

This review by Catarino and Stark discusses recent insights into mechanistic aspects of enhancer functions. It covers recent developments in the prediction of enhancers based on chromatin characteristics and their identification by functional reporter assays and endogenous DNA perturbations.
Keywords: enhancers, reporter assays, transcription regulation, enhancer prediction, gene expression, genetic screens
Abstract
Enhancers are important genomic regulatory elements directing cell type-specific transcription. They assume a key role during development and disease, and their identification and functional characterization have long been the focus of scientific interest. The advent of next-generation sequencing and clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9-based genome editing has revolutionized the means by which we study enhancer biology. In this review, we cover recent developments in the prediction of enhancers based on chromatin characteristics and their identification by functional reporter assays and endogenous DNA perturbations. We discuss that the two latter approaches provide different and complementary insights, especially in assessing enhancer sufficiency and necessity for transcription activation. Furthermore, we discuss recent insights into mechanistic aspects of enhancer function, including findings about cofactor requirements and the role of post-translational histone modifications such as monomethylation of histone H3 Lys4 (H3K4me1). Finally, we survey how these approaches advance our understanding of transcription regulation with respect to promoter specificity and transcriptional bursting and provide an outlook covering open questions and promising developments.
Throughout development, a single genome gives rise to different cell types with unique morphologies and functions. Each cell type differentially expresses a subset of genes, which defines its identity. Transcription begins with the recruitment of RNA polymerase II (Pol II) and auxiliary factors to core promoters, short DNA sequences around transcription start sites (TSSs). While core promoters are sufficient to recruit Pol II and drive basal levels of transcription (Orphanides et al. 1996; Roeder 1996; Blackwood and Kadonaga 1998), they require cis-regulatory elements (CREs) or enhancers for full activity (Banerji et al. 1981; Shlyueva et al. 2014). Enhancers bind transcription factors (TFs) and cofactors to recruit and activate Pol II at target gene promoters from both proximal and distal positions. The genomic positions of enhancers typically show characteristic chromatin properties, which are often used to predict enhancers (Fig. 1; Heintzman et al. 2009), but how enhancers function is still poorly understood, and their reliable identification in large genomes has been a major obstacle. In this review, we discuss recent technological advances in the field of regulatory genomics and novel insights into enhancer function and biology.
Figure 1.
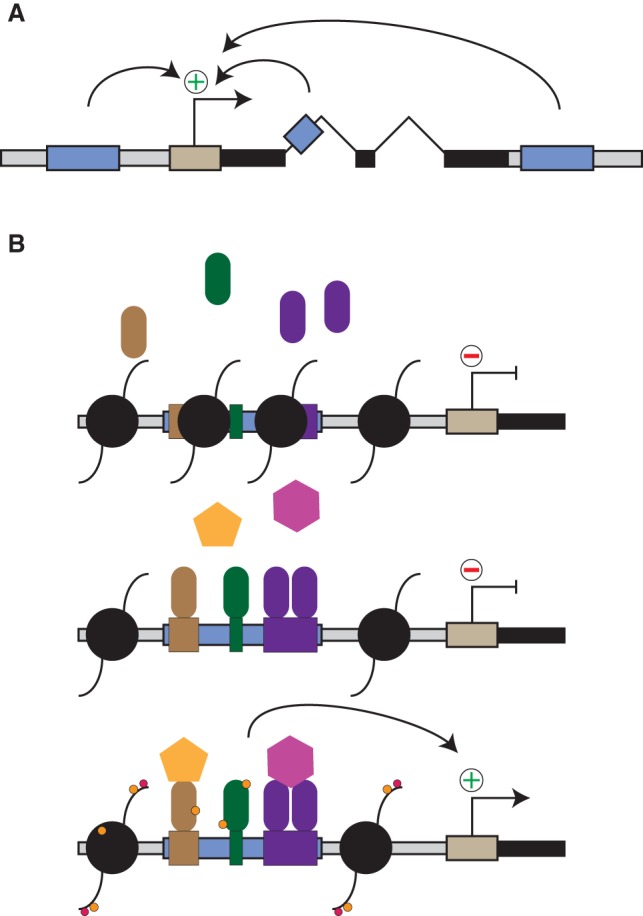
Overview: enhancers and (core) promoters. (A) Gene (black bars) transcription starts at the TSSs (straight arrow) within core promoter elements (light brown). Enhancers (blue boxes) are cis-regulatory DNA sequences that activate expression of their target genes and are often found in introns or distal intergenic regions both upstream and downstream. (B) Nucleosomes (black circles) bind to DNA and decrease accessibility to other proteins, such as TFs (colored rods). Enhancers contain TF-binding motifs (colored boxes), sequences specifically recognized by TFs and to which TFs bind in competition with nucleosomes. Enhancer-bound TFs recruit transcriptional cofactors (colored polygons) and activate gene expression from a distal gene. Cofactors often have catalytic activity and post-translationally modify TFs, histones, and other proteins in the vicinity of enhancers and promoters (small colored circles indicate such modifications).
Next-generation sequencing (NGS) enables the genome-wide profiling of gene expression and chromatin properties, allowing correlative enhancer predictions
Over the past decade, NGS has enabled studies of enhancers and their properties across entire genomes and, recently, with single-cell resolution (Adli and Bernstein 2011; Buenrostro et al. 2013; Nagano et al. 2013; Deng et al. 2014; Macosko et al. 2015). Chromatin immunoprecipitation (ChIP) coupled to NGS (Johnson et al. 2007; Robertson et al. 2007) is now used routinely to map TF binding and histone modifications across entire genomes, including histone H3 Lys4 monomethylation (H3K4me1) found at enhancers, H3K4 trimethylation (H3K4me3) at promoters, and H3K27 acetylation (H3K27ac) at active enhancers and promoters (for reviews, see Buecker and Wysocka 2012; Zentner and Scacheri 2012; Calo and Wysocka 2013). Together with other correlative traits such as DNA accessibility and (bidirectional) transcription, they allow genome-wide enhancer predictions (for reviews, see Buecker and Wysocka 2012; Maston et al. 2012; Shlyueva et al. 2014). Recent advances have explored the relationship between these correlative traits and enhancers, including their putative contributions to enhancer activity.
DNA accessibility is required for enhancer activity and is a good predictor of enhancers
Histone octamers compact genomic DNA into units termed nucleosomes, which inhibit the DNA accessibility of other proteins such as TFs (Svaren et al. 1994; Walter et al. 1995; Liu et al. 2006), creating a barrier for enhancer activation. Interestingly, nucleosomes have different affinities to different DNA sequences (Thåström et al. 1999; Bernstein et al. 2004; Segal et al. 2006; Kaplan et al. 2009; Brogaard et al. 2012), and enhancers appear to have a particularly high sequence-encoded nucleosome positioning preference. Inactive enhancers are therefore typically occupied by nucleosomes, which block TF-binding sites (TFBS), establishing a “default off” state (Lidor Nili et al. 2010; Charoensawan et al. 2012; Barozzi et al. 2014). Enhancer activation is thought to rely on specialized pioneer TFs that bind and displace nucleosomes (Perlmann and Wrange 1988; Imbalzano et al. 1994; Cirillo et al. 2002; Zaret and Carroll 2011; Soufi et al. 2015), presumably via ATP-dependent chromatin remodelers (for reviews, see Clapier and Cairns 2009; Hargreaves and Crabtree 2011), although alternative mechanisms are possible (for reviews, see Deplancke et al. 2016; Reiter et al. 2017).
Intriguingly, recent work has shown that nucleosomal DNA can be accessible to micrococcal nuclease (MNase) (Iwafuchi-Doi et al. 2016; Mieczkowski et al. 2016; Mueller et al. 2017), particularly at enhancers and promoters that have less stably bound nucleosomes, as determined by salt extraction fractionation (Henikoff et al. 2009). Decreased nucleosome stability and increased dynamics can be caused by TFs that alter nucleosome positioning (Iwafuchi-Doi et al. 2016), post-transcriptional modifications of histones (for review, see Tessarz and Kouzarides 2014), or the incorporation of histone variants (Dion et al. 2007; Henikoff et al. 2009; Mieczkowski et al. 2016; for review, see Talbert and Henikoff 2017). The H3.3 variant, for example, decreases nucleosome stability (Jin and Felsenfeld 2007), and its incorporation into nucleosomes at enhancers and promoters is mediated by the histone chaperone complex HIRA (Goldberg et al. 2010), recruited by the DNA-binding RPA complex (Zhang et al. 2017). Consistently, depletion of either RPA or HIRA impairs H3.3 incorporation and affects transcription, emphasizing the importance of destabilized nucleosomes for gene expression.
Given its importance for TF binding and enhancer activity, DNA accessibility has been used to predict enhancers and is one of the most predictive chromatin features (Boyle et al. 2008; Kwasnieski et al. 2014). However, other genomic regions such as insulators or promoters are also accessible (Boyle et al. 2008), and DNA accessibility of enhancers does not quantitatively reflect their activity; in fact, inactive enhancers can also be accessible (Arnold et al. 2013; Andersson et al. 2014b). Therefore, enhancer prediction approaches typically consider more features to increase the specificity toward active enhancers.
Certain histone modifications are predictive of enhancers yet are not required for enhancer activity
Some post-translational modifications (PTMs) of histones are frequently found at enhancers and are catalyzed by enzymes such as P300/CBP or Mll3/4 that function as transcriptional activators. While PTMs of histone residues that contact DNA can affect nucleosome stability (Neumann et al. 2009; Tropberger et al. 2013; Pradeepa et al. 2016; for review, see Tessarz and Kouzarides 2014), PTMs in the unstructured histone tails might recruit so-called reader proteins, but their requirement for enhancer function is less clear. The modifications of histone tail residues that are most often found at enhancers are H3K27ac and H3K4me1 (Heintzman et al. 2009; Creyghton et al. 2010; Rada-Iglesias et al. 2011; Zentner et al. 2011; Bonn et al. 2012; for reviews, see Buecker and Wysocka 2012; Shlyueva et al. 2014).
H3K4me1 occurs at most or all enhancers in both their active and inactive or primed states prior to activation (Creyghton et al. 2010; Rada-Iglesias et al. 2011; Zentner et al. 2011; Bonn et al. 2012; Arnold et al. 2013). Despite this extensive co-occurrence, two recent studies demonstrated that H3K4me1 is not required for enhancer activity: Even though rendering the mammalian H3K4 methyltransferases Mll3/Mll4 or their fly ortholog, Trithorax-related (Trr), catalytically inactive efficiently depleted H3K4me1, it had negligible effects on gene expression and resulted in viable and fertile flies (Dorighi et al. 2017; Rickels et al. 2017). The same mild impact on global gene expression was observed with a hyperactive mutant of Trr that increased H3K4me1 above physiological levels (Rickels et al. 2017). Even though H3K4me1 loss might affect individual genes, potentially relating to distal enhancer contacts and H3K4me1-bound proteins (Local et al. 2018; Yan et al. 2018), these observations suggest that H3K4me1 is not generally required for enhancer activity and does not seem to be able to cause ectopic gene expression. This is in stark contrast to the consequences of depleting the respective methyltransferases, which strongly perturbs gene expression and is lethal in flies and mice (Sedkov et al. 1999; Lee et al. 2013; Dorighi et al. 2017; Rickels et al. 2017; for reviews, see Shilatifard 2012; Herz et al. 2013). A similar finding applies to UTX, a H3K27 demethylase that recruits P300 and Mll4 and is required for enhancer activity, while its catalytic activity is not (Wang et al. 2017).
While the catalytic activity of Mll3/4/Trr methyltransferases is dispensable for transcription activation, the acetyltransferase activity of P300/CBP is required (Hilton et al. 2015; Boija et al. 2017). P300/CBP acetylates H3K27, a residue that can also be trimethylated to form H3K27me3 during Polycomb-repressive complex 2 (PRC2)-mediated silencing (Boyer et al. 2006). Indeed, H3K27 mutations can dominantly affect PRC2 target genes, as was shown experimentally by injecting mRNA of H3K27R (where R is arginine, a positively charged amino acid that cannot be acetylated or methylated) into mouse zygotes, which impaired heterochromatin establishment and transcriptional silencing (Santenard et al. 2010). More importantly, dominant PRC2 loss of function is also thought to underlie the cancer-causing effect of the H3K27M mutant that is observed in >70% of pediatric gliomas (Schwartzentruber et al. 2012; Wu et al. 2012; Bender et al. 2013; Lewis et al. 2013; Funato et al. 2014; Herz et al. 2014).
To assess the regulatory relevance of H3K27 in Drosophila, Pengelly et al. (2013) analyzed cells in which most of the canonical H3 was replaced with H3K27R. H3K27R mutant cells failed to silence PRC2 target genes and formed tissues that displayed developmental defects similar to PRC2 mutants. While enhancer activity was not directly assessed and noncanonical H3 variants were not mutated, the absence of a general loss of transcription suggests that H3K27ac might not be required for enhancer activity (Pengelly et al. 2013). This is consistent with independent observations that active enhancers in both flies (Bonn et al. 2012) and mice (Pradeepa et al. 2016) are not necessarily marked by H3K27ac and suggests that P300/CBP might target histone residues other than H3K27 (Pradeepa et al. 2016) or nonhistone proteins such as TFs (Edmunds and Mahadevan 2004; Ashwell 2006; Kim et al. 2006; Roe et al. 2015) or preinitiation complex (PIC) components, including Pol II (Schröder et al. 2013). However, even if not directly involved in transcription activation, H3K27ac could modulate other aspects of enhancer activity; for example, destabilizing nucleosomes or recruiting H3K27ac-binding proteins.
Histone 3 Ser10 and Ser28 phosphorylation at mitogen- and stress-activated protein kinase 1/2 (MSK1/2)-induced enhancers might also have an auxiliary role (Sawicka et al. 2014; Josefowicz et al. 2016). While the MSK1/2 kinases downstream from p38 and ERK signaling are mainly known to activate TFs by phosphorylation (Wiggin et al. 2002), nucleosomes that flank the TF-bound enhancers also become phosphorylated at H3S10 and H3S28 during kinase signaling (Sawicka et al. 2014). Mutation of H3S28, but not H3S10, to alanine reduced the recruitment of p300 to these enhancers, and in vitro transcription assays show that transcription output is reduced (Josefowicz et al. 2016). While the effects were small compared with the activation mediated by the MSK1/2 target TFs, this example shows that PTMs at secondary targets near enhancers can gain modulatory roles with potential benefits for the robustness or efficiency of enhancer function.
Altogether, recent data suggest that enhancer-associated histone modifications are not necessarily required for enhancer activity (Fig. 2); i.e., even a strong correlation does not imply causation (Pollex and Furlong 2017). We argue that the recent finding that H3K4me1 is entirely dispensable for transcription regulation warrants careful reconsideration of a putative role of other histone modifications such as H3K27ac. Among the possible functions of histone PTMs that could be more difficult to assess are, for example, indirect ones; i.e., PTMs that function by preventing others, such as H3K27ac, which may function to prevent H3K27 methylation and Polycomb-mediated silencing (Pengelly et al. 2013). Alternatively, it is possible that several PTMs contribute to enhancer function through parallel mechanisms such that the loss of one PTM may be obscured by the presence of others. However, it is also possible that some histone modifications in the vicinity of enhancers could be functionally neutral by-products: Perfect specificity does not exist in biological systems, and increasing specificity is typically costly, implies a trade-off in sensitivity or enzyme kinetics, and can evolve only if it confers selective advantage. If that were the case, evolution may still have made use of such by-products to modulate enhancer function or increase robustness to conditions not typically assessed in laboratory studies.
Figure 2.

Contributions of cofactors and histone tail modifications to enhancer activity. (A) The methyltransferases Mll3/4/Trr are required for enhancer activity and transcription, but their methyltransferase activity is not (Dorighi et al. 2017; Rickels et al. 2017). (Top) An active enhancer with methyltransferase and H3K4me1-marked flanking histones activates gene expression. (Middle) Mutation of the catalytic domain results in the loss of H3K4me1 but maintains enhancer activity and gene expression. (Bottom) Knockout of the methyltransferase leads to the loss of gene expression. (B) Enzymatic targets of acetyltransferases P300/CBP, which have been reported to acetylate many proteins, including TFs, cofactors, histones, and members of the PIC, including Pol II (for references, see the text). (C) H3K27ac may have an indirect role in preventing PRC2-mediated silencing. (Top) PRC2 (brown) catalyzes H3K27me3, which is blocked by H3K27ac, preventing PRC2-mediated silencing. (Bottom) Mutations of H3K27 to methionine (M; as observed frequently in pediatric gliomas) or arginine (R) prevent both acetylation and methylation. Both mutations induce changes in gene expression that mimic PRC2 loss-of-function H3K27M in a dominant fashion, as indicated by the dashed cross (for references, see the text).
Enhancer transcripts as predictors of enhancer activity
Enhancer transcription is also correlated with enhancer activity, as has been observed for individual genes (Tuan et al. 1992) and by genome-wide analysis of transcription and Pol II binding in mammals (Kim et al. 2010; Djebali et al. 2012), flies (De Santa et al. 2010; Kharchenko et al. 2011; Bonn et al. 2012), and nematodes (Chen et al. 2013; for review, see Li et al. 2016).
Transcription from enhancers has been reported to often be bidirectional (Kim et al. 2010; Andersson et al. 2014b), although it can also be unidirectional (Koch et al. 2011). The resulting enhancer RNAs (eRNAs) may be polyadenylated and stable (Koch et al. 2011; Andersson et al. 2014b), but, more typically, eRNAs are unstable and rapidly degraded by the exosome (Andersson et al. 2014b; Lubas et al. 2015). This instability hinders eRNA detection with RNA sequencing approaches that measure steady-state RNA levels (Rabani et al. 2014; Schwalb et al. 2016). Indeed, eRNA detection is improved by exosome inhibition (Andersson et al. 2014a) or methods that measure nascent RNA, such as global run-on (GRO) sequencing (GRO-seq) (Core et al. 2008), precision nuclear run-on (PRO) sequencing (PRO-seq) (Kwak et al. 2013), START-seq (Scruggs et al. 2015), native elongating transcript (NET) sequencing (NET-seq) (Churchman and Weissman 2011), and transient transcriptome sequencing (TT-seq) (Schwalb et al. 2016; Michel et al. 2017).
The correlation of eRNA transcription and enhancer activity has led to the proposal that enhancers and promoters might be more similar than traditionally assumed (Core et al. 2014; Andersson 2015; Kim and Shiekhattar 2015). It has also been used for enhancer prediction across different cell types and tissues (Melgar et al. 2011; Andersson et al. 2014a) and in inducible systems such as neuronal activation (Kim et al. 2010; Schaukowitch et al. 2014), immune response (De Santa et al. 2010; Kaikkonen et al. 2013; Michel et al. 2017), hormone signaling (Hah et al. 2011, 2013; Wang et al. 2011; Li et al. 2013; Lai et al. 2015), or the modulation of TF activity (Melo et al. 2013). Interestingly, the timing between enhancer and gene transcription seems to be locus-specific (or may depend on the approaches used), with eRNA transcription preceding mRNA production for some loci (De Santa et al. 2010; Arner et al. 2015), while, for others, both RNA species were transcribed synchronously (Kaikkonen et al. 2013; Michel et al. 2017).
Even though eRNAs seem to generally be good predictors of active enhancers (Melgar et al. 2011; Wang et al. 2011; Andersson et al. 2014a; Rennie et al. 2017; Henriques et al. 2018; Mikhaylichenko et al. 2018), not all predicted candidates function as enhancers. For example, while 70% of CAGE (cap analysis of gene expression)-defined enhancers validated in reporter assays (Andersson et al. 2014a), the remaining 30% may have other regulatory functions, and, indeed, bidirectional transcription has been reported for insulators (Melgar et al. 2011) and accessible DNA in general (Young et al. 2017). Similarly, active enhancers might show little or no eRNA transcription, outcomes that depend on the sensitivity of eRNA detection. For example, 20%–33% of the tested regions without detectable eRNA initiation yet with enhancer-associated histone marks showed enhancer activity in reporter assays in mammalian cells (Andersson et al. 2014a), and some active developmental enhancers in Drosophila did not initiate eRNAs to detectable levels (Mikhaylichenko et al. 2018). Enhancers that do or do not strongly initiate eRNA transcription seem to differ at the sequence level (i.e., the occurrence of core promoter elements) (Andersson et al. 2014b; Core et al. 2014; Arnold et al. 2017; Mikhaylichenko et al. 2018), and it will be interesting to see whether cell types or tissues—or their respective proliferation status—also influence eRNA abundance. Overall, the results obtained so far suggest that eRNAs associate with enhancer activity across different species but that they are not perfectly predictive and therefore may not be either required or sufficient for enhancer activity. This further suggests that enhancer and promoter elements can co-occur in the genome but that enhancer and promoter functionalities are distinct and not interdependent (Arnold et al. 2017; Catarino et al. 2017; Mikhaylichenko et al. 2018).
Proposed eRNA functions are diverse and context-dependent
Establishing a causal and potentially mechanistic relationship between eRNAs and enhancer activity has been difficult, presumably because of at least three reasons: (1) Enhancer activity might be influenced by the act of eRNA transcription or by the nascent or mature RNAs. (2) Genetic manipulations of eRNA sequences necessarily alter the enhancer sequence and thus, potentially, its activity. (3) Enhancer activity likely impacts eRNA transcription, creating a circularity that obscures directionality and causality.
Pol II binding and eRNA transcription have been reported to displace nucleosomes and establish DNA accessibility (Gilchrist et al. 2010; Mousavi et al. 2013) such that the act of eRNA transcription might have a role in enhancer activity. Alternatively, nascent eRNAs could be important, as they have been reported to perform diverse functions, including the stabilization of TF binding (Sigova et al. 2015), the recruitment and activation of cofactors (Kaikkonen et al. 2013; Gardini et al. 2014; Lai et al. 2015; Bose et al. 2017), the release of NELF from promoters (Schaukowitch et al. 2014), or the promotion of cohesin-mediated enhancer–promoter contacts (Li et al. 2013; Hsieh et al. 2015; Isoda et al. 2017). However, each of these functions seems to apply only to individual eRNAs rather than globally or may depend on the respective experimental models or approaches. A recent proposal might reconcile the diversity of eRNA functions, particularly regarding protein recruitment: If eRNAs mediated the formation of specialized membraneless compartments at active enhancers or promoters via phase transition (Muerdter and Stark 2016; Hnisz et al. 2017), these compartments could feature high local concentrations of diverse activators. RNA-mediated phase transition has indeed been reported for RNAs with tandem repeats and for nucleolar rRNAs (Berry et al. 2015; Jain and Vale 2017). However, it remains to be tested whether short unstable eRNAs with highly diverse sequences could drive phase transition and whether this could contribute to enhancer activity. It will also be interesting to learn how compartments around active gene loci can remain separated from compartments with repressive properties (Banani et al. 2017), such as those formed during heterochromatin protein 1 (HP1)-mediated phase transition (Larson et al. 2017; Strom et al. 2017).
The genetic manipulation of eRNAs is also challenging, as it necessarily affects the DNA sequence of the enhancers and thus, potentially, the enhancers’ activities. This has been highlighted by recent work that assessed the function of long noncoding RNAs (lncRNas) by polyA site insertion next to the lncRNA promoter, enforcing early transcription termination (Anderson et al. 2016; Engreitz et al. 2016; Paralkar et al. 2016). In many cases, the lncRNAs seemed to be dispensable, and transcription was activated by enhancers located proximally to the lncRNAs’ promoters (for discussion, see Bassett et al. 2014; Espinosa 2016). The lncRNA upperhand is one of the exceptions, as it seems to control Hand2 expression and heart development (Anderson et al. 2016). Similarly, the direct depletion of eRNAs by either RNAi or RNase H has been reported to abrogate the expression of individual genes (Hah et al. 2013; Lam et al. 2013; Li et al. 2013; Schaukowitch et al. 2014), yet this approach has not been used very frequently, presumably due to the inefficiency of targeting nascent RNAs by these approaches or the difficulty in further increasing the turnover of already short-lived eRNAs (De Santa et al. 2010; Andersson et al. 2014b; Lubas et al. 2015).
A final complication is that enhancer activity likely influences eRNA transcription: Active enhancers are characterized by accessible DNA and TFs that recruit activating cofactors such as P300/CBP, Mll3/4, or Mediator, which can bind and/or activate Pol II at target promoters. This established mechanism implies the existence of strongly activating cues at enhancers, making it likely that enhancers are transcribed even if this transcription was not functional; i.e., entirely neutral. This is because evolving a sequence specificity for transcription initiation that makes initiation perfectly specific for gene starts (or an active mechanism that prevents initiation in any other region) is energetically costly and can evolve only if there is a strong selective advantage. In addition, TSSs in enhancers contain sequences that weakly match to core promoter elements—short sequences that can occur by chance even in random sequences (Andersson et al. 2014a; Mikhaylichenko et al. 2018). While a sensible null hypothesis should therefore be that eRNAs are neutral by-products of accessible DNA in the vicinity of strong transcriptional activators (Young et al. 2017; for discussion, see Natoli and Andrau 2012), it is possible that evolution has taken advantage of Pol II binding, transcription, or eRNAs at enhancers and evolved means to modulate enhancer function.
Toward a functional definition of regulatory elements
Genomic traits used to predict enhancers can also be found in other genomic regions, making enhancer predictions imperfect. Thus, enhancer identification should include direct functional tests of enhancer activity, following the original definition of enhancers as DNA sequences that increase transcription from distal promoters (Banerji et al. 1981; for review, see Shlyueva et al. 2014). Along this paradigm, two approaches are being applied that measure either the ability of candidate sequences to drive transcription in standardized reporter assays or the requirement of candidate regions for endogenous gene expression; i.e., tests of sufficiency and necessity, respectively.
Enhancer DNA is sufficient for enhancer activity
One of the most fascinating properties of enhancers is their functional autonomy; i.e., their ability to retain their transcription-activating function outside their endogenous contexts even in combination with heterologous promoters and reporter genes (Banerji et al. 1981). Ectopic assays explore this property to test DNA sequences separated from their endogenous sequence and chromatin environments. This removes any regulatory cues that could confound the results, providing a fair comparison between the enhancer activities of different DNA sequences. Such activities can be quantified by measuring the abundance of reporter mRNAs or proteins; e.g., via the proteins’ enzymatic activities. In fact, the first enhancer was identified using such reporter assays, which also provided the functional definition of enhancers as DNA sequence elements that activate transcription irrespective of distance, orientation, and position (Banerji et al. 1981; for review, see Shlyueva et al. 2014). Classical reporter assays (e.g., those based on luciferase) enable systematic tests and have been widely used yet suffered from low throughput, as candidates needed to be tested one by one.
Assessing the enhancer potential of candidate DNA sequences genome-wide
Several methods have taken advantage of NGS to vastly increase the throughput of ectopic enhancer activity assays (Fig. 3). Massively parallel reporter assays (MPRAs) uniquely associate candidate enhancers with barcodes—short unique DNA sequences that are used instead of reporter genes (Kwasnieski et al. 2012; Melnikov et al. 2012; Patwardhan et al. 2012). Enhancers drive expression of their associated barcodes, and the abundance of each barcode among all reporter mRNAs reflects the activity of the associated enhancer, allowing the parallel testing of many candidates (for reviews, see Shlyueva et al. 2014; White 2015; Santiago-Algarra et al. 2017). Self-transcribing active regulatory region (STARR) sequencing (STARR-seq) (Arnold et al. 2013; Muerdter et al. 2018; for review, see Muerdter et al. 2015) tests enhancer candidates downstream from the TSS such that active enhancers drive their own transcription. The direct coupling of enhancer sequences and activities in cis (i.e., the use of each enhancer as its own barcode) simplifies library construction and allows millions of candidates to be tested at once, enabling genome-wide screens in fly and mammalian cells (Arnold et al. 2013; Muerdter et al. 2018).
Figure 3.

Ectopic reporter assays measure enhancer activities quantitatively. (A) Ectopic assays remove candidate sequences from their endogenous loci and test them in a heterologous setup using reporter genes (green) such as β-galactosidase (lacZ) or luciferase. Such reporter constructs can be used with nonintegrating (episomal) plasmids or can be integrated into the genome. (B) High-throughput reporter assays vastly increase the number of candidates per experiment by replacing the reporter gene with either a barcode or the enhancer itself. MPRAs uniquely assign each candidate to a barcode and define enhancer activities by quantifying the barcode-containing transcripts. STARR-seq uses each candidate as its own barcode, which simplifies library cloning and increases throughput. Both types of assays typically include ORFs (green) to stabilize the reporter mRNA.
The high-throughput testing of enhancer candidates and variants in multiplexed ectopic assays enables their application to many questions, including the importance of TF-binding motifs (Melnikov et al. 2012; Patwardhan et al. 2012; Kheradpour et al. 2013; Yanez-Cuna et al. 2014), their arrangements (Smith et al. 2013; Erceg et al. 2014; Fiore and Cohen 2016; White et al. 2016; Vierbuchen et al. 2017), and other sequence elements (White et al. 2013; Vockley et al. 2016; Grossman et al. 2017; Chaudhari and Cohen 2018) for enhancer activity or the functional impact of single-nucleotide polymorphisms (SNPs) (Kwasnieski et al. 2012; Reddy et al. 2012; Vockley et al. 2015; Tewhey et al. 2016; Ulirsch et al. 2016; for reviews, see Maston et al. 2012; Spitz and Furlong 2012; Yáñez-Cuna et al. 2013; Levine et al. 2014; Shlyueva et al. 2014; Zabidi and Stark 2016). It will be exciting to see such approaches advance our understanding of how enhancer activities are encoded in enhancer sequences.
Integrated reporters can recapitulate developmental enhancer activities but with differences from the endogenous enhancer loci
The study of cell type-specific enhancer activities across entire embryos and throughout development requires the reporter constructs to be integrated into the genome, which is typically achieved by microinjections (Kothary et al. 1989; Visel et al. 2008) or by integrating retroviruses and lentiviruses (Murtha et al. 2014). However, this leads to random integrations likely biased toward accessible DNA near active enhancer or promoter regions (Bushman 2003; Myers et al. 2005), which may strongly influence reporter gene transcription (Akhtar et al. 2013). To allow more controlled comparisons, enhancer candidates have been tested using reporters integrated into identical genomic positions (e.g., Dickel et al. 2014; Kvon et al. 2014) in a trade-off with throughput.
Genomically integrated reporter assays have been used to define enhancer activities throughout development in both flies (Kvon et al. 2014) and mice (Spitz et al. 2003; Visel et al. 2007; Marinić et al. 2013; Osterwalder et al. 2018), recapitulating dynamic and cell type-specific enhancer activities that often (82% for developmental enhancers in Drosophila embryos) (Kvon et al. 2014) match the endogenous activities as judged by the expression pattern of neighboring genes (Sagai et al. 2005; Kvon et al. 2014; Kvon 2015). Interestingly however, the activity patterns of some enhancers in such ectopic assays were broader than the enhancers’ endogenous activities (Spitz et al. 2003; Kvon et al. 2014). These discrepancies might stem from additional regulatory elements that regulate the enhancers’ target genes or from locus-specific transcriptional silencing of the enhancers, mediated, for example, by flanking sequences and chromatin features that differ between the endogenous loci and the ectopic site.
Episomal and integrated reporters in defined cell types
In contrast to the assays above, enhancer activities in individual defined cell types in culture are often tested using episomal plasmid-based reporters, particularly when many candidates are tested in highly parallelized assays (for review, see White 2015). Studies of simian virus 40 (SV40) DNA using electron microscopy suggest that episomal plasmid DNA is chromatinized and acquires nucleosomes similar to genomic DNA (Cremisi et al. 1975), although the chromatin likely differs from the enhancer candidates’ endogenous loci. Indeed, a substantial fraction of the sequences identified as strong enhancers in such assays is likely silenced in their endogenous contexts as judged by DNA accessibility (∼30% of enhancers active in ectopic assays in fly cells and 60% in human cells are closed) (Arnold et al. 2013; Muerdter et al. 2018). The prevalence of H3K27me3 and H3K9me3 marks at the endogenous loci of such closed enhancers suggests that they are silenced at the chromatin level by the Polycomb- and HP1-dependent pathways (Arnold et al. 2013; Muerdter et al. 2018). In human cells, many closed enhancers are retrotransposons, and, consistently, an LTR-derived mouse mammary tumor virus (MMTV) promoter was silenced only when stably integrated into the cellular chromatin but not on an episomal plasmid (Archer et al. 1992).
Genomic integration therefore has been assumed to provide a chromosomal environment similar to the enhancers’ natural chromatin state. However, this assumption is not generally true, as the insertion sites of the reporter constructs are typically different from the endogenous sites of the candidate enhancers (particularly when retroviruses or lentiviruses are used) (see above). Importantly, Drosophila S2 cell enhancers that were accessible in their endogenous contexts (i.e., open) and those that were silenced at the chromatin level (i.e., closed) were active in both episomal and genomically integrated assays (Arnold et al. 2013). Consistently, enhancer activities measured by reporter assays using integrating and nonintegrating lentiviruses were, overall, highly similar (Pearson's correlation coefficent = 0.85) (Inoue et al. 2017). These outcomes suggest that differences in enhancer activities may not result from episomal versus chromosomally integrated assays but from chromatin-mediated developmental silencing that is stablished during cell type differentiation and can be maintained in differentiated cells but not established de novo. In a given cell type, episomal and genomically integrated assays therefore should yield similar results, which, however, can differ from the candidates’ endogenous activities (Arnold et al. 2013; Muerdter et al. 2018).
Assessing the impact of enhancer candidates on gene expression by genetic enhancer perturbation
A key question not addressed by ectopic enhancer activity assays is whether—and how—a genomic enhancer affects cellular gene expression. It is known that mutations or deletions of genomic enhancers can cause gene misregulation and lead to different diseases, including developmental defects (e.g., Sagai et al. 2005) or cancer (e.g., Pomerantz et al. 2009; Wasserman et al. 2010; Sur et al. 2012; Mansour et al. 2014). In fact, many disease-associated genetic variants identified through genome-wide association studies (GWASs) map to noncoding regulatory sequences (Degner et al. 2012; Maurano et al. 2012; Schaub et al. 2012; Karczewski et al. 2013). The question of how different enhancers contribute to cellular gene expression motivated a second type of enhancer activity assay based on the genetic perturbation of genomic enhancers (Fig. 4).
Figure 4.

Clustered regularly interspaced short palindromic repeat (CRISPR)–Cas9-based approaches to assess endogenous enhancer activities. (A) Endogenous enhancer activities are typically assessed by genetic perturbation and assays that detect loss of enhancer function. Transcription of an endogenous target gene is driven by the enhancer (blue) and is lost upon enhancer mutation or deletion by Cas9 and guide RNAs (gRNAs). Deletions can be repaired through homologous recombination to insert exogenous sequences (purple), allowing essentially arbitrary manipulations such as the exchange of enhancers with homologous sequences from other species (e.g., Kvon et al. 2016). (B) Endogenous high-throughput screens rely on cell selection to enrich for gRNAs that perturb enhancer activities. In a typical screen, a pool of gRNAs is transfected into cells, which introduces mutations or deletions in candidate regions. gRNAs that target active enhancers (blue) disrupt target gene expression and can be enriched by selecting for a cellular phenotype (e.g., increased proliferation [left] or reporter gene expression [right]). The gRNAs enriched in the selected cells can identify the enhancers targeted (for references, see the text).
Versatile testing of endogenous enhancer activities by clustered regularly interspaced short palindromic repeat (CRISPR)–Cas9
Early experimental approaches disrupted endogenous enhancer activities as part of genetic screens via random insertional mutagenesis with transposons (e.g., P element or sleeping beauty) that randomly integrate in the genome (for reviews, see Kawakami et al. 2017; Kebriaei et al. 2017). This approach led, for example, to the identification of the extensive regulatory region of the Drosophila gene decapentaplegic (dpp) (St Johnston et al. 1990), which was further defined using ectopic enhancer activity assays (Blackman et al. 1991). Targeted genome engineering allows the direct testing of specific candidate sequences and was used, for example, to demonstrate the importance of the sonic hedgehog (shh) limb enhancer for shh expression and limb development (Sagai et al. 2005). Targeted editing of specific genomic regions has been revolutionized recently by CRISPR–Cas9, which introduces double-strand breaks in target DNA sequences defined by sequence-complementary guide RNAs (gRNAs) (Jinek et al. 2012).
The simultaneous use of two gRNAs to delete defined genomic regions (Fig. 4A) has been used, for example, to measure the regulatory contribution of an enhancer cluster (also called “superenhancer” [SE]) (Hnisz et al. 2013; Lovén et al. 2013; Whyte et al. 2013) in the Sox2 locus in embryonic stem cells (Li et al. 2014; Zhou et al. 2014) and in the Myc locus in blood cells (Bahr et al. 2018). In addition, the targeted deletion of individual constituent enhancers within SEs revealed that enhancer activity is mostly dependent on a few constituents that activate transcription predominately additively (Hnisz et al. 2015; Hay et al. 2016; Moorthy et al. 2017; Xie et al. 2017; for discussion, see Dukler et al. 2016). In Drosophila, enhancer deletion has been used to uncouple the tissue-specific contribution of different enhancers to the overall expression pattern of the rhomboid gene (Rogers et al. 2017).
Provided a DNA template to repair the double-strand breaks via homology-directed DNA repair, essentially arbitrary manipulations are possible, including the insertion of defined sequences. For example, to study the evolution of shh expression in vertebrates, the endogenous shh limb enhancer sequence was replaced by orthologous sequences from different species, including snakes (Kvon et al. 2016). Substitution of the mouse enhancer with the snake enhancer led to development of limbless (or “serpentized”) mice due to a 17-base-pair deletion in the snake enhancer that removed binding sites for the TF ETS1. Overall, CRISPR–Cas9 provides a powerful and flexible approach to assess the impact of endogenous enhancers on gene expression in vivo, including the functional impact of SNPs that are significantly associated with phenotypic traits according to GWASs (Smemo et al. 2014; Yao et al. 2014; Claussnitzer et al. 2015; Singh and Schimenti 2015; Cohen et al. 2017).
Multiplexed testing of endogenous enhancer functions by CRISPR–Cas9
CRISPR/Cas9-mediated loss-of-function studies of enhancers have been multiplexed to screen several regions via multiple gRNAs simultaneously (for review, see Lopes et al. 2016). While the approaches discussed above used CRISPR/Cas9 to introduce defined genetic manipulations and evaluated their impact on transcription, multiplexed screens introduce many DNA alterations via the use of complex gRNA pools, select cells based on a particular phenotype (e.g., increased proliferation), and identify gRNAs that are significantly enriched or depleted in the selected cells (Fig. 4B). Such screens have been used to identify functional enhancers among p53- and ERα-binding sites (Korkmaz et al. 2016) or identify important DNA motifs within regulatory regions (Canver et al. 2015; Sanjana et al. 2016). Since not all genes are associated with a selectable cellular phenotype, target genes can be tagged with GFP, allowing selection by FACS (Rajagopal et al. 2016; Diao et al. 2017). FACS-based CRISPR/Cas9 screens for genomic regulatory elements revealed regions required for the expression of the respective GFP-tagged genes, including “closed” regions without chromatin properties typically associated with enhancers and promoter regions that functioned as enhancers of distal genes (Rajagopal et al. 2016; Diao et al. 2017; for discussion, see Catarino et al. 2017).
Targeted recruitment of transcriptional repressors can identify enhancers
Catalytically dead Cas9 (dCas9) retains its DNA targeting ability, making it a flexible in vivo recruitment device. While DNA binding of dCas9 alone can inhibit gene expression through steric hindrance (Gilbert et al. 2013; Qi et al. 2013), dCas9-mediated recruitment of transcriptional repressors is more potent (Gilbert et al. 2013). For example, recruitment of KRAB (Thakore et al. 2015; Adamson et al. 2016; Dixit et al. 2016; Klann et al. 2017) or LSD1 (Kearns et al. 2015) has been used to repress enhancers, which can also be exploited for enhancer identification. For example, a pool of gRNAs targeting candidate regions in the GATA1 and MYC loci (essential genes for K562 survival and proliferation) was used to recruit KRAB to these candidates and identify enhancers (Fulco et al. 2016). Overall, such high-throughput endogenous assays are able to test thousands of genomic regions in a single screen, which is, however, typically centered on a particular target gene with a selectable phenotype. Recently, dCas9-KRAB recruitment has been coupled with single-cell RNA sequencing methods, enabling the combined targeting of multiple enhancers while assessing global effects on gene expression for many genes in parallel (Xie et al. 2017).
Endogenous and ectopic enhancer activity provides complementary insights into enhancer sufficiency and necessity
Ectopic and endogenous assays address characteristically different questions and provide complementary insights into gene regulation (Fig. 5): Ectopic assays assess the sufficiency of DNA sequences to activate transcription, while endogenous perturbations assess the necessity of genomic regions for the expression of a specific gene. The implications of these differences are interesting and important: As ectopic reporter assays measure the ability of DNA sequences to drive reporter gene transcription in a neutral context, active candidates can be inactive in their endogenous contexts, silenced at the chromatin level (Arnold et al. 2013; Muerdter et al. 2018).
Figure 5.

Ectopic enhancer activity assays and genetic perturbations of endogenous enhancers are complementary, and the outcomes need to be interpreted with care. Each row represents a different scenario in which the candidate (blue) is an active cellular enhancer or not (ground truth; left columns). The right columns indicate the respective outcomes of ectopic enhancer activity assays and genetic perturbations. (Green checkmark) Enhancer activity detected; (red cross) no enhancer activity detected; (yellow checkmark) outcome depends on degree of redundancy. See the text for details.
The results of endogenous enhancer perturbations also need to be interpreted with care: Regions required for the expression of certain genes are not necessarily enhancers, as perturbations might influence transcription by other means; e.g., when insulators, locus control elements, or promoters are affected. Conversely, regions that do not appear necessary for the expression of a particular gene can still be active: They might regulate a different gene or act redundantly with other enhancers. Key developmental genes are commonly regulated by multiple enhancers, which can act redundantly to assure robust gene regulation (Frankel et al. 2010; Perry et al. 2010; for review, see Barolo 2012). While full redundancy is thought to be rare, as the negative selection preserving such redundant sequences and functions would be low (Nowak et al. 1997), many enhancers seem to act partially redundantly, assuring transcription above a certain threshold (Bothma et al. 2015; Lam et al. 2015; Cannavò et al. 2016; Chatterjee et al. 2016; Osterwalder et al. 2018) or diverging in spatial activity patterns over development (Kvon et al. 2014; Bahr et al. 2018).
These considerations suggest that results from ectopic enhancer activity assays should be interpreted in combination with methods that assess DNA accessibility (Arnold et al. 2013; Muerdter et al. 2018) and that results from endogenous enhancer perturbations need to be tested for enhancer functionality by enhancer activity assays.
Functional enhancer activity assays define rules of promoter targeting within topologically associating domains (TADs)
Precise developmental gene regulation requires enhancers to specifically regulate their cognate promoters, and enhancer–promoter targeting is regulated at different levels, including the three-dimensional genome structure, DNA accessibility, and biochemical compatibilities (for review, see Zabidi and Stark 2016).
Distal enhancers come into close proximity with their target promoters (Wijgerde et al. 1995; Dillon et al. 1997), and some of these contacts are stable across developmental stages and appear to be independent of enhancer activity or gene transcription, while others seem to be dynamic and occur only during or after enhancer activation (Ghavi-Helm et al. 2014; Dixon et al. 2015; Fraser et al. 2015; Williamson et al. 2016; Dao et al. 2017; Rubin et al. 2017). Enhancer–promoter contacts occur typically within large chromosome domains, termed TADs (Dixon et al. 2012; Nora et al. 2012; Sexton et al. 2012), which are limited by boundaries that prevent interdomain contacts (Handoko et al. 2011; Schwartz et al. 2012; Narendra et al. 2015; for reviews, see Pombo and Dillon 2015; Merkenschlager and Nora 2016; Schmitt et al. 2016; Zabidi and Stark 2016). Within TADs, enhancer–promoter contacts appear to not be constrained (Symmons et al. 2014, 2016) yet may be facilitated or stabilized by factors such as YY1 (Weintraub et al. 2017). TADs may form by the extrusion of DNA loops (Nasmyth 2001; Alipour and Marko 2012; Sanborn et al. 2015; Fudenberg et al. 2016)—CTCF and cohesin being key factors in the formation of boundaries (Parelho et al. 2008; Wendt et al. 2008). Indeed, depletion of CTCF or cohesin disrupts TADs and seems to instead favor contacts within active or respressive compartments (Hou et al. 2012; Seitan et al. 2013; Zuin et al. 2014; Ing-Simmons et al. 2015; Ulianov et al. 2016; Haarhuis et al. 2017; Nora et al. 2017; Rao et al. 2017; Schwarzer et al. 2017; Wutz et al. 2017). Mutations of TAD boundaries highlight their importance in restricting enhancer–promoter contacts and gene activation, as they lead to the blurring of chromatin regions, improper gene expression, and developmental defects (Nora et al. 2012; Guo et al. 2015; Lupiáñez et al. 2015; Franke et al. 2016; Narendra et al. 2016; Hanssen et al. 2017).
Biochemical compatibilities between enhancers and promoters: different keys for different locks
Besides physical constraints imposed by the three-dimensional genome architecture, enhancers cannot regulate all promoters indiscriminately: Different promoters inserted at identical genomic positions were activated differentially (Butler and Kadonaga 2001), and housekeeping and developmental enhancers displayed a strong specificity toward housekeeping and developmental core promoters, respectively (Zabidi et al. 2015). This specificity appears to be encoded in the enhancer sequences and depends on specific TF motifs, suggesting that different enhancers recruit different TFs and cofactors to activate different promoters (Zabidi et al. 2015; Zabidi and Stark 2016). Indeed, directed recruitment of cofactors to minimal core promoters with the DNA-binding domain of Gal4 revealed that several cofactors were sufficient to activate transcription and showed preferences toward housekeeping versus developmental promoters or vice versa (Stampfel et al. 2015). These results suggest that enhancers and promoters need to be biochemically compatible (van Arensbergen et al. 2014; Zabidi et al. 2015; Zabidi and Stark 2016) and that different enhancers might use different cofactors and may respond differentially to cofactor inhibition or depletion.
Indeed, the inhibition or depletion of Brd4 in mammalian cells resulted, for example, in the selective down-regulation of certain genes, including Myc (Zuber et al. 2011), even though Brd4 appears to bind rather indiscriminately to most if not all active enhancers and promoters (Zhang et al. 2012; Kanno et al. 2014). Furthermore, in certain leukemic cells, some enhancers become activated upon Brd4 inhibition, indicating that Brd4-independent enhancers exist (Rathert et al. 2015). Likewise, inhibition of the fly ortholog of P300/CBP results in the deregulation of many genes that are both up-regulated and down-regulated (Boija et al. 2017). The inhibition of the cyclin-dependent kinases (CDKs) CDK7 (Chipumuro et al. 2014; Kwiatkowski et al. 2014; Ebmeier et al. 2017) and CDK8 (Lenstra et al. 2011; Kemmeren et al. 2014; Pelish et al. 2015; Jeronimo et al. 2016) also leads to differential gene expression effects, although this could result from differences at the transcription or post-transcriptional level; i.e., via differential RNA stabilities. In contrast, CDK9 seems to be required at virtually all promoters (Ni et al. 2008; Henriques et al. 2013; Jonkers et al. 2014; Gressel et al. 2017), presumably reflecting a universal requirement to release Pol II from a paused state after initiation into productive elongation (Adelman et al. 2005; Kwak et al. 2013; Henriques et al. 2018). Such promoter- and enhancer-specific requirements of different cofactors are consistent with the differential requirements for subunits of the large cofactor complex Mediator (Allen and Taatjes 2015), which has been assessed recently by rapid subunit depletion (Anandhakumar et al. 2016; Petrenko et al. 2017).
Our ability to rapidly inhibit or deplete cellular proteins (for review, see Housden et al. 2017) with technologies such as anchor away (Haruki et al. 2008) or degron-related methods (Dohmen et al. 1994; Nishimura et al. 2009) provides unprecedented opportunities to study the cofactor dependencies of gene transcription (Anandhakumar et al. 2016; Petrenko et al. 2017; Warfield et al. 2017; Winter et al. 2017; Xue et al. 2017). These developments promise exciting new insights into how enhancers activate transcription from target promoters and which cofactors—or the PTMs that they catalyze (see above)—might be mechanistically involved in this process.
Enhancers regulate transcriptional bursting frequency
New insights into enhancer-mediated activation also come from measuring transcription initiation kinetics at promoters; i.e., the pattern of transcription initiation events (for review, see Lenstra et al. 2016). Transcription is not a constant process but occurs in waves with bursts of transcription initiation that are separated by inactive intervals (Golding et al. 2005; Chubb et al. 2006; Raj et al. 2006; Dar et al. 2012). This bursting phenomenon means that the overall transcriptional output can be regulated by modulating the frequency of transcription bursts or the burst size; i.e., the number of mRNAs made per burst.
Fluorescent in situ hybridization (FISH) and the use of viral MS2 and PP7 RNA structures recognized by fluorescently tagged MCP and PCP proteins allow the imaging of RNA with single-molecule resolution (Bertrand et al. 1998; Janicki et al. 2004; Larson et al. 2011a; for reviews, see Larson et al. 2011b; Larson 2011; Chen and Larson 2016). Using single-molecule imaging of reporter genes in Drosophila embryos (Fukaya et al. 2016) or the β-globin and γ-globin genes in mouse and human cells (Bartman et al. 2016) showed that enhancers activate transcription predominantly via increasing burst frequency rather than burst size. Consistently, different enhancer strengths or the enhancer-blocking functions of insulators were reflected in the burst frequencies. At high transcription rates, individual bursts might merge (Fukaya et al. 2016), or a transition to a different mode might occur in which transcription is regulated only by increases of burst size (Skupsky et al. 2010; Dar et al. 2012).
Interestingly, burst size and the lengths of permissive and nonpermissive periods appear to be determined by the core promoter sequence. The presence of TATA-box motifs in promoters, particularly of inducible genes, is associated with large burst sizes (Basehoar et al. 2004; Hornung et al. 2012; Carey et al. 2013; Tantale et al. 2016), which are decreased upon mutating the TATA box (Hornung et al. 2012; Tantale et al. 2016). The dependency of burst size on the core promoter sequence and TATA boxes is reminiscent of “enhancer responsiveness”; i.e., the efficiency of core promoters to convert activating enhancer input into productive transcription events (Arnold et al. 2017). It is conceivable that highly enhancer-responsive core promoters show large burst sizes; i.e., produce a high number of mRNAs per burst. We are excited to see how new approaches such as single-molecule imaging shed new light on transcriptional regulation.
Discussion
In this review, we discussed recent progress in identifying and functionally characterizing enhancer elements in animal genomes, focusing on predictions via correlative features and functional assays that assess sufficiency or necessity. Using such assays, international consortia such as Encode and individual groups have compiled large compendia of enhancers and annotated genomic regions (e.g., Ernst and Kellis 2012; Hoffman et al. 2012), which allow the rough estimation of how many enhancers our genomes might contain. Work over the past years reported ∼10,000 enhancers for individual mammalian cells (e.g., ENCODE Project Consortium 2012; Muerdter et al. 2018) compared with ∼15,000 expressed genes (Ramsköld et al. 2009), between 200,000 and 300,000 across ∼20 mouse tissues (Shen et al. 2012; Yue et al. 2014), and ∼400,000 across a set of 127 cell lines (ENCODE Project Consortium 2012). For the much smaller Drosophila genome, estimates range from at least 50,000 to 100,000 enhancers (Kvon et al. 2014), which altogether suggests that the human genome might contain up to several million enhancers.
We anticipate that assays that assess the impact of endogenous enhancers on gene expression by genetic manipulations will be further improved by modifications and optimization of Cas9 (Kleinstiver et al. 2015, 2016; Chen et al. 2017) and improved rules of gRNA design (Fu et al. 2014; Sander and Joung 2014). By recruiting the transcription-activating or -repressing functions through extended gRNAs, such assays can be multiplexed, and different transcriptional regulators can be recruited simultaneously to different gene loci (Tak et al. 2017). Expanding CRISPR applications, Cas13a allows targeting of RNA molecules (Abudayyeh et al. 2017; Cox et al. 2017), potentially providing an alternative to RNase H or RNAi depletion, particularly for the study of ncRNAs in the nucleus.
In addition to these developments, over the past years, we have witnessed the characterization of the DNA-binding preferences for an increasingly complete set of TFs (e.g., Noyes et al. 2008; Badis et al. 2009; Jolma et al. 2013; Franco-Zorrilla et al. 2014; Weirauch et al. 2014; Hume et al. 2015; Narasimhan et al. 2015; Mathelier et al. 2016; Kulakovskiy et al. 2018; for reviews, see Deplancke et al. 2016; Inukai et al. 2017; Morgunova and Taipale 2017), their sensitivity to DNA methylation (Domcke et al. 2015; Yin et al. 2017), and how TF dimerization impacts DNA-binding affinities (Jolma et al. 2015; Isakova et al. 2016, 2017). This vocabulary and insights into the importance of binding site arrangement (Senger et al. 2004; Smith et al. 2013; Erceg et al. 2014; Farley et al. 2016; Fiore and Cohen 2016), the use of nonoptimal binding sites (Crocker et al. 2015; Farley et al. 2015), and how different types of TFs cooperate to control enhancer activity (Keung et al. 2014; Stampfel et al. 2015; for review, see Reiter et al. 2017) advance our understanding of how enhancer sequences encode and determine cell type-specific transcription.
Our increased understanding of TF–DNA interactions is complemented by the functional testing of enhancer candidates and mutant variants in ectopic and endogenous assays. Together with new technologies to rapidly deplete proteins and single-molecule live imaging of TFs (Chen et al. 2014) or nascent RNAs (Little et al. 2013; Levine et al. 2014), the upcoming years will provide not only novel insights into enhancer–promoter communication but the means to assess whether—and how—the different correlative traits are involved in enhancer function. We look forward to seeing how these developments allow an increasingly complete understanding of enhancer sequences and function and how our genomes encode gene expression.
Acknowledgments
We thank members of the Stark group (Research Institute of Molecular Pathology [IMP]) for helpful discussions and feedback on the manuscript. Research in the Stark group is supported by the Austrian Science Fund (FWF; P29613-B28 and F4303-B09) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 647320). Basic research at the IMP is supported by Boehringer Ingelheim GmbH and the Austrian Research Promotion Agency (FFG).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.310367.117.
References
- Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A, et al. 2017. RNA targeting with CRISPR–Cas13. Nature 550: 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. 2016. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167: 1867–1882.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman K, Marr MT, Werner J, Saunders A, Ni Z, Andrulis ED, Lis JT. 2005. Efficient release from promoter-proximal stall sites requires transcript cleavage factor TFIIS. Mol Cell 17: 103–112. [DOI] [PubMed] [Google Scholar]
- Adli M, Bernstein BE. 2011. Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-seq. Nat Protoc 6: 1656–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar W, de Jong J, Pindyurin AV, Pagie L, Meuleman W, de Ridder J, Berns A, Wessels LFA, van Lohuizen M, van Steensel B. 2013. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell 154: 914–927. [DOI] [PubMed] [Google Scholar]
- Alipour E, Marko JF. 2012. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res 40: 11202–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BL, Taatjes DJ. 2015. The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol 16: 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandhakumar J, Moustafa YW, Chowdhary S, Kainth AS, Gross DS. 2016. Evidence for multiple mediator complexes in yeast independently recruited by activated heat shock factor. Mol Cell Biol 36: 1943–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KM, Anderson DM, McAnally JR, Shelton JM, Bassel-Duby R, Olson EN. 2016. Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature 539: 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R. 2015. Promoter or enhancer, what's the difference? Deconstruction of established distinctions and presentation of a unifying model. BioEssays 37: 314–323. [DOI] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. 2014a. An atlas of active enhancers across human cell types and tissues. Nature 507: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Refsing Andersen P, Valen E, Core LJ, Bornholdt J, Boyd M, Heick Jensen T, Sandelin A. 2014b. Nuclear stability and transcriptional directionality separate functionally distinct RNA species. Nat Commun 5: 5336. [DOI] [PubMed] [Google Scholar]
- Archer TK, Lefebvre P, Wolford RG, Hager GL. 1992. Transcription factor loading on the MMTV promoter: a bimodal mechanism for promoter activation. Science 255: 1573–1576. [DOI] [PubMed] [Google Scholar]
- Arner E, Daub CO, Vitting-Seerup K, Andersson R, Lilje B, Drabløs F, Lennartsson A, Rönnerblad M, Hrydziuszko O, Vitezic M, et al. 2015. Gene regulation. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347: 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, Stark A. 2013. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339: 1074–1077. [DOI] [PubMed] [Google Scholar]
- Arnold CD, Zabidi MA, Pagani M, Rath M, Schernhuber K, Kazmar T, Stark A. 2017. Genome-wide assessment of sequence-intrinsic enhancer responsiveness at single-base-pair resolution. Nat Biotechnol 35: 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwell JD. 2006. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat Rev Immunol 6: 532–540. [DOI] [PubMed] [Google Scholar]
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. 2009. Diversity and complexity in DNA recognition by transcription factors. Science 324: 1720–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW, Murison A, Langenfeld K, Petretich M, Scognamiglio R, et al. 2018. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 553: 515–520. [DOI] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, Rosen MK. 2017. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18: 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji J, Rusconi S, Schaffner W. 1981. Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell 27: 299–308. [DOI] [PubMed] [Google Scholar]
- Barolo S. 2012. Shadow enhancers: frequently asked questions about distributed cis-regulatory information and enhancer redundancy. BioEssays 34: 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barozzi I, Simonatto M, Bonifacio S, Yang L, Rohs R, Ghisletti S, Natoli G. 2014. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol Cell 54: 844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartman CR, Hsu SC, Hsiung CCS, Raj A, Blobel GA. 2016. Enhancer regulation of transcriptional bursting parameters revealed by forced chromatin looping. Mol Cell 62: 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, Pugh BF. 2004. Identification and distinct regulation of yeast TATA box-containing genes. Cell 116: 699–709. [DOI] [PubMed] [Google Scholar]
- Bassett AR, Akhtar A, Barlow DP, Bird AP, Brockdorff N, Duboule D, Ephrussi A, Ferguson-Smith AC, Gingeras TR, Haerty W, et al. 2014. Considerations when investigating lncRNA function in vivo. eLife 3: e03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DTW, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. 2013. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24: 660–672. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Liu CL, Humphrey EL, Perlstein EO, Schreiber SL. 2004. Global nucleosome occupancy in yeast. Genome Biol 5: R62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry J, Weber SC, Vaidya N, Haataja M, Brangwynne CP. 2015. RNA transcription modulates phase transition-driven nuclear body assembly. Proc Natl Acad Sci 112: E5237–E5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, Long RM. 1998. Localization of ASH1 mRNA particles in living yeast. Mol Cell 2: 437–445. [DOI] [PubMed] [Google Scholar]
- Blackman RK, Sanicola M, Raftery LA, Gillevet T, Gelbart WM. 1991. An extensive 3′ cis-regulatory region directs the imaginal disk expression of decapentaplegic, a member of the TGF-β family in Drosophila. Development 111: 657–666. [DOI] [PubMed] [Google Scholar]
- Blackwood EM, Kadonaga JT. 1998. Going the distance: a current view of enhancer action. Science 281: 60–63. [DOI] [PubMed] [Google Scholar]
- Boija A, Mahat DB, Zare A, Holmqvist P-H, Philip P, Meyers DJ, Cole PA, Lis JT, Stenberg P, Mannervik M. 2017. CBP regulates recruitment and release of promoter-proximal RNA polymerase II. Mol Cell 68: 491–503.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonn S, Zinzen RP, Girardot C, Gustafson EH, Perez-Gonzalez A, Delhomme N, Ghavi-Helm Y, Wilczyński B, Riddell A, Furlong EEM. 2012. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet 44: 148–156. [DOI] [PubMed] [Google Scholar]
- Bose DA, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, Berger SL. 2017. RNA binding to CBP stimulates histone acetylation and transcription. Cell 168: 135–149.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothma JP, Garcia HG, Ng S, Perry MW, Gregor T, Levine M. 2015. Enhancer additivity and non-additivity are determined by enhancer strength in the Drosophila embryo. eLife 4: e07956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441: 349–353. [DOI] [PubMed] [Google Scholar]
- Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. 2008. High-resolution mapping and characterization of open chromatin across the genome. Cell 132: 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogaard K, Xi L, Wang J-P, Widom J. 2012. A map of nucleosome positions in yeast at base-pair resolution. Nature 486: 496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buecker C, Wysocka J. 2012. Enhancers as information integration hubs in development: lessons from genomics. Trends Genet 28: 276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman FD. 2003. Targeting survival: integration site selection by retroviruses and LTR-retrotransposons. Cell 115: 135–138. [DOI] [PubMed] [Google Scholar]
- Butler JE, Kadonaga JT. 2001. Enhancer-promoter specificity mediated by DPE or TATA core promoter motifs. Genes Dev 15: 2515–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Wysocka J. 2013. Modification of enhancer chromatin: what, how, and why? Mol Cell 49: 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavò E, Khoueiry P, Garfield DA, Geeleher P, Zichner T, Gustafson EH, Ciglar L, Korbel JO, Furlong EEM. 2016. Shadow enhancers are pervasive features of developmental regulatory networks. Curr Biol 26: 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, et al. 2015. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527: 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey LB, van Dijk D, Sloot PMA, Kaandorp JA, Segal E. 2013. Promoter sequence determines the relationship between expression level and noise. PLoS Biol 11: e1001528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catarino RR, Neumayr C, Stark A. 2017. Promoting transcription over long distances. Nat Genet 49: 972–973. [DOI] [PubMed] [Google Scholar]
- Charoensawan V, Janga SC, Bulyk ML, Babu MM, Teichmann SA. 2012. DNA sequence preferences of transcriptional activators correlate more strongly than repressors with nucleosomes. Mol Cell 47: 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Kapoor A, Akiyama JA, Auer DR, Lee D, Gabriel S, Berrios C, Pennacchio LA, Chakravarti A. 2016. Enhancer variants synergistically drive dysfunction of a gene regulatory network in Hirschsprung disease. Cell 167: 355–368.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari HG, Cohen BA. 2018. Local sequence features that influence AP-1 cis-regulatory activity. Genome Res 28: 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Larson DR. 2016. What have single-molecule studies taught us about gene expression? Genes Dev 30: 1796–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RA-J, Down TA, Stempor P, Chen QB, Egelhofer TA, Hillier LW, Jeffers TE, Ahringer J. 2013. The landscape of RNA polymerase II transcription initiation in C. elegans reveals promoter and enhancer architectures. Genome Res 23: 1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang Z, Li L, Chen B-C, Revyakin A, Hajj B, Legant W, Dahan M, Lionnet T, Betzig E, et al. 2014. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156: 1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, Sternberg SH, Joung JK, Yildiz A, Doudna JA. 2017. Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature 550: 407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, et al. 2014. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 159: 1126–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubb JR, Trcek T, Shenoy SM, Singer RH. 2006. Transcriptional pulsing of a developmental gene. Curr Biol 16: 1018–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. 2011. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469: 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. 2002. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell 9: 279–289. [DOI] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. 2009. The biology of chromatin remodeling complexes. Annu Rev Biochem 78: 273–304. [DOI] [PubMed] [Google Scholar]
- Claussnitzer M, Dankel SN, Kim K-H, Quon G, Meuleman W, Haugen C, Glunk V, Sousa IS, Beaudry JL, Puviindran V, et al. 2015. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med 373: 895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AJ, Saiakhova A, Corradin O, Luppino JM, Lovrenert K, Bartels CF, Morrow JJ, Mack SC, Dhillon G, Beard L, et al. 2017. Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nat Commun 8: 14400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. 2008. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, Lis JT. 2014. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet 46: 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, Zhang F. 2017. RNA editing with CRISPR–Cas13. Science 358: 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremisi C, Pignatti PF, Croissant O, Yaniv M. 1975. Chromatin-like structures in polyoma virus and simian virus 10 lytic cycle. J Virol 17: 204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker J, Abe N, Rinaldi L, McGregor AP, Frankel N, Wang S, Alsawadi A, Valenti P, Plaza S, Payre F, et al. 2015. Low affinity binding site clusters confer hox specificity and regulatory robustness. Cell 160: 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao LTM, Galindo-Albarrán AO, Castro-Mondragon JA, Andrieu-Soler C, Medina-Rivera A, Souaid C, Charbonnier G, Griffon A, Vanhille L, Stephen T, et al. 2017. Genome-wide characterization of mammalian promoters with distal enhancer functions. Nat Genet 49: 1073–1081. [DOI] [PubMed] [Google Scholar]
- Dar RD, Razooky BS, Singh A, Trimeloni TV, McCollum JM, Cox CD, Simpson ML, Weinberger LS. 2012. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc Natl Acad Sci 109: 17454–17459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Pai AA, Pique-Regi R, Veyrieras J-B, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, et al. 2012. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482: 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Ramsköld D, Reinius B, Sandberg R. 2014. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343: 193–196. [DOI] [PubMed] [Google Scholar]
- Deplancke B, Alpern D, Gardeux V. 2016. The genetics of transcription factor DNA binding variation. Cell 166: 538–554. [DOI] [PubMed] [Google Scholar]
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Müller H, Ragoussis J, Wei CL, Natoli G. 2010. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol 8: e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao Y, Fang R, Li B, Meng Z, Yu J, Qiu Y, Lin KC, Huang H, Liu T, Marina RJ, et al. 2017. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat Methods 14: 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickel DE, Zhu Y, Nord AS, Wylie JN, Akiyama JA, Afzal V, Plajzer-Frick I, Kirkpatrick A, Goettgens B, Bruneau BG, et al. 2014. Function-based identification of mammalian enhancers using site-specific integration. Nat Methods 11: 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon N, Trimborn T, Strouboulis J, Fraser P, Grosveld F. 1997. The effect of distance on long-range chromatin interactions. Mol Cell 1: 131–139. [DOI] [PubMed] [Google Scholar]
- Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, Rando OJ. 2007. Dynamics of replication-independent histone turnover in budding yeast. Science 315: 1405–1408. [DOI] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. 2016. Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167: 1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. 2015. Chromatin architecture reorganization during stem cell differentiation. Nature 518: 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohmen RJ, Wu P, Varshavsky A. 1994. Heat-inducible degron: a method for constructing temperature-sensitive mutants. Science 263: 1273–1276. [DOI] [PubMed] [Google Scholar]
- Domcke S, Bardet AF, Adrian Ginno P, Hartl D, Burger L, Schübeler D. 2015. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 528: 575–579. [DOI] [PubMed] [Google Scholar]
- Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N, Still CD II, Garcia BA, Adelman K, Wysocka J. 2017. Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol Cell 66: 568–576.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukler N, Gulko B, Huang Y-F, Siepel A. 2016. Is a super-enhancer greater than the sum of its parts? Nat Genet 49: 2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebmeier CC, Erickson B, Allen BL, Allen MA, Kim H, Fong N, Jacobsen JR, Liang K, Shilatifard A, Dowell RD, et al. 2017. Human TFIIH kinase CDK7 regulates transcription-associated chromatin modifications. Cell Rep 20: 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds JW, Mahadevan LC. 2004. MAP kinases as structural adaptors and enzymatic activators in transcription complexes. J Cell Sci 117: 3715–3723. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, Lander ES. 2016. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539: 452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erceg J, Saunders TE, Girardot C, Devos DP, Hufnagel L, Furlong EEM. 2014. Subtle changes in motif positioning cause tissue-specific effects on robustness of an enhancer's activity. PLoS Genet 10: e1004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. 2012. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9: 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa JM. 2016. Revisiting lncRNAs: how do you know yours is not an eRNA? Mol Cell 62: 1–2. [DOI] [PubMed] [Google Scholar]
- Farley EK, Olson KM, Zhang W, Brandt AJ, Rokhsar DS, Levine MS. 2015. Suboptimization of developmental enhancers. Science 350: 325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley EK, Olson KM, Zhang W, Rokhsar DS, Levine MS. 2016. Syntax compensates for poor binding sites to encode tissue specificity of developmental enhancers. Proc Natl Acad Sci 113: 6508–6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore C, Cohen BA. 2016. Interactions between pluripotency factors specify cis-regulation in embryonic stem cells. Genome Res 26: 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Zorrilla JM, López-Vidriero I, Carrasco JL, Godoy M, Vera P, Solano R. 2014. DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc Natl Acad Sci 111: 2367–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schöpflin R, Kraft K, Kempfer R, Jerković I, Chan W-L, et al. 2016. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538: 265–269. [DOI] [PubMed] [Google Scholar]
- Frankel N, Davis GK, Vargas D, Wang S, Payre F, Stern DL. 2010. Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature 466: 490–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser J, Ferrai C, Chiariello AM, Schueler M, Rito T, Laudanno G, Barbieri M, Moore BL, Kraemer DCA, Aitken S, et al. 2015. Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol Syst Biol 11: 852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. 2014. Improving CRISPR–Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32: 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. 2016. Formation of chromosomal domains by loop extrusion. Cell Rep 15: 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukaya T, Lim B, Levine M. 2016. Enhancer control of transcriptional bursting. Cell 166: 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, Engreitz JM. 2016. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354: 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, Tabar V. 2014. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346: 1529–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardini A, Baillat D, Cesaroni M, Hu D, Marinis JM, Wagner EJ, Lazar MA, Shilatifard A, Shiekhattar R. 2014. Integrator regulates transcriptional initiation and pause release following activation. Mol Cell 56: 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavi-Helm Y, Klein FA, Pakozdi T, Ciglar L, Noordermeer D, Huber W, Furlong EEM. 2014. Enhancer loops appear stable during development and are associated with paused polymerase. Nature 512: 96–100. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. 2013. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154: 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist DA, Santos Dos G, Fargo DC, Xie B, Gao Y, Li L, Adelman K. 2010. Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell 143: 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD, Banaszynski LA, Noh K-M, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. 2010. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140: 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding I, Paulsson J, Zawilski SM, Cox EC. 2005. Real-time kinetics of gene activity in individual bacteria. Cell 123: 1025–1036. [DOI] [PubMed] [Google Scholar]
- Gressel S, Schwalb B, Decker TM, Qin W, Leonhardt H, Eick D, Cramer P. 2017. CDK9-dependent RNA polymerase II pausing controls transcription initiation. eLife 6: e29736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Zhang X, Wang L, Engreitz J, Melnikov A, Rogov P, Tewhey R, Isakova A, Deplancke B, Bernstein BE, et al. 2017. Systematic dissection of genomic features determining transcription factor binding and enhancer function. Proc Natl Acad Sci 114: E1291–E1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, et al. 2015. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell 162: 900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarhuis JHI, van der Weide RH, Blomen VA, Yáñez-Cuna JO, Amendola M, van Ruiten MS, Krijger PHL, Teunissen H, Medema RH, van Steensel B, et al. 2017. The cohesin release factor WAPL restricts chromatin loop extension. Cell 169: 693–707.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, Kraus WL. 2011. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 145: 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Murakami S, Nagari A, Danko CG, Kraus WL. 2013. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res 23: 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CWH, Ye C, Ping JLH, Mulawadi F, et al. 2011. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet 43: 630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanssen LLP, Kassouf MT, Oudelaar AM, Biggs D, Preece C, Downes DJ, Gosden M, Sharpe JA, Sloane-Stanley JA, Hughes JR, et al. 2017. Tissue-specific CTCF–cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nat Cell Biol 19: 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, Crabtree GR. 2011. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 21: 396–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK. 2008. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell 31: 925–932. [DOI] [PubMed] [Google Scholar]
- Hay D, Hughes JR, Babbs C, Davies JOJ, Graham BJ, Hanssen L, Kassouf MT, Marieke Oudelaar AM, Sharpe JA, Suciu MC, et al. 2016. Genetic dissection of the α-globin super-enhancer in vivo. Nat Genet 48: 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459: 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG, Sakai A, Loeb GB, Ahmad K. 2009. Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res 19: 460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Gilchrist DA, Nechaev S, Bern M, Muse GW, Burkholder A, Fargo DC, Adelman K. 2013. Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals. Mol Cell 52: 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Scruggs BS, Inouye MO, Muse GW, Williams LH, Burkholder AB, Lavender CA, Fargo DC, Adelman K. 2018. Widespread transcriptional pausing and elongation control at enhancers. Genes Dev 32: 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz H-M, Garruss A, Shilatifard A. 2013. SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem Sci 38: 621–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz H-M, Morgan M, Gao X, Jackson J, Rickels R, Swanson SK, Florens L, Washburn MP, Eissenberg JC, Shilatifard A. 2014. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 345: 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton IB, D'Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA. 2015. Epigenome editing by a CRISPR–Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33: 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. 2013. Super-enhancers in the control of cell identity and disease. Cell 155: 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, Bradner JE, Young RA. 2015. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell 58: 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, Sharp PA. 2017. A phase separation model for transcriptional control. Cell 169: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MM, Buske OJ, Wang J, Weng Z, Bilmes JA, Noble WS. 2012. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods 9: 473–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung G, Bar-Ziv R, Rosin D, Tokuriki N, Tawfik DS, Oren M, Barkai N. 2012. Noise-mean relationship in mutated promoters. Genome Res 22: 2409–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou C, Li L, Qin ZS, Corces VG. 2012. Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol Cell 48: 471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housden BE, Muhar M, Gemberling M, Gersbach CA, Stainier DYR, Seydoux G, Mohr SE, Zuber J, Perrimon N. 2017. Loss-of-function genetic tools for animal models: cross-species and cross-platform differences. Nat Rev Genet 18: 24–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh T-HS, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ. 2015. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell 162: 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume MA, Barrera LA, Gisselbrecht SS, Bulyk ML. 2015. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein–DNA interactions. Nucleic Acids Res 43: D117–D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbalzano AN, Zaret KS, Kingston RE. 1994. Transcription factor (TF) IIB and TFIIA can independently increase the affinity of the TATA-binding protein for DNA. J Biol Chem 269: 8280–8286. [PubMed] [Google Scholar]
- Ing-Simmons E, Seitan VC, Faure AJ, Flicek P, Carroll T, Dekker J, Fisher AG, Lenhard B, Merkenschlager M. 2015. Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res 25: 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue F, Kircher M, Martin B, Cooper GM, Witten DM, McManus MT, Ahituv N, Shendure J. 2017. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res 27: 38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inukai S, Kock KH, Bulyk ML. 2017. Transcription factor-DNA binding: beyond binding site motifs. Curr Opin Genet Dev 43: 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakova A, Berset Y, Hatzimanikatis V, Deplancke B. 2016. Quantification of cooperativity in heterodimer-DNA binding improves the accuracy of binding specificity models. J Biol Chem 291: 10293–10306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakova A, Groux R, Imbeault M, Rainer P, Alpern D, Dainese R, Ambrosini G, Trono D, Bucher P, Deplancke B. 2017. SMiLE-seq identifies binding motifs of single and dimeric transcription factors. Nat Methods 14: 316–322. [DOI] [PubMed] [Google Scholar]
- Isoda T, Moore AJ, He Z, Chandra V, Aida M, Denholtz M, Piet van Hamburg J, Fisch KM, Chang AN, Fahl SP, et al. 2017. Non-coding transcription instructs chromatin folding and compartmentalization to dictate enhancer-promoter communication and T cell fate. Cell 171: 103–119.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi-Doi M, Donahue G, Kakumanu A, Watts JA, Mahony S, Pugh BF, Lee D, Kaestner KH, Zaret KS. 2016. The pioneer transcription factor FoxA maintains an accessible nucleosome configuration at enhancers for tissue-specific gene activation. Mol Cell 62: 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Vale RD. 2017. RNA phase transitions in repeat expansion disorders. Nature 546: 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH, et al. 2004. From silencing to gene expression: real-time analysis in single cells. Cell 116: 683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo C, Langelier M-F, Bataille AR, Pascal JM, Pugh BF, Robert F. 2016. Tail and kinase modules differently regulate core mediator recruitment and function in vivo. Mol Cell 64: 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Felsenfeld G. 2007. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev 21: 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. 2007. Genome-wide mapping of in vivo protein–DNA interactions. Science 316: 1497–1502. [DOI] [PubMed] [Google Scholar]
- Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei G, et al. 2013. DNA-binding specificities of human transcription factors. Cell 152: 327–339. [DOI] [PubMed] [Google Scholar]
- Jolma A, Yin Y, Nitta KR, Dave K, Popov A, Taipale M, Enge M, Kivioja T, Morgunova E, Taipale J. 2015. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527: 384–388. [DOI] [PubMed] [Google Scholar]
- Jonkers I, Kwak H, Lis JT. 2014. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife 3: e02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Shimada M, Armache A, Li CH, Miller RM, Lin S, Yang A, Dill BD, Molina H, Park H-S, et al. 2016. Chromatin kinases act on transcription factors and histone tails in regulation of inducible transcription. Mol Cell 64: 347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, Chun HB, Tough DF, Prinjha RK, Benner C, et al. 2013. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell 51: 310–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T, Kanno Y, LeRoy G, Campos E, Sun H-W, Brooks SR, Vahedi G, Heightman TD, Garcia BA, Reinberg D, et al. 2014. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol 21: 1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. 2009. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458: 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Dudley JT, Kukurba KR, Chen R, Butte AJ, Montgomery SB, Snyder M. 2013. Systematic functional regulatory assessment of disease-associated variants. Proc Natl Acad Sci 110: 9607–9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, Largaespada DA, Ivics Z. 2017. Transposons as tools for functional genomics in vertebrate models. Trends Genet 33: 784–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, Maehr R. 2015. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods 12: 401–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebriaei P, Izsvák Z, Narayanavari SA, Singh H, Ivics Z. 2017. Gene therapy with the sleeping beauty transposon system. Trends Genet 33: 852–870. [DOI] [PubMed] [Google Scholar]
- Kemmeren P, Sameith K, van de Pasch LAL, Benschop JJ, Lenstra TL, Margaritis T, O'Duibhir E, Apweiler E, van Wageningen S, Ko CW, et al. 2014. Large-scale genetic perturbations reveal regulatory networks and an abundance of gene-specific repressors. Cell 157: 740–752. [DOI] [PubMed] [Google Scholar]
- Keung AJ, Bashor CJ, Kiriakov S, Collins JJ, Khalil AS. 2014. Using targeted chromatin regulators to engineer combinatorial and spatial transcriptional regulation. Cell 158: 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al. 2011. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 471: 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradpour P, Ernst J, Melnikov A, Rogov P, Wang L, Zhang X, Alston J, Mikkelsen TS, Kellis M. 2013. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res 23: 800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T-K, Shiekhattar R. 2015. Architectural and functional commonalities between enhancers and promoters. Cell 162: 948–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Woo EM, Chong YTE, Homenko DR, Kraus WL. 2006. Acetylation of estrogen receptor α by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol Endocrinol 20: 1479–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T-K, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. 2010. Widespread transcription at neuronal activity-regulated enhancers. Nature 465: 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann TS, Black JB, Chellappan M, Safi A, Song L, Hilton IB, Crawford GE, Reddy TE, Gersbach CA. 2017. CRISPR–Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat Biotechnol 35: 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales APW, Li Z, Peterson RT, Yeh J-RJ, et al. 2015. Engineered CRISPR–Cas9 nucleases with altered PAM specificities. Nature 523: 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. 2016. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529: 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch F, Fenouil R, Gut M, Cauchy P, Albert TK, Zacarias-Cabeza J, Spicuglia S, de la Chapelle AL, Heidemann M, Hintermair C, et al. 2011. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat Struct Mol Biol 18: 956–963. [DOI] [PubMed] [Google Scholar]
- Korkmaz G, Lopes R, Ugalde AP, Nevedomskaya E, Han R, Myacheva K, Zwart W, Elkon R, Agami R. 2016. Functional genetic screens for enhancer elements in the human genome using CRISPR–Cas9. Nat Biotechnol 34: 192–198. [DOI] [PubMed] [Google Scholar]
- Kothary R, Clapoff S, Darling S, Perry MD, Moran LA, Rossant J. 1989. Inducible expression of an hsp68-lacZ hybrid gene in transgenic mice. Development 105: 707–714. [DOI] [PubMed] [Google Scholar]
- Kulakovskiy IV, Vorontsov IE, Yevshin IS, Sharipov RN, Fedorova AD, Rumynskiy EI, Medvedeva YA, Magana-Mora A, Bajic VB, Papatsenko DA, et al. 2018. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-seq analysis. Nucleic Acids Res 46: D252–D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvon EZ. 2015. Using transgenic reporter assays to functionally characterize enhancers in animals. Genomics 106: 185–192. [DOI] [PubMed] [Google Scholar]
- Kvon EZ, Kazmar T, Stampfel G, Yáñez-Cuna JO, Pagani M, Schernhuber K, Dickson BJ, Stark A. 2014. Genome-scale functional characterization of Drosophila developmental enhancers in vivo. Nature 512: 91–95. [DOI] [PubMed] [Google Scholar]
- Kvon EZ, Kamneva OK, Melo US, Barozzi I, Osterwalder M, Mannion BJ, Tissières V, Pickle CS, Plajzer-Frick I, Lee EA, et al. 2016. Progressive loss of function in a limb enhancer during snake evolution. Cell 167: 633–642.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, Lis JT. 2013. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339: 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasnieski JC, Mogno I, Myers CA, Corbo JC, Cohen BA. 2012. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc Natl Acad Sci 109: 19498–19503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasnieski JC, Fiore C, Chaudhari HG, Cohen BA. 2014. High-throughput functional testing of ENCODE segmentation predictions. Genome Res 24: 1595–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. 2014. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511: 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Gardini A, Zhang A, Shiekhattar R. 2015. Integrator mediates the biogenesis of enhancer RNAs. Nature 525: 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MTY, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, et al. 2013. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 498: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam DD, de Souza FSJ, Nasif S, Yamashita M, López-Leal R, Otero-Corchon V, Meece K, Sampath H, Mercer AJ, Wardlaw SL, et al. 2015. Partially redundant enhancers cooperatively maintain mammalian Pomc expression above a critical functional threshold. PLoS Genet 11: e1004935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DR. 2011. What do expression dynamics tell us about the mechanism of transcription? Curr Opin Genet Dev 21: 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DR, Zenklusen D, Wu B, Chao JA, Singer RH. 2011a. Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science 332: 475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson MH, Landick R, Block SM. 2011b. Single-molecule studies of RNA polymerase: one singular sensation, every little step it takes. Mol Cell 41: 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, Narlikar GJ. 2017. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547: 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-E, Wang C, Xu S, Cho Y-W, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, et al. 2013. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife 2: e01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenstra TL, Benschop JJ, Kim T, Schulze JM, Brabers NACH, Margaritis T, van de Pasch LAL, van Heesch SAAC, Brok MO, Groot Koerkamp MJA, et al. 2011. The specificity and topology of chromatin interaction pathways in yeast. Mol Cell 42: 536–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenstra TL, Rodriguez J, Chen H, Larson DR. 2016. Transcription dynamics in living cells. Annu Rev Biophys 45: 25–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Cattoglio C, Tjian R. 2014. Looping back to leap forward: transcription enters a new era. Cell 157: 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. 2013. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340: 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. 2013. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498: 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Rivera CM, Ishii H, Jin F, Selvaraj S, Lee AY, Dixon JR, Ren B. 2014. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS ONE 9: e114485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, Rosenfeld MG. 2016. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet 17: 207–223. [DOI] [PubMed] [Google Scholar]
- Lidor Nili E, Field Y, Lubling Y, Widom J, Oren M, Segal E. 2010. p53 binds preferentially to genomic regions with high DNA-encoded nucleosome occupancy. Genome Res 20: 1361–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little SC, Tikhonov M, Gregor T. 2013. Precise developmental gene expression arises from globally stochastic transcriptional activity. Cell 154: 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Lee C-K, Granek JA, Clarke ND, Lieb JD. 2006. Whole-genome comparison of Leu3 binding in vitro and in vivo reveals the importance of nucleosome occupancy in target site selection. Genome Res 16: 1517–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Local A, Huang H, Albuquerque CP, Singh N, Lee AY, Wang W, Wang C, Hsia JE, Shiau AK, Ge K, et al. 2018. Identification of H3K4me1-associated proteins at mammalian enhancers. Nat Genet 50: 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes R, Korkmaz G, Agami R. 2016. Applying CRISPR–Cas9 tools to identify and characterize transcriptional enhancers. Nat Rev Mol Cell Biol 17: 597–604. [DOI] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. 2013. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153: 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubas M, Andersen PR, Schein A, Dziembowski A, Kudla G, Jensen TH. 2015. The human nuclear exosome targeting complex is loaded onto newly synthesized RNA to direct early ribonucleolysis. Cell Rep 10: 178–192. [DOI] [PubMed] [Google Scholar]
- Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. 2015. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161: 1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. 2015. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161: 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, et al. 2014. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346: 1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinić M, Aktas T, Ruf S, Spitz F. 2013. An integrated holo-enhancer unit defines tissue and gene specificity of the Fgf8 regulatory landscape. Dev Cell 24: 530–542. [DOI] [PubMed] [Google Scholar]
- Maston GA, Landt SG, Snyder M, Green MR. 2012. Characterization of enhancer function from genome-wide analyses. Annu Rev Genom Human Genet 13: 29–57. [DOI] [PubMed] [Google Scholar]
- Mathelier A, Fornes O, Arenillas DJ, Chen C-Y, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley-Hunt R, et al. 2016. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 44: D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. 2012. Systematic localization of common disease-associated variation in regulatory DNA. Science 337: 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melgar MF, Collins FS, Sethupathy P. 2011. Discovery of active enhancers through bidirectional expression of short transcripts. Genome Biol 12: R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG, Kinney JB, et al. 2012. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol 30: 271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JAF, Elkon R, Melo SA, Léveillé N, Kalluri R, et al. 2013. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell 49: 524–535. [DOI] [PubMed] [Google Scholar]
- Merkenschlager M, Nora EP. 2016. CTCF and cohesin in genome folding and transcriptional gene regulation. Annu Rev Genomics Hum Genet 17: 17–43. [DOI] [PubMed] [Google Scholar]
- Michel M, Demel C, Zacher B, Schwalb B, Krebs S, Blum H, Gagneur J, Cramer P. 2017. TT-seq captures enhancer landscapes immediately after T-cell stimulation. Mol Syst Biol 13: 920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieczkowski J, Cook A, Bowman SK, Mueller B, Alver BH, Kundu S, Deaton AM, Urban JA, Larschan E, Park PJ, et al. 2016. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat Commun 7: 11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhaylichenko O, Bondarenko V, Harnett D, Schor IE, Males M, Viales RR, Furlong EEM. 2018. The degree of enhancer or promoter activity is reflected by the levels and directionality of eRNA transcription. Genes Dev 32: 42–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorthy SD, Davidson S, Shchuka VM, Singh G, Malek-Gilani N, Langroudi L, Martchenko A, So V, Macpherson NN, Mitchell JA. 2017. Enhancers and super-enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res 27: 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgunova E, Taipale J. 2017. Structural perspective of cooperative transcription factor binding. Curr Opin Struct Biol 47: 1–8. [DOI] [PubMed] [Google Scholar]
- Mousavi K, Zare H, Dell'Orso S, Grøntved L, Gutierrez-Cruz G, Derfoul A, Hager GL, Sartorelli V. 2013. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol Cell 51: 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller B, Mieczkowski J, Kundu S, Wang P, Sadreyev R, Tolstorukov MY, Kingston RE. 2017. Widespread changes in nucleosome accessibility without changes in nucleosome occupancy during a rapid transcriptional induction. Genes Dev 31: 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muerdter F, Stark A. 2016. Gene regulation: activation through space. Curr Biol 26: R895–R898. [DOI] [PubMed] [Google Scholar]
- Muerdter F, Boryń ŁM, Arnold CD. 2015. STARR-seq—principles and applications. Genomics 106: 145–150. [DOI] [PubMed] [Google Scholar]
- Muerdter F, Boryń ŁM, Woodfin AR, Neumayr C, Rath M, Zabidi MA, Pagani M, Haberle V, Kazmar T, Catarino RR, et al. 2018. Resolving systematic errors in widely used enhancer activity assays in human cells. Nat Methods 15: 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtha M, Tokcaer-Keskin Z, Tang Z, Strino F, Chen X, Wang Y, Xi X, Basilico C, Brown S, Bonneau R, et al. 2014. FIREWACh: high-throughput functional detection of transcriptional regulatory modules in mammalian cells. Nat Methods 11: 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. 2005. A fine-scale map of recombination rates and hotspots across the human genome. Science 310: 321–324. [DOI] [PubMed] [Google Scholar]
- Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. 2013. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502: 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan K, Lambert SA, Yang AWH, Riddell J, Mnaimneh S, Zheng H, Albu M, Najafabadi HS, Reece-Hoyes JS, Fuxman Bass JI, et al. 2015. Mapping and analysis of Caenorhabditis elegans transcription factor sequence specificities. eLife 4: e06967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, Reinberg D. 2015. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347: 1017–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra V, Bulajić M, Dekker J, Mazzoni EO, Reinberg D. 2016. CTCF-mediated topological boundaries during development foster appropriate gene regulation. Genes Dev 30: 2657–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K. 2001. Disseminating the genome: joining, resolving, and separating sister chromatids during mitosis and meiosis. Annu Rev Genet 35: 673–745. [DOI] [PubMed] [Google Scholar]
- Natoli G, Andrau J-C. 2012. Noncoding transcription at enhancers: general principles and functional models. Annu Rev Genet 46: 1–19. [DOI] [PubMed] [Google Scholar]
- Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. 2009. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol Cell 36: 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z, Saunders A, Fuda NJ, Yao J, Suarez J-R, Webb WW, Lis JT. 2008. P-TEFb is critical for the maturation of RNA polymerase II into productive elongation in vivo. Mol Cell Biol 28: 1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. 2009. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6: 917–922. [DOI] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. 2012. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485: 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton A-L, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG. 2017. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169: 930–944.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak MA, Boerlijst MC, Cooke J, Smith JM. 1997. Evolution of genetic redundancy. Nature 388: 167–171. [DOI] [PubMed] [Google Scholar]
- Noyes MB, Christensen RG, Wakabayashi A, Stormo GD, Brodsky MH, Wolfe SA. 2008. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell 133: 1277–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides G, Lagrange T, Reinberg D. 1996. The general transcription factors of RNA polymerase II. Genes Dev 10: 2657–2683. [DOI] [PubMed] [Google Scholar]
- Osterwalder M, Barozzi I, Tissières V, Fukuda-Yuzawa Y, Mannion BJ, Afzal SY, Lee EA, Zhu Y, Plajzer-Frick I, Pickle CS, et al. 2018. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paralkar VR, Taborda CC, Huang P, Yao Y, Kossenkov AV, Prasad R, Luan J, Davies JOJ, Hughes JR, Hardison RC, et al. 2016. Unlinking an lncRNA from its associated cis element. Mol Cell 62: 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, et al. 2008. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132: 422–433. [DOI] [PubMed] [Google Scholar]
- Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee S-I, Cooper GM, et al. 2012. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol 30: 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelish HE, Liau BB, Nitulescu II, Tangpeerachaikul A, Poss ZC, Da Silva DH, Caruso BT, Arefolov A, Fadeyi O, Christie AL, et al. 2015. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 526: 273–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pengelly AR, Copur Ö, Jäckle H, Herzig A. 2013. A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science 339: 698–699. [DOI] [PubMed] [Google Scholar]
- Perlmann T, Wrange O. 1988. Specific glucocorticoid receptor binding to DNA reconstituted in a nucleosome. EMBO J 7: 3073–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry MW, Boettiger AN, Bothma JP, Levine M. 2010. Shadow enhancers foster robustness of Drosophila gastrulation. Curr Biol 20: 1562–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrenko N, Jin Y, Wong KH, Struhl K. 2017. Evidence that Mediator is essential for Pol II transcription, but is not a required component of the preinitiation complex in vivo. eLife 6: e28447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollex T, Furlong EEM. 2017. Correlation does not imply causation: histone methyltransferases, but not histone methylation, SET the stage for enhancer activation. Mol Cell 66: 439–441. [DOI] [PubMed] [Google Scholar]
- Pombo A, Dillon N. 2015. Three-dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol 16: 245–257. [DOI] [PubMed] [Google Scholar]
- Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, Doddapaneni H, Beckwith CA, Chan JA, Hills A, Davis M, et al. 2009. The 8q24 cancer risk variant rs6983267 shows long-range interaction with. Nat Genet 41: 882–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradeepa MM, Grimes GR, Kumar Y, Olley G, Taylor GCA, Schneider R, Bickmore WA. 2016. Histone H3 globular domain acetylation identifies a new class of enhancers. Nat Genet 48: 681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152: 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabani M, Raychowdhury R, Jovanovic M, Rooney M, Stumpo DJ, Pauli A, Hacohen N, Schier AF, Blackshear PJ, Friedman N, et al. 2014. High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell 159: 1698–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470: 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. 2006. Stochastic mRNA synthesis in mammalian cells. PLoS Biol 4: e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal N, Srinivasan S, Kooshesh K, Guo Y, Edwards MD, Banerjee B, Syed T, Emons BJM, Gifford DK, Sherwood RI. 2016. High-throughput mapping of regulatory DNA. Nat Biotechnol 34: 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsköld D, Wang ET, Burge CB, Sandberg R. 2009. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol 5: e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang S-C, Hilaire BGS, Engreitz JM, Perez EM, Kieffer-Kwon K-R, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. 2017. Cohesin loss eliminates all loop domains. Cell 171: 305–309.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P, Roth M, Neumann T, Muerdter F, Roe J-S, Muhar M, Deswal S, Cerny-Reiterer S, Peter B, Jude J, et al. 2015. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525: 543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy TE, Gertz J, Pauli F, Kucera KS, Varley KE, Newberry KM, Marinov GK, Mortazavi A, Williams BA, Song L, et al. 2012. Effects of sequence variation on differential allelic transcription factor occupancy and gene expression. Genome Res 22: 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter F, Wienerroither S, Stark A. 2017. Combinatorial function of transcription factors and cofactors. Curr Opin Genet Dev 43: 73–81. [DOI] [PubMed] [Google Scholar]
- Rennie S, Dalby M, Lloret-Llinares M, Bakoulis S, Dalager Vaagenso C, Jensen TH, Andersson R. 2017. Transcription start site analysis reveals widespread divergent transcription in D. melanogaster and core promoter-encoded enhancer activities. bioRxiv 10.1101/221952. [DOI] [PMC free article] [PubMed]
- Rickels R, Herz H-M, Sze CC, Cao K, Morgan MA, Collings CK, Gause M, Takahashi Y-H, Wang L, Rendleman EJ, et al. 2017. Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat Genet 49: 1647–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. 2007. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods 4: 651–657. [DOI] [PubMed] [Google Scholar]
- Roe J-S, Mercan F, Rivera K, Pappin DJ, Vakoc CR. 2015. BET bromodomain inhibition suppresses the function of hematopoietic transcription factors in acute myeloid leukemia. Mol Cell 58: 1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder RG. 1996. The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem Sci 21: 327–335. [PubMed] [Google Scholar]
- Rogers WA, Goyal Y, Yamaya K, Shvartsman SY, Levine MS. 2017. Uncoupling neurogenic gene networks in the Drosophila embryo. Genes Dev 31: 634–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin AJ, Barajas BC, Furlan-Magaril M, Lopez-Pajares V, Mumbach MR, Howard I, Kim DS, Boxer LD, Cairns J, Spivakov M, et al. 2017. Lineage-specific dynamic and pre-established enhancer–promoter contacts cooperate in terminal differentiation. Nat Genet 512: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagai T, Hosoya M, Mizushina Y, Tamura M, Shiroishi T. 2005. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development 132: 797–803. [DOI] [PubMed] [Google Scholar]
- Sanborn AL, Rao SSP, Huang S-C, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. 2015. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci 112: E6456–E6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Joung JK. 2014. CRISPR–Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32: 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Wright J, Zheng K, Shalem O, Fontanillas P, Joung J, Cheng C, Regev A, Zhang F. 2016. High-resolution interrogation of functional elements in the noncoding genome. Science 353: 1545–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santenard A, Ziegler-Birling C, Koch M, Tora L, Bannister AJ, Torres-Padilla M-E. 2010. Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nat Cell Biol 12: 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Algarra D, Dao LTM, Pradel L, España A, Spicuglia S. 2017. Recent advances in high-throughput approaches to dissect enhancer function. F1000Res 6: 939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicka A, Hartl D, Goiser M, Pusch O, Stocsits RR, Tamir IM, Mechtler K, Seiser C. 2014. H3S28 phosphorylation is a hallmark of the transcriptional response to cellular stress. Genome Res 24: 1808–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. 2012. Linking disease associations with regulatory information in the human genome. Genome Res 22: 1748–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaukowitch K, Joo J-Y, Liu X, Watts JK, Martinez C, Kim T-K. 2014. Enhancer RNA facilitates NELF release from immediate early genes. Mol Cell 56: 29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AD, Hu M, Ren B. 2016. Genome-wide mapping and analysis of chromosome architecture. Nat Rev Mol Cell Biol 17: 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder S, Herker E, Itzen F, He D, Thomas S, Gilchrist DA, Kaehlcke K, Cho S, Pollard KS, Capra JA, et al. 2013. Acetylation of RNA polymerase II regulates growth-factor-induced gene transcription in mammalian cells. Mol Cell 52: 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb B, Michel M, Zacher B, Frühauf K, Demel C, Tresch A, Gagneur J, Cramer P. 2016. TT-seq maps the human transient transcriptome. Science 352: 1225–1228. [DOI] [PubMed] [Google Scholar]
- Schwartz YB, Linder-Basso D, Kharchenko PV, Tolstorukov MY, Kim M, Li H-B, Gorchakov AA, Minoda A, Shanower G, Alekseyenko AA, et al. 2012. Nature and function of insulator protein binding sites in the Drosophila genome. Genome Res 22: 2188–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang D-AK, Tönjes M, et al. 2012. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482: 226–231. [DOI] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, Haering CH, Mirny L, et al. 2017. Two independent modes of chromatin organization revealed by cohesin removal. Nature 551: 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scruggs BS, Gilchrist DA, Nechaev S, Muse GW, Burkholder A, Fargo DC, Adelman K. 2015. Bidirectional transcription arises from two distinct hubs of transcription factor binding and active chromatin. Mol Cell 58: 1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedkov Y, Benes JJ, Berger JR, Riker KM, Tillib S, Jones RS, Mazo A. 1999. Molecular genetic analysis of the Drosophila trithorax-related gene which encodes a novel SET domain protein. Mech Dev 82: 171–179. [DOI] [PubMed] [Google Scholar]
- Segal E, Fondufe-Mittendorf Y, Chen L, Thåström A, Field Y, Moore IK, Wang J-PZ, Widom J. 2006. A genomic code for nucleosome positioning. Nature 442: 772–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitan VC, Faure AJ, Zhan Y, McCord RP, Lajoie BR, Ing-Simmons E, Lenhard B, Giorgetti L, Heard E, Fisher AG, et al. 2013. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res 23: 2066–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger K, Armstrong GW, Rowell WJ, Kwan JM, Markstein M, Levine M. 2004. Immunity regulatory DNAs share common organizational features in Drosophila. Mol Cell 13: 19–32. [DOI] [PubMed] [Google Scholar]
- Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. 2012. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148: 458–472. [DOI] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. 2012. A map of the cis-regulatory sequences in the mouse genome. Nature 488: 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. 2012. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81: 65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G, Stark A. 2014. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet 15: 272–286. [DOI] [PubMed] [Google Scholar]
- Sigova AA, Abraham BJ, Ji X, Molinie B, Hannett NM, Guo YE, Jangi M, Giallourakis CC, Sharp PA, Young RA. 2015. Transcription factor trapping by RNA in gene regulatory elements. Science 350: 978–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Schimenti JC. 2015. The genetics of human infertility by functional interrogation of SNPs in mice. Proc Natl Acad Sci 112: 10431–10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skupsky R, Burnett JC, Foley JE, Schaffer DV, Arkin AP. 2010. HIV promoter integration site primarily modulates transcriptional burst size rather than frequency. PLoS Comput Biol 6: e1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, Gómez-Marín C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, et al. 2014. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507: 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RP, Taher L, Patwardhan RP, Kim MJ, Inoue F, Shendure J, Ovcharenko I, Ahituv N. 2013. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat Genet 45: 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, Zaret KS. 2015. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161: 555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EEM. 2012. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13: 613–626. [DOI] [PubMed] [Google Scholar]
- Spitz F, Gonzalez F, Duboule D. 2003. A global control region defines a chromosomal regulatory landscape containing the HoxD cluster. Cell 113: 405–417. [DOI] [PubMed] [Google Scholar]
- Stampfel G, Kazmar T, Frank O, Wienerroither S, Reiter F, Stark A. 2015. Transcriptional regulators form diverse groups with context-dependent regulatory functions. Nature 528: 147–151. [DOI] [PubMed] [Google Scholar]
- St Johnston RD, Hoffmann FM, Blackman RK, Segal D, Grimaila R, Padgett RW, Irick HA, Gelbart WM. 1990. Molecular organization of the decapentaplegic gene in Drosophila melanogaster. Genes Dev 4: 1114–1127. [DOI] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH. 2017. Phase separation drives heterochromatin domain formation. Nature 547: 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur IK, Hallikas O, Vähärautio A, Yan J, Turunen M, Enge M, Taipale M, Karhu A, Aaltonen LA, Taipale J. 2012. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science 338: 1360–1363. [DOI] [PubMed] [Google Scholar]
- Svaren J, Klebanow E, Sealy L, Chalkley R. 1994. Analysis of the competition between nucleosome formation and transcription factor binding. J Biol Chem 269: 9335–9344. [PubMed] [Google Scholar]
- Symmons O, Uslu VV, Tsujimura T, Ruf S, Nassari S, Schwarzer W, Ettwiller L, Spitz F. 2014. Functional and topological characteristics of mammalian regulatory domains. Genome Res 24: 390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons O, Pan L, Remeseiro S, Aktas T, Klein F, Huber W, Spitz F. 2016. The Shh topological domain facilitates the action of remote enhancers by reducing the effects of genomic distances. Dev Cell 39: 529–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak YE, Kleinstiver BP, Nuñez JK, Hsu JY, Horng JE, Gong J, Weissman JS, Joung JK. 2017. Inducible and multiplex gene regulation using CRISPR-Cpf1-based transcription factors. Nat Methods 14: 1163–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S. 2017. Histone variants on the move: substrates for chromatin dynamics. Nat Rev Mol Cell Biol 18: 115–126. [DOI] [PubMed] [Google Scholar]
- Tantale K, Mueller F, Kozulic-Pirher A, Lesne A, Victor J-M, Robert M-C, Capozi S, Chouaib R, Bäcker V, Mateos-Langerak J, et al. 2016. A single-molecule view of transcription reveals convoys of RNA polymerases and multi-scale bursting. Nat Commun 7: 12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessarz P, Kouzarides T. 2014. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol 15: 703–708. [DOI] [PubMed] [Google Scholar]
- Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, Andersen KG, Mikkelsen TS, Lander ES, Schaffner SF, et al. 2016. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 165: 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakore PI, D'Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, Reddy TE, Crawford GE, Gersbach CA. 2015. Highly specific epigenome editing by CRISPR–Cas9 repressors for silencing of distal regulatory elements. Nat Methods 12: 1143–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thåström A, Lowary PT, Widlund HR, Cao H, Kubista M, Widom J. 1999. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J Mol Biol 288: 213–229. [DOI] [PubMed] [Google Scholar]
- Tropberger P, Pott S, Keller C, Kamieniarz-Gdula K, Caron M, Richter F, Li G, Mittler G, Liu ET, Bühler M, et al. 2013. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell 152: 859–872. [DOI] [PubMed] [Google Scholar]
- Tuan D, Kong S, Hu K. 1992. Transcription of the hypersensitive site HS2 enhancer in erythroid cells. Proc Natl Acad Sci 89: 11219–11223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulianov SV, Khrameeva EE, Gavrilov AA, Flyamer IM, Kos P, Mikhaleva EA, Penin AA, Logacheva MD, Imakaev MV, Chertovich A, et al. 2016. Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains. Genome Res 26: 70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulirsch JC, Nandakumar SK, Wang L, Giani FC, Zhang X, Rogov P, Melnikov A, McDonel P, Do R, Mikkelsen TS, et al. 2016. Systematic functional dissection of common genetic variation affecting red blood cell traits. Cell 165: 1530–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Arensbergen J, van Steensel B, Bussemaker HJ. 2014. In search of the determinants of enhancer-promoter interaction specificity. Trends Cell Biol 24: 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, Greenberg ME. 2017. AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol Cell 68: 1067–1082.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA. 2007. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res 35: D88–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Prabhakar S, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Afzal V, Rubin EM, Pennacchio LA. 2008. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat Genet 40: 158–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vockley CM, Guo C, Majoros WH, Nodzenski M, Scholtens DM, Hayes MG, Lowe WL, Reddy TE. 2015. Massively parallel quantification of the regulatory effects of noncoding genetic variation in a human cohort. Genome Res 25: 1206–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vockley CM, D'Ippolito AM, McDowell IC, Majoros WH, Safi A, Song L, Crawford GE, Reddy TE. 2016. Direct GR binding sites potentiate clusters of TF binding across the human genome. Cell 166: 1269–1281.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter PP, Owen-Hughes TA, Côté J, Workman JL. 1995. Stimulation of transcription factor binding and histone displacement by nucleosome assembly protein 1 and nucleoplasmin requires disruption of the histone octamer. Mol Cell Biol 15: 6178–6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. 2011. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 474: 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S-P, Tang Z, Chen C-W, Shimada M, Koche RP, Wang L-H, Nakadai T, Chramiec A, Krivtsov AV, Armstrong SA, et al. 2017. A UTX–MLL4–p300 transcriptional regulatory network coordinately shapes active enhancer landscapes for eliciting transcription. Mol Cell 67: 308–321.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfield L, Ramachandran S, Baptista T, Devys D, Tora L, Hahn S. 2017. Transcription of nearly all yeast RNA polymerase II-transcribed genes is dependent on transcription factor TFIID. Mol Cell 68: 118–129.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman NF, Aneas I, Nobrega MA. 2010. An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res 20: 1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL, et al. 2017. YY1 is a structural regulator of enhancer-promoter loops. Cell 171: 1573–1588.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. 2014. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158: 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, et al. 2008. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451: 796–801. [DOI] [PubMed] [Google Scholar]
- White MA. 2015. Understanding how cis-regulatory function is encoded in DNA sequence using massively parallel reporter assays and designed sequences. Genomics 106: 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Myers CA, Corbo JC, Cohen BA. 2013. Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proc Natl Acad Sci 110: 11952–11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Kwasnieski JC, Myers CA, Shen SQ, Corbo JC, Cohen BA. 2016. A simple grammar defines activating and repressing cis-regulatory elements in photoreceptors. Cell Rep 17: 1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. 2013. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153: 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JSC. 2002. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol 22: 2871–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijgerde M, Grosveld F, Fraser P. 1995. Transcription complex stability and chromatin dynamics in vivo. Nature 377: 209–213. [DOI] [PubMed] [Google Scholar]
- Williamson I, Lettice LA, Hill RE, Bickmore WA. 2016. Shh and ZRS enhancer colocalisation is specific to the zone of polarising activity. Development 143: 2994–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, et al. 2017. BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol Cell 67: 5–18.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. 2012. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44: 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz G, Várnai C, Nagasaka K, Cisneros DA, Stocsits RR, Tang W, Schoenfelder S, Jessberger G, Muhar M, Hossain MJ, et al. 2017. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J 36: 3573–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S, Duan J, Li B, Zhou P, Hon GC. 2017. Multiplexed engineering and analysis of combinatorial enhancer activity in single cells. Mol Cell 66: 285–299.e5. [DOI] [PubMed] [Google Scholar]
- Xue Y, Pradhan SK, Sun F, Chronis C, Tran N, Su T, Van C, Vashisht A, Wohlschlegel J, Peterson CL, et al. 2017. Mot1, Ino80C, and NC2 function coordinately to regulate pervasive transcription in yeast and mammals. Mol Cell 67: 594–607.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Chen S-AA, Local A, Liu T, Qiu Y, Dorighi KM, Preissl S, Rivera CM, Wang C, Ye Z, et al. 2018. Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res 28: 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez-Cuna JO, Kvon EZ, Stark A. 2013. Deciphering the transcriptional cis-regulatory code. Trends Genet 29: 11–22. [DOI] [PubMed] [Google Scholar]
- Yanez-Cuna JO, Arnold CD, Stampfel G, Boryn LM, Gerlach D, Rath M, Stark A. 2014. Dissection of thousands of cell type-specific enhancers identifies dinucleotide repeat motifs as general enhancer features. Genome Res 24: 1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Tak YG, Berman BP, Farnham PJ. 2014. Functional annotation of colon cancer risk SNPs. Nat Commun 5: 5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Das PK, Kivioja T, Dave K, Zhong F, et al. 2017. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356: eaaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RS, Kumar Y, Bickmore WA, Taylor MS. 2017. Bidirectional transcription initiation marks accessible chromatin and is not specific to enhancers. Genome Biol 18: 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, et al. 2014. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515: 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabidi MA, Stark A. 2016. Regulatory enhancer–core–promoter communication via transcription factors and cofactors. Trends Genet 32: 801–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabidi MA, Arnold CD, Schernhuber K, Pagani M, Rath M, Frank O, Stark A. 2015. Enhancer–core–promoter specificity separates developmental and housekeeping gene regulation. Nature 518: 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, Carroll JS. 2011. Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25: 2227–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Scacheri PC. 2012. The chromatin fingerprint of gene enhancer elements. J Biol Chem 287: 30888–30896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Tesar PJ, Scacheri PC. 2011. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 21: 1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Prakash C, Sum C, Gong Y, Li Y, Kwok JJT, Thiessen N, Pettersson S, Jones SJM, Knapp S, et al. 2012. Bromodomain-containing protein 4 (BRD4) regulates RNA polymerase II serine 2 phosphorylation in human CD4+ T cells. J Biol Chem 287: 43137–43155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Gan H, Wang Z, Lee J-H, Zhou H, Ordog T, Wold MS, Ljungman M, Zhang Z. 2017. RPA interacts with HIRA and regulates H3.3 deposition at gene regulatory elements in mammalian cells. Mol Cell 65: 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HY, Katsman Y, Dhaliwal NK, Davidson S, Macpherson NN, Sakthidevi M, Collura F, Mitchell JA. 2014. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev 28: 2699–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. 2011. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478: 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuin J, Dixon JR, van der Reijden MIJA, Ye Z, Kolovos P, Brouwer RWW, van de Corput MPC, van de Werken HJG, Knoch TA, van IJcken WFJ, et al. 2014. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci 111: 996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]