = 42). The analysis of gene expression revealed prodynorphin (PDYN) mRNA levels significantly reduced in subjects with BD-II but not in those with BD-I, when compared to CT. Other target genes (i.e. catechol-O-methyltransferase (COMT), glutamate decarboxylase (GAD67), serotonin transporter (SERT) mRNA levels remained unaltered. Consistently, an increase in DNA methylation at PDYN gene promoter was observed in BD-II patients vs CT. After stratifying data on the basis of pharmacotherapy, patients on mood-stabilizers (i.e., lithium and anticonvulsants) were found to have lower DNA methylation at PDYN gene promoter. A significantly positive correlation in promoter DNA methylation was observed in all subjects between PDYN and brain derived neurotrophic factor (BDNF), whose methylation status had been previously found altered in BD. Moreover, among key genes relevant for DNA methylation establishment here analysed, an up-regulation of DNA Methyl Transferases 3b (DNMT3b) and of the methyl binding protein MeCP2 (methyl CpG binding protein 2) mRNA levels was also observed again just in BD-II subjects. A clear selective role of DNA methylation involvement in BD-II is shown here, further supporting a role for BDNF and its possible interaction with PDYN. These data might be relevant in the pathophysiology of BD, both in relation to BDNF and for the improvement of available treatments and development of novel ones that modulate epigenetic signatures.
Keywords: Bipolar disorder, Epigenetics, DNA methylation, Brain derived neurotrophic factor, Prodynorphin
1. Introduction
Bipolar Disorder (BD) comprises different prevalent, comorbid and disabling conditions, characterized by severe and pervasive mood episodes (American Psychiatric Association, APA, 2013). Some forms of BD are likely underdiagnosed and, in many cases, misdiagnosed with unipolar depression (Major Depressive Disorder), particularly for patients affected by BD type II (BD-II) (Singh and Rajput, 2006; Dell’Osso et al., 2015). In addition, BD can be misdiagnosed with other Major Psychoses, such as Schizophrenia – particularly for patients with psychotic BD type I (BD-I) – and, in some cases, with Personality Disorders (e.g., Borderline Personality Disorder), with major clinical and economic consequences (Meyer and Meyer, 2009; Ruggero et al., 2010; John and Sharma, 2009).
Therefore, over the last years research efforts were focused to better characterize the diagnosis of BD from a biological perspective, in order to establish clinically distinct patterns of illness as well as novel and more specific treatments. To this aim, the relevance of identification of molecular markers that go beyond classical diagnostic criteria and clinical observations is apparent.
To date, despite evidence supporting a role for many genes in the pathophysiology of BD (MacQueen et al., 2005), the precise molecular bases of this disorder remain unclear, and no specific gene has been incontrovertibly related to disease development. Thus, BD is a complex condition with multiple contributing factors triggered by the interplay of numerous susceptibility genes with many environmental factors (Uher, 2014; Ludwig and Dwivedi, 2016).
Gene-environment interactions are mediated by epigenetic mechanisms, which evoke transient changes in gene expression, through chemical modifications that do not modify DNA sequence (Feinberg and Fallin, 2015). The renewed interest in epigenetics has opened new perspectives for the understanding of biological determinants of psychiatric disorders, as well as of the impact that environment may have on their progression (Costa et al., 2004; Nestler et al., 2016).
DNA methylation has been the characterized as the most stable epigenetic mechanism, also in the context of psychiatric disorders (Jirtle and Skinner, 2007). It consists of the transfer of a methyl group to position 5 of the pyrimidine ring in a cytosine-guanine dinucleotide (CpG), which, at gene promoters level, ultimately blocks the binding of transcription factors and causes chromatin compaction and gene silencing (Pidsley and Mill, 2011; Klose and Bird, 2006). The role of epigenetic factors in patients with Major Psychoses has been mainly investigated through the assessment of DNA methylation changes in the promoter regions of candidate targets (Abdolmaleky et al., 2006) as well as changes in the expression of DNA methyltransferases (DNMTs) (Veldic et al., 2004; Higuchi et al., 2011).
Among genes potentially implicated in the pathophysiology of BD, brain-derived neurotrophic factor (BDNF) has been extensively investigated over the last few years (Castrén and Rantamäki, 2010; Grande et al., 2010; Hashimoto et al., 2004; Henikoff and Matzke, 1997; Shirayama et al., 2002). In particular, our group has recently reported a selective down-regulation of BDNF gene expression in BD-II, compared with BD-I patients and healthy controls, paralleled by a consistent increase in DNA methylation at gene promoter in BD-II subjects only (D’Addario et al., 2012).
In order to gain further insights on the epigenetic regulation of BDNF in peripheral blood mononuclear cells (PBMCs) of patients suffering from BD, in the present study we evaluated the transcriptional regulation of key genes that might interact with BDNF to determine the phenotype of psychotic disorders: catechol-O-methyltransferase (COMT) (Zanoni et al., 2011; Lee et al., 2013), glutamate decarboxylase (GAD67) (Hashimoto and Lewis, 2006), serotonin transporter (SERT) (Szapacs et al., 2004; Kronenberg et al., 2016), and prodynorphin (PDYN) (Logrip et al., 2008). In line with this, evidence that abnormal BDNF signalling leads to abnormalities in GABAergic neurons has been reported in studies showing that BDNF induces the expression of GAD67 (Bolton et al., 2000; Hashimoto and Lewis, 2006; Marty et al., 2000; Yamada et al., 2002), and that both GAD67 and BDNF are down-regulated in mood disorders (Thompson Ray et al., 2011). Also synergies between serotonin and BDNF systems were suggested to contribute to the regulation of the development and plasticity of neural circuits involved in the pathophysiology of affective disorders (Martinowich and Lu, 2008).
Moreover, we also investigated the regulation of PDYN, a precursor of the dynorphin peptide, that has been already associated with dysphoria and negative mood states (Chavkin, 2013), and that has been suggested as a downstream effector of striatal BDNF regulation of ethanol intake (Logrip et al., 2008).
Finally, we evaluated the expression of key genes in the establishment of DNA methylation patterns, such as the different isoforms of DNMTs, and the methyl binding protein MeCP2 (methyl CpG binding protein 2).
The aim of the present study was thus to evaluate possible transcriptional regulation of target genes interacting with BDNF in BD and the role of DNA methylation in these effects.
2. Methods
2.1. Subjects
Ninety-nine patients diagnosed with BD (54 with BD-I and 45 with BD-II) of either gender and any age (Table 1), treated and followed up at the mood disorders outpatient Clinic of the University Department of Psychiatry of Milan Policlinico, were included in the study.
Table 1.
Demographical and clinical characteristics of patients enrolled for DNA methylation studies.
| Sample | Total (n) | M | F | Mean age ± SD | Current phase |
|---|---|---|---|---|---|
| BD-I | 54 | 27 | 28 | 49,94 ± 13,25 | Eu = 8 |
| Dep = 16 | |||||
| M/H = 27 | |||||
| Mix = 3 | |||||
| BD-II | 45 | 15 | 30 | 53,10 ± 12,10 | Eu = 19 |
| Dep = 19 | |||||
| M/H = 5 | |||||
| Mix = 2 | |||||
| CT | 42 | 17 | 25 | 64,5 ± 18,94 | – |
CT = controls; BD-I = bipolar disorder type I; BD-II = bipolar disorder type II. Eu = euthymia; Dep = depression; M/H = mania/hypomania; Mix = mixed state
Diagnoses were assessed by the administration of semi-structured interviews based on DSM-5 criteria (SCID I and II, research version) (First et al., 2015a,b). In case of psychiatric comorbidity, BD had to be the primary disorder, causing the most significant distress and dysfunction and representing the primary motivation to seek treatment. Patients were excluded from the study if they had recent or current alcohol or substance abuse (last 3 months), as well as medical conditions including autoimmune diseases due to their potential influence over BDNF expression (Linker et al., 2009). For the same reason, a positive lifetime history of trauma (according to DSM-5), as well as the current presence of relevant psychological stress, were considered exclusion criteria.
Socio-demographic and clinical assessment included the collection of the following main variables: gender, age, subtype of BD, current pharmacological treatment, and mood state (i.e., euthymic, depressed, manic, hypomanic, and mixed), defined through the administration of the following psychometric scales: Hamilton Depression Rating Scale, Montgomery Asberg Depression Rating Scale, and Young Mania Rating Scale (Hamilton, 1960; Montgomery and Asberg, 1979; Young et al., 1978).
Patients had maintained their current pharmacological treatment stable for at least one month in order to be enrolled in the study. Control subjects (n = 42) were volunteers matched for gender, age and ethnicity, without any psychiatric diagnosis, as determined by the SCID-I and without positive family history for major psychiatric disorders in the first-degree relatives (as assessed by the Family Interview for Genetic Studies) (Maxwell, 1992). All subjects had given their written informed consent to participate to the study, which included the use of personal and clinical data as well as blood drawing for genotyping and methylation analysis. The study protocol had been previously approved by the local Ethics Committee.
2.2. Real-time qPCR (RT-qPCR)
Total RNA was isolated from PBMCs according to the method of Chomczynski and Sacchi (1987). RT-PCR reactions were performed using the RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA, USA). The relative abundance was assessed by RT-qPCR using iQ SYBR Green Supermix (Hercules, CA, USA) on a DNA Engine Opticon 2 Continuous Fluorescence Detection System (MJ Research, Waltham, MA, USA). To provide precise quantification of the initial target in each PCR reaction, the amplification plot was examined and the point of early log phase of product accumulation defined by assigning a fluorescence threshold above background, defined as the threshold cycle number or Ct. Differences in threshold cycle number were used to quantify the relative amount of the PCR targets contained within each tube. After PCR, a dissociation curve (melting curve) was constructed in the range of 60 to 95 °C (Lyon, 2001) to evaluate the specificity of the amplification products. The relative expression of different amplicons was calculated by the delta-delta Ct (DDCt) method and converted to relative expression ratio (2−DDCt) for statistical analysis (Livak and Schmittgen, 2001). All data were normalized to the endogenous reference genes β-ACTIN and GAPDH combined. The primers used for PCR amplification are reported in Table 2.
Table 2.
Primer sequences used for reverse transcription–polymerase chain reaction.
| Human gene | Forward (5′ → 3′) | Reverse (3′ → 5′) |
|---|---|---|
| SERT | CAGCGTGTGAAGATGGAGAAG | TGGGATAGAGTGCCGTGTGT |
| COMT | CTGCTTTGCTGCCGAGCTCAGAGGAGAC | GCCCAGCAACACAGCTGCCAACAG |
| PDYN | GCCTGCCTCCTCATGTTCC | CCTTCCCCAACCGACTTGC |
| GAD67 | GCCAGACAAGCAGTATGATGT | CCAGTTCCAGGCATTTGTTGA |
| DNMT1 | CCCCTGAGCCCTACCGAAT | CTCGCTGGAGTGGACTTGTG |
| DNMT3A | TATTGATGAGCGCACAAGAGAGC | GGGTGTTCCAGGGTAACATTGAG |
| DNMT3B | GGCAAGTTCTCCGAGGTCTCTG | TGGTACATGGCTTTTCGATAGGA |
| MeCP2 | ACTCCTCAGAATACACCTTGCTT | TGAGGCCCTGGAGGTCCT |
| β-ACTIN | TGACCCAGATCATGTTTGAG | TTAATGTCACGCACGATTTCC |
| GAPDH | CAGCCTCAAGATCATCAGCA | TGTGGTCATGAGTCCTTCCA |
SERT = serotonin transporter; COMT = catechol-O-methyltransferase; PDYN = prodynorphin; GAD67 = glutamate decarboxylase; β-ACTIN = beta-actin; GAPDH = glyceraldehyde 3-phosphate dehydrogenase; DNMT1, 3A, 3B = DNA methyltransferases; MeCP2 = methyl CpG binding protein 2.
2.3. Analysis of DNA methylation
Genomic DNA was extracted by the salting-out method as described previously (Arosio et al., 2010). First, DNA underwent bisulfite modification to convert unmethylated cytosine residues to uracil, using the CpGenome DNA Modification Kit (Chemicon International, Purchase, NY), according to the manufacturer’s instructions. Methylation analysis was performed by fluorescence-based real-time PCR using MS Opticon 2 Light Cycler Instrument (Roche, Germany). PDYN sequence amplified contained 12 CpG sites, and was located within the promoter region of the gene (see Fig. 1 for sequence details). PCR was also performed for non-CpG-containing region of myoD, which served as control gene. Bisulfite-modified CpGenome universal unmethylated DNA (Chemicon International, Temecula CA, USA) was used as negative control. The percentage of methylation was calculated by the 2−DDCt method (Livak and Schmittgen, 2001), where DDCt (Ct, Target-Ct, myoD) sample - (Ct, Target-Ct, myoD) fully methylated DNA, multiplied by 100. For relative quantification, standard curves were generated separately for each gene and myoD from serial dilutions of bisulfite-modified CpGenome universal methylated DNA (Chemicon International). To confirm our result, we also used in selected DNA bisulfite-converted samples primers for the unmethylated DNA sequence calculating the % of methylation, as reported previously (Lu et al., 2008). These latter data are not shown to avoid redundancy. The primers for bisulfite-converted DNA are herein reported:
Primers of bisulfite-converted DNA.
M_DYN:
Forward 5′-ATTAAATTGTAGTTTTTGGTTGTCG-3′
Reverse: 5′-ACTAAAACAATCTTCTAATCACGAA-3′
Product size: 250 bp
U_DYN:
Forward: 5′-TTAAATTGTAGTTTTTGGTTGTTGG-3′
Reverse: 5′-ACTAAAACAATCTTCTAATCACAAA-3′
Product size: 249 bp
myoD:
Forward 50-TGATTAATTTAGATTGGGTTTAGAGAAGGA-30
Reverse 50-CCAACTCCAAATCCCCTCTCTAT-30
Product size: 162 bp.
Fig. 1. Sequence of the human prodynorphin (PDYN) exon I promoter (chr11: 27,743,605–27,744,379).
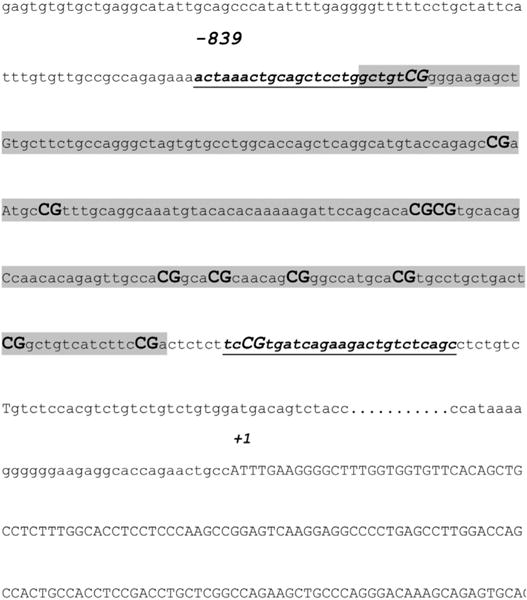
Amplicon includes 11 cytosine guanine dinucleotide (CpG) sites (bold) and primer sequences (methylated) are underlined. The transcriptional start site (+1) is indicated and the predicted CpG island is highlighted (criteria used: island size 200, Obs/Exp = 0.62 and %GC = 59.50).
2.4. Statistical analysis
All results are expressed as mean ± SEM. Statistical differences of genes expression and DNA methylation changes at PDYN promoter of BD patients vs control subjects were determined by analysis of variance (ANOVA) followed by Dunnett’s test. Changes in mRNA levels of DNMTs and MeCP2 were analysed using the non-parametric Mann-Whitney test since data in pooled samples were not normally distributed. In Fig. 5, data are compared by Spearman’s rank correlation coefficient. The P-values < 0.05 were considered to be statistically significant. All tests were performed using GraphPad Prism version 6.00 (GraphPad Software, San Diego, CA, USA).
Fig. 5. Correlation between DNA methylation levels at PDYN and BDNF gene promoters.
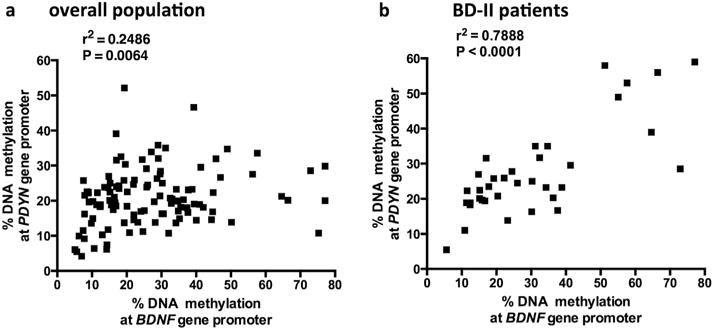
Correlation between BDNF and PDYN percentage change in DNA methylation at gene promoters in the overall population (a) as well as selectively in bipolar disorders type 2 (BD II) subjects (b). Data are compared by Spearman’s rank correlation coefficient.
3. Results
Demographic and clinical characteristics of the study sample and related subgroups (BD-I and BD-II individuals, and healthy controls) are shown in Table 1.
No changes were observed between BD-I, BD-II patients and healthy controls in relation to GAD67, SERT and COMT (Fig. 2a, b, c). PDYN gene expression was found to be significantly decreased in BD-II patients (0.67 ± 0.09; P < 0.05 Dunnett’s post-hoc test; ANOVA: P = 0.0188; F = 4.330), but not in those affected by BD-I (0.84 ± 0.13) compared to healthy controls (1.2 ± 0.2; Fig. 2d).
Fig. 2. Gene expression changes in BD.
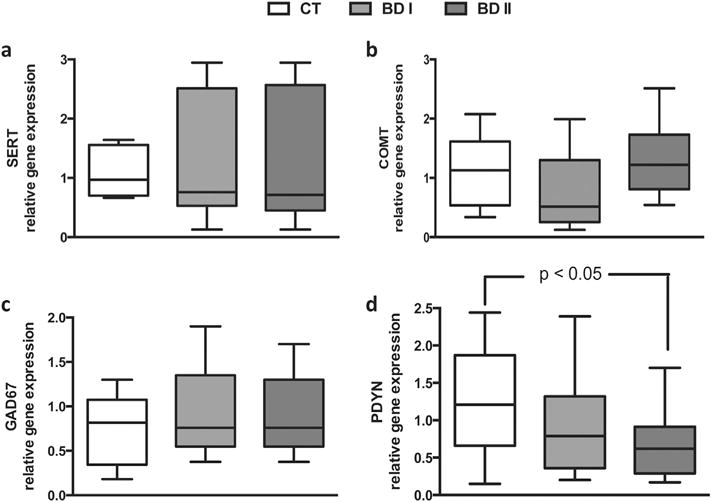
Levels of SERT, GAD67, COMT, PDYN mRNA in peripheral blood mono-nuclear cells from patients diagnosed with bipolar disorders type 1 (BD I) and BD type 2 (BD II). Box plots with whiskers from minimum to maximum represent 2−DDCt values calculated by the delta-delta Ct (DDCt) method. Means of mRNA levels are expressed relative to control subjects.
An increase of DNA methylation at PDYN gene promoter was observed in BD-II (but not in BD-I) patients compared to healthy controls (CT: 17.02 ± 1.20%; BD-I: 18.67 ± 1.16%; BD-II: 21.78 ± 1.02%; P < 0.05 Dunnett’s post-hoc test; ANOVA: P = 0.0168; F = 4.212; Fig. 3).
Fig. 3. DNA methylation levels at PDYN gene promoter layered according to diagnosis.
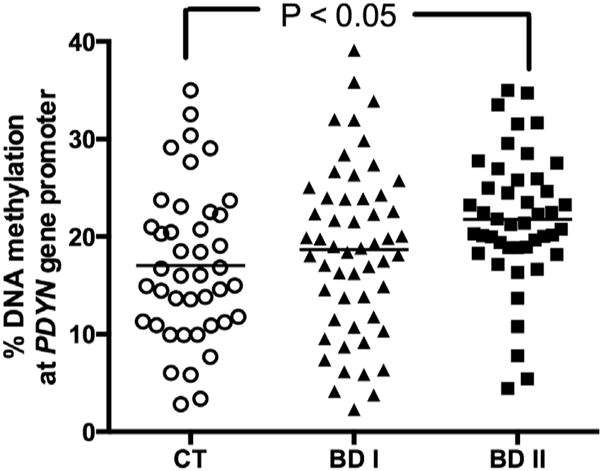
Amount of methylated DNA in the promoter region of PDYN in controls (CT), patients diagnosed with bipolar disorders type 1 (BD I) and BD type 2 (BD II). Scatter dot plots with mean values are shown.
The regression analysis conducted to adjust for age differences showed that the observed alterations were independent of age for DNA methylation levels at PDYN promoter (r2 = 0.01082; P = 0.2147). Moreover, when data were stratified on the basis of the different pharmacological treatments taken by the patients, those on mood stabilizers (e.g., lithium or valproate) (16.16 ± 1.34%) showed a lower level of DNA methylation at PDYN promoter, when compared to those treated with other drugs (21.01 ± 1.23%; P = 0.0147, Mann-Whitney test; Fig. 4). Data stratification based on gender showed no differences in DNA methylation levels between male and females in both controls and BD subjects. Figs. S1 and S2 report in more details these changes based on age and gender.
Fig. 4. DNA methylation levels at PDYN gene promoter layered according to treatment.
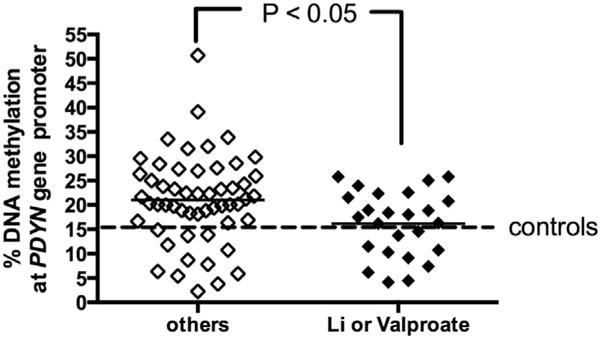
Amount of methylated DNA in the promoter region of PDYN in patients diagnosed with BD (bipolar disorders type 1 (BD I) + BD type 2 (BD II) treated with lithium, valproate or other drugs. Scatter dot plots with mean values are shown.
A significantly positive correlation was observed between changes in DNA methylation at BDNF, as previously reported (D’Addario et al., 2012), and PDYN promoters in all subjects under study (P = 0.0064, Spearman’s r2 = 0.2486; Fig. 5a). This effect was even stronger when considering just BD II subjects (P < 0.0001, Spearman’s r2 = 0.7888; Fig. 5a).
Finally, in pooled samples of BD-II subjects a selective marginally significant up-regulation of DNMT3b (1.79 ± 0.35, P = 0.0618, Mann-Whitney test) and a significant up-regulation for MeCP2 (1.89 ± 0.25, P = 0.0036, Mann-Whitney test) gene expression were also observed (Table 3), when compared to pooled control samples.
Table 3.
DNMTs and MeCP2 genes expression in pooled samples from BD patients relative to CT subjects.
| CT | BD-I | BD-II | |
|---|---|---|---|
| DNMT1 | 1.04 ± 0.09 | 0.97 ± 0.20 | 0.90 ± 0.16 |
| DNMT3a | 1.14 ± 0.21 | 1.39 ± 0.33 | 0.97 ± 0.25 |
| DNMT3b | 1.04 ± 0.13 | 1.05 ± 0.27 | 1.78 ± 0.35; P = 0.0618 vs CT |
| MeCP2 | 1.06 ± 0.11 | 0.91 ± 0.14 | 1.89 ± 0.25; P = 0.0036 vs CT |
CT = controls; BD-I = bipolar disorder type I; BD-II = bipolar disorder type; Values in bold indicate data statistical significant or relevant.
4. Discussion
An improved understanding of epigenetic mechanisms has boosted investigations into their roles in many fields, including the pathophysiology of psychiatric disorders. In this context, we conducted previous studies by selecting a panel of genes known to be relevant for BD pathophysiology. The same genes were also connected with BDNF activity and regulation, that we have extensively shown to be epigenetically modulated in patients with BD and Major Depressive Disorder (D’Addario et al., 2012, 2013; Dell’Osso et al., 2014).
A major outcome of this study is the selective decrease of PDYN gene expression in PBMCs of BD-II patients, but not in those of BD-I subjects, when compared to healthy controls. These data extend a previous report showing a significant reduction (~38%) of PDYN expression in the amygdalo-hippocampal area and in the parvicellular division of the accessory basal area of BD patients (Hurd, 2002). Furthermore, we found an increased DNA methylation at PDYN gene promoter level, consistent with the down-regulation of gene expression (Figs. 2d and 3).
Overall, our findings suggest that PBMCs (easily accessible cells in circulating blood) might be useful to better understand disease conditions within the brain (Arosio et al., 2014). These peripheral cells appear a reliable model also for the investigation of epigenetic mechanisms (Gavin and Sharma, 2009; Arosio et al., 2014). Our data together with prior studies using whole blood (Domschke et al., 2012, 2014) seem to support the “mirror-site model” whereby methylation status of blood might mirror that of brain (Aberg et al., 2013). Of relevance, an early study showed similarity of CpG methylation patterns in brain tissues and PBMCs in the human promoter region of PDYN (Yuferov et al., 2011).
Dynorphins are ligands of kappa opioid receptors (KOP), and KOP antagonists have been explored for the possible treatment of different diseases (Kuzmin et al., 1998; Jewett et al., 2001), and in particular of psychotic disorders (Roth et al., 2002). Indeed, depressogenic effects have been observed in both animals (Carlezon et al., 2006) and humans (Barber and Gottschlich, 1997) upon KOP activation, whereas KOP antagonism induced antidepressant-like effects in animals (Mague et al., 2003; Reindl et al., 2008). Taken together, accumulated evidence supports the potential of KOP agonists as antimanic therapeutic agents (Zarate and Manji, 2008). In line with this hypothesis, in a previous clinical study administration of KOP partial agonist pentazocine to BD subjects was associated with a transient but substantial and statistically significant reduction in manic symptoms (Cohen and Murphy, 2008). However, the psychotomimetic and dysphoric effects of KOP agonists to the central nervous system are also well-documented, clearly limiting their use for human therapy (Rimoy et al., 1994; Walsh et al., 2001).
In this context, the epigenetic regulation of PDYN reported here appears relevant when attempting to develop new strategies to treat BD-II patients, by indirectly targeting KOP and avoiding the possible side-effects associated with the use of direct KOP agonists.
It is important to note that, in contrast to genetic marks that cannot be restored, epigenetic marks are reversible and might be targets for preventive and/or therapeutic modifications based on a variety of agents, such as enzymes, hormones, vitamins, and nutrients (Choi and Friso, 2010). An additional option is, for instance, the development of zinc finger proteins to obtain PDYN sequence-targeted DNA binding (Grover et al., 2010) and more recently the application of CRISPR-Cas9 system for the epigenetic editing at specific loci (Komor et al., 2017; Vojta et al., 2016).
Thus, new approaches that take advantage of the reversible nature of epigenetic regulation, and (based on present results) drugs targeting the PDYN gene, might hold promise to identify novel medications for BD-II treatment that could be rapidly moved into the clinical practice.
Another major outcome of the present study, even if not necessarily playing a role in BD, is the correlation between alteration of DNA methylation at PDYN and BDNF gene promoters in the investigated population.
BDNF is an important CREB target (Lonze and Ginty, 2002), that modulates PDYN expression in a brain region-specific manner (Nair and Vaidya, 2006). Incidentally, previous studies have already reported that BDNF and PDYN are epigenetically regulated through the acetylation of histone proteins in hyperalgesia (Liang et al., 2014). Moreover, our data support a role for DYN as a downstream effector of BDNF regulation, already suggested in a previous study on alcohol intake (Logrip et al., 2008). Here, additional support to this hypothesis is provided also in BD, where BDNF changes are possibly mediated via dynorphin signalling.
The latter hypothesis is also supported by reduced DNA methylation of PDYN, much alike BDNF (D’Addario et al., 2012), in patients under treatment with lithium and valproic acid, two of the most widely used mood-stabilizing compounds for the acute and maintenance therapy of BD, when compared with any other treatment (D’Addario et al., 2012).
4.1. Study limitations
In this study the relatively small change differences in DNA methylation between controls and BD subjects might be of concern. Yet, it should be pointed out that available evidence suggests that complex phenotypes are actually influenced by many factors, each contributing a small effect (Lupski et al., 2011; Wellcome Trust Case Control C, 2007). At the epigenetic level, these small differences would be responsible for phenotypic outcomes in the general population (Docherty et al., 2012).
From a clinical perspective, our findings provide further evidence for distinct mechanisms underlying the pathophysiology of the two main clinical subtypes of BD. Indeed, BD-I and BD-II can not be simplistically conceptualized as similar expressions of illness which only differ for a clinical severity (Dell’Osso et al., 2014). Instead, they need to be better characterized in the light of genetic and epigenetic phenotypes, neuroimaging profiles and therapeutic approaches (D’Addario et al., 2012; Zarate and Manji, 2008; MacQueen et al., 2005).
It seems relevant that the expression of additional target genes investigated herein remains unaffected under our experimental conditions, despite previous studies showing their involvement in BD pathophysiology (see Introduction section). However, many different factors could explain this discrepancy, for instance use of different drugs and pharmacological treatments, different disease phases and others. Moreover, further studies are needed to address the role of other possible genes not considered in the present study and interacting with BDNF, such as the calcium voltage-gated channel subunit alpha1 C (CACNA1C) gene, already found to be hypermethylated in BD (Starnawska et al., 2016) and associated to BD by GWAS studies (Ferreira et al., 2008).
Another possible limitation of the present study might be the use of whole blood, that contains different cell types with potentially different DNA methylation profiles (Jaffe and Irizarry, 2014). However any tissue (brain included) is made of different cell types (Bakulski et al., 2012), and remarkably isolation of distinct cell populations involves manipulations that may influence gene expression profiles (Debey et al., 2004; Aberg et al., 2013). Thus, new epigenetic methods based on cell-sorting or tissue microdissection need to be developed, in order to extend present findings.
Finally, it should be taken into consideration the limited amount of clinical information of the patients’ sample. Indeed, the group of bipolar patients belongs to a larger epigenetic investigation that includes also patients with different psychiatric disorders. Therefore, other and more specific variables for BD, such as for instance prevalent polarity, presence of rapid cycling and psychotic symptoms, were not collected and should be investigated in future studies.
5. Conclusions
Our results further support the relevance of selective changes in DNA methylation in BD, and the importance of target genes such as PDYN and BDNF.
Also our data on DNMTs appear rather relevant, because they show again selectivity among two BD subtypes with an involvement of DNMT3b. Altered expression of the latter enzyme has been already suggested as a driver of changes in promoter DNA methylation of neurobiological genes that are important for suicidal behaviour (Poulter et al., 2008).
Moreover, we show selective alteration in MeCP2 mRNA levels in BD-II subjects. Of note, already in 2003 it was observed that BDNF transcription involves MeCP2 interaction with its promoter (Martinowich et al., 2003), and a more recent report has confirmed that indeed MeCP2 plays a key role in BDNF gene regulation (Martínez-Levy and Cruz-Fuentes, 2014).
Understanding gene-environment interactions through epigenetic regulation seems extremely important in the context of psychiatric disorders like BD, and holds potential for improving disease treatment through innovative epi-treating drugs.
5.1. Future plans
Further studies are needed to discover novel targets, as well as possible biomarkers of the disease, with the aim of developing novel therapies for BD that eventually modulate DNA methylation via environmental cues (i.e., diet or physical activity) and aid classical pharmacological interventions.
It is also known that genetic variations influence the inter-individual variation in DNA methylation (Zhang et al., 2010). However, how genetic variations might impact DNA methylation pattern of most genes is not fully understood. It would be of great relevance that further studies address the genetic and epigenetic regulation of target genes like PDYN and BDNF in BD by analysing single nucleotide polymorphisms (SNPs) associated with BD and the correlation with DNA methylation changes.
In Fig. 6 we propose a possible mechanism triggered by DNMT inhibitors that might rescue expression changes in relevant genes for BD. This strategy has been put forward also by a recent clinical trial with green tea extracts, able to inhibit DNMTs and to act as adjunct to maintenance antipsychotic medication (Loftis et al., 2013).
Fig. 6.
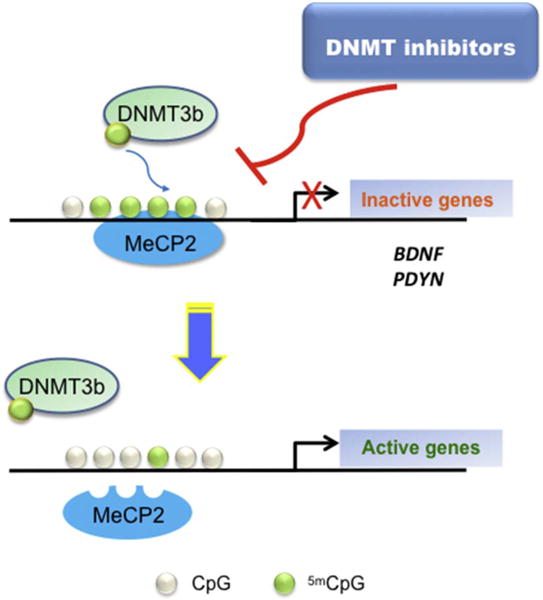
A proposed model for future studies on the role of DNMT inhibitors on target genes regulation in BD.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
This research was supported in part by the Italian Ministry of University and Research under grants FIRB-RBFR12DELS to CD and by the Intramural research Program of the NIH (grant number: AG000362-02), National institute on Aging (fellowship to ADF). We would like to thank Laura Cremaschi, M.D., Chiara Arici, M.D., Matteo Vismara, M.D., Vera De Carlo, M.D for helping in data collection.
BD was speaker for Astra Zeneca, Bristol Myers Squibb, Jansen-Cilag, Eli Lilly, Pfizer, Cyberonics, Glaxo Smith Klein, Lundbeck, Italfarmaco.
DG reports grants from Italian Ministry of University (FIRB-RBFR12DELS), and from Italian Ministry of Health (5X1000 grant 2012).
ES reports grants from the Italian Ministry of Health (RC2015).
CA was speaker of Sanofi, Eli Lilly and Pfizer. CA was in the advisory boards for Roche, Merck, Astra Zeneca, Bristol Myers Squibb.
Abbreviations
- CT
controls
- BD-I
bipolar disorder type I
- BD-II
bipolar disorder type II
- SERT
serotonin transporter
- GAD 67
glutamic acid decarboxylase
- COMT
catechol-O-methyltransferase
- PDYN
prodynorphin
- Li
lithium
- BDNF
brain derived neurotrophic factor
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.pnpbp.2017.08.011.
Contributors
CD and BD conceived and designed the experiments; CD, MCP, BB, MP, ADF, GC performed the experiments; CD, BD, MM analysed the data; CD, BD, DG contributed reagents/material/analysis tools; CD, BD, MM wrote the paper; ES, ACA revised critically the manuscript.
Statement of interest
MCP, BB, GC, ADF, MP, CD, MM declare to have nothing to disclose.
Ethical statement
All subjects had given their written informed consent to participate to the study, which included the use of personal and clinical data as well as blood drawing for genotyping and methylation analysis. The study protocol had been previously approved by the local Ethics Committee.
References
- Abdolmaleky HM, Cheng KH, Faraone SV, Wilcox M, Glatt SJ, Gao F, et al. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum Mol Genet. 2006;15:3132–3145. doi: 10.1093/hmg/ddl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aberg KA, Xie LY, McClay JL, Nerella S, Vunck S, Snider S, et al. Testing two models describing how methylome-wide studies in blood are informative for psychiatric conditions. Epigenomics. 2013;5:367–377. doi: 10.2217/epi.13.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th. Author; Washington, DC: 2013. [Google Scholar]
- Arosio B, Viazzoli C, Mastronardi L, Bilotta C, Vergani C, Bergamaschini L. Adenosine A2A receptor expression in peripheral blood mononuclear cells of patients with mild cognitive impairment. J Alzheimers Dis. 2010;20:991–996. doi: 10.3233/JAD-2010-090814. [DOI] [PubMed] [Google Scholar]
- Arosio B, D’Addario C, Gussago C, Casati M, Tedone E, Ferri E, et al. Peripheral blood mononuclear cells (PBMCs) as a laboratory to study dementia in the elderly. Biomed Res Int. 2014:169203. doi: 10.1155/2014/169203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakulski KM, Dolinoy DC, Sartor MA, Paulson HL, Konen JR, Lieberman AP, et al. Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J Alzheimers Dis. 2012;29:571–588. doi: 10.3233/JAD-2012-111223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber A, Gottschlich R. Novel developments with selective, non-peptidic kappa-opioid receptor agonists. Expert Opin Investig Drugs. 1997;6:1351–1368. doi: 10.1517/13543784.6.10.1351. [DOI] [PubMed] [Google Scholar]
- Bolton MM, Pittman AJ, Lo DC. Brain-derived neurotrophic factor differentially regulates excitatory and inhibitory synaptic transmission in hippocampal cultures. J Neurosci. 2000;20:3221–3232. doi: 10.1523/JNEUROSCI.20-09-03221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Béguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, et al. Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther. 2006;316:440–447. doi: 10.1124/jpet.105.092304. [DOI] [PubMed] [Google Scholar]
- Castrén E, Rantamäki T. The role of BDNF and its receptors in depression and antidepressant drug action: reactivation of developmental plasticity. Dev Neurobiol. 2010;70:289–297. doi: 10.1002/dneu.20758. [DOI] [PubMed] [Google Scholar]
- Chavkin C. Dynorphin-still an extraordinarily potent opioid peptide. Mol Pharmacol. 2013;83:729–736. doi: 10.1124/mol.112.083337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, Friso S. Epigenetics: a new bridge between nutrition and health. Adv Nutr. 2010;1:8–16. doi: 10.3945/an.110.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Cohen BM, Murphy B. The effects of pentazocine, a kappa agonist, in patients with mania. Int J Neuropsychopharmacol. 2008;11:243–247. doi: 10.1017/S1461145707008073. [DOI] [PubMed] [Google Scholar]
- Costa E, Grayson DR, Veldic M, Guidotti A. Neurochemical basis for an epigenetic vision of synaptic organization. Int Rev Neurobiol. 2004;59:73–91. doi: 10.1016/S0074-7742(04)59004-9. [DOI] [PubMed] [Google Scholar]
- D’Addario C, Dell’Osso B, Palazzo MC, Benatti B, Lietti L, Cattaneo E, et al. Selective DNA methylation of BDNF promoter in bipolar disorder: differences among patients with BDI and BDII. Neuropsychopharmacology. 2012;37:1647–1655. doi: 10.1038/npp.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Addario C, Dell’Osso B, Galimberti D, Palazzo MC, Benatti B, Di Francesco A, et al. Epigenetic modulation of BDNF gene in patients with major depressive disorder. Biol Psychiatry. 2013;73:e6–7. doi: 10.1016/j.biopsych.2012.07.009. [DOI] [PubMed] [Google Scholar]
- Debey S, Schoenbeck U, Hellmich M, Gathof BS, Pillai R, Zander T, et al. Comparison of different isolation techniques prior gene expression profiling of blood derived cells: impact on physiological responses, on overall expression and the role of different cell types. Pharm J. 2004;4:193–207. doi: 10.1038/sj.tpj.6500240. [DOI] [PubMed] [Google Scholar]
- Dell’Osso B, D’Addario C, Palazzo MC, Benatti B, Camuri G, Galimberti D, et al. Epigenetic modulation of BDNF gene: differences in DNA methylation between unipolar and bipolar patients. J Affect Disord. 2014;166:330–333. doi: 10.1016/j.jad.2014.05.020. [DOI] [PubMed] [Google Scholar]
- Dell’Osso B, Holtzman JN, Goffin KC, Portillo N, Hooshmand F, Miller S, et al. American tertiary clinic-referred bipolar II disorder compared to bipolar I disorder: more severe in multiple ways, but less severe in a few other ways. J Affect Disord. 2015;188:257–262. doi: 10.1016/j.jad.2015.09.001. [DOI] [PubMed] [Google Scholar]
- Docherty SJ, Davis OS, Haworth CM, Plomin R, D’Souza U, Mill J. A genetic association study of DNA methylation levels in the DRD4 gene region finds associations with nearby SNPs. Behav Brain Funct. 2012;8:31. doi: 10.1186/1744-9081-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domschke K, Tidow N, Kuithan H, Schwarte K, Klauke B, Ambree O, et al. Monoamine oxidase A gene DNA hypomethylation - a risk factor for panic disorder? Int J Neuropsychopharmacol. 2012;15:1217–1228. doi: 10.1017/S146114571200020X. [DOI] [PubMed] [Google Scholar]
- Domschke K, Tidow N, Schwarte K, Deckert J, Lesch KP, Arolt V, et al. Serotonin transporter gene hypomethylation predicts impaired antidepressant treatment response. Int J Neuropsychopharmacol. 2014;17:1167–1176. doi: 10.1017/S146114571400039X. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Fallin MD. Epigenetics at the crossroads of genes and the environment. JAMA. 2015;314:1129–1133. doi: 10.1001/jama.2015.10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MA, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- First MB, Williams JBW, Benjamin LS, Spitzer RL. User’s Guide for the SCID-5-PD (Structured Clinical Interview for DSM-5 Personality Disorder) American Psychiatric Association; Arlington, VA: 2015a. [Google Scholar]
- First MB, Williams JBW, Karg RS, Spitzer RL. Structured Clinical Interview for DSM-5—Research Version (SCID-5 for DSM-5, Research Version; SCID-5-RV) American Psychiatric Association; Arlington, VA: 2015b. [Google Scholar]
- Gavin DP, Sharma RP. Chromatin from peripheral blood mononuclear cells as biomarkers for epigenetic abnormalities in schizophrenia. Cardiovasc Psychiatry Neurol. 2009;2009:409562. doi: 10.1155/2009/409562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande I, Fries GR, Kunz M, Kapczinski F. The role of BDNF as a mediator of neuroplasticity in bipolar disorder. Psychiatry Investig. 2010;7:243–250. doi: 10.4306/pi.2010.7.4.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover A, Pande A, Choudhary K, Gupta K, Sundar D. Re-programming DNA-binding specificity in zinc finger proteins for targeting unique address in a genome. Syst Synth Biol. 2010;4:323–329. doi: 10.1007/s11693-011-9077-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Lewis DA. BDNF Val66Met polymorphism and GAD67 mRNA expression in the prefrontal cortex of subjects with schizophrenia. Am J Psychiatry. 2006;163:534–537. doi: 10.1176/appi.ajp.163.3.534. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Shimizu E, Iyo M. Critical role of brain-derived neurotrophic factor in mood disorders. Brain Res Brain Res Rev. 2004;45:104–114. doi: 10.1016/j.brainresrev.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Matzke MA. Exploring and explaining epigenetic effects. Trends Genet. 1997;13:293–295. doi: 10.1016/s0168-9525(97)01219-5. [DOI] [PubMed] [Google Scholar]
- Higuchi FL, Uchida S, Yamagata H, Otsuki K, Hobara T, Abe N, Shibata T, Watanabe Y. State-dependent changes in the expression of DNA methyltransferases in mood disorder patients. J Psychiatr Res. 2011;45:1295–1300. doi: 10.1016/j.jpsychires.2011.04.008. [DOI] [PubMed] [Google Scholar]
- Hurd YL. Subjects with major depression or bipolar disorder show reduction of prodynorphin mRNA expression in discrete nuclei of the amygdaloid complex. Mol Psychiatry. 2002;7:75–81. doi: 10.1038/sj.mp.4000930. [DOI] [PubMed] [Google Scholar]
- Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epi-genome-wide association studies. Genome Biol. 2014;15:R31. doi: 10.1186/gb-2014-15-2-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett DC, Grace MK, Jones RM, Billington CJ, Portoghese PS, Levine AS. The kappa-opioid antagonist GNTI reduces U50,488-, DAMGO-, and deprivation-induced feeding, but not butorphanol- and neuropeptide Y-induced feeding in rats. Brain Res. 2001;909:75–80. doi: 10.1016/s0006-8993(01)02624-5. [DOI] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John H, Sharma V. Misdiagnosis of bipolar disorder as borderline personality disorder: clinical and economic consequences. World J Biol Psychiatry. 2009;10:612–615. doi: 10.1080/15622970701816522. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Komor AC, Badran AH, Liu DR. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;168:20–36. doi: 10.1016/j.cell.2016.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg G, Mosienko V, Gertz K, Alenina N, Hellweg R, Klempin F. Increased brain-derived neurotrophic factor (BDNF) protein concentrations in mice lacking brain serotonin. Eur Arch Psychiatry Clin Neurosci. 2016;266:281–284. doi: 10.1007/s00406-015-0611-3. [DOI] [PubMed] [Google Scholar]
- Kuzmin AV, Gerrits MA, Van Ree JM. Kappa-opioid receptor blockade with nor-binaltorphimine modulates cocaine self-administration in drug-naive rats. Eur J Pharmacol. 1998;358:197–202. doi: 10.1016/s0014-2999(98)00637-2. [DOI] [PubMed] [Google Scholar]
- Lee SY, Chen SL, Wang YS, Chang YH, Huang SY, Tzeng NS, et al. COMT and BDNF interacted in bipolar II disorder not comorbid with anxiety disorder. Behav Brain Res. 2013;237:243–248. doi: 10.1016/j.bbr.2012.09.039. [DOI] [PubMed] [Google Scholar]
- Liang DY, Sun Y, Shi XY, Sahbaie P, Clark JD. Epigenetic regulation of spinal cord gene expression controls opioid-induced hyperalgesia. Mol Pain. 2014;10:59. doi: 10.1186/1744-8069-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linker R, Gold R, Luhder F. Function of neurotrophic factors beyond the nervous system: inflammation and autoimmune demyelination. Crit Rev Immunol. 2009;29:43–68. doi: 10.1615/critrevimmunol.v29.i1.20. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-delta delta C(T) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Loftis JM, Wilhelm CJ, Huckans M. Effect of epigallocatechin gallate supplementation in schizophrenia and bipolar disorder: an 8-week, randomized, double-blind, placebo-controlled study. Ther Adv Psychopharmacol. 2013;3:21–27. doi: 10.1177/2045125312464103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logrip ML, Janak PH, Ron D. Dynorphin is a downstream effector of striatal BDNF regulation of ethanol intake. FASEB J. 2008;22:2393–2404. doi: 10.1096/fj.07-099135. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Lu X, Freund JN, Muller M, Ravey J, Nicolas JP, Gueant JL, et al. Differential regulation of CDX1 and CDX2 gene expression by deficiency in methyl group donors. Biochimie. 2008;90:697–704. doi: 10.1016/j.biochi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Ludwig B, Dwivedi Y. Dissecting bipolar disorder complexity through epigenomic approach. Mol Psychiatry. 2016;21:1490–1498. doi: 10.1038/mp.2016.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell. 2011;147:32–43. doi: 10.1016/j.cell.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon E. Mutation detection using fluorescent hybridization probes and melting curve analysis. Expert Rev Mol Diagn. 2001;1:92–101. doi: 10.1586/14737159.1.1.92. [DOI] [PubMed] [Google Scholar]
- MacQueen GM, Hajek T, Alda M. The phenotypes of bipolar disorder: relevance for genetic investigations. Mol Psychiatry. 2005;10:811–812. doi: 10.1038/sj.mp.4001701. [DOI] [PubMed] [Google Scholar]
- Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr, et al. Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323–330. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- Martínez-Levy GA, Cruz-Fuentes CS. Genetic and epigenetic regulation of the brain-derived neurotrophic factor in the central nervous system. Yale J Biol Med. 2014;87:173–186. [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Lu B. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology. 2008;33:73–83. doi: 10.1038/sj.npp.1301571. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Marty S, Wehrle R, Sotelo C. Neuronal activity and brain-derived neurotrophic factor regulate the density of inhibitory synapses in organotypic slice cultures of postnatal hippocampus. J Neurosci. 2000;20:8087–8095. doi: 10.1523/JNEUROSCI.20-21-08087.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell ME. The Family Interview for Genetic Studies Manual. National Institute of Mental Health, Intramural Research Program, Clinical Neurogenetics Branch; Washington: 1992. [Google Scholar]
- Meyer F, Meyer TD. The misdiagnosis of bipolar disorder as a psychotic disorder: some of its causes and their influence on therapy. J Affect Disord. 2009;112:174–183. doi: 10.1016/j.jad.2008.04.022. [DOI] [PubMed] [Google Scholar]
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–389. doi: 10.1192/bjp.134.4.382. [DOI] [PubMed] [Google Scholar]
- Nair A, Vaidya VA. Cyclic AMP response element binding protein and brain-derived neurotrophic factor: molecules that modulate our mood? J Biosci. 2006;31:423–434. doi: 10.1007/BF02704114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Peña CJ, Kundakovic M, Mitchell A, Akbarian S. Epigenetic basis of mental illness. Neuroscientist. 2016;22:447–463. doi: 10.1177/1073858415608147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidsley R, Mill J. Epigenetic studies of psychosis: current findings, methodological approaches, and implications for postmortem research. Biol Psychiatry. 2011;69:146–156. doi: 10.1016/j.biopsych.2010.03.029. [DOI] [PubMed] [Google Scholar]
- Poulter MO, Du L, Weaver IC, Palkovits M, Faludi G, Merali Z, et al. GABAA receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biol Psychiatry. 2008;64:645–652. doi: 10.1016/j.biopsych.2008.05.028. [DOI] [PubMed] [Google Scholar]
- Reindl JD, Rowan K, Carey AN, Peng X, Neumeyer JL, MC Laughlin JP, et al. Antidepressant-like effects of the novel kappa opioid antagonist MCL-144B in the forced-swim test. Pharmacology. 2008;81:229–235. doi: 10.1159/000112867. [DOI] [PubMed] [Google Scholar]
- Rimoy GH, Wright DM, Bhaskar NK, Rubin PC. The cardiovascular and central nervous system effects in the human of U-62066E. A selective opioid receptor agonist. Eur J Clin Pharmacol. 1994;46:203–207. doi: 10.1007/BF00192549. [DOI] [PubMed] [Google Scholar]
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, et al. Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc Natl Acad Sci U S A. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero CJ, Zimmerman M, Chelminski I, Young D. Borderline personality disorder and the misdiagnosis of bipolar disorder. J Psychiatr Res. 2010;44:405–408. doi: 10.1016/j.jpsychires.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T, Rajput M. Misdiagnosis of bipolar disorder. Psychiatry (Edgmont) 2006;3:57–63. [PMC free article] [PubMed] [Google Scholar]
- Starnawska A, Demontis D, Pen A, Hedemand A, Nielsen AL, Staunstrup NH, Grove J, Als TD, Jarram A, O’Brien NL, Mors O, McQuillin A, Børglum AD, Nyegaard M. CACNA1C hypermethylation is associated with bipolar disorder. Transl Psychiatry. 2016;6:e831. doi: 10.1038/tp.2016.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szapacs ME, Mathews TA, Tessarollo L, Ernest Lyons W, Mamounas LA, Andrews AM. Exploring the relationship between serotonin and brain-derived neurotrophic factor: analysis of BDNF protein and extraneuronal 5-HT in mice with reduced serotonin transporter or BDNF expression. J Neurosci Methods. 2004;140:81–92. doi: 10.1016/j.jneumeth.2004.03.026. [DOI] [PubMed] [Google Scholar]
- Thompson Ray M, Weickert CS, Wyatt E, Webster MJ. Decreased BDNF, trkB-TK+ and GAD67 mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J Psychiatry Neurosci. 2011;36:195–203. doi: 10.1503/jpn.100048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uher R. Gene-environment interactions in severe mental illness. Front Psychiatry. 2014;5:48. doi: 10.3389/fpsyt.2014.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldic ML, Caruncho HJ, Liu WS, Davis J, Satta R, Grayson DR, Guidotti A, Costa E. DNA-methyltransferase 1 mRNA is selectively overexpressed in telencephalic GABAergic interneurons of schizophrenia brains. Proc Natl Acad Sci U S A. 2004;101:348–353. doi: 10.1073/pnas.2637013100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojta A, Dobrinić P, Tadić V, Bočkor L, Korać P, Julg B, Klasić M, Zoldoš V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016;44:5615–5628. doi: 10.1093/nar/gkw159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh SL, Strain EC, Abreu ME, Bigelow GE. Enadoline, a selective kappa opioid agonist: comparison with butorphanol and hydromorphone in humans. Psychopharmacology. 2001;157:151–162. doi: 10.1007/s002130100788. [DOI] [PubMed] [Google Scholar]
- Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada MK, Nakanishi K, Ohba S, Nakamura T, Ikegaya Y, Nishiyama N, et al. Brain-derived neurotrophic factor promotes the maturation of GABAergic mechanisms in cultured hippocampal neurons. J Neurosci. 2002;22:7580–7585. doi: 10.1523/JNEUROSCI.22-17-07580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RC, Biggs JT, Ziegler VE, Meyer DA. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133:429–435. doi: 10.1192/bjp.133.5.429. [DOI] [PubMed] [Google Scholar]
- Yuferov V, Nielsen DA, Levran O, Randesi M, Hamon S, Ho A, Morgello S, Kreek MJ. Tissue-specific DNA methylation of the human prodynorphin gene in post-mortem brain tissues and PBMCs. Pharmacogenet Genomics. 2011;21:185–196. doi: 10.1097/FPC.0b013e32833eecbc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni M, Tosato S, Bonetto C, Lasalvia A, Bissoli S, Elisa I, et al. BDNF and COMT interaction in determining clinical presentation of first-episode psychosis: data from the PICOS-Veneto study. Psychiatr Prax. 2011;38:P70. [Google Scholar]
- Zarate CA, Jr, Manji HK. Bipolar disorder: candidate drug targets. Mt Sinai J Med. 2008;75:226–447. doi: 10.1002/msj.20042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Cheng L, Badner JA, Chen C, Chen Q, Luo W, et al. Genetic control of individual differences in gene-specific methylation in human brain. Am J Hum Genet. 2010;86:411–419. doi: 10.1016/j.ajhg.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.