

Abstract
Patient: Male, 36
Final Diagnosis: Levofloxacin-induced hepatotoxicity
Symptoms: Cellulitis • pain
Medication: Levofloxacin
Clinical Procedure: —
Specialty: Infectious Diseases
Objective:
Unusual clinical course
Background:
Levofloxacin covers a broad spectrum of pathogens and is readily prescribed by clinicians. Hepatotoxicity is a known but unusual complication of levofloxacin use. Here, we present a case of severe transaminitis caused by levofloxacin.
Case Report:
A young man in his thirties with a history of asthma, chronic alcoholism, methamphetamine intravenous drug abuse (IVDA), and non-compliant insulin-dependent diabetes mellitus (IDDM) presented to an emergency department with suicidal ideation. Vital signs were stable and the patient was noted to have cellulitis of the right forearm, for which cultures were drawn, and he received IV clindamycin. He was admitted to behavioral medicine for further care. Blood cultures were positive for gram-negative rods and he was transferred to the medicine ward. Cultures eventually grew Brevundimonas diminuta. Clindamycin was discontinued and he was started on levofloxacin. Transaminase levels measured soon after levofloxacin administration showed aminotransferase levels raised to approximately 50 times baseline within a few days. Levofloxacin was discontinued due to concern about drug-induced hepatotoxicity. After discontinuation, transaminase levels decreased immediately. Work-up for other causes of transaminitis revealed no other etiology.
Conclusions:
Clinicians should remain mindful that levofloxacin can induce hepatotoxicity in rare cases. In patients presenting with acute liver injury who have recently taken levofloxacin, it would be wise to remain cognizant of the possibility of levofloxacin-induced hepatotoxicity.
MeSH Keywords: Drug-Induced Liver Injury; Fluoroquinolones; Liver Failure, Acute
Background
Levofloxacin covers a broad spectrum of pathogens and is readily prescribed by clinicians. Hepatotoxicity is a known but unusual complication of levofloxacin use. Clinical trial data of over 7000 subjects suggests a remarkably low frequency (0.3%) of associated liver enzyme elevation, and data collected since public debut estimates the rate of hepatotoxicity to be less than 1 per 1 million prescriptions [1,2]. Some data has even estimated this rate to be less than 1 per 5 million prescriptions [3]. In comparison, it has been estimated that upwards of 40 000 Americans experience drug-induced liver injury (DILI) each year, and of those cases, it is posited that nearly 50% are due to antimicrobial therapies [4]. Here, we present a rare case of severe transaminitis in a patient taking levofloxacin to treat gram-negative bacillary bacteremia.
Case Report
A young man in his thirties with a history of asthma, meth-amphetamine intravenous drug abuse (IVDA), chronic alcohol abuse, and non-compliant insulin-dependent diabetes mellitus (IDDM) presented to an emergency department with suicidal ideation. Vital signs were stable and upon symptomatic review, the patient reported having body aches, chills, sinus congestion, and rhinorrhea. The patient was noted to have cellulitis of the right forearm, for which cultures were drawn, and he received IV clindamycin. Urine toxicology was negative for any recent illicit drug use, and the acetaminophen level was also normal. Lab test results showed hyperglycemia with borderline diabetic ketoacidosis (DKA). The patient was given IV fluids and IV insulin for treatment of DKA, and blood glucose improved to 180 mg/dL. He was then admitted under the psychiatry service to the behavioral medicine unit (BMU) for medical treatment. Antibiotic therapy was switched from intravenous to oral clindamycin and he was followed by the medicine team while in the BMU. Blood cultures were positive for gram-negative rods. Oral levofloxacin was added to the antibiotic regimen on day 2 of admission, pending type and sensitivity of the culture results. The patient was transferred to the medicine floor for further evaluation and to rule out endocarditis in light of the IVDA. Cultures eventually grew Brevundimonas diminuta. The clinical significance of this result was uncertain due to the possibility of contamination. Ultrasound showed thrombophlebitis of the basilic vein and a 2D echocardiogram showed mild thickening of the mitral valve anterior leaflet, with no obvious valvular vegetations. This was discussed with the cardiology team and the findings were not suggestive of endocarditis, so no TEE (transesophageal echocardiogram) was recommended. After evaluation by the infectious disease team, clindamycin was discontinued on day 4 and the patient remained on oral levofloxacin.
Liver enzyme levels measured soon after levofloxacin administration showed significant transaminitis (AST 665 IU/L, ALT 364 IU/L, and ALP 207 IU/L). Levofloxacin was discontinued on day 5 due to concern about drug-induced hepatotoxicity. Transaminases increased to approximately 50 times that of baseline and peaked at AST 1626 IU/L, ALT 1100 IU/L, and ALP 269 IU/L on the next day (Table 1). Extensive testing was performed, including transglutaminase, ceruloplasmin, alpha fetoprotein, antimitochondrial M2 antibody, ANA, alpha-1 anti-trypsin, and a hepatitis panel, and results were unremarkable. An ultrasound of the abdomen showed a partially contracted gallbladder and the common bile duct was 7 mm, near the upper limit of normal. Fatty infiltration of the liver was noted, compatible with a history of alcohol use. Repeat blood cultures 5 days after admission showed no growth, and thrombophlebitis had improved.
Table 1.
Hepatic function panel results before and after levofloxacin administration.
| Test | Unit | Reference | Days after admission | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| Albumin | mg/dL | 3.5–5.7 | 3.9 | – | – | 3.3 | 3.3 | 3.8 | 3.7 | 3.4 | 3.7 |
| Protein, Total | mg/dL | 6.0–8.9 | 6.7 | – | – | 5.4 | 5.7 | 6.4 | 6.4 | 5.7 | 6.0 |
| Alkaline Phos. | IU/L | 34–104 | 175 | – | – | 129 | 167 | 207 | 269 | 233 | 232 |
| ALT | IU/L | 7–52 | 24 | – | – | 76 | 171 | 364 | 1100 | 773 | 619 |
| AST | IU/L | 13–39 | 59 | – | – | 188 | 314 | 665 | 1626 | 632 | 305 |
| Bilirubin, Total | mg/dL | 0.3–1.0 | 0.5 | – | – | 0.3 | 0.3 | 0.4 | 0.6 | 0.7 | 0.6 |
| Bilirubin, Direct | mg/dL | 0.0–0.2 | – | – | – | 0.1 | 0.1 | 0.1 | 0.2 | 0.2 | 0.2 |
| Bilirubin, Indirect | mg/dL | 0.0–0.8 | – | – | – | 0.2 | 0.2 | 0.3 | 0.4 | 0.5 | 0.4 |
| Antibiotics administered | |||||||||||
| Levofloxacin Administered days 2–5 | |||||||||||
| Clindamycin Administered days 0–4 | |||||||||||
ALT – alanine transaminase; AST – aspartate transaminase (AST).
After levofloxacin was discontinued, transaminase levels decreased to AST 305 IU/L, ALT 619 IU/L, and ALP 232 IU/L by day 8. The patient was discharged and arrangements were made to stay at an outpatient facility for detoxification and to ensure transaminases returned to baseline levels (Figure 1).
Figure 1.
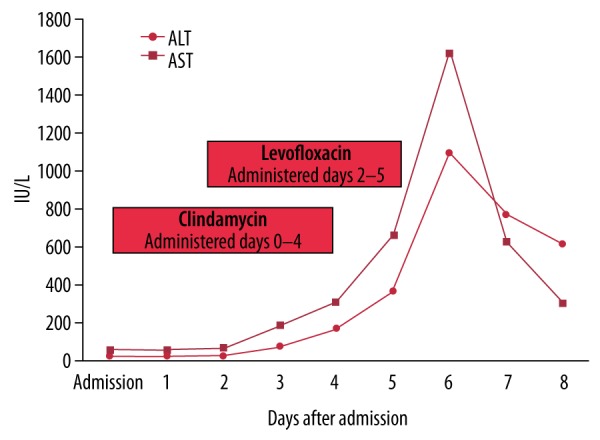
Transaminase levels before and after levofloxacin administration from day 2 to 5.
Discussion
Fluoroquinolones such as levofloxacin act principally through the inhibition of bacterial DNA gyrase, resulting in DNA strand breakage and consequent genomic damage. Although they do not target mammalian isoforms of nucleic DNA gyrase, fluoroquinolone antibiotics have been shown to damage and deplete mitochondrial DNA (mtDNA) in mammalian cells [5]. In one study, ciprofloxacin was found to cause a loss of mtDNA, resulting in decreased ATP production and a subsequent arrest in cell division and growth [6]. Another study of ciprofloxacin found site-specific mtDNA strand breaks, suggesting damage was due to the inhibition of DNA gyrase [7]. Fluoroquinolone antibiotics at clinically relevant levels have also been shown to cause mitochondrial dysfunction through the production of reactive oxygen species [8,9]. Although the precise mechanism of levofloxacin-induced hepatotoxicity remains unknown, pre-existing hepatic mitochondrial damage in our patient from chronic alcohol abuse, methamphetamine IVDA, and non-compliant IDDM may have decreased the ability of hepatocytes to utilize ATP and initiate cellular repair. Mitochondrial damage as a causative agent in extensive hepatic injury has also been described in hepatitis and cirrhosis [10,11]. Processes such as these predispose hepatocytes to acute toxicity. The addition of levofloxacin may have further damaged the remaining mtDNA and prevented hepatocellular repair mechanisms from offsetting cellular injury, ultimately leading to rapid and acute liver toxicity. To elucidate the true pathogenesis of levofloxacin-induced hepatotoxicity, further investigation is required.
Fluoroquinolones have been previously linked to hepatotoxicity. A retrospective case-control study of 7862 patients without a history of prior liver disease found a significant association between treatment with fluoroquinolones and an increased risk of hepatotoxicity, most notably with ciprofloxacin and moxifloxacin usage [12]. A retrospective population-based study of 746 subjects also identified an increased risk of acquiring hepatotoxicity in older patients who had recently been prescribed either levofloxacin or moxifloxacin [13]. A review of 12 patients who experienced hepatotoxicity related to fluoroquinolones found similar features in cases presenting after ciprofloxacin, moxifloxacin, levofloxacin, and gatifloxacin administration, including a short latency period (2–9 days) and abrupt onset of injury with rapid recovery following discontinuation [14]. These predominant features mirror the case presented here. In a case report of hepatotoxicity in a hemodialysis patient, after resolution of liver enzyme elevation following levofloxacin discontinuation, successive treatment with ciprofloxacin prompted a recurrent episode of transaminitis [15]. Similar presentations across a spectrum of individual cases indicates a probable class effect in certain patients who have fluoroquinolone-induced hepatotoxicity. Therefore, the administration of a divergent fluoroquinolone antibiotic following transaminitis attributed to a single-class agent should generally be avoided. While data on levofloxacin gathered from clinical trials and post-marketing surveillance have demonstrated a largely consistent hepatic risk profile, fulminant liver injury with death shortly thereafter has occurred in several reported cases of levofloxacin-induced hepatotoxicity [16–18]. Hepatotoxicity precipitated by fluoroquinolone antibiotics is atypical, but characteristic liver injury in identifiable cases is paroxysmal and severe.
Although normal liver enzyme levels vary among laboratories, generally acceptable ranges for both AST and ALT fall within 10–50 IU/L, while ALP ranges are normally set within 20–140 IU/L. Our laboratory reference ranges are AST 13–39 U/L, ALT 7–52 IU/L, and ALP 34–104 IU/L. In the present case, AST and ALT levels increased to nearly 50 times the baseline level within 72 h, with a noticeably higher increase in AST, a scenario consistent with drug-induced toxicity. Toxic injury, ischemic injury, and acute viral hepatitis are most commonly associated with aminotransferase deviations greater than 10 times the upper limit of normal (ULN). Aminotransferase levels greater than 75×ULN indicate ischemic or toxic injury in more than 90% of cases [19]. In our case, hepatitis panel results were negative. Although this patient did not exhibit aminotransferase levels over 75×ULN, the toxic agent was removed early enough to prevent a transient increase of this magnitude. Hy’s law is a simple algorithm used by the US Food and Drug Administration (FDA) to assess hepatic safety in clinical drug trials, and has 3 main components: (1) AST or ALT elevation >3×ULN; (2) total bilirubin (TBL) elevation >2×ULN; and (3) absence of initial findings of cholestasis (i.e., ALP elevation <2×ULN) [20]. FDA guidance states that any 2 cases meeting these parameters during a clinical trial is “highly predictive that the drug has the potential to cause severe drug-induced liver injury when given to a patient population” [20]. In 3 separate studies encompassing over 1500 patients, approximately 10% of patients who met these 3 criteria experienced liver failure culminating in either transplantation or mortality [21–23]. In our case, transaminase elevation was far above 3×ULN, TBL elevation was 0.7×ULN, and ALP elevation was 2.6×ULN. Although TBL elevation did not meet Hy’s law criteria in our case, the patient presented with a TBL at the lower end of the reference range, which increased to double the baseline measurements soon after levofloxacin administration and was trending upward. Had levofloxacin therapy not been terminated so quickly, additional toxicity may have quickly increased TBL levels to >2×ULN. Furthermore, the patient in this case did not progress to liver failure. Hy’s law criteria also requires ALP elevation to be <2×ULN in order to exclude cholestasis as an etiology. In our case, ALP elevation was 2.6 × ULN. The patient presented with a high baseline ALP, which never fell below the ULN throughout his stay. In comparison to peak serum ALP, the lowest measured ALP increased by a factor of only 2.08. Additionally, cholestatic injury usually presents as a disproportionate elevation in ALP in comparison to serum aminotransferases [24]. Data from a study of over 750 patients with drug-induced liver injury suggest that the risk of developing acute liver failure in subjects with ALP elevation >2×ULN is equal to that of subjects who meet Hy’s law criteria [25]. If true, further studies could make the third criterion of Hy’s law subsidiary in the determination of the capacity of an agent to produce severe liver injury.
Drug-induced liver injury is a diagnosis of exclusion. There are 3 fundamental elements used to make the diagnosis: (1) drug exposure preceding the onset of liver injury; (2) exclusion of underlying liver disease as a cause; and (3) discontinuation of the drug, leading to improvement in the liver injury [26]. Clindamycin discontinuation and levofloxacin administration took place prior to the observed liver enzyme elevations. Despite a patient history of IV drug abuse, urine toxicology on admission was negative for any illicit drugs, substantially lowering the probability that methamphetamine abuse produced the slightly elevated liver enzyme levels seen on admission, and later prompted further progression to acute liver injury. DKA resolved approximately 36 h prior to levofloxacin administration, and blood glucose levels remained stable until discharge. Moreover, transaminitis did not begin to evolve until 2 days after admission. These observations indicate that DKA was not responsible for the considerable elevation in serum transaminase levels. It is far more likely that pre-existing liver damage constrained hepatic function to a threshold at which additional noxious insult from levofloxacin administration resulted in abrupt hepatocellular failure. Once levofloxacin was discontinued, transaminase levels began to rapidly decrease, supporting levofloxacin as the cause of hepatotoxicity.
Conclusions
Clinicians should remain mindful that levofloxacin can induce hepatotoxicity in rare cases. Patients with pre-existing liver damage may be especially susceptible, and levofloxacin may not be the best antibiotic to prescribe in questionable cases. Although rare, current recommendations for levofloxacin mandate therapy be discontinued immediately if a patient develops signs and symptoms of hepatitis. Likewise, if transaminase levels begin to increase rapidly in a patient receiving levofloxacin for antimicrobial therapy, we recommend prompt removal because it is most likely the offending agent. In patients presenting with acute liver injury who have recently taken levofloxacin, it would be wise for clinicians to remain cognizant to the possibility of levofloxacin-induced hepatotoxicity.
Acknowledgments
We would like to thank the IRB committee at Southeast Alabama Medical Center for careful review of this article, as well as Daniel Fuchs for his encouragement and assistance.
References:
- 1.Yagawa K. Latest industry information on the safety profile of levofloxacin in Japan. Chemotherapy. 2001;47:38–43. doi: 10.1159/000057843. [DOI] [PubMed] [Google Scholar]
- 2.Lipsky BA, Baker CA. Fluoroquinolone toxicity profiles: A review focusing on newer agents. Clin Infect Dis. 1999;28:352–64. doi: 10.1086/515104. [DOI] [PubMed] [Google Scholar]
- 3.Carbon C. Levofloxacin adverse effects, data from clinical trials and pharmacovigilance. Therapie. 2001;56:35–40. [PubMed] [Google Scholar]
- 4.Bell LN, Chalasani N. Epidemiology of idiosyncratic drug-induced liver injury. Semin Liver Dis. 2009;29:337–47. doi: 10.1055/s-0029-1240002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sobek S, Boege F. DNA topoisomerases in mtDNA maintenance and ageing. Exp Gerontol. 2014;56:135–41. doi: 10.1016/j.exger.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Lawrence JW, Claire DC, Weissig V, Rowe TC. Delayed cytotoxicity and cleavage of mitochondrial DNA in ciprofloxacin-treated mammalian cells. Mol Pharmacol. 1996;50:1178–88. [PubMed] [Google Scholar]
- 7.Lawrence JW, Darkin-Rattray S, Xie F, et al. 4-Quinolones cause a selective loss of mitochondrial DNA from mouse L1210 leukemia cells. J Cell Biochem. 1993;51:165–74. doi: 10.1002/jcb.240510208. [DOI] [PubMed] [Google Scholar]
- 8.Kalghatgi S, Spina CS, Costello JC. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in mammalian cells. Sci Transl Med. 2013;5 doi: 10.1126/scitranslmed.3006055. 192ra85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowes DA, Wallace C, Murphy MP, et al. The mitochondria targeted anti-oxidant MitoQ protects against fluoroquinolone-induced oxidative stress and mitochondrial membrane damage in human Achilles tendon cells. Free Radic Res. 2009;43:323–28. doi: 10.1080/10715760902736275. [DOI] [PubMed] [Google Scholar]
- 10.Nishikawa T, Bellance N, Damm A, et al. A switch in the source of ATP production and a loss in capacity to perform glycolysis are hallmarks of hepatocyte failure in advance liver disease. J Hepatol. 2014;60:1203–11. doi: 10.1016/j.jhep.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang T, Weinman S. Interactions between hepatitis C virus and mitochondria: Impact on pathogenesis and innate immunity. Curr Pathobiol Rep. 2013;1:179–87. doi: 10.1007/s40139-013-0024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alshammari TM, Larrat EP, Morrill HJ, et al. Risk of hepatotoxicity associated with fluoroquinolones: A national case-control safety study. Am J Health Syst Pharm. 2014;71:37–43. doi: 10.2146/ajhp130165. [DOI] [PubMed] [Google Scholar]
- 13.Paterson JM, Mamdani MM, Manno M, Juurlink DN, Canadian Drug Safety and Effectiveness Research Network Fluoroquinolone therapy and idiosyncratic acute liver injury: A population-based study. CMAJ. 2012;184(14):1565–70. doi: 10.1503/cmaj.111823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orman ES, Conjeevaram HS, Vuppalanchi R, et al. Clinical and histopathologic features of fluoroquinolone-induced liver injury. Clin Gastroenterol Hepatol. 2011;9:517–23. doi: 10.1016/j.cgh.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Airey K, Koller E. Acute hepatitis associated with levofloxacin in a patient with renal insufficiency. CMAJ. 2003;169:755. [PMC free article] [PubMed] [Google Scholar]
- 16.Gulen M, Ay MO, Avci A, et al. Levofloxacin-induced hepatotoxicity and death. Am J Ther. 2015;22(3):e93–96. doi: 10.1097/MJT.0b013e3182a44055. [DOI] [PubMed] [Google Scholar]
- 17.Karim A, Ahmed S, Rosshoff LJ, et al. Possible levofloxacin-induced hepatocellular injury in a patient with chronic obstructive lung disease. Clin Infect Dis. 2001;33:2088–90. doi: 10.1086/338156. [DOI] [PubMed] [Google Scholar]
- 18.Schwalm J-D, Lee CH. Acute hepatitis associated with oral levofloxacin therapy in a hemodialysis patient. CMAJ. 2003;168(7):847–48. [PMC free article] [PubMed] [Google Scholar]
- 19.Giannini EG, Testa R, Savarino V. Liver enzyme alteration: A guide for clinicians. CMAJ. 2005;172:367–79. doi: 10.1503/cmaj.1040752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Regev A, Björnsson ES. Drug-induced liver injury: morbidity, mortality, and Hy’s law. Gastroenterology. 2014;147:20–24. doi: 10.1053/j.gastro.2014.05.027. [DOI] [PubMed] [Google Scholar]
- 21.Andrade RJ, Lucena MI, Fernandez MC, et al. Drug-induced liver injury: An analysis of 461 incidences submitted to the Spanish registry over a 10-year period. Gastroenterology. 2005;129:512–21. doi: 10.1016/j.gastro.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 22.Chalasani N, Fontana RJ, Bonkovsky HL, et al. Causes, clinical features, and outcomes from a Prospective Study of drug-induced liver injury in the United States. Gastroenterology. 2008;135:1924–34. doi: 10.1053/j.gastro.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Björnsson E, Olsson R. Outcome and prognostic markers in severe drug-induced liver disease. Hepatology. 2005;42:481–89. doi: 10.1002/hep.20800. [DOI] [PubMed] [Google Scholar]
- 24.Batt AM, Ferrari L. Manifestations of chemically induced liver damage. Clin Chem. 1995;4:1882–87. [PubMed] [Google Scholar]
- 25.Robles-Diaz M, Lucena MI, Kaplowitz N, et al. Use of Hy’s law and a new composite algorithm to predict acute liver failure in patients with drug-induced liver injury. Gastroenterology. 2014;147:109–18. doi: 10.1053/j.gastro.2014.03.050. [DOI] [PubMed] [Google Scholar]
- 26.Navarro VJ, Senior JR. Drug-related hepatotoxicity. N Engl J Med. 2006;353:731–39. doi: 10.1056/NEJMra052270. [DOI] [PubMed] [Google Scholar]