

Abstract
Protein homeostasis (or proteostasis) within the endoplasmic reticulum (ER) is regulated by the unfolded protein response (UPR). The UPR consists of three integrated signaling pathways activated by the accumulation of misfolded proteins within the ER lumen. Activation of the UPR alters ER proteostasis through translational attenuation of new protein synthesis and transcriptional remodeling of ER proteostasis pathways, providing a mechanism to adapt ER proteostasis in response to cellular stress. The capacity of the UPR to alter ER proteostasis suggests that exogenous manipulation of UPR signaling pathways offers therapeutic promise to alter the fate of pathologic proteins associated with human protein misfolding diseases. Here, we discuss the therapeutic potential of exogenous UPR activation to treat human disease and highlight specific small molecule approaches for regulating UPR signaling that could be beneficial to treat protein misfolding diseases.
Endoplasmic reticulum proteostasis and protein misfolding diseases
Nearly one-third of the human proteome is targeted to the endoplasmic reticulum (ER) for folding and trafficking to downstream environments of the secretory pathway, such as the plasma membrane and the extracellular space. These proteins are involved in numerous essential biological processes including cell–cell communication, inflammatory signaling, and immunological signaling. Thus, the maintenance of ER protein homeostasis (or proteostasis) is a critical factor in organismal physiology.
ER proteostasis is maintained by the partitioning of polypeptide clients between biological pathways involved in ER-assisted folding (ERAF) and ER-associated degradation (ERAD) [1–3]. Newly synthesized proteins, cotranslationally translocated into the ER lumen as unfolded polypeptides, interact with an ER-localized network of chaperones and folding enzymes (ERAF pathways) that facilitate the proper folding of proteins into their native three-dimensional structures. Once folded, proteins are incorporated into COPII vesicles for trafficking to downstream environments of the secretory pathway. Proteins unable to obtain a folded conformation in the ER are recognized by components of the ERAD pathway and retro-translocated to the cytosol where they are degraded by the cytosolic proteasome. Thus, the partitioning of proteins between ERAF and ERAD is a critical determinant in defining the integrity of proteostasis in the ER and throughout the secretory pathway.
Imbalances in the partitioning of destabilized mutant proteins between ERAD and ERAF can result in pathology associated with numerous human protein misfolding diseases [4,5]. Loss-of-function protein misfolding diseases, such as cystic fibrosis and the lysosomal storage diseases, can be attributed to the excessive degradation of destabilized proteins, preventing their proper folding and trafficking (Figure 1a). Thus, pathology results directly from the low activity of these proteins in their downstream functional environments. Alternatively, gain-of-toxicity protein misfolding diseases, including the amyloidoses, often result from the efficient folding and trafficking of destabilized, aggregation-prone protein variants (Figure 1b). This efficient trafficking leads to high extracellular concentrations of misfolding-prone proteins, facilitating extracellular formation of large insoluble aggregates and amyloid fibrils that appear to cause post-mitotic tissue loss in these disorders.
Figure 1.
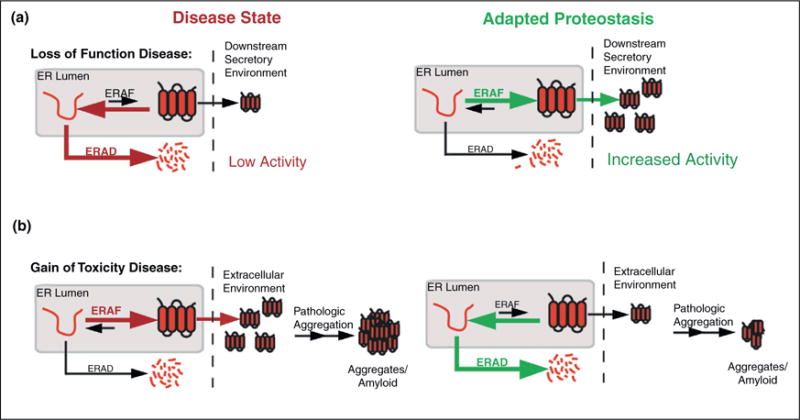
Adapting the proteostasis capacity of ERAF and ERAD pathways can attenuate the aberrant ER protein folding, trafficking or degradation processes involved in human protein misfolding disease pathology. (a) Left, illustration showing the excessive ERAD for destabilized, mutant proteins involved in loss-of-function protein misfolding diseases. Premature ERAD reduces trafficking of these mutants to their downstream functional environments, resulting in pathology that stems from low protein activities in their native environments. Right, illustration showing that increasing ERAF activity could attenuate the premature degradation of destabilized mutant proteins and increase trafficking to their downstream functional environment, allowing for increased protein activity. (b) Left, illustration showing the efficient folding and trafficking of destabilized, mutant proteins involved in gain-of-toxicity protein misfolding diseases. Efficient trafficking leads to high extracellular concentrations that facilitate pathologic concentration-dependent aggregation. Right, illustration showing that increasing ERAD activity could attenuate the secretion of these destabilized mutant proteins, reducing extracellular concentrations and decreasing concentration-dependent aggregation.
These imbalances have prompted many researchers to speculate about whether exogenous manipulation of ER proteostasis would offer a potential therapeutic strategy to prevent the aberrant degradation or ER protein folding/trafficking intricately involved in these disorders [4,5]. For example, increasing the pro-folding ERAF pathway/activity(ies) offers a potential strategy to prevent the aberrant ERAD of disease-associated mutants involved in loss-of-function protein misfolding diseases (Figure 1a). Alternatively, increasing ERAD activity could potentially reduce the secretion of destabilized, aggregation-prone proteins linked to degenerative disorders (Figure 1b). While many strategies to target specific aspects of ER proteostasis have been proposed, here we focus on evaluating the potential for activating the unfolded protein response (UPR), the endogenous stress-responsive signaling pathway responsible for regulating ER proteostasis, to attenuate the aberrant ER protein folding, trafficking or degradation associated with protein misfolding pathology.
UPR activation and ER proteostasis
The UPR comprises three integrated signaling pathways that emanate from the ER-localized transmembrane proteins: inositol-requiring protein-1 (IRE1), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (Figure 2) [6–9]. These three proteins act as sensors of the ER proteostasis environment and are activated by the accumulation of misfolded proteins within the ER lumen. Misfolding can be induced by environmental, genetic, developmental or aging-related stress. Activation of the UPR adapts ER proteostasis through translational attenuation and transcriptional remodeling of the ER proteostasis network. While many good reviews have highlighted the mechanisms of UPR activation and the involvement of UPR activation in the context of human disease (e.g. [6–9]), here we focus on highlighting the functional implications for UPR activation to adapt ER proteostasis in the context of human protein misfolding diseases.
Figure 2.
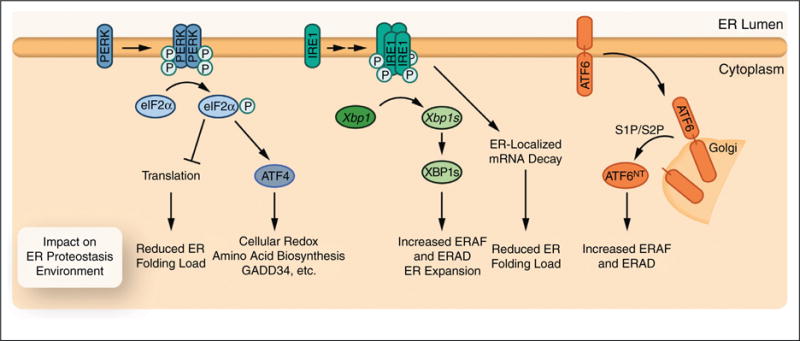
Activation of the three UPR signaling arms differentially influences ER proteostasis capacity. Illustration of downstream consequences of PERK, IRE1, or ATF6 activation on ER proteostasis. PERK activation leads to a reduced protein folding load mediated through translational attenuation and an increase in global proteostasis capacity mediated by the ATF4-dependent induction of genes involved in general cellular proteostasis maintenance including cellular redox regulation, amino acid biosynthesis, and negative feedback regulation of translational attenuation (i.e. GADD34). IRE1 activation increases ERAD/ERAF activity and induces ER expansion through the downstream activation of XBP1s. Furthermore, IRE1 can potentially attenuate ER protein folding load through the degradation of ER-localized mRNA. ATF6 activation leads to the cleavage of the active ATF6NT transcription factor that primarily induces genes involved in ERAF and ERAD pathways.
PERK-mediated translational attenuation promotes ER proteostasis by reducing the protein folding load
UPR-dependent translational attenuation is primarily mediated through the activation of PERK, by a well-described mechanism involving dimerization and trans-autophosphorylation [6–9] (Figure 2). Once activated, a cytosolic PERK kinase domain selectively phosphorylates the α subunit of eukaryotic initiation factor 2 (eIF2α). Phosphorylated eIF2α inhibits the activity of the eIF2B translation initiation complex, attenuating ribosomal translation and reducing the population of newly synthesized proteins entering the ER [6–9]. eIF2α phosphorylation also leads to the translation of a selective set of stress-responsive transcription factors such as activating transcription factor 4 (ATF4) that increases expression of genes involved in basic aspects of cellular proteostasis, including cellular redox regulation, amino acid biosynthesis, and the eIF2α phosphatase regulatory subunit GADD34 — a protein involved in the suppression of eIF2α phosphorylation in a negative feedback signaling loop [6–9].
PERK-dependent translational attenuation directly impacts ER proteostasis by reducing the concentration of newly synthesized, unfolded protein clients entering the ER (Figure 2). This reduced translation in turn attenuates the burden on ERAF and ERAD pathways, freeing components of these pathways to influence the folding, trafficking and degradation of unfolded or misfolded proteins in the ER by increasing the proteostasis network component/polypeptide client ratio. This capacity to reduce the protein folding and degradation load offers a potential mechanism to restore the aberrant ER proteostasis associated with human protein misfolding diseases. For example, preemptive PERK activation increases secretion of a destabilized fibulin-3 mutant involved in the pathology of the rare macular dystrophy malattia leventinese [10]. Presumably this results from increasing the ERAF component/client ratio in the ER lumen, although the specific molecular mechanism(s) remains to be determined. Activation of PERK also appears to be required for the UPR-dependent increased secretion of destabilized mutants of β-glucocerebrosidase involved in Gaucher disease, a loss-of-function disorder [11•]. These results and others indicate that promoting PERK-mediated translation attenuation could be beneficial for adapting ER proteostasis in the context of ER protein misfolding diseases. However, there is one significant drawback to therapeutic PERK activation, as eIF2α phosphorylation also leads to activation of CHOP — a pro-apoptotic transcription factor that increases the probability of cellular apoptosis following prolonged ER stress [6–9].
Activation of IRE1 and/or ATF6 results in distinct ER proteostasis network environments that can be therapeutically accessed to ameliorate ER misfolding diseases
Activating the IRE1 and/or ATF6 stress sensors primarily influences ER proteostasis through the activation of downstream transcription factors that remodel the ER proteostasis network via a transcriptional program. Activation of IRE1, the most evolutionarily conserved arm of the UPR, involves oligomerization and trans-autophosphorylation, resulting in the activation of a cytosolic IRE1 endoribonuclease domain that is required for the splicing of Xbp1 mRNA [6–9] (Figure 2). Spliced Xbp1 encodes the bZIP transcription factor, XBP1s, that induces the expression of genes involved in nearly all aspects of ER proteostasis, including translocation, ERAF, COPII-trafficking, and ERAD [12,13]. IRE1-dependent XBP1s splicing is also required for other aspects of ER proteostasis maintenance, including the ER expansion essential for the development of plasma cells [13–15]. IRE1 activation can also lead to regulated IRE1-dependent decay (RIDD), a promiscuous mRNA ribonuclease activity that targets mRNAs encoding proteins directed to the ER [16–19]. RIDD activity may provide an alternative mechanism to reduce the folding and degradation load in the ER, analogous to that observed for PERK-mediated translational attenuation. While the mechanism of this divergent, multi-tiered IRE1 response requires further study, it has been suggested that IRE1 phosphorylation and oligomerization states are important for the relative XBP1 splicing and RIDD activities [16,19].
ATF6 also influences ER proteostasis through direct remodeling of the ER proteostasis network. ATF6 is activated by trafficking full-length ATF6 from the ER to the Golgi, where it is proteolytically processed by the Site 1 and Site 2 proteases [6–9] (Figure 2). This processing releases the N-terminal, cytosolic domain of ATF6 (ATF6NT) comprising an active bZIP transcription factor, which induces expression of ER proteostasis genes primarily involved in ERAF and ERAD [20,21]. While both XBP1s and ATF6NT function to remodel ER proteostasis pathways, the gene sets induced by these transcription factors are distinct, suggesting that activation of XBP1s or ATF6NT uniquely impacts the activity of ER proteostasis pathways [12,13,20,21]. These transcription factors can also heterodimerize. The heterodimer is predicted to selectively increase expression of genes involved in specific aspects of ER proteostasis maintenance, such as ERAD [22]. Thus, the independent or combined activation of IRE1–XBP1s and/or ATF6 could result in three distinct ER proteostasis environments that could be therapeutically accessed to ameliorate the aberrant ER protein folding, trafficking or degradation of destabilized mutant proteins involved in human protein misfolding diseases.
A challenge for evaluating the potential of IRE1–XBP1s and/or ATF6 activation to influence ER proteostasis is that each disease-associated protein has a unique dependence on specific ERAF and ERAD pathways, sometimes differentially influenced by arm-selective UPR signaling. Thus, the impact of activating IRE1–XBP1s and/or ATF6 on ER proteostasis for a specific disease-associated protein client cannot be easily determined a priori. Regardless, significant evidence is emerging that demonstrates targeting IRE1–XBP1s or ATF6 influences the ER proteostasis of mutant disease-associated proteins. Activation of either IRE1–XBP1s or ATF6 reduces the intracellular aggregation of disease-associated rhodopsin mutants, potentially indicating that activating these pathways can attenuate the photoreceptor cell death associated with autosomal dominant retinitis pigmentosa [23••,24••]. IRE1, but not ATF6, is required for the UPR-dependent increase in β-glucocerebrosidase mutant trafficking in cellular models of Gaucher disease [11•]. Furthermore, ATF6NT overexpression increases ERAD of the destabilized α1-antitrypsin (A1AT) mutant, PiZZ, and attenuates PiZZ aggregation, indicating that ATF6 activation could be an approach to ameliorate the pathologic aggregation of this protein involved in A1AT-related liver disease [25••]. Collectively, these results highlight the unique sensitivity of destabilized mutant proteins to XBP1s-dependent and/or ATF6NT-dependent remodeling of the ER proteostasis environment and demonstrate the potential for activating these transcription factors to influence ER proteostasis in the context of protein misfolding diseases.
Therapeutic targeting of UPR signaling arms to ameliorate protein misfolding diseases
Evidence for the therapeutic relevance of arm-selective UPR activation has resulted in a significant effort to identify small molecule regulators of the UPR signaling arms as potential therapeutic strategies to treat protein misfolding and other human diseases. We highlight some of the most relevant pharmacologic or chemical biological approaches to modulate UPR signaling pathways below.
Targeting PERK-mediated translational attenuation
To date, no small molecule activator of PERK has been identified, although small molecule PERK inhibitors have been recently described [26,27]. Small molecule regulation of PERK-dependent translational attenuation has been achieved by targeting the two eIF2α phosphatase complexes, which are composed of protein phosphatase 1 (PP1) and either the constitutively expressed regulatory subunit, CreP, or the stress-induced regulatory subunit, GADD34 [7,9] (Figure 3). The first described inhibitor was salubrinal, which was found to inhibit both PP1–CreP and PP1–GADD34 phosphatase complexes [28•]. Inhibition of the constitutive PP1–CreP complex allows salubrinal to induce eIF2α-mediated translational attenuation in the absence of stress. Salubrinal-dependent continuance of eIF2α phosphorylation decreases viral replication, suggesting an increased capacity to regulate ER proteostasis and viral production through translational attenuation.
Figure 3.
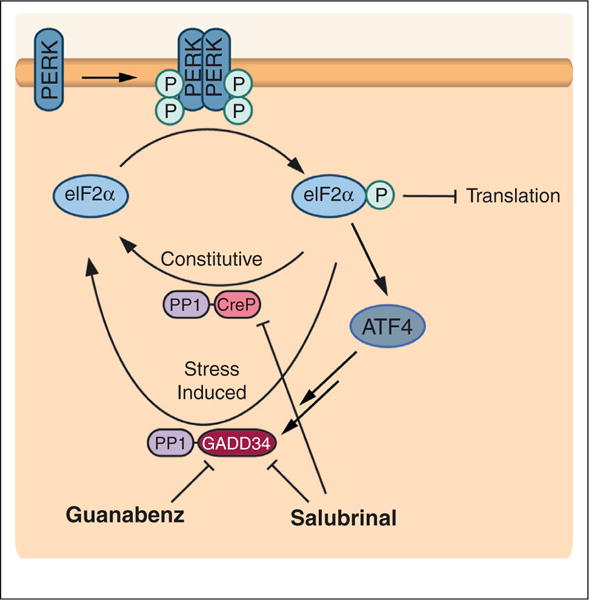
Small molecule modulation of PERK signaling can be mediated by targeting eIF2α phosphatase complexes. PERK activation increases eIF2α phosphorylation, which in turn attenuates translation and increases expression of stress-responsive transcription factors. This pathway is negatively regulated by a phosphatase complex between protein phosphatase 1 and the constitutively expressed regulatory subunit CreP (PP1–CreP) and/or a stress-induced regulatory subunit GADD34 (PP1–GADD34). Small molecules that target these complexes can modulate eIF2α-dependent signaling, effectively mimicking PERK activation. Salubrinal inhibits both the PP1–CreP and PP1–GADD34 phosphatase complexes, allowing for increased eIF2α phosphorylation in the absence of stress. Guanabenz selectively targets PP1–GADD34, providing a mechanism to prolong PERK-dependent eIF2α phosphorylation signaling activation in response to ER stress. Figure adapted from Wiseman et al. [39].
A potential limitation of the increase in eIF2α phosphorylation afforded by salubrinal is the potential for prolonged eIF2α phosphorylation to increase expression of pro-apoptotic genes. Interestingly, guanabenz, an α2-adrenergic receptor agonist used in the treatment of hypertension, avoids this potential pitfall by selectively inhibiting the stress-induced PP1–GADD34 eIF2α phosphatase complex [29••]. The selective inhibition of PP1–GADD34 means that guanabenz alone does not induce eIF2α phosphorylation in the absence of stress, but instead prolongs the translational attenuation observed following stress-dependent activation of PERK or other eIF2α kinases. This allows guanabenz to selectively modulate stress-responsive PERK signaling, potentially providing a mechanism to manipulate ER proteostasis in the context of ER stress-induced UPR activation. For example, guanabenz treatment increased survival of Min6 and Ins1 cells overexpressing the Akita insulin mutation, suggesting that modulating stress-induced translational attenuation may provide a therapeutic approach to attenuate pancreatic beta cell death involved in diabetes [29••].
Therapeutic targeting of the IRE1–XBP1s or ATF6 signaling arms
The influence of IRE1 signaling on numerous human diseases has resulted in a significant focus on identifying small molecule modulators of IRE1 signaling, both activators and inhibitors, the latter having the potential to ameliorate the clonal expansion of plasma B cells pathologically linked to cancers such as multiple myeloma [30•]. Much of this work has focused on targeting the IRE1 nucleotide binding pocket to regulate IRE1 activity. Historically, focus on the nucleotide binding pocket stems from experiments showing that mutating the pocket using ‘bump-hole’ technology allowed the identification of an IRE1 mutant that could be activated by the addition of an exogenous complementary nucleotide analog, clearly demonstrating that IRE1 activation could be induced by binding to the kinase binding site [31••,32••]. Kinase inhibitors that bind to the nucleotide binding pocket can activate the IRE1 endoribonuclease domain, although the selectivity of these kinase inhibitors for IRE1 remains to be clarified [19,33,34••]. Interestingly, recent evidence suggests that type I and type II kinase inhibitors that stabilize different active site kinase conformations have distinct effects on IRE1 activity [34••], although the underlying molecular mechanism of these diverse effects remains to be determined. Type I kinase inhibitors appear to activate the IRE1 RNAse activity while type II inhibitors inhibit IRE1 RNAse activity, indicating that therapeutic targeting of the IRE1 nucleotide binding pocket could allow for either the activation or inhibition of IRE1.
Apart from the nucleotide binding pocket, other sites on IRE1 have been identified that could be targeted to influence IRE1 RNAse activity. The small molecule quercetin activates yeast Ire1 by binding to a small molecule allosteric regulatory site on the dimeric interface of the IRE1 RNAse domain [35•]. This unique small molecule binding pocket could be used to modulate IRE1 activity in vivo, although a similar site has yet to be characterized in mammalian IRE1. Alternatively, small molecule inhibitors of IRE1 have been found to bind directly to the RNase active site through the formation of reversible covalent interactions [30•,36••,37•]. The capacity to target the RNAse active site or the quercetin binding sites offers the potential for developing highly selective small molecule modulators of IRE1 signaling — a difficult challenge when targeting the more widely conserved nucleotide binding pocket.
The lack of identifiable small molecule binding sites on ATF6 has significantly challenged the pharmacologic inhibition or activation of ATF6. Currently, we are aware of only one small molecule that appears to activate the ATF6 arm of the UPR, BIX [38]. The addition of BIX increases expression of ATF6 target genes in an ATF6-dependent manner and does not appear to activate the IRE1–XBP1s or PERK arms of the UPR. However, the mechanism of BIX-dependent induction of ATF6 target genes remains unknown and the potential for this molecule to adapt ER proteostasis through ATF6 activation has not been explored. We anticipate that as more studies demonstrate the therapeutic potential of ATF6 activation to adapt ER proteostasis of disease associated proteins, more effort will be directed toward identifying selective, small molecule ATF6 activators.
Concluding remarks
While there is emerging evidence suggesting that arm-selective UPR activation is a viable therapeutic approach to alter the aberrant ER protein folding, trafficking or degradation involved in a variety of human protein misfolding diseases, many critical questions still remain that must be addressed before aggressively applying this therapeutic strategy to treat these maladies. For example, how does activation of IRE1, ATF6, or PERK influence the folding and trafficking of the endogenous secreted proteome? While destabilized misfolding-prone proteins appear to be sensitive to activation of these stress-responsive signaling pathways, it is unclear how activation of these pathways will influence the folding and trafficking of the nearly one-third of the endogenous human proteome targeted to the ER. Furthermore, additional studies need to be focused on characterizing the transcriptional output of these pathways on the activity of the ER proteostasis network. While transcriptional profiling has identified specific transcriptional targets induced by these pathways, how this transcriptional remodeling alters the composition, and thus activity, of ER proteostasis pathways remains to be determined. Finally, more studies are required to explore the advantages of IRE1, ATF6 or PERK activation for altering the folding, trafficking or degradation of destabilized pathologic proteins in the ER lumen. Identifying classes of protein misfolding diseases that are potentially treatable by IRE1, PERK, and/or ATF6 activation will further motivate the identification of small molecule activators of these pathways, as one pharmacologic agent could be used for treating multiple maladies of similar origin. Currently, we are just beginning to understand the power of activating individual UPR signaling arms in the context of protein misfolding diseases, and we believe that in the coming years more studies will continue to emerge that highlight the potential for this approach to treat these devastating disorders.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
••of outstanding interest
- 1.Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- 3.Wiseman RL, et al. An adaptable standard for protein export from the endoplasmic reticulum. Cell. 2007;131:809–821. doi: 10.1016/j.cell.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 4.Powers ET, et al. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 5.Balch WE, et al. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 6.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 8.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 9.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 10.Hulleman JD, Balch WE, Kelly JW. Translational attenuation differentially alters the fate of disease-associated fibulin proteins. FASEB J. 2012;26:4548–4560. doi: 10.1096/fj.11-202861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11•.Mu TW, et al. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134:769–781. doi: 10.1016/j.cell.2008.06.037. This manuscript demonstrates that global activation of the UPR increases the folding and trafficking of destabilized β-glucocerebrosidase mutants through a mechanism requiring the activity of IRE1 and PERK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaffer AL, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 14.Lee AH, et al. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005;24:4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sriburi R, et al. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol. 2004;167:35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hollien J, et al. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaddam D, Stevens N, Hollien J. Comparison of mRNA localization and regulation during endoplasmic reticulum stress in Drosophila cells. Mol Biol Cell. 2013;24:14–20. doi: 10.1091/mbc.E12-06-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 19.Han D, et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu J, et al. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13:351–364. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 21.Okada T, et al. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J. 2002;366(Pt 2):585–594. doi: 10.1042/BJ20020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamamoto K, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13:365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 23••.Chiang WC, Messah C, Lin JH. IRE1 directs proteasomal and lysosomal degradation of misfolded rhodopsin. Mol Biol Cell. 2012;23:758–770. doi: 10.1091/mbc.E11-08-0663. This manuscript highlights the potential for chemical biologic activation of IRE1–XBP1s to increase the degradation of destabilized rhodopsin mutants and reduce mutant rhodopsin aggregation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24••.Chiang WC, et al. Selective activation of ATF6 and PERK endoplasmic reticulum stress signaling pathways prevent mutant rhodopsin accumulation. Invest Ophthalmol Vis Sci. 2012;53:7159–7166. doi: 10.1167/iovs.12-10222. In this manuscript, the activation of the ATF6 transcriptional program mediated through tetracycline-inducible ATF6NT is shown to attenuate intracellular aggregation of mutant rhodopsins. With Ref. [23••], this work highlights the therapeutic potential for arm-selective UPR activation to attenuate the pathologic, intracellular aggregation of mutant rhodopsin associated with photoreceptor cell death in autosomal dominant retinitis pigmentosa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25••.Smith SE, et al. Activating transcription factor 6 limits intracellular accumulation of mutant alpha(1)-antitrypsin Z and mitochondrial damage in hepatoma cells. J Biol Chem. 2011;286:41563–41577. doi: 10.1074/jbc.M111.280073. This work demonstrates that overexpression of the active ATF6NT transcription factor ameliorates the pathologic intracellular aggregation and increases ERAD of the destabilized, Z-variant of α1-antitrypsin (A1AT), but does not influence the folding and trafficking of wild-type A1AT. These results suggest that ATF6 activation may provide a mechanism to attenuate the pathologic, intracellular accumulation of the destabilized Z-variant of A1AT and reduce the hepatic loss observed in some A1AT-related diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Axten JM, et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) J Med Chem. 2012;55:7193–7207. doi: 10.1021/jm300713s. [DOI] [PubMed] [Google Scholar]
- 27.Harding HP, Zyryanova AF, Ron D. Uncoupling proteostasis and development in vitro with a small molecule inhibitor of the pancreatic endoplasmic reticulum kinase, PERK. J Biol Chem. 2012;287:44338–44344. doi: 10.1074/jbc.M112.428987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28•.Boyce M, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. Through a high-throughput screen to identify small molecules that protect PC12 cells from ER stress, the authors identify and characterize salubrinal as a small molecule inhibitor of eIF2α phosphatase complexes containing PP1 and either the CreP or GADD34 regulatory subunits. [DOI] [PubMed] [Google Scholar]
- 29••.Tsaytler P, et al. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science. 2011;332:91–94. doi: 10.1126/science.1201396. This work describes the identification and characterization of guanabenz as a selective inhibitor of the stress-induced eIF2α phosphatase complex composed of PP1 and GADD34. Importantly, this work highlights the potential to selectively modulate stress-induced eIF2α phosphorylation without the stress-independent activation of this pathway, effectively enhancing the capacity for PERK-dependent eIF2α phosphorylation to regulate ER proteostasis by reducing protein folding load by delaying translational recovery. [DOI] [PubMed] [Google Scholar]
- 30•.Papandreou I, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117:1311–1314. doi: 10.1182/blood-2010-08-303099. Using a high-throughput, cell based screening approach, the authors identify STF-08310 as a small molecule that inhibits IRE1 RNAse activity but does not influence IRE1 autophosphorylation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31••.Papa FR, et al. Bypassing a kinase activity with an ATP-competitive drug. Science. 2003;302:1533–1537. doi: 10.1126/science.1090031. Using a chemical biologic approach that modifies the Ire1 active site to selectively bind the small molecule 1NM-PP1, this work demonstrates that yeast Ire1 activation can be achieved through small molecule binding to the nucleotide binding pocket. [DOI] [PubMed] [Google Scholar]
- 32••.Lin JH, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. This work demonstrates that the chemical biologic approach to regulate yeast Ire1 described in Ref. [31••] can similarly be applied to mammalian IRE1 and demonstrates that prolonged activation of IRE1 is beneficial to increase cellular survival during persistent ER stress. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korennykh AV, et al. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34••.Wang L, et al. Divergent allosteric control of the IRE1alpha endoribonuclease using kinase inhibitors. Nat Chem Biol. 2012;8:982–989. doi: 10.1038/nchembio.1094. This work shows that type I and type II kinase inhibitors differentially influence IRE1 activity, with type I inhibitors activating IRE1 and type II inhibitors inhibiting IRE1. This suggests that targeting the IRE1 kinase binding pocket could potentially be applied to activate or inhibit IRE1 signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35••.Wiseman RL, et al. Flavonol activation defines an unanticipated ligand-binding site in the kinase-RNase domain of IRE1. Mol Cell. 2010;38:291–304. doi: 10.1016/j.molcel.2010.04.001. This manuscript identifies a previously unanticipated ligand binding pocket on yeast Ire1 that can potentially be therapeutically accessed to selectively activate IRE1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36••.Cross BC, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A. 2012;109:E869–E878. doi: 10.1073/pnas.1115623109. Using a high-throughput screening approach, this manuscript describes the identification and characterization of a small molecule IRE1 inhibitor that forms a Schiff base with K907 in IRE1, blocking substrate accessibility to the IRE1 RNAse active site. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37•.Volkmann K, et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J Biol Chem. 2011;286:12743–12755. doi: 10.1074/jbc.M110.199737. This manuscript identifies salicylaldehydes as potent and selective IRE1 inhibitors that non-competitively inhibit stress-induced IRE1 RNAse activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kudo T, et al. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008;15:364–375. doi: 10.1038/sj.cdd.4402276. [DOI] [PubMed] [Google Scholar]
- 39.Wiseman RL, Kelly JW. Cell biology. Phosphatase inhibition delays translational recovery. Science. 2011;332:44–45. doi: 10.1126/science.1204505. [DOI] [PubMed] [Google Scholar]