

Abstract
Ectodomain shedding of cell-surface precursor proteins by metalloproteases generates important cellular signaling molecules. Of importance for disease is the release of ligands that activate the epidermal growth factor receptor (EGFR), such as transforming growth factor-alpha (TGFA), which is mostly carried out by ADAM17 [a member of the A-disintegrin and metalloprotease (ADAM) domain family]. EGFR ligand shedding has been linked to many diseases, in particular cancer development, growth and metastasis, as well as resistance to cancer therapeutics. Excessive EGFR ligand release can outcompete therapeutic EGFR inhibition or the inhibition of other growth factor pathways by providing bypass signaling via EGFR activation. Drugging metalloproteases directly have failed clinically because it indiscriminately affected shedding of numerous substrates. It is therefore essential to identify regulators for EGFR ligand cleavage. Here, integration of a functional shRNA genomic screen, computational network analysis, and dedicated validation tests succeeded in identifying several key signaling pathways as novel regulators of TGFα shedding in cancer cells. Most notably, a cluster of genes with NF-kB pathway regulatory functions was found to strongly influence TGFα release, albeit independent of their NF-kB regulatory functions. Inflammatory regulators thus also govern cancer cell growth promoting ectodomain cleavage, lending mechanistic understanding to the well-known connection between inflammation and cancer.
Keywords: gene-interference, pathways, cancer, growth-factor, computational networks
Introduction
Epidermal-growth-factor-receptor (EGFR) activation generates signals for cell proliferation, migration, differentiation, or survival (reviewed in (1)), cellular phenotypes that are often dysregulated in cancer. A-disintegrin-and–metalloprotease-17 (ADAM17) — releases most ligands that activate the EGFR, e.g. transforming-growth-factor-alpha (TGFα) (2-4). ADAM17, EGFR ligands and EGFR are indeed upregulated in numerous different cancer types, driving EGFR activity that promotes cancer growth and metastasis (reviewed in (5) and (1)). The same components, ADAM17 and EGFR/EGFR ligands, have also been associated e.g. with the development of organ fibrosis in lung, liver or kidney (6-9) and have important roles in inflammation (reviewed in (10); (11)). Broad-spectrum metalloprotease inhibition has been unsuccessful in the clinic due to significant side effects resulting from indiscriminate suppression of the cleavage of many metalloprotease substrates and their dependent physiological functions (reviewed in (12),(13)). Even recently developed selective ADAM10 or 17 inhibitors still affect the cleavage of many substrates (14,15). Existing strategies to targeting EGFR itself would be more specific, but they also have had limited therapeutic success in cancer and other disease applications due to development of resistance to the inhibitors (16),(17).
Excessive release of EGFR ligands is an important mechanism of therapeutic resistance in cancer treatment. It can outcompete attempts to block ligand binding to EGFR or to block EGFR kinase activity (18-20), and it can confer resistance to inhibition of other oncogenic driver pathways by establishing “by-pass signaling” via the EGFR pathway (21). As examples for the latter, EGF can induce resistance to cMet inhibition (crizotinib) in lung cancer patients (22,23), EGF or NRG1 can induce resistance to cMet inhibition in cMet-amplified gastric cancer cells and BRaf (V600E) inhibition in melanoma cells (21), and TGFα causes resistance to ALK inhibition in lung cancer cells (24). Development of strategies to target EGFR ligand release is therefore of great importance for successful inhibition of the EGFR pathway and for effective targeting of other growth pathways under current clinical investigation in cancer and other diseases.
Ectodomain shedding by ADAMs can be induced by activation of intracellular signaling pathways involving e.g. calcium influx, the activation of G protein-coupled receptors, and the release of diacylglycerol, an activator of protein kinase C (PKC) (25). Several mechanisms that regulate cleavage on the level of ADAM17 have been reported, including regulation of expression, maturation, trafficking to the cell surface (reviewed in(12)), posttranslational modifications of ADAM17´s ectodomain (26-29) or C-terminus (by the mitogen-activated protein kinases p38 or ERK) (30,31), and interaction with other transmembrane proteins, such as iRhom1/2 (32,33) and annexins (34,35). In addition, a small domain contained in the ectodomain of ADAM17 has been identified that interacts with and regulates the cleavage of a small subset of ADAM17 substrates (36,37). Specific regulation of substrate cleavage also involves intracellular signal-induced C-terminal modifications of the substrate (4,25,38,39) that lead to protease accessibility changes of the substrate’s ectodomain (40), but it has remained mostly unknown which intracellular signaling components and pathways are involved (reviewed in (12)).
To identify such signaling components and pathways that regulate induced TGFα cleavage, we carried out an shRNA screen of all human kinases and phosphatases. An early limited screen analysis revealed that protein-kinase-C-alpha PKCα and the PKC-regulated protein-phosphatase-1-inhibitor-14D (PPP1R14D) are required for specific regulation of phorbol ester (TPA; a diacylglycerol mimic and PKC activator)- or angiotensin II-induced shedding of TGFα and that this regulation indeed occurred on the substrate and not the protease level (4).
Here we report a combined experimental and computational approach for discovering novel regulators of TGFα cleavage. First, using stringent analysis of our primary shRNA screen carried out in acute T cell leukemia-derived Jurkat cells, we validated a significant number of positive and negative regulators of TGFα cleavage in triple negative breast cancer cells. We then modeled and validated signaling pathways identified in an integrative network modeling approach. This network incorporated experimental genes measured in the screen and predicted genes that regulate TGFα cleavage in different cancer cells. We discovered and validated a cluster of TGFα regulatory genes that are also known to influence NF-κB signaling and show that NF-κB-regulatory functions of these proteins are not required for their cleavage regulatory functions.
Our results thus define targets that might allow therapeutic control of EGFR ligand ectodomain shedding and thus avoid EGFR ligand-mediated therapeutic resistance. More broadly, our results highlight quite generally how a network approach can improve shRNA screen validation, provide new opportunities for existing therapies, and identify genes relevant for EGFR-driven diseases.
Materials and Methods
The initial screen was conducted in Jurkat cells, as is fully described in (4). An important feature of this screen is that it uses a Jurkat-TE cell line which expresses TGFα with an intracellular GFP and extracellular HA tag.
shEnrich method
The shEnrich method analyzes redundant shRNAs against a given gene to select for consistency and strength of biological effect (in our case, projected effect on TGFα shedding). The method rank-orders all shRNAs and calculates a moving enrichment score (ES) based on summing the probability of finding an shRNA within the gene’s family (probability of a hit, “p_hit”) and the probability of finding an shRNA outside the family (probability of a miss, “p_miss”). For each gene’s shRNA cohort, there is a total effect represented by the sum of all shRNAs z-scores; the p_hit is a fractional representation of how much of this total effect is captured at each rank. The p_miss is a fractional penalty equivalent to the inverse of the number of nom-family shRNAs in the screen.
A phosphorylation-site specific interactome
We used the set of human interactions contained in version 13 of the iRefIndex database (41) as our source for protein-protein interactions, which consolidates information from a variety of source databases. We used the MIscore system (42) to assign confidence scores (ranging from 0 to 1) to these interactions; this scoring system considers the number of publications (publication score), the type of interaction (type score), and the experimental method used to find the interaction (method score). We extracted the relevant scoring information for interactions from the iRefIndex MITAB2.6 file, using the redundant interaction group identifier (RIGID) to consolidate interactions between the same two proteins reported by multiple databases, and used a java implementation of MIscore (version 1.3.2, available at https://github.com/EBI-IntAct/miscore/blob/wiki/api.md) with default parameters for individual score weights. We only considered interactions between two human proteins (i.e. we excluded human-viral interactions) and converted protein identifiers, generally provided as UniProt or RefSeq accessions, to valid HUGO gene nomenclature committee (HGNC) symbols. Once converted, we removed redundant interactions (generally arising from isoform-specific interactions that map to the same protein/gene symbols) and retained the maximum observed score. This produced a total of 175,854 unique protein-protein interactions.
To this interaction set, we added predicted and experimental interactions for kinase and phosphatase sites. We collected the kinase-site interactions from Phosphosite (43) and phosphatase-site interactions from DEPOD (44,45) Where site-specific information existed, we created edges in the interactome, first from the kinase/phosphatase to a substrate-site node (represented in the network as ‘PROTEIN_SITE’) and second from the substrate-site to the substrate (represented as ‘PROTEIN’). The kinase/phosphatase to substrate-site interactions were scored using a modified MIscore framework (42). This scoring method is a normalized, weighted sum of an interaction type score(SP), evidence type score(St), and publication value score(Sp):
Using the MIscore framework as a guide, all interaction type scores (St) were set to 1 (because this interaction information was ‘direct’). Publication scores were set using the MIscore scale (Sp=0.00/0.33/0.53/0.67/0.77/0.86/0.94/1.00 for 0/1/2/3/4/5/6/7 publications). The method scores (Sm) were set based on the evidence available for each data set:
|
| |||
| DEPOD | Phosphosite | ||
|
| |||
| in vitro reaction or in vivo in one lab | 0.33 | in vitro evidence | 0.33 |
|
| |||
| in vitro and in vivo or evidence in multiple labs | 0.66 | in vivo evidence | 0.66 |
|
| |||
| in vitro and in vivo in multiple labs | 1.0 | both | 1.0 |
|
| |||
All K values were set to 1. The interactome uses Uniprot identifiers. A text version of this interactome is hosted on the Fraenkel Lab website (http://fraenkel-nsf.csbi.mit.edu/tgfashedding/)
Prize-collecting Steiner Forest (PCSF)
We used the genes selected by the shEnrich method and the interactome network described above as inputs to the Prize Collecting Steiner Forest algorithm (46). The genes from shEnrich were all assigned prize values of 1 and converted to Uniprot identifiers. We used the parameters β=10, D=10, ω=1, and μ=0.006 (parameter optimization explained in supplemental methods). After finding an optimal network of genes, we augmented this forest and added back all possible interactions among these genes from our initial interactome. We converted to gene identifiers back to gene symbol and visualized the network in Cytoscape. The PCSF algorithm is available online as part of our ‘Omics Integrator suite of tools (http://fraenkel-nsf.csbi.mit.edu/omicsintegrator/). An archived version of PCSF that was used for this analysis is hosted on the Fraenkel Lab website (http://fraenkel-nsf.csbi.mit.edu/tgfashedding/).
Sensitivity, Specificity, and Centrality Metrics
To determine a gene’s sensitivity within our network solution, we created a family of 100 networks using the original prize values and genes selected from the shEnrich method. In each of these simulations, we added 0.5% noise to the interaction edge scores in our interactome by randomly increasing or decreasing these edge scores by 0.5%. For a given gene, we measured sensitivity by counting the gene’s representation in the family of noisy networks. A gene that shows up in all of the networks is insensitive to noise and thus robust to our method. Because the prize nodes are constant inputs to the algorithm, we expect them to be insensitive to edge noise. To measure specificity, we again created a family of 100 networks. This time, we hold the interactome constant and randomly select gene targets from our initial library as inputs. We run the algorithm at the same parameters as above and count a gene’s representation in the family of random networks. We calculated 1-specificity to rank genes that are specific to the real experimental data and eliminate biases in selection due to the initial library construction. For all centrality metrics we used the networkX module in Python.
For aggregate scoring, we used the following equation to rank experimental and predicted genes:
In this equation, sens, spec, and norm_cl refer to a gene’s sensitivity, specificity, and normalized closeness, and σsens,σspec, and σnorm_cl are the standard deviation of these measurements for the entire network.
Analysis of NF-κB related cluster in model cell lines
We pulled RMA-normalized cell line expression data from CCLE (47) and plotted these values using the Seaborn package in Python.
Analysis of network-selected genes in cancer patient samples
We pulled patient mRNA expression data from www.cbioportal.org (RNA Seq. V2 RSEM, z-scores) for the time frame January 11, 2016 to February 11, 2016. The data for IRAK1, CALM1, OBSCN, PPEF1, and PPEF2 across cancer types histograms comparison was pulled from the same website on June 6, 2016. The data that was investigated was only from data using z-scores with a 0.0 threshold used for analysis (in order to have the data clearly represented without bias).
We further explored differential expression of human cancer cell lines using RNA-seq from EMBL expression Atlas (48). We selected for all instances of ‘cancer’ vs. respective ‘normal’ tissue. We plotted the log2 fold-change expression as reported from the Expression Atlas Downloads and used Seaborn package in Python to group cancers based on their differential expression of NF-κB -related cluster genes. All differentially expressed genes are P<0.05 using a t-test provided by the Expression Atlas web interface.
Cell lines
Kato-III and MDA-MB-231 cells were purchased from ATCC (Manassas, VA) in March, 2016. Both cells lines were tested for mycloplasma using the Lonza MycoAlert Mycloplasma Detection Kit. We tested cells within one month of arrival and expanded the initial population into aliquots for all further experiments.
shRNA knockdown experiments
MDA-MB-231 or Jurkat cells were transduced with lentiviral particles used in the original screen or, for newly identified regulators, obtained from Sigma Aldrich (Mission shRNA Transduction Particles). The clone IDs of the particles were: PRKCA: TRCN0000001693, TRCN0000001692; PPP1R14D: TRCN000000192, TRCN0000001924; DUSP9: TRCN0000002426, TRCN0000002427; INPP5D: TRCN0000039894, TRCN0000039896; ENPP1: TRCN0000002538, TRCN0000002537; OBSCN: TRCN0000021599, TRCN0000021602; PPAP2A: TRCN0000002579, TRCN0000002577; PPEF1: TRCN0000002551, TRCN0000002550; PKIB: TRCN0000002817, TRCN0000002815; PPP4R1: TRCN0000052763, TRCN0000052766; PTPRE: TRCN0000002893, TRCN0000002895; SSH3: TRCN0000002612, TRCN0000002613; SH2D1A: TRCN0000360148, TRCN0000367940 TRCN0000360149; PTPN22: TRCN0000355586, TRCN0000355534, TRCN0000355533; TAB1: TRCN0000381913, TRCN0000380746, TRCN0000380345; XIAP: TRCN0000231575, TRCN0000231576, TRCN0000231577. MDA-MB-231 or Jurkat cells were plated at 2.5×105 cells per well in 96 well plates, and infected with 1 uL of virus in media with 4ug/mL polybrene. After 24 hours (day 1), cells were switched to media with of puromycin (2ug/mL or 2.5ug/mL for MDA-MB-231 or Jurkat cells respectively). On day 3, fresh media with puromycin was added. On day 5, cells were switched to regular growth media and allowed to recover before stimulation. On day 7, cells were either stimulated with 100 nM PMA or left in media. At 1h (MDA-MB-231, ELISA measurements) or 5 minutes (Jurkat, FACS analysis), released or surface bound TGFα was measured.
Measuring surface bound TGFα in Jurkat cells
For all measurements in Jurkat-TE cells, we used the following staining and washing procedure: Jurkat-TEs were spun out of media and re-suspended in 25 uL of 1:100 mouse anti-HA (Covance #MMS-101) on ice for 1 hour. Cells were washed 3 times with 200 uL cold PBS with 3% FCS, and resuspended in 25 uL 1:100 APC-coupled anti-mouse (BD Pharmigen #550826) on ice for 2 hours. They were resuspended with propidium iodide (PI) and relative TGFα at the surface was quantified using FACS. We analyzed all FACS data using FlowJo. We measured the FITC and APC-A geometric means in the PI-negative (live) population. Relative TGFα is determined from the ratio of red:green fluorescence and represents the relative amounts of extracellular:intracellular TGFα.
Measuring TGFα shedding in Kato-III and MDA-MB-231 cell lines
Validation of screen hits was conducted using MDA-MB-231 cells plated in 96 well plates. 90% confluent cells were stimulated with 100 nM PMA for 2 hours after which time supernatant was collected. A microsphere-based Luminex Technology and ELISA kits (DY239, R&D Systems) were used to measure a panel of shed growth factors, as previously described (49).
To measure changes in TGFα shedding with inhibitor treatments, 40,000 cells per well were plated in a 96 well plate, allowed to adhere overnight and then incubated with serum-free medium for 3 hours. Cells were then pre-treated for 1 hour with DMSO control or IRAK4 inhibitor (5uM AS2444697, TOCRIS) or IRAK1/4 inhibitor (5 uM IRAK1/4 Inhibitor I, TOCRIS) or IKKb inhibitor (2 uM BI605906, TOCRIS) or metalloprotease inhibitor (10uM Batimistat, TOCRIS) after which medium was replaced with fresh media containing the respective inhibitor and 100nM PMA (or vehicle). After 1 hour incubation, TGFα shedding in the cell culture medium was quantified using the DuoSet ELISA Development System (R&D systems). Cell viability was immediately assessed using CellTiter-Glo Luminescent Cell Viability Assay (Promega). TGFα shedding values (pg/mL) were normalized to cell viability measurements.
Statistical analysis
For Jurkat experiments, we conducted unpaired t-tests using Prism software to assess significance. For all control shRNAs (denoted “shCtl” in each figure), n=6 and for all other shRNAs, n=3. In all cases, error bars are standard error of the mean. For the ELISA experiments in KATOIII or MDA-MB-231 cell lines, a one way ANOVA test was performed using Prism software to determine significance of treatment conditions compared to the untreated control (n=5). In all cases, error bars are standard error of the mean.
Results
A kinome/phosphatome screen for regulators of TGFα shedding
We conducted a screen for signaling regulators of TGFα shedding, comprising a library of 750 human kinases and phosphatases, using a FACS-based assay (Figure 1A) in acute T cell leukemia-derived Jurkat cells. Most genes were targeted by 4–5 redundant shRNAs (Figure 1B). After gene knockdown cells were stimulated for 2 min with phorbol ester (TPA), a commonly used cleavage stimulus that activates protein kinase C (PKC). We then measured mean geometric extracellular TGFα (APC) to intracellular TGFα (GFP) fluorescence by FACS and normalized this red:green ratio for all shRNAs to control shRNAs targeting lacZ . Most individual shRNAs showed no effect or had a normalized ratio with an absolute value less than +/− 1 z-score from the distribution mean. Z-scores < 0 correspond to shRNAs with low red:green ratio, indicating enhanced TGFα shedding, and z-scores > 0 correspond to shRNAs with high red:green ratio and reduced TGFα shedding (ranked distribution in upper and middle panel Figure 1C). A limited analysis validated the screen in principal by revealing that PKCα and the PKC-regulated protein-phosphatase-1-inhibitor-14D (PPP1R14D) are required for specific regulation of TGFα cleavage (4).
Figure 1. An shRNA screen measured kinase and phosphatase effects on phorbol ester (TPA) induced TGFα shedding.
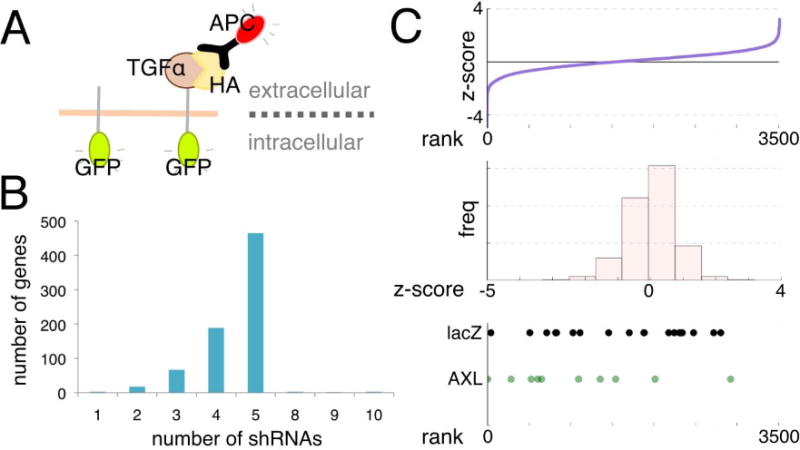
(A) Jurkat cells expressing HA-TGFα-GFP and lentiviral vectors either expressing shRNAs targeting the human kinome and phosphatome or lacZ targeting control shRNAs were treated with TPA, stained with APC-coupled anti-HA antibody, and subjected to FACS. TGFα shedding was detected by determination of the mean red:green fluorescent ratio of the cells. (B) shRNA coverage for most genes was 4-5 individual shRNAs/gene. (C) Z-score normalized red:green ratios for all independent shRNAs relative to shlacZ controls plotted as a ranked distribution (upper panel) and as a histogram of scores (middle panel). Z-scores < 0 correspond to shRNAs which had a low red:green ratio and enhanced TPA-induced shedding, and z-scores > 0 correspond to shRNAs which had a relatively high red:green ratio and prevented TPA-induced shedding. The lower panel shows where all lacZ shRNAs (20 total) and all AXL shRNAs (10 total) fell within this distribution, and highlights the difficulty in determining which genes are consistently affecting shedding.
Within a given family of redundant shRNAs targeting the same gene, and also within samples targeted by the same control shRNA, resulting z-scores varied over a considerable range and overlapped significantly; shown as example for one targeted gene, Axl, and shLacZ controls (lower panel Figure 1C). This demonstrates inherent difficulties similar to other shRNA screens in determining which shRNAs consistently produce a phenotype. We explored multiple ranking strategies before pursuing a computational network modeling approach to significantly improve upon these inherent problems.
A very stringent two-best shRNA scoring method used the average of multiple measurements for the same shRNA for z-score calculation and required a gene’s top two shRNAs contain one hairpin that scored 2 z-scores and one hairpin that scored 1.5 z-scores above or below the mean; this method selected 5 genes. In contrast, a less stringent approach which calculated z-scores based on the median measurements of all redundant shRNAs targeting a given gene identified 22 genes with a z-score of 1 above/below the mean (in the positive/negative direction). The shEnrich method, explained in the next paragraph, selected the most targets of all the methods (Table 1). Each scoring method selected different and partially-overlapping gene lists (comparison of the different scoring methods shown in Supplemental Table 1). PRKCA and PPP1R14D published in the original screen validation (4) scored in the 2 best shRNA and shEnrich methods, whereas other genes only scored in shEnrich.
Table 1. Genes selected by the shEnrich method.
The shEnrich method selected genes that had consistent shRNA effects in the forward direction (shRNAs increased TPA-induced TGFα shedding) and in the reverse direction (shRNAs decreased TPA-induced TGFα shedding). Asterisks indicate genes that are included in following network analysis.
| shEnrich Method |
| Forward shEnrich |
| shRNA family size 2 |
| PPP2R1A |
| PTBP1 |
| RIOK2 |
| shRNA family size 3 |
| CDC25A |
| PTPN18 |
| TP53RK |
| shRNA family size 4 |
| BLK |
| CKM |
| DUSP5 |
| EGLN1 |
| FLJ16518 |
| GALK2 |
| GMFB |
| HYPB |
| INPP5B |
| KIAA2002 |
| LOC400687 |
| LOC441868 |
| LOC90353 |
| LOC91461 |
| NEK7 |
| NUDT10 |
| PHKA2 |
| PIN1 |
| PMVK |
| PTPN22 |
| PTPN5 |
| PTPRE |
| RIPK5 |
| RXRB |
| SACM1L |
| TEX14 |
| TRPM7 |
| shRNA family size 5 |
| ABL1 |
| AKAP11 |
| CKS2 |
| DDR2 |
| DKFZP566K0524 |
| DUSP13 |
| EAT2 |
| EEF2K |
| EIF2AK3 |
| EPM2A |
| FBP2 |
| FLJ25449 |
| Gpr109b |
| GUCY2C |
| IMPA2 |
| INPP5F |
| IRAK1 |
| ITPKA |
| LOC283871 |
| LOC401313 |
| LPPR4 |
| MAP3K11 |
| MAP3K7IP1 |
| MAPK4 |
| NR1I3 |
| NT5E |
| NUDT11 |
| OBSCN |
| PCTK3 |
| PIB5PA |
| PIP5K1A |
| PLCB4 |
| PPAPDC1 |
| PPAPDC1A |
| PPEF1 |
| PPEF2 |
| PPFIA1 |
| PPM1J |
| PPP1R12B |
| PPP2CB |
| PPP3CA |
| PPP4R1 |
| PPP4R1L |
| PPP5C |
| PRG-3 |
| PRKWNK3 |
| PTPRM |
| RAF1 |
| RIMS4 |
| SGPP2 |
| SH2D1A |
| SMG1 |
| SNRK |
| SPAP1 |
| SYT14L |
| WWP2 |
| shRNA family size 8 |
| CIB2 |
| ERBB4 |
| Reverse shEnrich |
| shRNA family size 2 |
| CALM1 |
| PPP2R1A |
| DUSP9 |
| shRNA family size 3 |
| PPAP2A |
| SSH3 |
| shRNA family size 4 |
| GPR109A |
| LCK |
| PPP1R14D |
| SIK2 |
| TRPV5 |
| shRNA family size 5 |
| CDC14A |
| ENPP1 |
| EPB41L4A |
| NME5 |
| PIM1 |
| PKIB |
| PRKCA |
Selection of gene candidates using the shEnrich method
To improve our scoring of genes identified in the original shRNA screen, we developed the shEnrich method to measure consistency of redundant shRNAs and strength of their effect. This enrichment scoring method is conceptually modeled after the original Gene Set Enrichment Method (GSEA), and is representative of other rank-order methods for RNAi scoring (e.g. RIGER and dRIGER) (50-53). These methods all developed ranking statistics that look for consistency of shRNA effects compared to the remainder of the tested shRNA population and control shRNAs.
Our method most closely resembles that of Kampmann et al (53), in that our screening readout was a single measurement (z-score normalized red:green fluorescent ratios) and that we derived our null distributions from a cohort of non-targeting controls. However, that earlier approach does not correct for the number of shRNAs targeting a gene, which in our case ranged from 1 to 10. We therefore calculated expected distributions for each gene based on its shRNA family size; e.g. for a gene with 4 shRNAs, we calculated 1000 permutations using 4 shLacZs from our control set. Our enrichment method resulted in a projected z-score and a maximal normalized enrichment score (NES). This represents the rank at which maximal enrichment occurs for a gene’s shRNA family. Figure 2A represents the shEnrich score for Axl in the forward direction (highest rank reflects that an shRNA increased shedding) and for PPP1R14D in the reverse direction (highest rank indicates an shRNA decreased shedding). Both AXL and PPP1R14D show high NES relative to the lacZ controls indicating high consistency. We repeated this process for all genes of all shRNA family sizes in both the forward (shRNAs increase shedding) and reverse (shRNAs decrease shedding) ranking directions. Within each family size (in forward and reverse direction), we compared gene shEnrich NES against 1000 permutations calculated using subsets of the shlacZ controls. Figure 2B shows the distribution of shEnrich NES for all genes relative to the distribution of shlacZ scores for shRNA family size 5 (all other family sizes plotted in Supplemental Fig. 1 and 2). Most genes did not show an effect that was as consistent or more consistent than the shlacZ controls. To additionally filter gene candidates for strength of effect, we plotted gene shEnrich NES against the projected z-score (Figure 2C). Many genes had a low shEnrich NES and a low projected z-score (low referring to a score with a value < −1 in the forward direction or > +1 in the reverse direction). Only a few genes had a high shEnrich NES and a relatively high projected z-score; these genes fell in the upper-left/upper-right quadrants (highlighted with orange squares) in the forward/reverse directions (scatter plots for all other family sizes shown in Supplemental Fig. 3). For our example genes, AXL did not make the projected z-score cut-off of < −1, but PPP1R14D did make the > +1 z-score cut-off. The full list of genes that passed these stringent criteria is shown in Table 1.
Figure 2. shEnrich selects genes for consistency and effect size.
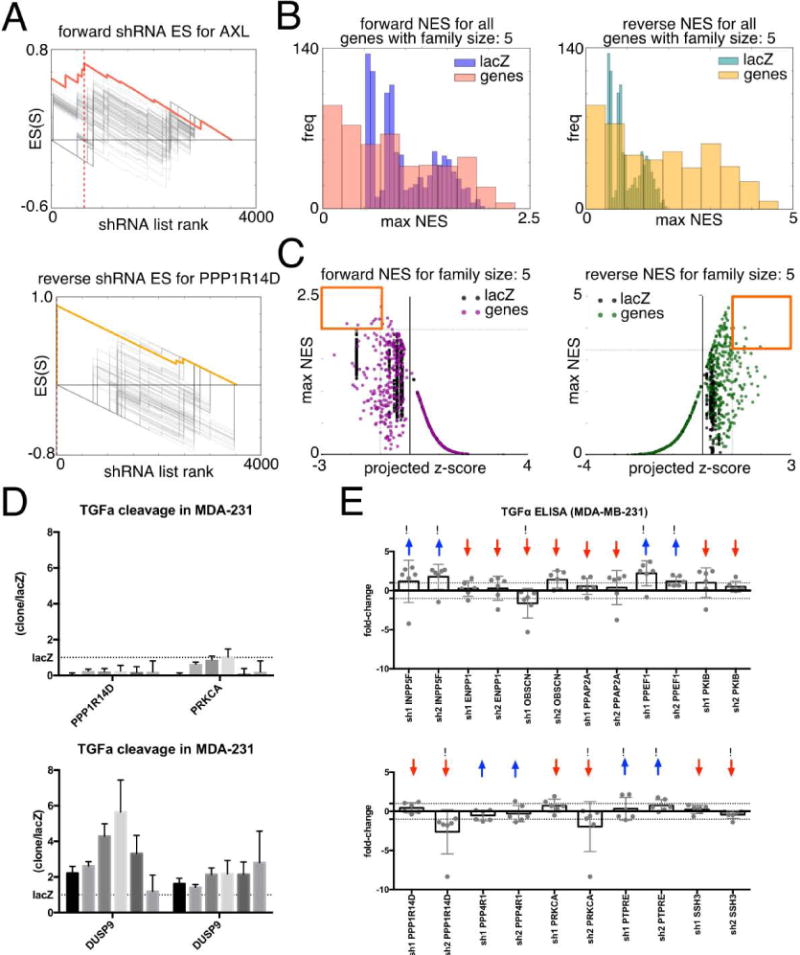
(A) Lightening plots show the enrichment score for AXL in the forward direction (red, upper) and PPP1R14D (gold, lower) in the reverse direction. Grey lines represent 100/1000 enrichment scores calculated for shlacZ. Dashed line represents position of maximal enrichment for the gene of interest. (B) Normalized enrichment scores (NES) for all genes with 5 redundant shRNAs (family size 5) plotted against 1000 NES for lacZ in the forward direction (left) and reverse direction (right). (C) Maximal NES score for all genes with 5 redundant shRNAs and lacZ controls plotted against the z-score at which maximal enrichment occurs (‘projected z-score’). Normalized distributions and maximal enrichment plots for all other gene family sizes are included in supplemental figures. (D) Supernatant ELISA detection of cleaved TGFα in MDA-MB-231 cells with PPP1R14D and PRKCA knockdown (upper panel), or DUSP9 knockdown (lower panel). Each bar represents redundant tests of the same shRNA; error bars are standard errors of the ratio of shRNA gene:shlacZ control. Dotted line indicates measurements in respective shlacZ control (E) TGFα ELISA of additional genes identified by shEnrich method. Two shRNAs were selected for each gene and tested in sextuplicate in MDA-MB-231 cells. Bars represent average log2 fold-change relative to a non-targeting control, and each dot represents an individual replicate. Red/blue arrows indicate the shEnrich prediction of directionality of effect (increased/decreased TGFα shedding) for each gene tested, and exclamation marks indicate where the experimental shRNA effect agreed with the shEnrich prediction.
We first used a traditional strategy to validate a portion of shEnrich-identified gene targets in MDA-MB-231 cells, a triple-negative breast cancer cell line and well-studied cancer model, using an ELISA assay that detected cleaved TGFα ectodomain in cell culture supernatants. PPP1R14D or PRKCA knockdown blocked TGFα cleavage, again confirming our previously published results (4) and DUSP9 knockdown increased shedding; results from 6 independent experiments are shown (Figure 2D) using one shRNA for PPP1R14D and PRKCA, and (lower panel, Figure 2D) using two shRNAs for DUSP9. Knockdown of 11 other shEnrich-identified genes that induced strong effects on TGFα cleavage in the initial shRNA screen revealed that for 8 of the 11 gene targets, shRNAs showed consistent directionality of effect for both shRNAs tested in the MDA_MB-231 context (INPP5F, ENPP1, PPAP2A, PPEF1, PKIB, PPP4R1, and PTPRE), though effect size varied (Figure 2E). Of the individual shRNAs, 10 out of 22 had a directionality of effect consistent with the shEnrich prediction for their gene target (up or down arrows highlighted with exclamation points in Figure 2E); the other shRNAs with shEnrich predictions showed opposite effects from the shEnrich predictions (arrows, but no exclamation points in Figure 2E).
These effects may result from differences in effectiveness of knockdown depending on the cell type studied, as has been described in other reports (51) highlighting the importance of investigating a larger set of shRNAs per gene and using aggregate statistics such as shEnrich to analyze their effects. Detected switches in directionality suggest that contextual factors might affect the shedding phenotype, making it difficult to incorporate shRNA hits into signaling pathways from enrichment scores or targeted validation alone. We therefore performed additional analysis by network modeling to identify a more complete TGFα cleavage regulatory pathway.
Prize-Collecting Steiner Forest (PCSF) identifies a TGFα shedding pathway
We previously described the role of off-target effects in RNAi or shRNA screens, specifically that both reagent-based and biology-based effects determine whether a gene can be identified as part of a pathway based on gene knockdown data (43). Interpreting RNAi or shRNA results in a network framework, in contrast to an individual “hits” or “targets” framework, leverages contributions from all hit/target contributions to pathways via their relationships with other network genes. This interpretation can ameliorate dependence upon individual reagent performance and increase confidence in biological validation.
Our previous work demonstrated that data integration into a network context could construct novel pathways despite noise from RNAi screens (54). Starting with results obtained from shEnrich, we used the Prize-Collecting Steiner Forest (PCSF) method (46,55) to construct a pathway for TGFα cleavage; the method predicted additional pathway genes not identified in the initial experimental set in this process. The Prize Collecting Steiner Forest (PCSF) acted as a filter, and required genes to have interaction associations with other genes identified from the screen to be included in the final pathway. To define all sets of possible associations, we derived an interactome from iRefWeb and added predicted kinase and phosphatase site-specific interactions from Networkin and DEPOD (45,56) (details of the interactome construction are explained in methods; optimization of network node selection is shown in Supplemental Fig. 4). The algorithm identified an optimal sub-network from this interactome by selecting edges that captured genes from the shEnrich method (“experimental genes”) without over-fitting. In this process, the algorithm added predicted genes that connected experimental genes (Figure 3, top panel). We used a set of parameters to tune the algorithm to ensure an appropriate selection of experimental and predicted genes (see methods). This optimized network contained 86 genes, and 97 edges. Of the original 108 genes selected from the shEnrich method, 43 were retained. Of note, the network contained interaction evidence for the experimental genes PPEF1, OBSCN, DUSP9, PRKCA, and PPP4R1; these genes were selected by shEnrich and confirmed with ELISA in MDA-MB-231 cells. PPP1R14D was not included despite experimental validation, suggesting that there is not sufficient interaction evidence to include this gene in the pathway. The remaining 43 genes were previously unidentified genes predicted by the algorithm to be relevant to TGFα cleavage. These predicted genes also included gene targets contained in the original screen that were refractory to knockdown using available shRNAs.
Figure 3. PCSF identifies TGFα shedding regulatory network.
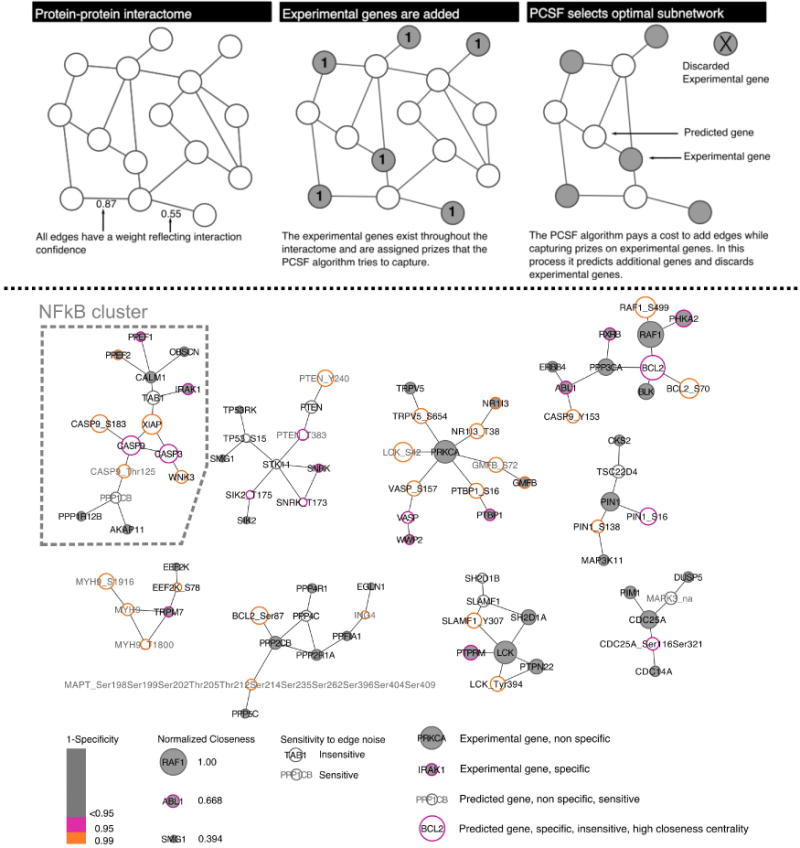
(Top panel) The cartoon schematic represents how PCSF selects an optimal sub-network. Interaction edges are aggregated from multiple databases and all edges are scored (two shown for simplicity). Prizes are assigned from experimental data; in our case prizes were assigned to genes selected by shEnrich. The algorithm weights the cost of adding edges with capturing prizes in selecting an optimal network. (Bottom panel) The optimal, clustered network contains 86 genes and 83 edges. Grey face coloring indicates genes selected from the original experimental data set; white face represents predicted gene (genes and phospho-sites) selected by the algorithm. Gene border represents specificity to randomization (pink<=0.1; orange<=0.05), and gene size represents closeness centrality (larger genes are more central and more robust). Grey gene labels indicate genes that are sensitive to edge noise. Genes on the far right of the legend below the networks are annotated examples to help interpret all of their network properties. The cluster associated with NFkB signaling is highlighted.
We hypothesized that the optimized network gene list contained subsets of functionally related genes. To find these subsets, we leveraged edges in the network solution. Genes with a high number of inter-neighbor edges are more likely to have functional similarities. We used the GLay Community clustering Cytoscape plug-in (57) to subdivide the optimized network into a final target set of 9 network sub-modules. This process removed 14 edges and retained all genes (Figure 3, bottom panel; full network shown in Supplemental Fig. 5).
Robustness analysis prioritizes predicted pathway genes
To prioritize candidates selected by PCSF, we performed a series of robustness and connectivity analyses. Here we explain the metrics for gene prioritization and the corresponding quantification as presented in Table 2. We measured the sensitivity of each gene in the solution by counting representation in 100 runs of PCSF with noise added to the edges. Genes that are insensitive to noise show up in all networks and are assigned a score of 0.00, whereas genes that are sensitive appear in a few networks only and are assigned a score equal to 1-fraction of networks in which they are represented. A score of 0.99 indicates that the gene was present in only one network. None of the experimental genes identified by shEnrich were sensitive to edge noise (high aggregate score) (left column Table 2), however 12 of the newly predicted genes were highly sensitive and showed up only in the solution without edge noise (genes with low aggregate score bottom of right column Table 2). This suggests that low probability edges connected these predicted genes to the network; varying the edge value caused the algorithm to remove the predicted gene in some simulations.
Table 2. Experimental and predicted genes ranked by multiple robustness metrics.
Experimental genes (left) and predicted genes (right) were ranked by the following metrics: (a) Sensitivity represents fractional representation in a family of 100 networks created with 0.5% noise added to the interactome edges; (b) Specificity represents the fractional representation in a family of 100 networks created with random inputs and 1-Specificty is used for ranking since low specificity indicates robustness to this type of randomization; (c) Closeness centrality reflects a node’s robustness as this metric indicates association to other nodes within the network. (d) Aggregate score was calculated by weighting each metric by the standard deviation for that metric and normalized to the maximum score possible for all experimental genes. Shading highlights the highest scoring nodes from this analysis.
| Experimental Genes |
Sensitivity | 1-Specificity | Normalized Closeness |
Aggregate Score |
Predicted genes | Sensitivity | 1-Specificity | Normalized Closeness |
Aggregate Score |
|---|---|---|---|---|---|---|---|---|---|
| LCK | 0.00 | 0.891 | 0.920 | 0.963 | BCL2 | 0.00 | 0.901 | 0.964 | 0.975 |
| RAF1 | 0.00 | 0.723 | 1.000 | 0.956 | RAF1_S499 | 0.00 | 0.950 | 0.876 | 0.961 |
| PHKA2 | 0.00 | 0.941 | 0.780 | 0.936 | BCL2_S70 | 0.00 | 0.970 | 0.849 | 0.958 |
| PIN1 | 0.00 | 0.871 | 0.821 | 0.935 | XIAP | 0.00 | 0.950 | 0.851 | 0.955 |
| PRKCA | 0.00 | 0.713 | 0.923 | 0.935 | CASP9 | 0.00 | 0.941 | 0.835 | 0.950 |
| PTPRM | 0.00 | 0.921 | 0.730 | 0.921 | CASP3 | 0.00 | 0.931 | 0.821 | 0.945 |
| PPP3CA | 0.00 | 0.851 | 0.774 | 0.920 | CASP9_S183 | 0.00 | 0.960 | 0.799 | 0.944 |
| BLK | 0.00 | 0.861 | 0.758 | 0.918 | BCL2_Ser87 | 0.00 | 0.950 | 0.795 | 0.941 |
| SH2D1A | 0.00 | 0.851 | 0.738 | 0.911 | PIN1_S16 | 0.00 | 0.941 | 0.786 | 0.938 |
| PTPN22 | 0.00 | 0.832 | 0.732 | 0.907 | SLAMF1_Y307 | 0.00 | 0.990 | 0.738 | 0.933 |
| ABL1 | 0.00 | 0.901 | 0.668 | 0.902 | NR1I3_T38 | 0.00 | 0.990 | 0.736 | 0.933 |
| NR1I3 | 0.00 | 0.960 | 0.609 | 0.897 | PTBP1_S16 | 0.00 | 0.970 | 0.736 | 0.930 |
| RXRB | 0.00 | 0.911 | 0.635 | 0.895 | LCK_Tyr394 | 0.00 | 0.970 | 0.732 | 0.929 |
| GMFB | 0.00 | 0.950 | 0.609 | 0.895 | VASP_S157 | 0.00 | 0.950 | 0.739 | 0.928 |
| PIM1 | 0.00 | 0.881 | 0.654 | 0.895 | TRPV5_S654 | 0.00 | 0.950 | 0.736 | 0.927 |
| PPP2CB | 0.00 | 0.842 | 0.673 | 0.894 | CASP9_Y153 | 0.00 | 0.990 | 0.688 | 0.921 |
| PTBP1 | 0.00 | 0.901 | 0.609 | 0.887 | PIN1_S138 | 0.00 | 0.950 | 0.670 | 0.910 |
| TRPV5 | 0.00 | 0.891 | 0.609 | 0.886 | WNK3 | 0.00 | 0.950 | 0.667 | 0.909 |
| IRAK1 | 0.00 | 0.911 | 0.587 | 0.883 | CDC25A_Ser116Ser321 | 0.00 | 0.931 | 0.656 | 0.904 |
| CKS2 | 0.00 | 0.881 | 0.563 | 0.873 | TAB1 | 0.00 | 0.842 | 0.703 | 0.901 |
| CDC14A | 0.00 | 0.891 | 0.554 | 0.872 | TSC22D4 | 0.00 | 0.871 | 0.670 | 0.898 |
| DUSP5 | 0.00 | 0.891 | 0.554 | 0.872 | VASP | 0.00 | 0.941 | 0.614 | 0.895 |
| TRPM7 | 0.00 | 0.911 | 0.540 | 0.872 | SLAMF1 | 0.00 | 0.891 | 0.616 | 0.887 |
| CDC25A | 0.00 | 0.505 | 0.801 | 0.872 | PPP4C | 0.00 | 0.881 | 0.572 | 0.875 |
| MAP3K11 | 0.00 | 0.871 | 0.563 | 0.871 | SH2D1B | 0.00 | 0.891 | 0.525 | 0.865 |
| WWP2 | 0.00 | 0.931 | 0.524 | 0.871 | PTEN | 0.00 | 0.683 | 0.656 | 0.864 |
| PPEF2 | 0.00 | 0.950 | 0.509 | 0.870 | EEF2K_S78 | 0.00 | 0.950 | 0.470 | 0.861 |
| PPEF1 | 0.00 | 0.941 | 0.509 | 0.869 | SIK2_T175 | 0.00 | 0.921 | 0.442 | 0.849 |
| PPP2R1A | 0.00 | 0.822 | 0.574 | 0.866 | SNRK_T173 | 0.00 | 0.911 | 0.441 | 0.847 |
| ERBB4 | 0.00 | 0.802 | 0.562 | 0.860 | TP53_S15 | 0.00 | 0.644 | 0.443 | 0.805 |
| OBSCN | 0.00 | 0.881 | 0.509 | 0.859 | STK11 | 0.00 | 0.535 | 0.503 | 0.803 |
| AKAP11 | 0.00 | 0.891 | 0.497 | 0.858 | LCK_S42 | 0.99 | 0.990 | 0.842 | 0.373 |
| CALM1 | 0.00 | 0.733 | 0.594 | 0.857 | PTEN_Y240 | 0.99 | 0.970 | 0.768 | 0.351 |
| PPP5C | 0.00 | 0.891 | 0.489 | 0.856 | MYH9_S1916 | 0.99 | 0.980 | 0.751 | 0.348 |
| PPP4R1 | 0.00 | 0.881 | 0.493 | 0.855 | GMFB_S72 | 0.99 | 0.980 | 0.736 | 0.345 |
| PPP1R12B | 0.00 | 0.871 | 0.497 | 0.855 | CASP9_Thr125 | 0.99 | 0.960 | 0.685 | 0.329 |
| SNRK | 0.00 | 0.911 | 0.441 | 0.847 | MYH9 | 0.99 | 0.980 | 0.630 | 0.318 |
| PPFIA1 | 0.00 | 0.802 | 0.497 | 0.844 | MAPT_Ser198Ser199Ser202Thr205Thr212Ser214Ser235Ser262Ser396Ser404Ser409 | 0.99 | 0.950 | 0.568 | 0.298 |
| EEF2K | 0.00 | 0.851 | 0.415 | 0.831 | MYH9_T1800 | 0.99 | 0.990 | 0.538 | 0.297 |
| SIK2 | 0.00 | 0.871 | 0.393 | 0.829 | PTEN_T383 | 0.99 | 0.921 | 0.571 | 0.294 |
| EGLN1 | 0.00 | 0.851 | 0.390 | 0.825 | MAPK3_na | 0.99 | 0.782 | 0.656 | 0.293 |
| TP53RK | 0.00 | 0.822 | 0.394 | 0.821 | ING4 | 0.99 | 0.960 | 0.438 | 0.268 |
| SMG1 | 0.00 | 0.802 | 0.394 | 0.818 | PPP1CB | 0.99 | 0.723 | 0.579 | 0.265 |
We then tested specificity of the network solution using random inputs from the targets in the original shRNA library. This process involved selecting random sets of genes from the original library (regardless of whether they scored using our shEnrich method), rerunning the PCSF routine at the optimal parameters and counting gene representation in this family of 100 random networks. Gene border color indicates fractional representation in these 100 random networks (Figure 3, bottom panel). Of the 43 experimental genes, 30 genes showed low specificity, indicating general association among targets from the original library. These targets may still have a role in shedding regulation (such as PRKCA), however, we could not distinguish these effects from general library association.
We further used centrality metrics to quantify the robustness of our network selections. Centrality did not imply a biological centrality per se, but reflected how the genes were selected in this network solution. Having a high centrality implied a greater resilience to experimental noise from the input set; mathematically, centrality reflects how interaction edges contributed to a gene’s presence in the final network. We calculated the degree, betweenness, page-rank, and closeness centrality, and an aggregate score (Supplemental Fig. 6A) based on interactions in the original, unclustered network. We created a normalized histogram of centrality scores to evaluate the relative distribution of values (Supplemental Fig. 6B). From this histogram, we observed that closeness centrality best discerns connectivity, while other metrics are dominated by a few high-value nodes (i.e. Page-Rank, Betweenness, and Degree centrality). Using closeness centrality, we visualized these genes within the network solution by adjusting their symbol size to reflect extent of connectivity (Figure 3; quantification in Table 2). Lastly, we created a normalized, aggregate score (Table 2) reflecting an experimental gene’s performance across these three metrics. We adjusted specificity/sensitivity to be (1-specificity)/(1-sensitivity), and normalized closeness centrality to the max value to scale all metrics from 0-1. We weighted each metric by the standard deviation for that metric and normalized to the maximum score possible for all experimental genes (Table 2, left panel). We completed a similar analysis for predicted genes in the network, and again measured sensitivity to edge noise, specificity using random input terminals, and closeness centrality as a metric for connectivity (Table 2, right panel).
Experimental validation of PCSF-selected network genes
First, we validated PCSF-selected experimental gene hits from the original screen. SLAM-associated protein (SAP) (SHD2D1A) and Protein Tyrosine Phosphatase, Non-Receptor Type 22 (PTPN22), using our FACS based TGFα shedding assay in Jurkat cells with or without TPA stimulation. We used TPA stimulation as a benchmark to explore if our gene perturbations were of similar magnitude to a known modulator of TGFα shedding. Knockdown of SH2D1A was monitored by qPCR, demonstrating 70-80% decrease of SH2D1A mRNA levels for all three individual shRNAs (Figure 4A). Knockdown of SH2D1A with three individual shRNAs enhanced TPA-stimulated TGFα cleavage from the cell surface as determined by decreased red:green fluorescent ratio compared to TPA-stimulated control shRNA expressing cells (Figure 4B). Again, mRNA knockdown for each individual shRNA against PTPN22 was monitored by qPCR and varied between 40-90% (Figure 4C). In contrast to SH2D1A, three shRNAs targeting PTPN22 decreased TPA-stimulated TGFα cleavage (increased red:green ratio) as compared to control shRNA (Figure 4D); Three other genes contained in the original screen (OBSCN, PPEF1 and PRKCA) and present in our network model were already validated in MDA-MB-231 triple negative breast cancer cells by ELISA measurements in the context of our shEnrich method validation (Figure 2E). Knockdown of SH2D1A or PTPN22 in MDA-MB-231 cells confirmed their effects on TPA-induced TGFα cleavage in this cell type (enhanced cleavage after SH2D1A and reduced cleavage after PTPN22 knockdown), as measured by ELISA (Supplemental Fig. 7A, knockdown efficiency shown in Supplemental Fig. 7B).
Figure 4. Validation of effect of experimental genes on TGFα cleavage in Jurkat cells.
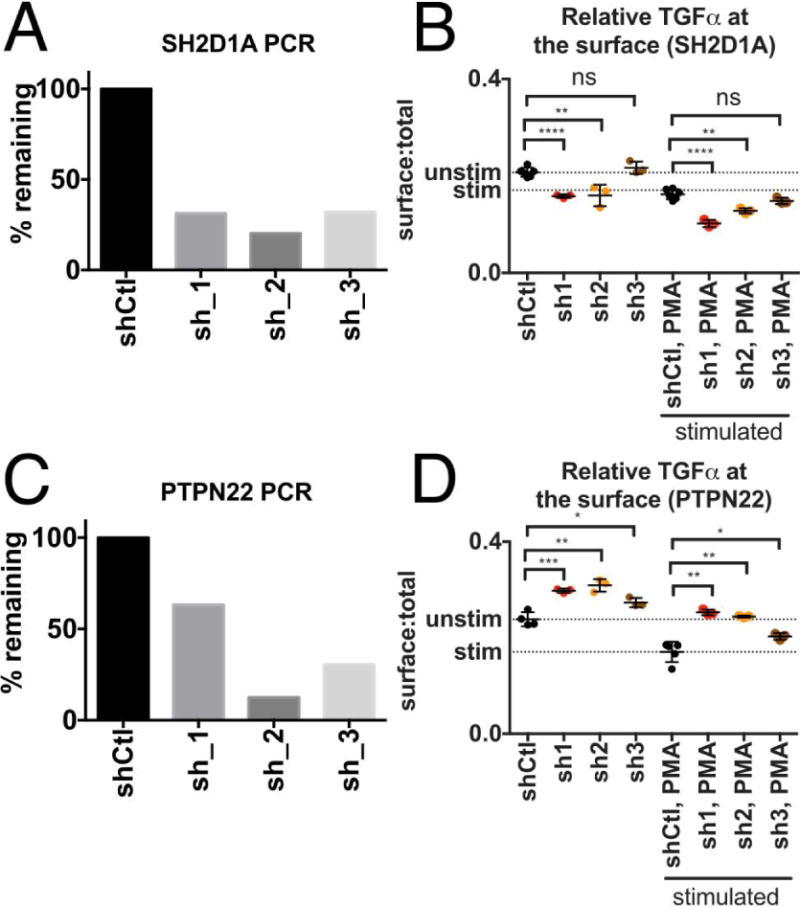
(A) Knockdown of SH2D1A was monitored by qPCR. (B) Knockdown of SH2D1A with three individual shRNAs enhanced TPA-stimulated TGFα cleavage from the cell surface compared to control shRNA expressing cells (indicated by lower red:green fluorescent ratio as compared to control). (C) Knockdown of PTPN22 was monitored by qPCR. (D) Knockdown of PTPN22 with three individual shRNAs decreased TPA-stimulated TGFα cleavage as compared to control shRNA (indicated by higher red:green fluorescent ratio as compared to control). Error bars are standard error of the mean. We used an unpaired t-test to test significance. *p≤0.05; **p≤0.01;***p≤0.001; ****p<0.0001.
We next validated PCSF-selected predicted genes not found in the original screen. Our optimized network contained a 15-gene cluster with high centrality, specificity, and low sensitivity (Figure 3). Of these genes 7 were from the original screen: PPEF1, PPEF2, OBSCN, CALM1, IRAK1, PPP1R12B, and AKAP11. By shEnrich analysis six of these genes decreased TGFα shedding, except for AKAP11 which induced shedding (lightening plots in Supplemental Fig. 8, B–H). We then tested the effect of knockdown of two predicted genes contained in this cluster, X-linked-inhibitor-of-apoptosis (XIAP1) and TGF-Beta-Activated-Kinase-1-Binding-Protein-1 (TAB1) for their effect on TGFα shedding. We selected these predicted genes because they were robust among this cluster (right column Table 2,). As we constructed this pathway using a tissue non-specific interactome, we also explored the expression of this regulatory cluster in Jurkat, MDA-MB-231, Kato-III, and MKN-45 cell lines (Supplemental Fig. 8I) using CCLE data and found that all components were expressed (47). We monitored mRNA knockdown by qPCR in the presence of three shRNAs against TAB1 (Figure 5A). TAB1 knockdown decreased TPA-stimulated TGFα cleavage from the cell surface (increased red:green fluorescent ratio) as compared to TPA-stimulated control shRNA expressing cells (Figure 5B). XIAP mRNA knockdown (Figure 5C) decreased surface-bound TGFα (decreased red fluorescence) as compared to control shRNA (Figure 5D). Additional experiments evaluating the effect of TAB1 or XIAP knockdown on TGFα shedding in Jurkat cells are shown in Supplemental Fig 9A,B,C,D. We also confirmed the same effects of TAB1 and XIAP knockdown on TGFα cleavage in MDA-MB-231 triple negative breast cancer cells (Supplemental Fig 9E) and monitored mRNA expression after shRNA perturbation (Supplemental Fig 9F). Taken together, these results confirmed the relevance of PCSF-identified experimental and predicted network genes in TGFα cleavage regulation.
Figure 5. Validation of predicted genes on TGFα cleavage in Jurkat cells.
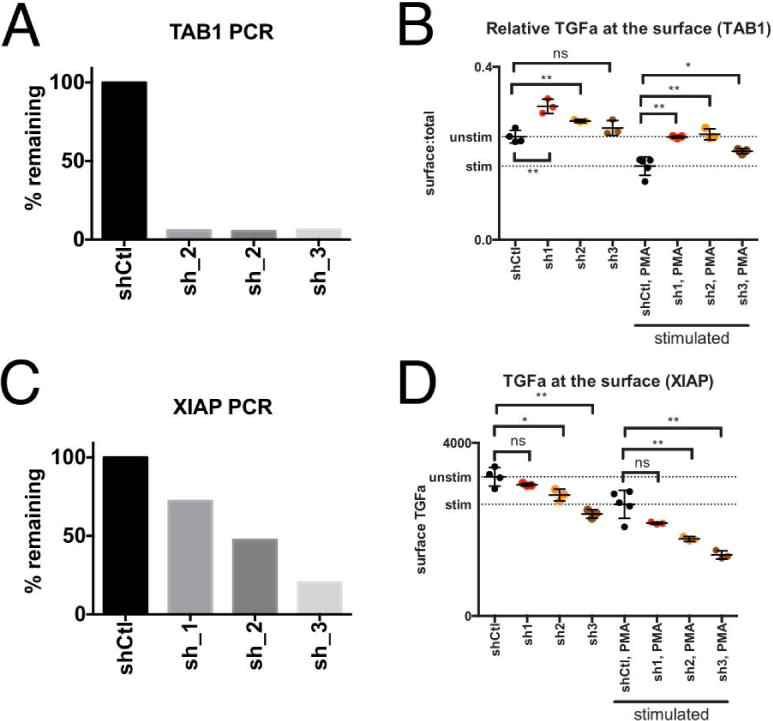
(A) Knockdown of TAB1 was monitored by qPCR. (B) Knockdown of TAB1 with three individual shRNAs decreased TPA-stimulated TGFα cleavage from the cell surface as compared to control shRNA expressing cells (indicated by higher red:green fluorescent ratio as compared to control) (C) Knockdown of XIAP was monitored by qPCR. (D) Knockdown of XIAP with three individual shRNAs decreased TGFα at the cell surface as compared to control shRNA (indicated by lower red fluorescence as compared to control). Error bars are standard error of the mean. We used an unpaired t-test to test significance. *p≤0.05; **p≤0.01;***p≤0.001; ****p<0.0001.
Network modeling identifies intersection between genes that regulate inflammation and ectodomain cleavage
Since it is widely accepted that inflammation contributes significantly to cancer pathogenesis (reviewed in (58)), it is interesting to note that most genes contained in our PCSF-identified 15-gene TGFα-regulatory cluster are associated with the NF-κB pathway, suggesting a mechanistic link between the release of tumor growth factors such as TGFα and inflammation. Here we hypothesized that our gene cluster was relevant for understanding cancer pathology and that testing of an existing chemical inhibitor against the predicted gene, IRAK1, could lend mechanistic understanding of how this inhibitor could be applied clinically.
To explore the relevance of genes contained in our 15-gene “TGFα/NF-κB regulatory cluster” and growth factor release in cancer, we first examined their normalized expression in various cancers by analyzing cBioPortal data (59) and differential expression in human cancer cell lines from Expression Atlas (48). We focused on the upper subset of already validated “TGFα/NFκB regulatory cluster” genes, PPEF1, PPEF2, OBSCN, CALM1, XIAP, TAB1. We observed that the genes in this subset are expressed in multiple cancer types, including in gastric cancer, but none of them at significantly different levels when compared between cancers (Figure 6 A–E). However, some subsets of components were differentially expressed in multiple cancer types (log2 fold-change in Figure 6F, p-values in Supplemental Fig 10, and data accession numbers in Supplemental Table 2). IRAK1 is most overexpressed in glioblastoma multiforme, astrocytoma, hepatocellular carcinoma, and liver cancer from cell line data (Figure 6F).
Figure 6. Analysis of genes in the “TGFα/NF-κB regulatory cluster”.
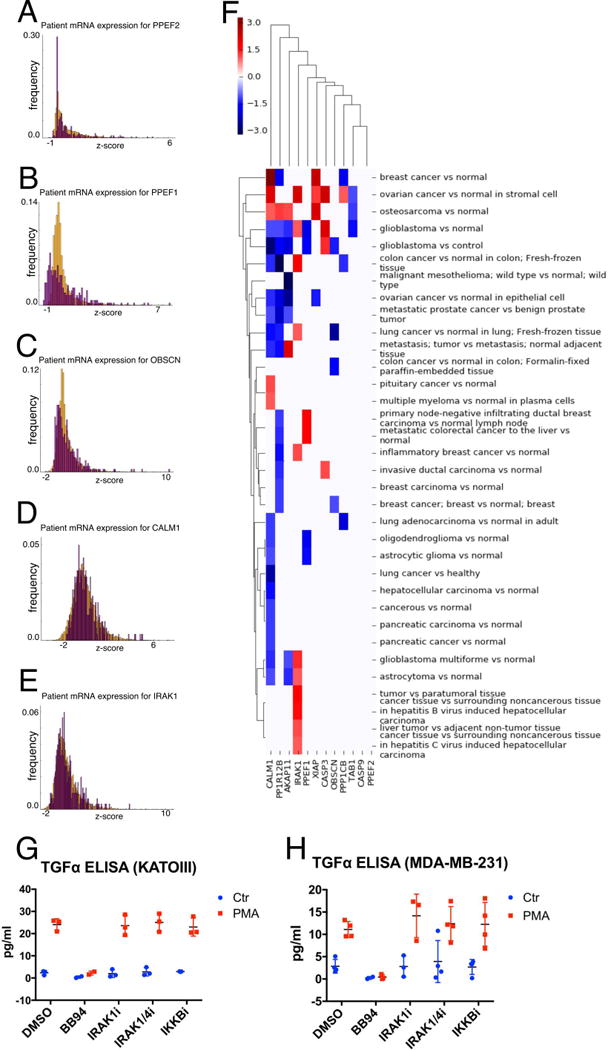
(A–E) mRNA expression of network genes in human cancer samples. Histograms represent distribution of mRNA z-scores for (A) PPEF2, (B) PPEF1, (C) OBSCN, (D) CALM1, and (E) IRAK1 in gastric cancer (orange; 302 samples) and ‘other’ cancers (purple; 10,550 samples) (also see methods). (F) Differential expression of genes of the TGFα/NF-κB regulatory cluster in multiple cancer types from Expression Atlas. Colorbar indicates log2 fold-change as reported from Expression Atlas. Kato-III (G) or MDA-MB-231 (H) cells were treated with vehicle (DMSO), the metalloprotease inhibitor BB94, IRAK4 inhibitor, IRAK1/4 inhibitor or IKKB inhibitor for 1 hour and exposed to control treatment or TPA (100nM) for 30 min. TGFα release was measured in cell supernatants by ELISA. Error bars are standard error of the mean.
Although we validated the effect of some of these NF-κB regulatory genes on TGFα cleavage (Figure 5 and Supplemental Fig 9), two inhibitors of IRAK1 had no effect on TGFα cleavage. Further, an inhibitor of IKKβ (upstream of NF-κB; controls the phosphorylation and subsequent degradation of one important direct regulator of NF-κB (IκB)) did not affect TGFα cleavage (Figure 6 G–H). This suggests that the NFκB regulatory function of this gene cluster is not required for their effect on TGFα cleavage. In addition, these results suggest that IRAK1 inhibitors do not modulate TGFα cleavage.
In summary, these results suggest that tumor associated inflammation may enhance tumor growth by also enhancing growth factor release due to overlap in their regulatory pathways and suggests unexpected novel applications for inhibition of NF-κB regulatory genes in combination targeted therapies in cancer.
Discussion
Here we report a putative TGFα shedding pathway through a combined computational and experimental approach and demonstrated the relevance of this pathway for targeted therapy design. We identified a cohort of genes organized in subnetworks that affect TGFα ectodomain release, vetted these selections computationally, and validated genes using targeted experiments in several cancer cell types. In the process, we identified an unexpected connection between regulators of the NfκB pathway and the release of cancer growth factors of the EGFR ligand family by ectodomain cleavage.
While the identified genes tune TGFα shedding and thus EGFR activation, many of them are not predicted oncogenes by traditional expression, mutation, or activation metrics. Our approach thus further highlights a trend toward selecting therapeutic targets based on functional role rather than over-expression of a particular gene in diseased over normal tissue. It is important to note that our network modeling approach yielded predicted genes not previously tested in our shRNA screen and that we could not have predicted these genes using traditional metrics. The directionality of their effect on TGFα regulation cannot be predicted, and discovering directionality requires dedicated experimental validation (see also discussion of the predicted gene XIAP below). Further, we made these predictions in a tissue, non-specific manner, but were able to validate these genes in multiple cell lines. The results show that we can discover alternative control mechanisms for growth factor pathway activation regardless of tissue type and that these targets require novel methods for discovery.
We explored the relevance of this pathway for cancer therapeutics and specifically investigated whether there was a link between the NF-κB pathway and TGFα shedding. The literature supports the role of these genes in regulating NF-κB signaling and there are several observations that suggest that more indirect connections and co-regulation of the EGFR and NF-κB pathways indeed exist. In head and neck squamous cell carcinoma, EGFR and TGFα genes are up-regulated concurrently with NF-κB signaling components. The NF-κB activators, IKKα and IKKβ enhance EGFR signaling, and activation of both IKKs can induce EGFR, TGFα, and Jun expression, a downstream effector of EGFR (60). Gastric H. pylori infection induces activation of EGFR and NFκB activity and both are thought to be associated with gastric cancer progression (61,62). NF-κB has been associated with resistance to EGFR therapies in cancer and inhibition of NFκB is able to sensitize cells to erlotinib treatment. Anti-EGFR therapies were indeed able to reduce NF-κB activation (reviewed in (10)). Further, it is well established that stimulation of EGFR can activate NF-κB through proteasome-mediated degradation of IkBα (63,64). Patient and cell line expression data validated our first hypothesis that these network components are expressed in cancer samples, albeit at varying levels in different cancers, and possibly relevant to pathology. Our experimental validation confirmed the network-based hypothesis that TAB1 and XIAP affect TGFα shedding. Our experiments with chemical inhibitors of IRAK1 or of IκB suggest that the NFκB regulatory function of these genes is not needed for their cleavage regulatory function, and thus, we were unable to confirm a mechanistic hypothesis about inhibitor mode of action. We sought to validate the role of IRAK1 in tuning TGFα shedding in gastric cancer because these cancers are growth factor-driven and exhibit EGFR pathway dependencies. However, from our cancer cell line analysis, it appears that our NF-κB-related cluster is differentially expressed in brain and liver cancers, which are not traditionally EGFR-pathway driven. Further analysis may explore the role of IRAK1 inhibitors in these cancers as opposed to gastric cancers.
Our network model predicts new mechanistic information about XIAP, a gene that is currently a focus for therapeutic development. XIAP-TAB1 binding is an upstream regulator of NF-κB signaling (65) and overexpression of XIAP is associated with loss of apoptotic signaling in cancer (66). XIAP binds and inhibits caspases 3 and 9, and also TAK1; binding TAK1 causes ubiquitination and degradation of TAK1 to prevent its activation of JNK (67). NF023, a new compound, inhibits XIAP-TAB1 binding, and thus could act as a pro-apoptotic therapeutic in cancer (65). Our work complements these efforts by predicting an additional functional role for XIAP: XIAP targeting can also affect EGFR signaling by modulating TGFα cleavage. This information could indicate when XIAP intervention might be clinically useful. Although XIAP1-TAB1 interaction is necessary for activation of NF-κB signaling, knockdown of XIAP1 and TAB1 proteins individually have opposite effects on TGFα cleavage. This suggests that their cleavage regulatory function is independent of their functions in NF-κB signaling. Experimental validation determined the directionality of predicted genes’ effect on TGFα shedding because our model did not describe this directionality.
Our focus here has been on signaling pathway nodes, as a category generally offering potential for therapeutic targeting. This focus differs from an earlier study in which the Kveiborg laboratory undertook a genome-wide screen for regulators of HB-EGF shedding (like TGFα also an EGFR ligand ADAM17 substrate) (68). Based on selection criteria requiring that at least three of four individual siRNAs inhibited HB-EGF shedding after TPA treatment, they validated 81 genes, including ADAM17 and PKCα. The signaling nodes obtained in their study corresponded well with the findings in our screen. Further stringent re-testing of these initial hits confirmed that 24 hits mimicked the effects of ADAM17 knockdown on HB-EGF cleavage, including the multifunctional sorting protein PACS2 as a top hit. PACS2 was shown to co-localize with ADAM17 on early endosomes and to regulate recycling and stability of internalized ADAM17, thereby sustaining ADAM17 cell-surface activity by diverting ADAM17 away from degradation. The fact that the main finding of this screen represents an ADAM17 trafficking factor might be related to the significantly longer time frame of induced shedding stimulation in the screen (30min vs 2-5min in our screen). Shedding can occur within seconds to minutes, while trafficking usually requires more time. This suggests that the genes we identified might act on regulatory components that do not require trafficking (or transcription). Similar to our screen results, hits were segregated into multiple functional categories but no enrichment in particular signaling pathways was found. This further emphasized the need for novel network modeling approaches as we performed and validated them in the present work. Future work will have to address whether the regulators we identified act on the protease, the substrate (TGFα) or other proteins that interact with ADAM, substrate or both (12), and by what mechanism they affect cleavage. In this context, particularly if they act on the protease, we need to explore the possibility that our regulators also affect the cleavage of other ADAM substrates beyond TGFα.
In summary, our work identified and validated numerous regulatory components of TGFα shedding that could be leveraged for multiple treatment and disease contexts. These genes may be relevant as novel therapeutic targets in contexts where EGFR signaling is hyperactive or where EGFR is a source of therapeutic resistance. While we focus primarily on cancer, this pathway model is relevant for other diseases that are strongly affected by EGFR signaling, such as organ fibrosis or inflammation (8). Our network modeling approach proves useful for identifying how signaling pathways intersect or overlap and can thus affect targeted therapies. Our pathway analysis lays the ground-work for identifying signaling intermediates that bridge multiple pathways to affect the release of growth factors in the tumor environment. While discovering how these pathways intersect remains a difficult challenge, our analysis demonstrates a path toward target selection and considering mechanistic implications of therapeutic interventions.
Supplementary Material
Implications.
Using genomic screens and network analysis, this study defines targets that regulate ectodomain shedding and suggests new treatment opportunities for EGFR-driven cancers.
Acknowledgments
The authors would like to thank Brian Joughin for helpful discussions in designing and troubleshooting the shEnrich method.
Financial Support
This work was partially supported by NIH grant U01-CA155758 (D.A.L., M.H.), NIH grant R01-CA096504 (D.A.L.), U01-CA184898 (E.F.), NIDDK R00DK077731 (A.H., E.K.), and R01 DK108947 (A.H., E.K.). Computing resources were partially supported by the National Science Foundation Grant DB1-0821391.
Footnotes
The authors declare no conflicts of interest.
Author Contributions
J.L.W., A.H., E.F., and D.A.L. designed the project. J.L.W., L.S., C.H., E.K, V.S. and A.H. acquired the data. J.L.W., L.S., and C.H. analyzed the data. J.L.W., D.A.L. and A.H. wrote the manuscript. A.H., E.F., and D.A.L. provided materials and reagents. A.H. and D.A.L. supervised the research.
References
- 1.Chen J, Zeng F, Forrester SJ, Eguchi S. Expression and function of the epidermal growth factor receptor in physiology and disease. Physiological …. 2016 doi: 10.1152/physrev.00030.2015. [DOI] [PubMed] [Google Scholar]
- 2.Borrell-Pagès M, Rojo F, Albanell J, Baselga J, Arribas J. TACE is required for the activation of the EGFR by TGF-alpha in tumors. The EMBO Journal. 2003;22(5):1114–24. doi: 10.1093/emboj/cdg111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horiuchi K, Le Gall S, Schulte M, Yamaguchi T, Reiss K, Murphy G, et al. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Molecular biology of the cell. 2007;18(1):176–88. doi: 10.1091/mbc.E06-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dang M, Armbruster N, Miller MA, Cermeno E, Hartmann M, Bell GW, et al. Regulated ADAM17-dependent EGF family ligand release by substrate-selecting signaling pathways. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(24):9776–81. doi: 10.1073/pnas.1307478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duffy MJ, Mullooly M, O’Donovan N, Sukor S, Crown J, Pierce A, et al. The ADAMs family of proteases: new biomarkers and therapeutic targets for cancer? Clinical proteomics. 2011;8(1):9. doi: 10.1186/1559-0275-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuchs BC, Hoshida Y, Fujii T, Wei L, Yamada S, Lauwers GY, et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology (Baltimore, Md) 2014;59(4):1577–90. doi: 10.1002/hep.26898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Chen J-K, Nagai K, Plieth D, Tan M, Lee T-C, et al. EGFR signaling promotes TGFβ-dependent renal fibrosis. Journal of the American Society of Nephrology. 2012;23(2):215–24. doi: 10.1681/ASN.2011070645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kefaloyianni E, Muthu ML, Kaeppler J, Sun X, Sabbisetti V, Chalaris A, et al. ADAM17 substrate release in proximal tubule drives kidney fibrosis. JCI insight. 2016;1(13) doi: 10.1172/jci.insight.87023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Y, Lee J-Y, Lee C-M, Cho W-K, Kang M-J, Koff JL, et al. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-β-induced pulmonary fibrosis. The Journal of biological chemistry. 2012;287(50):41991–2000. doi: 10.1074/jbc.M112.356824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shostak K, Chariot A. EGFR and NF-κB: partners in cancer. Trends in molecular medicine. 2015;21(6):385–93. doi: 10.1016/j.molmed.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 11.Lichtenberger BM, Gerber PA, Holcmann M, Buhren BA, Amberg N, Smolle V, et al. Epidermal EGFR controls cutaneous host defense and prevents inflammation. Science translational medicine. 2013;5(199):199ra11–ra11. doi: 10.1126/scitranslmed.3005886. [DOI] [PubMed] [Google Scholar]
- 12.Hartmann M, Herrlich A, Herrlich P. Who decides when to cleave an ectodomain? Trends in biochemical sciences. 2013;38(3):111–20. doi: 10.1016/j.tibs.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 13.Ferrini S, Rossello A, Nuti E, Fabbi M. Targeting ADAM17 Sheddase Activity In Cancer. Current Drug Targets. 2016 doi: 10.2174/1389450117666160727143618. [DOI] [PubMed] [Google Scholar]
- 14.Le Gall SM, Maretzky T, Issuree PDA, Niu X-D, Reiss K, Saftig P, et al. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. Journal of Cell Science. 2010;123(Pt 22):3913–22. doi: 10.1242/jcs.069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman S, Levy R, Garrett W, Doval D, Bondarde S, Sahoo T, et al. Clinical Benefit of INCB7839, a Potent and Selective Inhibitor of ADAM10 and ADAM17, in Combination with Trastuzumab in Metastatic HER2 Positive Breast Cancer Patients. Cancer research. 2010;69(24 Supplement):5056. doi: 10.1158/0008-5472.SABCS-09-5056. [DOI] [Google Scholar]
- 16.Zhu AX, Rosmorduc O, Evans TRJ, Ross PJ, Santoro A, Carrilho FJ, et al. SEARCH: a phase III, randomized, double-blind, placebo-controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(6):559–66. doi: 10.1200/JCO.2013.53.7746. [DOI] [PubMed] [Google Scholar]
- 17.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Science translational medicine. 2011;3(75):75ra26–75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hobor S, Van Emburgh BO, Crowley E, Misale S, Di Nicolantonio F, Bardelli A. TGFα and amphiregulin paracrine network promotes resistance to EGFR blockade in colorectal cancer cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(24):6429–38. doi: 10.1158/1078-0432.CCR-14-0774. [DOI] [PubMed] [Google Scholar]
- 19.Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S, et al. Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(24):6751–65. doi: 10.1158/1078-0432.CCR-13-0423. [DOI] [PubMed] [Google Scholar]
- 20.Minakata K, Takahashi F, Nara T, Hashimoto M, Tajima K, Murakami A, et al. Hypoxia induces gefitinib resistance in non-small-cell lung cancer with both mutant and wild-type epidermal growth factor receptors. Cancer Science. 2012;103(11):1946–54. doi: 10.1111/j.1349-7006.2012.02408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–9. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanizaki J, Okamoto I, Okabe T, Sakai K, Tanaka K, Hayashi H, et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(22):6219–26. doi: 10.1158/1078-0432.CCR-12-0392. [DOI] [PubMed] [Google Scholar]
- 23.Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer research. 2011;71(18):6051–60. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tani T, Yasuda H, Hamamoto J, Kuroda A, Arai D, Ishioka K, et al. Activation of EGFR Bypass Signaling by TGFα Overexpression Induces Acquired Resistance to Alectinib in ALK-Translocated Lung Cancer Cells. Molecular cancer therapeutics. 2016;15(1):162–71. doi: 10.1158/1535-7163.MCT-15-0084. [DOI] [PubMed] [Google Scholar]
- 25.Dang M, Dubbin K, D’Aiello A, Hartmann M, Lodish H, Herrlich A. Epidermal growth factor (EGF) ligand release by substrate-specific a disintegrin and metalloproteases (ADAMs) involves different protein kinase C (PKC) isoenzymes depending on the stimulus. The Journal of biological chemistry. 2011;286(20):17704–13. doi: 10.1074/jbc.M110.187823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willems SH, Tape CJ, Stanley PL, Taylor NA, Mills IG, Neal DE, et al. Thiol isomerases negatively regulate the cellular shedding activity of ADAM17. The Biochemical journal. 2010;428(3):439–50. doi: 10.1042/BJ20100179. [DOI] [PubMed] [Google Scholar]
- 27.Stawikowska R, Cudic M, Giulianotti M, Houghten RA, Fields GB, Minond D. Activity of a disintegrin and metal0loprotease 17 (ADAM17) is Regulated by its non-catalytic domains and secondary structure of its substrates. The Journal of biological chemistry. 2013 doi: 10.1074/jbc.M113.462267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu P, Liu J, Sakaki-Yumoto M, Derynck R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Science signaling. 2012;5(222):ra34. doi: 10.1126/scisignal.2002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chavaroche A, Cudic M, Giulianotti M, Houghten RA, Fields GB, Minond D. Glycosylation of a disintegrin and metalloprotease 17 affects its activity and inhibition. Analytical Biochemistry. 2013;449C:68–75. doi: 10.1016/j.ab.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu P, Derynck R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Molecular Cell. 2010;37(4):551–66. doi: 10.1016/j.molcel.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Díaz-Rodríguez E, Montero JC, Esparís-Ogando A, Yuste L, Pandiella A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor alpha-converting enzyme at threonine 735: a potential role in regulated shedding. Molecular biology of the cell. 2002;13(6):2031–44. doi: 10.1091/mbc.01-11-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maney SK, McIlwain DR, Polz R, Pandyra AA, Sundaram B, Wolff D, et al. Deletions in the cytoplasmic domain of iRhom1 and iRhom2 promote shedding of the TNF receptor by the protease ADAM17. Science signaling. 2015;8(401):ra109. doi: 10.1126/scisignal.aac5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maretzky T, McIlwain DR, Issuree PDA, Li X, Malapeira J, Amin S, et al. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(28):11433–8. doi: 10.1073/pnas.1302553110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakayama H, Fukuda S, Inoue H, Nishida-Fukuda H, Shirakata Y, Hashimoto K, et al. Cell surface annexins regulate ADAM-mediated ectodomain shedding of proamphiregulin. Molecular biology of the cell. 2012;23(10):1964–75. doi: 10.1091/mbc.E11-08-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsukamoto H, Tanida S, Ozeki K, Ebi M, Mizoshita T, Shimura T, et al. Annexin A2 Regulates A Disintegrin and Metalloproteinase 17-mediated Ectodomain Shedding of Pro-Tumor Necrosis Factor-α in Monocytes and Colon Epithelial Cells. Inflammatory bowel diseases. 2013;19(7):1365–73. doi: 10.1097/MIB.0b013e318281f43a. [DOI] [PubMed] [Google Scholar]
- 36.Düsterhöft S, Michalek M, Kordowski F, Oldefest M, Sommer A, Röseler J, et al. Extracellular Juxtamembrane Segment of ADAM17 Interacts with Membranes and Is Essential for Its Shedding Activity. Biochemistry. 2015;54(38):5791–801. doi: 10.1021/acs.biochem.5b00497. [DOI] [PubMed] [Google Scholar]
- 37.Düsterhöft S, Höbel K, Oldefest M, Lokau J, Waetzig GH, Chalaris A, et al. A disintegrin and metalloprotease 17 dynamic interaction sequence, the sweet tooth for the human interleukin 6 receptor. The Journal of biological chemistry. 2014;289(23):16336–48. doi: 10.1074/jbc.M114.557322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartmann M, Parra LM, Ruschel A, Böhme S, Li Y, Morrison H, et al. Tumor Suppressor NF2 Blocks Cellular Migration by Inhibiting Ectodomain Cleavage of CD44. Molecular cancer research : MCR. 2015;13(5):879–90. doi: 10.1158/1541-7786.MCR-15-0020-T. [DOI] [PubMed] [Google Scholar]
- 39.Parra LM, Hartmann M, Schubach S, Li Y, Herrlich P, Herrlich A. Distinct ICD substrate modifications selectively regulate ectodomain cleavage of NRG1 or CD44. Molecular and Cellular Biology. 2015 doi: 10.1128/MCB.00500-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parra LM, Hartmann M, Schubach S, Ma J, Herrlich P, Herrlich A. Growth factor and co-receptor release by structural regulation of substrate metalloprotease accessibility. Scientific reports. 2016;6:37464. doi: 10.1038/srep37464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Razick S, Mora A, Michalickova K, Boddie P, Donaldson IM. iRefScape. A Cytoscape plug-in for visualization and data mining of protein interaction data from iRefIndex. BMC bioinformatics. 2011;12(1):388. doi: 10.1186/1471-2105-12-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Villaveces JM, Jiménez RC, Porras P, Del-Toro N, Duesbury M, Dumousseau M, et al. Merging and scoring molecular interactions utilising existing community standards: tools, use-cases and a case study. Database : the journal of biological databases and curation. 2015;2015 doi: 10.1093/database/bau131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B. PhosphoSite: A bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics. 2004;4(6):1551–61. doi: 10.1002/pmic.200300772. [DOI] [PubMed] [Google Scholar]
- 44.Li X, Wilmanns M, Thornton J, Köhn M. Elucidating human phosphatase-substrate networks. Science signaling. 2013;6(275):rs10. doi: 10.1126/scisignal.2003203. [DOI] [PubMed] [Google Scholar]
- 45.Duan G, Li X, Köhn M. The human DEPhOsphorylation database DEPOD: a 2015 update. Nucleic acids research. 2015;43:D531–5. doi: 10.1093/nar/gku1009. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tuncbag N, Gosline SJC, Kedaigle A, Soltis AR, Gitter A, Fraenkel E. Network-Based Interpretation of Diverse High-Throughput Datasets through the Omics Integrator Software Package. PLoS computational biology. 2016;12(4):e1004879. doi: 10.1371/journal.pcbi.1004879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petryszak R, Keays M, Tang YA. Expression Atlas update—an integrated database of gene and protein expression in humans, animals and plants. Nucleic acids …. 2015 doi: 10.1093/nar/gkv1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sabbisetti VS, Ito K, Wang C, Yang L, Mefferd SC, Bonventre JV. Novel assays for detection of urinary KIM-1 in mouse models of kidney injury. Toxicological sciences : an official journal of the Society of Toxicology. 2013;131(1):13–25. doi: 10.1093/toxsci/kfs268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, et al. Highly parallel identification of essential genes in cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(51):20380–5. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rameseder J, Krismer K, Dayma Y, Ehrenberger T, Hwang MK, Airoldi EM, et al. A Multivariate Computational Method to Analyze High-Content RNAi Screening Data. Journal of biomolecular screening. 2015;20(8):985–97. doi: 10.1177/1087057115583037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kampmann M, Bassik MC, Weissman JS. Integrated platform for genome-wide screening and construction of high-density genetic interaction maps in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(25):E2317–26. doi: 10.1073/pnas.1307002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilson JL, Hemann MT, Fraenkel E, Lauffenburger DA. Integrated network analyses for functional genomic studies in cancer. Seminars in cancer biology. 2013;23(4):213–8. doi: 10.1016/j.semcancer.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tuncbag N, Braunstein A, Pagnani A, Huang S-SC, Chayes J, Borgs C, et al. Simultaneous reconstruction of multiple signaling pathways via the prize-collecting steiner forest problem. Journal of computational biology : a journal of computational molecular cell biology. 2013;20(2):124–36. doi: 10.1089/cmb.2012.0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Linding R, Jensen LJ, Pasculescu A, Olhovsky M, Colwill K, Bork P, et al. NetworKIN: a resource for exploring cellular phosphorylation networks. Nucleic acids research. 2008;36:D695–9. doi: 10.1093/nar/gkm902. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Su G, Kuchinsky A, Morris JH, States DJ, Meng F. GLay: community structure analysis of biological networks. Bioinformatics (Oxford, England) 2010;26(24):3135–7. doi: 10.1093/bioinformatics/btq596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Todoric J, Antonucci L, Karin M. Targeting Inflammation in Cancer Prevention and Therapy. Cancer prevention research (Philadelphia, Pa) 2016;9(12):895–905. doi: 10.1158/1940-6207.CAPR-16-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6(269):pl1–pl. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nottingham LK, Yan CH, Yang X, Si H, Coupar J, Bian Y, et al. Aberrant IKKα and IKKβ cooperatively activate NF-κB and induce EGFR/AP1 signaling to promote survival and migration of head and neck cancer. Oncogene. 2014;33(9):1135–47. doi: 10.1038/onc.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Mestier L, Lardière-Deguelte S, Volet J, Kianmanesh R, Bouché O. Recent insights in the therapeutic management of patients with gastric cancer. Digestive and liver disease : official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver. 2016;48(9):984–94. doi: 10.1016/j.dld.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 62.Wroblewski LE, Peek RM. Helicobacter pylori in gastric carcinogenesis: mechanisms. Gastroenterology clinics of North America. 2013;42(2):285–98. doi: 10.1016/j.gtc.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allen CT, Conley B, Sunwoo JB, Van Waes C. CCR 20th anniversary commentary: Preclinical study of proteasome inhibitor bortezomib in head and neck cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(5):942–3. doi: 10.1158/1078-0432.CCR-14-2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Allen C, Saigal K, Nottingham L, Arun P, Chen Z, Van Waes C. Bortezomib-induced apoptosis with limited clinical response is accompanied by inhibition of canonical but not alternative nuclear factor-{kappa}B subunits in head and neck cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(13):4175–85. doi: 10.1158/1078-0432.CCR-07-4470. [DOI] [PubMed] [Google Scholar]
- 65.Cossu F, Milani M, Grassi S, Malvezzi F, Corti A, Bolognesi M, et al. NF023 binding to XIAP-BIR1: searching drugs for regulation of the NF-κB pathway. Proteins. 2015;83(4):612–20. doi: 10.1002/prot.24766. [DOI] [PubMed] [Google Scholar]
- 66.Obexer P, Ausserlechner MJ. X-linked inhibitor of apoptosis protein - a critical death resistance regulator and therapeutic target for personalized cancer therapy. Frontiers in oncology. 2014;4:197. doi: 10.3389/fonc.2014.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaur S, Wang F, Venkatraman M, Arsura M. X-linked inhibitor of apoptosis (XIAP) inhibits c-Jun N-terminal kinase 1 (JNK1) activation by transforming growth factor beta1 (TGF-beta1) through ubiquitin-mediated proteosomal degradation of the TGF-beta1-activated kinase 1 (TAK1) The Journal of biological chemistry. 2005;280(46):38599–608. doi: 10.1074/jbc.M505671200. [DOI] [PubMed] [Google Scholar]
- 68.Dombernowsky SL, Samsøe-Petersen J, Petersen CH, Instrell R, Hedegaard A-MB, Thomas L, et al. The sorting protein PACS-2 promotes ErbB signalling by regulating recycling of the metalloproteinase ADAM17. Nature communications. 2015;6:7518. doi: 10.1038/ncomms8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.