

In this study, Reich et al. researched the functions of Caenorhabditis elegans adenosine deaminases that act on RNA (ADARs), which catalyze A-to-I RNA editing in dsRNA. Using dsRNA immunoprecipitation (dsRIP) and RNA-seq, they identified 1523 regions of clustered A-to-I editing, termed editing-enriched regions (EERs), in four stages of C. elegans development, often with highest expression in embryos.
Keywords: self–nonself, RNA editing, siRNA, Dicer, deaminase
Abstract
Cellular dsRNAs are edited by adenosine deaminases that act on RNA (ADARs). While editing can alter mRNA-coding potential, most editing occurs in noncoding sequences, the function of which is poorly understood. Using dsRNA immunoprecipitation (dsRIP) and RNA sequencing (RNA-seq), we identified 1523 regions of clustered A-to-I editing, termed editing-enriched regions (EERs), in four stages of Caenorhabditis elegans development, often with highest expression in embryos. Analyses of small RNA-seq data revealed 22- to 23-nucleotide (nt) siRNAs, reminiscent of viral siRNAs, that mapped to EERs and were abundant in adr-1;adr-2 mutant animals. Consistent with roles for these siRNAs in silencing, EER-associated genes (EAGs) were down-regulated in adr-1;adr-2 embryos, and this was dependent on associated EERs and the RNAi factor RDE-4. We observed that ADARs genetically interact with the 26G endogenous siRNA (endo-siRNA) pathway, which likely competes for RNAi components; deletion of factors required for this pathway (rrf-3 or ergo-1) in adr-1;adr-2 mutant strains caused a synthetic phenotype that was rescued by deleting antiviral RNAi factors. Poly(A)+ RNA-seq revealed EAG down-regulation and antiviral gene induction in adr-1;adr-2;rrf-3 embryos, and these expression changes were dependent on rde-1 and rde-4. Our data suggest that ADARs restrict antiviral silencing of cellular dsRNAs.
Adenosine deaminases that act on RNA (ADARs) catalyze conversion of adenosine (A) to inosine (I) within double-stranded regions of cellular RNAs (Hundley and Bass 2010; Nishikura 2016). Like guanosine (G), inosine prefers to pair with cytosine, and thus A-to-I RNA editing can alter mRNA-coding capacity. Recoding events are critical for normal function of the nervous system in vertebrates, squid, and Drosophila (Deffit and Hundley 2016; Nishikura 2016). However, in all organisms examined to date, the majority of A-to-I editing is outside of coding sequences, mostly in introns and 3′ untranslated regions (UTRs) (Whipple et al. 2015; Blango and Bass 2016; Walkley and Li 2017). In some cases, noncoding A-to-I changes in pre-mRNAs impact splicing and mRNA processing (Nishikura 2016). However, the function of most editing in noncoding regions is not well understood.
In mammals, ADARs are essential to prevent aberrant immune signaling by antiviral dsRNA sensor proteins (Mannion et al. 2014; Liddicoat et al. 2015; Pestal et al. 2015; George et al. 2016). Loss of mouse ADAR1 causes embryonic lethality characterized by interferon (IFN) overproduction and up-regulation of IFN-inducible transcripts (Hartner et al. 2004; Mannion et al. 2014; Liddicoat et al. 2015). IFN-stimulated gene expression in ADAR1−/−;p53−/− mutant fibroblasts is partially rescued by an editing-deficient ADAR1 point mutant but more completely rescued by a catalytically active ADAR1, suggesting that both binding and editing contribute to ADAR1 antagonism of IFN signaling (Mannion et al. 2014; O'Connell et al. 2015). Strikingly, mutations in the immune signaling genes Mavs or Ifih1 (MDA5) rescue ADAR1−/− mutant embryonic lethality and IFN hyperactivation (Mannion et al. 2014; Pestal et al. 2015). Thus, mammalian ADAR1 is thought to prevent cellular dsRNAs from activating RIG-I-like receptors, although it is unclear whether this is a conserved ADAR function.
The nematode Caenorhabditis elegans lacks an IFN response and instead uses RNAi to sense viral dsRNA and silence viral transcripts (Ashe et al. 2013; Guo et al. 2013). C. elegans ADARs inhibit RNAi-mediated transgene silencing (Knight and Bass 2002) and small RNA biogenesis (Warf et al. 2012), suggesting that, by analogy to vertebrates, ADARs could mark cellular dsRNA as self. C. elegans have two genes encoding ADARs, adr-1 and adr-2, and deletion of either gene causes chemotaxis defects, transgene silencing, and shortened life span (Knight and Bass 2002; Tonkin et al. 2002; Sebastiani et al. 2009). Consistent with the notion that these phenotypes relate to altered dsRNA-mediated silencing, these adr mutant phenotypes are rescued by additional loss of function of RNAi factors (Knight and Bass 2002; Tonkin and Bass 2003; Sebastiani et al. 2009).
The sole C. elegans Dicer enzyme, DCR-1, is required for biogenesis of microRNAs (miRNAs) as well as viral and endogenous siRNAs (endo-siRNAs) (Supplemental Fig. S1). Studies in C. elegans extracts indicate that DCR-1 cleaves dsRNA to produce siRNAs that are predominantly 23 nucleotides (nt) in length with a 5′ monophosphate (Welker et al. 2011). While such products arise during viral infection, they have not been observed among endo-siRNAs in wild-type C. elegans (Ruby et al. 2006; Ashe et al. 2013; Billi et al. 2014). Rather, characterized DCR-1-dependent endo-siRNAs are 26 nt, have a 5′ guanosine monophosphate (26G siRNAs), and are produced by DCR-1 acting in concert with the RNA-dependent RNA polymerase (RdRP) RRF-3 (Thivierge et al. 2012; Blumenfeld and Jose 2016). 26G siRNAs occur in embryos and germline tissues and exist in two classes bound to distinct Argonaute proteins. In sperm, 26G siRNAs bind ALG-3 and ALG-4, while ERGO-1 binds 26G siRNAs in oocytes and embryos. To effect silencing, 26G siRNAs and siRNAs from exogenous dsRNAs, such as those introduced by feeding, injection, or viral infection, trigger the RdRP-mediated production of 22-nt siRNAs with a 5′ triphosphorylated guanosine (22G siRNAs) antisense to target transcripts (Ruby et al. 2006; Pak and Fire 2007; Sijen et al. 2007; Vasale et al. 2010; Ashe et al. 2013; Billi et al. 2014).
Here we define the editing-enriched regions (EERs), ADAR-edited long dsRNAs, expressed in four stages of C. elegans development. EERs and their associated genes show highest expression in embryos and give rise to 23-nt 5′ monophosphorylated siRNAs that are abundant in adr-1;adr-2 mutant animals. Using quantitative RT–PCR (qRT–PCR), we show that EER-associated genes (EAGs) are down-regulated in adr-1;adr-2 mutant embryos but not L3 larvae. Down-regulation of EAGs in adr-1;adr-2 embryos requires both RDE-4 and an associated double-stranded EER. Analysis of a siRNA-sensitive GFP::NRDE-3 transgene indicates that ADARs antagonize siRNA biogenesis independent of the 26G pathway. However, in adr-1;adr-2 mutant backgrounds, 26G loss of function causes a synthetic phenotype dependent on the antiviral RNAi pathway. Transcriptomes of adr-1;adr-2;rrf-3 mutant embryos reveal robust EAG down-regulation and increased expression of genes induced during Orsay virus infection, both of which are rescued by rde-1 and rde-4 deletion. Together, our results suggest a conserved role for C. elegans ADARs in antagonizing the antiviral response to self dsRNAs.
Results
Clustered ADAR-editing sites define dsRNAs expressed during C. elegans development
To identify dsRNAs expressed during C. elegans development, we performed high-throughput RNA sequencing (RNA-seq) on total and dsRNA-enriched rRNA-depleted RNA samples from embryos, early larval stages (L1 and L2), late larval stages (L3 and L4), and young adults. We used a previously developed pipeline (Whipple et al. 2015) to define dsRNAs by scanning for clusters of A-to-I RNA editing (Supplemental Fig. S2). From combined dsRNA immunoprecipitation (dsRIP) and input data sets of all stages, we defined 1523 EERs in total (Fig. 1A; Supplemental File S1). We found more than twice as many EERs in dsRIP samples than input, suggesting that the dsRIP enriched for dsRNA. Across developmental stages, we defined the greatest number of EERs in embryos, in contrast to a previous study that observed more clusters in L1 larvae (Zhao et al. 2015). This discrepancy may reflect differences in populations sequenced, since the previous study used L1s aged 4 h after diapause, while we sequenced mixed L1–L2 larvae aged 6–20 h after diapause (Materials and Methods). Many EERs were defined in multiple stages (Fig. 1B), with 81 EERs common to all four stages and 406 unique to one stage. Stage-specific EERs generally were most highly expressed at the stage in which they were defined (Supplemental Fig. S3). Since our pipeline required at least five reads to call an EER, expression primarily determined the stages where an EER was found.
Figure 1.

EER abundance, but not editing, changes during development. (E) Early; (L) late; (Y) young. (A) The number of EERs defined from each group of data sets. (B) Venn diagram of EERs defined in each developmental stage. (C) Heat map of relative abundance in input RNA-seq samples for the 250 EERs with the greatest number of edited windows. (D) Distribution of mean EER abundance in each stage and treatment. (****) P < 0.0001, Mann-Whitney U-test. (E) Fraction of all EER-editing sites that appeared as guanosines in each stage and treatment. While individual sites ranged from 1% to 99% edited, all sites together were ∼15% edited in each sample. Error bars show standard deviation (SD) of three biological replicates. (*) P < 0.05, Student's t-test.
The EERs defined in our study had properties similar to previously defined C. elegans editing clusters (Wu et al. 2011; Whipple et al. 2015; Zhao et al. 2015). EER loci were enriched on autosome distal arms (Supplemental Fig. S4) and largely derived from repetitive elements, particularly transposons (Supplemental Fig. S5). EERs mostly overlapped gene-associated noncoding sequences, while a control set of random expressed regions mapped to more exonic and intergenic sequences (Supplemental Fig. S6A). Specifically, 72.3% of EER nucleotides mapped to introns and 10.2% mapped to 5′ UTRs, 3′ UTRs, or regions within 1 kb of a gene on the same strand. Many EERs were predicted by UNAFold to form long stable intramolecular structures, in contrast to random regions (Supplemental Fig. S6B; Markham and Zuker 2008). Previously, we found significant overlap between human EERs and circular RNAs (circRNAs) (Blango and Bass 2016), and, similarly, in C. elegans, 78 EERs overlapped circRNAs (78 observed and 56 expected; P = 0.0033 by χ2 test) (http://www.circbase.org; Supplemental File S2).
EER abundance, but not editing, varies during development
We next examined developmental patterns of EER abundance and editing. The heat map in Figure 1C shows relative abundance in each stage for the 250 most highly edited EERs. Of the 1523 EERs, 1336 overlapped or were within 1 kb of one of 955 unique EAGs (Supplemental File S3). For each gene-associated EER, we calculated a Pearson correlation coefficient (PCC) comparing EER abundance with EAG expression across all stages. We found that the mean EER–EAG PCC was 0.396 and the median PCC was 0.591, demonstrating that EER and EAG expression between developmental stages is correlated.
Strikingly, more than half of EERs (50.4%; 768 out of 1523) and EAGs (53.1%; 507 out of 955) displayed highest expression in embryos. We plotted the abundance of all 1523 EERs in each developmental stage and treatment (Fig. 1D), observing the highest collective expression in embryos, with decreased expression in subsequent stages. We compared EERs with length-matched random regions (Supplemental Fig. S7A) and observed greater differences in EER abundance between developmental stages than random regions (Supplemental Fig. S7B–D), suggesting that EER expression patterns are distinct from most transcripts.
Next, we assessed EER-editing levels during development. We made a list of adenosines within EERs edited >1% in all input and dsRIP samples pooled (referred to here as EER-editing sites). For each individual RNA-seq replicate, we determined the total number of A-to-G changes (#Ed) observed at EER-editing sites (Supplemental Fig. S8). In both input and dsRIP samples, we observed the greatest #Ed in embryos. For each replicate, we then counted the total reads covering each EER-editing site (#Tot) and calculated fraction editing as #Ed/#Tot (Materials and Methods). Finally, we calculated the average fraction editing across the three replicates in each stage and treatment (Fig. 1E). Editing frequency changed only minorly across development, in agreement with previous work (Zhao et al. 2015). We conclude that EER abundance changes during normal development, while editing within EERs remains stable.
We tested whether ADARs impacted development or viability by determining the fraction of wild-type and adr-1;adr-2 mutant embryos that developed to adulthood over 3 d. We used three independently derived sets of adr-1;adr-2 deletion alleles: two sets of previously described EMS-induced mutants (Tonkin et al. 2002; Hundley et al. 2008) and one that we created by injection of CRISPR/Cas9 ribonucleoparticles (Cho et al. 2013; Paix et al. 2015). Although the CRISPR mutant (adr-1(uu49);adr-2(uu28)) and adr-1(tm668);adr-2(ok735) strains were no different from wild type, the adr-1(gv6);adr-2(gv42) strain displayed a low-penetrance larval arrest phenotype (Supplemental Fig. S9), possibly due to background mutations. Since two mutant lines did not differ from wild type, we conclude that development occurs normally in adr-1;adr-2 mutants, consistent with previous work (Tonkin et al. 2002; Sebastiani et al. 2009).
Abundant siRNAs mapping to EERs in adr-1;adr-2 mutants suggest EER processing by DCR-1
Since ADARs edit only dsRNAs, EERs must be double-stranded, and we hypothesized that EERs would also be substrates for other dsRNA-binding proteins (dsRBPs), particularly in the absence of ADARs. Like other Dicer enzymes, C. elegans DCR-1 cleaves dsRNA to produce primary siRNAs that have 5′ monophosphates and 3′ hydroxyls (Ruby et al. 2006). To distinguish primary siRNAs from secondary siRNAs that have 5′ triphosphates, small RNAs are typically sequenced using 5′ phosphate (5′P)-dependent protocols that capture only primary siRNAs and 5′P-independent protocols that capture both primary and secondary siRNAs (Ruby et al. 2006; Pak and Fire 2007). To determine whether EERs were DCR-1 substrates, we analyzed published 5′P-dependent small RNA-seq data sets from mixed-stage wild-type or adr-1(tm668);adr-2(ok735) worms (Warf et al. 2012). We found that siRNAs mapped sense to 74.1% (1128 out of 1523) of EERs. Strikingly, 94.0% (1060 out of 1128) of these EERs showed increased siRNA abundance in adr-1;adr-2 mutant animals compared with wild type (Fig. 2A,B; Supplemental File S1). Length-matched random regions did not show similar siRNA enrichment in adr-1;adr-2 samples.
Figure 2.

EER-23H siRNAs are abundant in adr-1;adr-2 double mutants. (A) Genome browser view of 5′P-dependent small RNA-seq reads from mixed-stage wild-type (WT) and adr-1(tm668);adr-2(ok735) mutant animals mapping sense to EER1380. (B) EER-23H siRNA enrichment in adr-1(tm668);adr-2(ok735) mutants. Plots show the log2 ratio of siRNA abundance in adr-1;adr-2 mutants over wild type for EERs (black solid line) and control regions (gray dashed line). (C) Analysis of 5′ nucleotide and length distribution of all EER-23H siRNAs from adr-1;adr-2 mutant and wild-type animals.
To determine the origin of EER-mapped 5′ monophosphate siRNAs, we analyzed their length distribution and 5′ nucleotide preferences (Fig. 2C). In wild-type worms, EER-mapped siRNAs showed a small peak at 21 nt with a preference for 5′ U, typical of C. elegans 21U-RNAs/piRNAs (Ruby et al. 2006). We suspect that we observed this 21U peak because 18 21U-RNA loci overlap EERs (Supplemental File S2), 21U-RNAs are abundantly expressed in embryonic and germline tissue, and 5′P-dependent protocols include these small RNAs (Batista et al. 2008). In contrast to wild type, EER-siRNAs from adr-1;adr-2 animals showed a peak at 23 nt with a 5′ nucleotide bias against G (abbreviated H) (Cornish-Bowden 1985). This 5′ nucleotide preference is also seen in C. elegans miRNAs (Warf et al. 2011) and primary antiviral siRNAs (Ashe et al. 2013), suggesting that EER-mapped siRNAs in adr-1;adr-2 mutants, which we refer to as EER-23H siRNAs, are produced by direct DCR-1 cleavage of EERs.
A previous study identified 454 regions (termed ADAR-modulated RNA loci) that give rise to abundant 23- to 24-nt primary siRNAs and 22-nt secondary siRNAs in adr-1(gv6);adr-2(gv42) animals (Wu et al. 2011). Of these 454 loci, 93 overlapped EERs (85 EERs observed and four expected; P < 0.0001 by χ2 test) (Supplemental File S2), suggesting that EERs and ADAR-modulated loci represent related but mostly distinct regions. ADAR-modulated RNA loci display markedly lower coverage than EERs in all stages (Supplemental Fig. S10), suggesting that these regions have different rates of transcription or degradation.
Like the 23- to 24-nt primary siRNAs from ADAR-modulated loci, we reasoned that EER-23H siRNAs may promote the production of secondary siRNAs. We thus analyzed siRNAs antisense to EERs from published 5′P-independent small RNA-seq data sets from wild-type and adr-1(gv6);adr-2(gv42) embryos and L4 larvae (Supplemental Fig. S11; Wu et al. 2011). Although 5′P-independent siRNAs often mapped both sense and antisense to EERs when allowed to map multiple locations, we found that uniquely aligned reads mostly mapped antisense. In all samples, siRNAs antisense to EERs were primarily 22 nt with a 5′ G, suggesting an RdRP-dependent origin (Billi et al. 2014). We refer to these antisense secondary siRNAs as EER-22G siRNAs. Like EER-23H siRNAs, most EERs showed increased EER-22G siRNA abundance in adr-1;adr-2 mutants relative to wild type in both embryo and L4 larval samples (Supplemental Fig. S12A,B; Supplemental File S1). Control random regions did not show similar enrichment. The RNAi genes rde-1 and rde-4 promote secondary and primary siRNA biogenesis, respectively, in response to viral dsRNA (Ashe et al. 2013). In embryos, most EERs showed reduced EER-22G siRNA abundance in adr-1(gv6);adr-2(gv42);rde-1(ne219) and adr-1(gv6);adr-2(gv42);rde-4(ne299) triple mutants relative to adr-1(gv6);adr-2(gv42) double mutants (Supplemental Fig. S12C–F). However, EER-22G siRNA abundance in L4 larvae was reduced only marginally in adr-1;adr-2;rde-4 and was no different from control regions in adr-1;adr-2;rde-1. Thus, rde-1 and rde-4 mediate EER-22G siRNA accumulation in embryos but not in L4 animals. These data suggest that abundant EER-23H siRNAs in adr-1;adr-2 mutants promote the production of antisense secondary siRNAs.
In embryos, ADARs prevent down-regulation of EAGs via RNAi against EERs
We next sought to determine whether EER-siRNAs regulate expression of EAGs and first identified EAGs that were the best candidates for such regulation. Since adr-1, adr-2, and most EAGs are maximally expressed in embryos, we used input RNA-seq data to identify 452 EAGs with ≥50% higher gene expression in embryos than late stage larvae (Supplemental File S4). Using existing data sets from mixed-stage animals (Warf et al. 2012), we selected genes with more EER-23H siRNAs in adr-1(tm668);adr-2(ok735) mutants than wild type and finally narrowed our list to genes down-regulated in adr-1(tm668);adr-2(ok735) mutants relative to wild type by microarray analyses. This analysis revealed 53 EAGs as strong candidates for ADAR-mediated gene regulation.
From our 53 candidates, we assayed expression of eight EAGs in three adr-1;adr-2 double-mutant strains relative to wild-type embryos (Supplemental Fig. S13). For all genes, we observed a modest reduction in at least two strains tested. The adr-1(gv6);adr-2(gv42) strain had more substantial differences compared with others tested, again suggesting that it harbors additional mutations. All further experiments were performed using adr-1(uu49);adr-2(uu28) deletions created for this study.
We next tested EAG expression in embryos and L3 larvae of adr-1;adr-2 mutants with or without additional mutations in rde-1 or rde-4. Six EAGs displayed significantly decreased expression in adr-1;adr-2 embryos relative to wild type (Fig. 3A). Although the differences were small (∼20% below wild type on average), we observed these differences reproducibly across many independent biological replicates (n ≥ 8). A recent study observed small but reproducible down-regulation of pseudogenes and genes with edited 3′ UTRs in adr-1;adr-2 embryos (Goldstein et al. 2017). When we measured EAG expression in L3 larvae, we observed no significant difference in expression between wild type and adr-1;adr-2 mutants for all genes down-regulated in embryos (Fig. 3B). This suggests that ADARs promote EAG expression early in development but not at later stages. Importantly, deleting rde-4 in adr-1;adr-2 mutants with CRISPR protocols (Cho et al. 2013; Paix et al. 2015) completely or partially rescued the reduced gene expression in embryos for four of six EAGs, suggesting that these EAGs are down-regulated by RNAi in adr-1;adr-2 embryos. Curiously, deleting rde-1 did not strongly affect the expression of the EAGs tested. C. elegans encode 27 Argonaute proteins, and we speculate that some of these act redundantly with RDE-1 (Billi et al. 2014). Altogether, these data suggest that ADARs antagonize RNAi activity in embryos to promote normal EAG expression.
Figure 3.

EAG expression decreases in adr-1(uu49);adr-2(uu28) embryos in an RNAi- and EER-dependent manner. Expression of EAGs in embryos (A; n ≥ 8) and L3 larvae (B; n = 5) of four genotypes, measured by qRT–PCR. Expression levels for three EAGs in embryos (C; n ≥ 6) and L3 larvae (D; n = 5) in strains where each EAG's sole EER was deleted by CRISPR (ΔEER). All panels show expression as mean. Error bars show SD. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001, Student's t-test.
To confirm that EERs are required for EAG regulation by ADARs, we used CRISPR protocols (Cho et al. 2013; Paix et al. 2015) to remove intronic EER sequences (ΔEER) in three EAGs: efa-6, ccb-1, and egl-8 (Fig. 3C,D; Supplemental Fig. S14). We chose genes with a single intronic EER that gave rise to abundant siRNAs in adr-1;adr-2 strains (Supplemental File S4). All ΔEER mutants were viable without obvious morphological and developmental abnormalities, and we observed no expression differences in ΔEER mutants compared with wild type. If EERs are required to down-regulate associated EAGs in adr-1;adr-2 mutant embryos, we predicted that EER deletion would abrogate this expression change. Indeed, for two of the three genes tested, EER deletion rescued EAG down-regulation in adr-1;adr-2 embryos (Fig. 3C). We did not observe rescue of ccb-1 expression upon EER deletion, although ccb-1 expression was only slightly affected in adr-1;adr-2 mutants, making it more difficult to establish significance. In L3 larvae, EAG mRNA levels in adr-1;adr-2 double mutants and adr-1;adr-2;ΔEER triple mutants did not differ significantly from wild type (Fig. 3D). We conclude that ADARs antagonize RNAi-mediated down-regulation of EAGs via their EERs.
ADARs regulate a siRNA-sensitive reporter independent of the 26G endo-siRNA pathway
While ADARs impacted EAG expression, we were puzzled about why expression differences were so minor. Since EAG down-regulation in adr-1;adr-2 mutants is RNAi-dependent, we considered that a parallel RNAi pathway might restrict EAG silencing by competing for common factors. The 26G endo-siRNA pathway was a prime candidate for such competition, since it uses factors required for robust RNAi, and its loss of function causes enhanced RNAi phenotypes (Vasale et al. 2010; Billi et al. 2014). In the 26G pathway, the ERI complex (containing DCR-1, RDE-4, and the RdRP RRF-3) couples dsRNA synthesis and cleavage to produce 26G siRNAs that promote secondary 22G siRNA biogenesis and silence target genes (Supplemental Fig. S1; Thivierge et al. 2012).
We first determined whether the 26G pathway was required for ADAR-antagonized siRNA biogenesis using a GFP::NRDE-3 reporter whose localization depends on 22G secondary siRNA binding (Guang et al. 2008). When the 26G pathway is active, 26G siRNAs stimulate 22G siRNA production, causing NRDE-3 to localize to the nucleus. However, in rrf-3 mutants, 26G siRNAs are absent, precluding downstream 22G synthesis, and NRDE-3 localizes to the cytoplasm. To test interactions between ADARs and the 26G pathway, we introduced the GFP::NRDE-3 transgene into adr-1;adr-2, rrf-3, and adr-1;adr-2;rrf-3 deletion strains. While GFP::NRDE-3 was primarily cytoplasmic in the rrf-3 background, it localized to the nucleus in adr-1;adr-2;rrf-3 animals (Fig. 4A), suggesting that ADARs antagonize siRNA production independent of the 26G pathway. Furthermore, these data support the conclusion that ADARs antagonize production of both primary and secondary siRNAs.
Figure 4.
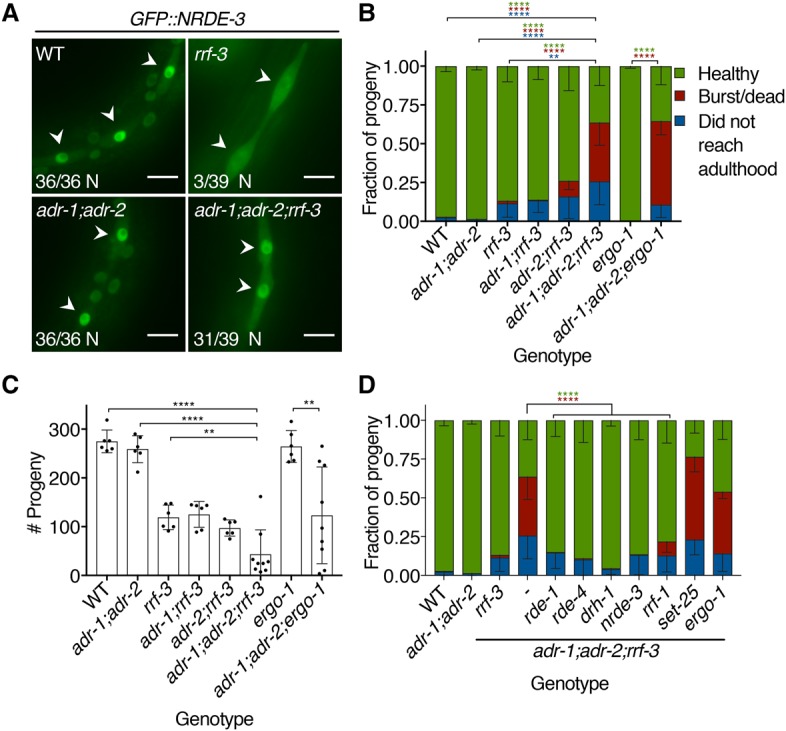
The 26G endo-siRNA pathway genetically interacts with ADARs. (A) GFP::NRDE-3 visualized in L3 larvae seam cells (arrowheads) of the indicated mutant genotypes. Numbers in the bottom left of each panel report the fraction of worms with nuclear-enriched (N) GFP::NRDE-3 in seam cells. Bar, 10 µm. (B) Bursting assay shows the fate of embryos laid by each genotype 5 d after egg lay. Error bars show SD. n ≥ 6 assays. (**) P < 0.01; (****) P < 0.0001. Asterisk colors show categories compared by two-way ANOVA with Tukey's multiple comparisons correction. (C) Average brood size for each genotype in B, with individual broods shown as dots. Error bars show SD. n ≥ 6 assays. (**) P < 0.01; (****) P < 0.0001, Student's t-test. (D) Developmental fates, as in B, of adr-1(uu49);adr-2(uu28);rrf-3(uu56) mutant strains with additional mutations in genes encoding RNAi-related factors. Error bars show SD. n ≥ 6 assays. (****) P < 0.0001. Asterisk colors show categories compared by two-way ANOVA with Tukey's multiple comparisons correction.
ADARs genetically interact with the 26G pathway in a manner dependent on antiviral RNAi
In testing GFP::NRDE-3 subcellular localization, we noticed that adr-1;adr-2;rrf-3 mutants displayed defects not present in adr-1;adr-2 and rrf-3 mutants, suggesting a synthetic genetic interaction. While adr-1;adr-2 double mutants are healthy, and rrf-3 single mutants show temperature-sensitive sterility and reduced brood size (Simmer et al. 2002), adr-1;adr-2;rrf-3 triple mutants displayed a phenotype marked by frequent adult bursting (Fig. 4B; Supplemental Fig. S15A). In addition, adr-1;adr-2;rrf-3 mutants had markedly lower brood sizes than rrf-3 single mutants (Fig. 4C). We confirmed the adr-1;adr-2;rrf-3 genetic interaction using three independent rrf-3 deletions and two sets of adr-1;adr-2 deletions (Supplemental Fig. S15B,C), implying that it is specific to our genes of interest. Both adr-1 and adr-2 contributed to the bursting phenotype, as 37.9% of adr-1;adr-2;rrf-3 worms burst by day 5 after egg lay compared with 10.0% of adr-2;rrf-3 and 0.4% of adr-1;rrf-3 worms. Since ADR-1 binds dsRNA but lacks catalytic activity, while ADR-2 catalyzes A-to-I editing (Tonkin et al. 2002; Washburn et al. 2014), our observations suggest that both binding and editing contribute to ADAR functions in vivo. We observed the same phenotypes in adr-1;adr-2;ergo-1 mutants lacking both ADARs and the ERGO-1 Argonaute required in the oocyte/embryo arm of the 26G pathway (Fig. 4B,C), suggesting that C. elegans ADARs genetically interact with the 26G pathway broadly, not with rrf-3 alone.
Since ADARs limit RNAi against EAGs and since 26G loss of function causes enhanced RNAi, we hypothesized that the adr-1;adr-2;rrf-3 synthetic phenotype resulted from increased RNAi activity against EAGs. To test this, we crossed rde-1 and rde-4 deletions into the adr-1;adr-2;rrf-3 mutant background (Fig. 4D; Supplemental Fig. S15D). Loss of either rde-1 or rde-4 rescued the frequent bursting and reduced brood sizes of adr-1;adr-2;rrf-3 mutants, suggesting that these genes are required for the adr-1;adr-2;rrf-3 synthetic defects. Both RDE-1 and RDE-4 function in RNAi-mediated antiviral immunity (Ashe et al. 2013), so we tested additional antiviral components by deleting them with CRISPR in an adr-1;adr-2;rrf-3 triple-mutant background. Intriguingly, adr-1;adr-2;rrf-3 defects were fully rescued by mutating drh-1, which encodes a RIG-I-like helicase required for processing viral dsRNA into primary siRNAs, and largely rescued by deleting rrf-1, which encodes a somatic RdRP that makes secondary siRNAs (Ashe et al. 2013; Guo et al. 2013). Deleting nrde-3, the nucleocytoplasmic shuttling Argonaute, also rescued adr-1;adr-2;rrf-3 bursting and brood size defects. However, loss of set-25, which encodes a histone H3 Lys9 methylase, had no effect. Deleting ergo-1 in the adr-1;adr-2;rrf-3 mutant background also had no effect, consistent with ergo-1 acting downstream from rrf-3 in the 26G pathway. We conclude that antiviral RNAi activity causes bursting and small brood size when ADARs and the 26G pathway are inactive.
EAGs and virus-induced genes are misregulated in adr-1;adr-2;rrf-3 mutants
To gain insight into gene expression changes underlying adr-1;adr-2;rrf-3 mutant phenotypes, we sequenced poly(A)+ RNA from four biological replicates of embryos of six genotypes: wild type, adr-1;adr-2, rrf-3, adr-1;adr-2;rrf-3, adr-1;adr-2;rrf-3;rde-1, and adr-1;adr-2;rrf-3;rde-4. Using DESeq2 (Love et al. 2014), we analyzed differential gene expression between genotypes (Supplemental File S5).
Consistent with our predictions, collective EAG expression decreased in adr-1;adr-2 mutant embryos and decreased further in adr-1;adr-2;rrf-3 embryos in a manner dependent on both rde-1 and rde-4 (Fig. 5A). As observed by qRT–PCR (Fig. 3A), rde-1 mutation did not rescue EAG expression as robustly as loss of rde-4. EAG expression also decreased in the rrf-3 mutant, similar to the adr-1;adr-2 mutant, indicating that the 26G pathway influences EAG expression even with ADARs present. Expression differences of most individual EAGs were small, <20% below wild type, and we hypothesize that the collective down-regulation of many EAGs—rescued by loss of rde-1 or rde-4—causes adr-1;adr-2;rrf-3 mutant phenotypes. Indeed, gene ontology (GO) analysis on EAGs revealed enrichment for terms associated with morphogenesis and development, suggesting that EAG misregulation could result in developmental defects that cause bursting (Supplemental File S6). A recent RNA-seq analysis revealed expression changes in isolated neurons from adr-1;adr-2 animals that are not observed in whole worms (Deffit et al. 2017), suggesting that EAG misregulation could be more substantial in specific tissues.
Figure 5.

EAGs and Orsay virus-induced genes are misregulated when ADARs and the 26G pathway are disrupted. (A) Tukey box plots show distributions of log2(expression fold change compared with wild type) for EAGs in each mutant genotype analyzed by RNA-seq. (*) P < 0.05; (****) P < 0.0001, Mann-Whitney U-test. (B) Venn diagram showing the overlap between differentially expressed genes (DEGs) up-regulated and down-regulated in adr-1(uu49);adr-2(uu28);rrf-3(uu56) mutants compared with wild type as well as EAGs expressed in RNA-seq samples (>10 reads total). Tukey box plots as in A show expression fold change in mutant genotypes for significantly down-regulated EAGs (down) (C), significantly up-regulated EAGs (up) (D), and EAGs not significantly changed (NS) (E) in adr-1;adr-2;rrf-3 mutant embryos relative to wild type. Adjusted P-value cutoff was 0.05. (ns) P > 0.05; (*) P < 0.05; (**) P < 0.01; (****) P < 0.0001, Mann-Whitney U-test. (F) Genes analyzed by poly(A)+ RNA-seq are plotted by log2(expression fold change compared with wild type) against −10log10(adjusted P-value) in adr-1;adr-2;rrf-3 mutants compared with wild type (i.e., higher y-values indicate more significant differences). The horizontal dotted line designates the adjusted P-value cutoff of 0.05 used to define DEGs. (G) Tukey box plots as in A showing Orsay-induced gene expression fold change in mutants. (**) P < 0.01; (****) P < 0.0001, Mann-Whitney U-test.
Our analyses involved 920 EAGs, and we considered the possibility that this large number of genes might mask certain trends. Thus, we divided EAGs into three groups: genes that were significantly down-regulated (231 genes), significantly up-regulated (50 genes), or not significantly changed (639 genes) in adr-1;adr-2;rrf-3 triple mutants relative to wild type (Fig. 5B). Compared with all EAGs together, down-regulated EAGs displayed more robust silencing in all mutant genotypes relative to wild type (Fig. 5C). Up-regulated EAGs showed rrf-3-dependent increased expression but were largely unchanged in adr-1;adr-2 double mutants (Fig. 5D). EAGs that were not significantly misexpressed in adr-1;adr-2;rrf-3 mutants showed modest down-regulation in all mutant strains (Fig. 5E) in a pattern resembling that in Figure 5A. Potentially informing why down-regulated EAGs were robustly silenced, we found that these genes often had more than one EER-associated intron or UTR and had longer EERs than other genes (Supplemental Fig. S16A,B). In contrast, up-regulated EAGs tended to have a single shorter EER that was less likely to occur in an intron (Supplemental Fig. S16A–C).
We next determined differentially expressed genes (DEGs) in pairwise comparisons between genotypes. We observed 2269 genes significantly up-regulated and 2797 down-regulated in adr-1;adr-2;rrf-3 triple-mutant embryos compared with wild type (Fig. 5B,F). EAGs were significantly enriched among down-regulated DEGs and depleted from up-regulated DEGs (P < 0.0001, χ2 test). In adr-1;adr-2 double mutants compared with wild type, we observed only nine DEGs up and 15 down, excluding adr-1 and adr-2 (Supplemental File S5). The 15 down-regulated DEGs included four EAGs and other genes with properties suggesting that they form dsRNA: Two were transposons, three were antisense to other genes, and two were edited in introns covered by too few reads in our original analysis to be defined as EERs. We observed only three DEGs comparing adr-1;adr-2;rrf-3 triple-mutant embryos with adr-1;adr-2;rrf-3;rde-1 or adr-1;adr-2;rrf-3;rde-4 quadruple mutants, and only one gene, the TURMOIL1 transposon Y48G1BL.4, was rescued by both rde-1 and rde-4 deletion (Supplemental Fig. S17A,B). As Y48G1BL.4 is silenced in adr-1;adr-2 mutants that do not burst, we conclude that it does not mediate adr-1;adr-2;rrf-3 bursting. We speculated that the reason that we identified so few DEGs in adr-1;adr-2 samples was that expression differences were too small or variable to achieve statistical significance. Thus, we performed gene set enrichment analysis (GSEA) (Subramanian et al. 2005) to find overrepresented gene classes altered in rde-1- and rde-4-rescued quadruple mutants relative to adr-1;adr-2;rrf-3. In both cases, the most enriched class of up-regulated genes was the 231 EAGs down in adr-1;adr-2;rrf-3 relative to wild type (Supplemental Fig. S17C,D; Supplemental File S7), supporting the idea that rde-1- and rde-4-dependent EAG silencing mediates adr-1;adr-2;rrf-3 defects.
We hypothesized that ADARs prevent antiviral pathways from recognizing self EERs as viral dsRNAs, so we next examined whether genes induced during viral infection were changed in adr-1;adr-2;rrf-3 triple mutants. A previous study identified 320 genes (many with predicted functions in degradation and innate immunity) differentially expressed during Orsay virus infection, 298 of which increased in expression (Chen et al. 2017). We analyzed expression of these Orsay virus-induced genes and found that, of 268 genes expressed, 157 were significantly up-regulated in adr-1;adr-2;rrf-3 mutant embryos (Fig. 5F). These differences were primarily rrf-3-dependent, as expression in adr-1;adr-2 mutants was largely unchanged (Fig. 5G). Thus, unlike mammals, where unedited dsRNAs are thought to induce IFN-stimulated genes (Liddicoat et al. 2015), we did not see strong evidence that unedited dsRNAs drive Orsay virus-induced gene expression, as rrf-3 deletion is not predicted to increase dsRNA levels. However, expression of virus-induced genes in rrf-3 mutants was further increased in adr-1;adr-2;rrf-3 mutants, suggesting that ADARs still impact their regulation. Loss of rde-1 and rde-4 in adr-1;adr-2;rrf-3 mutants reduced expression of antiviral genes. Furthermore, by GSEA, Orsay-induced genes were the most enriched gene class down-regulated in quadruple mutants relative to adr-1;adr-2;rrf-3 (Supplemental Fig. S17E,F; Supplemental File S7), suggesting that RNAi contributes to antiviral gene induction in adr-1;adr-2;rrf-3 mutants.
Discussion
Here we present evidence that C. elegans ADARs prevent silencing of self dsRNAs by antiviral RNAi (Fig. 6). We identify 1523 EERs (edited structures in gene introns and 3′ UTRs) that give rise to abundant 23H siRNAs and promote RDE-4-dependent gene silencing when ADARs are absent. Genetic analyses suggest that the 26G endo-siRNA pathway restricts antiviral RNAi activity in adr-1;adr-2 mutants. Robust EAG silencing and antiviral gene induction seen in adr-1;adr-2;rrf-3 mutants emphasize that ADARs and the 26G pathway limit antiviral activity in the absence of viral infection.
Figure 6.
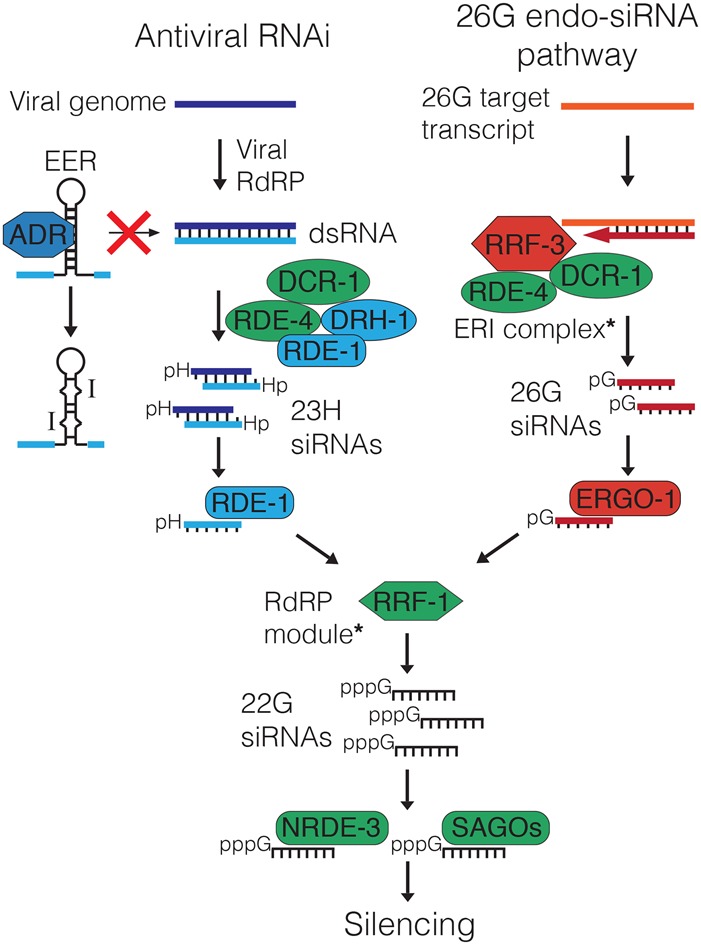
ADARs and the 26G pathway prevent antiviral RNAi-mediated silencing of self dsRNAs. During viral infection, viral replication generates dsRNAs that are processed into 23H siRNAs by a complex of DCR-1, RDE-1, RDE-4, and DRH-1. RDE-1 binds 23H siRNAs and stimulates 22G siRNA production by RRF-1. 22G siRNAs bind NRDE-3 and SAGO Argonautes to effect silencing. ADARs bind and edit EERs to prevent recognition as viral dsRNA and processing to 23H siRNAs by the antiviral DCR-1 complex. In the 26G pathway, the ERI complex (containing RRF-3, DCR-1, and RDE-4) generates 26G siRNAs, which bind ERGO-1, promote 22G siRNA synthesis, and silence targets through NRDE-3 and SAGO proteins. Thus, absent viral infection, antiviral RNAi is kept inactive by ADARs binding and editing self dsRNAs and by 26G pathway sequestration of common RNAi factors (green). For simplicity, we show only factors relevant to this study (see Supplemental Fig. S1); complexes containing additional components are noted with asterisks.
ADARs and the 26G pathway limit antiviral RNAi responses to self dsRNAs
Antiviral RNAi is initiated when viral dsRNA is cleaved into primary siRNAs by DCR-1 in association with the dsRBP RDE-4 and the RIG-I-like helicase DRH-1 (Ashe et al. 2013). The Argonaute RDE-1 uses primary siRNAs to promote production of antisense secondary 22G siRNAs that silence viral transcripts (Pak and Fire 2007; Sijen et al. 2007; Guo et al. 2013). By analyzing small RNAs in wild-type and adr-1;adr-2 mutant worms, we found that primary and secondary siRNAs mapping to EERs are more abundant in the absence of ADARs. Like primary siRNAs from viral dsRNA, sense primary EER-siRNAs are 22–23 nt with a bias against 5′ G (Ashe et al. 2013), suggesting that they result from DCR-1 cleavage of EERs. EER-23H siRNAs promote secondary siRNA production, as evidenced by increased EER-22G siRNAs in adr-1;adr-2 mutants and nuclear GFP::NRDE-3 in adr-1;adr-2;rrf-3 mutants. ADAR-antagonized EER-siRNAs are functional, since EAGs are down-regulated in adr-1;adr-2 embryos, and this requires RDE-4 and EERs. Thus, ADARs prevent EER cleavage and EAG silencing by an RNAi mechanism analogous to the processing and silencing of viral dsRNAs.
The 26G pathway further limits antiviral RNAi activity. While adr-1;adr-2, rrf-3, and ergo-1 mutant strains are largely healthy, deleting either rrf-3 or ergo-1 in the adr-1;adr-2 deletion strain caused bursting and reduced brood size. These phenotypes required rde-1, rde-4, drh-1, and rrf-1, components also required for antiviral RNAi during Orsay virus infection, suggesting that antiviral RNAi activity causes adr-1;adr-2;rrf-3 mutant phenotypes. The 26G and antiviral pathways compete for common sets of proteins (Vasale et al. 2010; Thivierge et al. 2012; Ashe et al. 2013), including the Argonautes NRDE-3, SAGO-1, and SAGO-2 that are limiting factors in RNAi (Yigit et al. 2006; Zhuang et al. 2013). The bursting of adr-1;adr-2;ergo-1 triple mutants suggests that the 26G pathway primarily restricts antiviral RNAi activity by competing for these downstream factors rather than DCR-1 and RDE-4, which would still act upstream in 26G biogenesis in adr-1;adr-2;ergo-1 mutants.
ADAR antagonism of C. elegans antiviral RNAi clearly parallels mammalian ADAR1 antagonism of MDA5-dependent IFN signaling (Mannion et al. 2014; Liddicoat et al. 2015; Pestal et al. 2015). We conclude that ADARs perform a conserved role in preventing antiviral responses to self dsRNAs. The role of the 26G pathway in further limiting antiviral RNAi underscores the importance of restricting immune signaling to appropriate contexts.
How do ADARs prevent recognition of cellular dsRNAs as nonself?
Editing converts A–U base pairs in dsRNA to I–U mismatches (Hundley and Bass 2010; Nishikura 2016), making dsRNAs less “double-stranded” and less ideal substrates for Dicer and other dsRBPs. Indeed, edited dsRNAs are poorly processed into siRNAs in Drosophila extracts (Scadden and Smith 2001), while, in HeLa cells, dsRNAs containing I–U mismatches fail to activate MDA5 like control dsRNA (Vitali and Scadden 2010). We found that ADR-1, which binds but does not edit dsRNA (Washburn et al. 2014), restricts antiviral RNAi, since adr-2;rrf-3 double mutants that lack all editing burst less frequently than adr-1;adr-2;rrf-3 triple mutants. Still, adr-1;rrf-3 double mutants retain ADR-2 editing and rarely burst, suggesting that the loss of editing is more deleterious than losing ADR-1 binding. Abundant intron editing suggests that ADARs act in the nucleus, consistent with nuclear localization of most mammalian ADARs (Nishikura 2016) and A-to-I editing of nascent transcripts (Rodriguez et al. 2012). Nuclear localization likely allows ADARs to bind and edit EERs soon after transcription to preempt processing by DCR-1.
How does EER-mediated silencing occur?
EERs are predominantly intronic and thus likely nuclear, while DCR-1 and RRF-1 act primarily in the cytoplasm (Aoki et al. 2007; Drake et al. 2014). However, we observed primary and secondary siRNAs mapping to intronic EERs, and our expression analyses indicate that EAGs can be silenced via intronic EERs (Fig. 3C). Thus, either DCR-1 and RRF-1 act in the nucleus or EERs go to the cytoplasm. In somatic and germline tissues, secondary siRNA production occurs in perinuclear foci (Phillips et al. 2012; Yang et al. 2014) that conceivably could facilitate nucleocytoplasmic exchange of RNAi factors and/or EERs. Alternatively, mitotic nuclear breakdown may provide a window for EER processing and secondary amplification. A mitosis-dependent mechanism may explain why EAGs are down-regulated in adr-1;adr-2 mutant embryos but not L3 larvae, since nonproliferative larval cells would not generate EER-siRNAs. Still, EER-22G siRNAs are more abundant in adr-1;adr-2 mutants than wild type at both the embryo and L4 stages. Perhaps adr-1;adr-2 L3 larvae do not silence EAGs because Argonautes at this stage bind fewer EER-siRNAs either because they comprise a smaller proportion of total siRNAs or because Argonautes become less abundant or tissue-restricted. Our future work aims to establish precise mechanisms of EER silencing.
What causes adr-1;adr-2;rrf-3 mutant phenotypes?
Like bursting and brood size defects, EAG misregulation in adr-1;adr-2;rrf-3 mutants is rde-1- and rde-4-dependent. GO terms associated with development and morphogenesis are enriched among EAGs, and we suspect that EAG misexpression compromises vulval morphogenesis, although we did not test this. Bursting is a frequent phenotype of miRNA mutants, attributed to lin-41 misregulation by let-7 (Parry et al. 2007; Ecsedi et al. 2015), and adr-1;adr-2 mutants show altered miRNA networks (Warf et al. 2012). Still, we saw no evidence of lin-41 misexpression in adr-1;adr-2;rrf-3 mutant embryos, although we did not test later stages. Importantly, the fact that disrupting RNAi factors rescued adr-1;adr-2;rrf-3 bursting suggests that siRNA, not miRNA, regulation is perturbed.
Orsay virus-induced genes are robustly induced in adr-1;adr-2;rrf-3 triple mutants, partly through rde-1 and rde-4, suggesting that activation of an antiviral transcriptional program could contribute to bursting. While we predicted that unedited EERs in adr-1;adr-2 mutants might induce antiviral gene transcription, we instead saw Orsay-induced genes induced in rrf-3 but not adr-1;adr-2 mutants, suggesting that 26G inactivity is associated with antiviral gene expression. Still, expression of virus-induced genes further increases in adr-1;adr-2;rrf-3 mutants, suggesting that ADARs limit their induction. Although the 26G pathway has not been implicated in the antiviral response, we suspect that its inhibition could help combat viral infection by relieving competition with antiviral RNAi.
What triggers the virus-induced transcriptional program remains unclear. Generalized stresses such as heat shock, oxidative stress, and translation inhibition trigger innate immune signaling (Kim and Ewbank 2015), so adr-1;adr-2;rrf-3 mutant antiviral gene induction could indicate nonspecific cellular dysfunction. However, a subset of genes expressed during Orsay infection is not induced in drh-1 mutant JU1580 worms (Sarkies et al. 2013), suggesting the intriguing possibility that DRH-1 may activate antiviral gene transcription in response to viral dsRNA.
Materials and methods
C. elegans maintenance and strains used in this study
All C. elegans strains were cultured at 20°C under standard conditions (Brenner 1974). Strains used in the study are listed in Supplemental Table S1.
Sample collection and RNA isolation
Embryos were obtained by sodium hypochlorite treatment (Emmons et al. 1979) of well-fed worms grown 4–5 d in S-Complete liquid medium, washed three times in M9 buffer, and then collected or hatched overnight without food. Synchronized L1 larvae were filtered over two layers of Miracloth (Calbiochem) and cultured in S-Complete liquid medium to mid-L1 (6–8 h), L1/L2 molt (12–14 h), mid-L2 (18–20 h), mid-L3 (28–30 h), L3/L4 molt (34–36 h), mid-L4 (40–42 h), or young adulthood (50–54 h). Early larval samples were prepared by mixing mid-L1, mid-L2, and L1/L2 molt populations at a 5:5:2 volumetric ratio, respectively. Late larval samples were similarly prepared with mid-L3, mid-L4, and L3/L4 molt populations.
Samples were lysed by three freeze–thaw cycles in Trizol reagent. RNA was extracted with chloroform, ethanol-precipitated, treated for 1 h at 37°C with TURBO DNase, and isolated using Zymo Research RNA clean and concentrator columns.
RNA-seq and data preparation
For developmental RNA-seq, we prepared three biological replicates of each stage as described above. For each sample, we treated 40 µg of total RNA with Ribo-Zero human–mouse–rat rRNA removal kit and took 10% of the output for input cDNA libraries. The remaining RNA was incubated overnight at 4°C with 10 µg of J2 anti-dsRNA antibody (English Scientific and Consulting Kft.) in 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, and 1% NP-40. J2-bound RNA was collected with Protein-A/G agarose beads for 4 h at 4°C and isolated with Trizol. cDNA libraries were prepared with the Illumina TruSeq stranded total RNA sample preparation LS protocol modified by addition of 2.5% DMSO to reverse transcription reactions. cDNA libraries were sequenced by paired-end 101-cycle sequencing on an Illumina HiSeq 2000 platform by the Microarray and Genome Analysis Core Facility at the University of Utah Huntsman Cancer Institute.
For poly(A)+ RNA-seq of the adr-1;adr-2;rrf-3 triple-mutant and related strains, we collected four biological replicates of embryos of each genotype. Each library was prepared from 1 µg of total RNA by the University of Utah Microarray and Genome Analysis Core Facility using the Illumina TruSeq stranded mRNA library preparation kit. cDNA libraries were sequenced by paired-end 125-cycle sequencing on an Illumina HiSeq 2500 platform.
The Novoalign alignment package (http://www.novocraft.com) was used to trim adaptor sequences and align reads to the C. elegans genome (ce10/WS220). Reads were filtered to allow up to four mismatches with the USeq (http://useq.sourceforge.net) application SamTranscriptomeParser using parameters “-a 120 -n 1 –p –r –b.” The USeq AligmentEndTrimmer application was used to remove reads with more than one non-A-to-G mismatch and trim read ends of low-quality bases.
Detection of EERs
We detected EERs as described in Whipple et al. (2015). Sequence variants were called with SAMtools mpileup (http://samtools.sourceforge.net). USeq applications RNAEditingPileupParser, RNAEditingScanSeqs, and EnrichedRegionMaker were used to define 50-nt strand-specific windows covered by five or more sequencing reads that contained three or more adenosines edited in >1% of reads. Overlapping edited windows and windows within 1 kb were merged to define EERs. EERs comprised of a single 50-nt window were excluded.
RNA-seq expression and editing analyses
A RefFlat table of C. elegans genes (ce10) was downloaded from the University of California at Santa Cruz (UCSC) genome browser (https://genome.ucsc.edu), and EER coordinates were added. Expression was quantified for RefFlat table entries using the USeq application DefinedRegionDifferentialSeq, which also identified DEGs via the R package DESeq2 (Love et al. 2014).
For editing analyses, we determined A-to-G mismatches from .bar files outputted by the USeq application RNAEditingPileupParser. Restricting our analyses to adenosines edited >1% and <99% within EERs, for each RNA-seq replicate, we calculated total A-to-G mismatches (Supplemental Fig. S8) and divided by the total reads covering each base (i.e., if a read covered X edited bases, it was counted X times, once for each base).
Small RNA-seq analyses
Published small RNA-seq data sets, alignment parameters, read filtering, and analyses are described in the Supplemental Material.
qRT–PCR
For each sample, 2 µg of total RNA was reverse-transcribed with the Applied Biosystems high-capacity cDNA reverse transcription kit. qPCR was performed with Roche LightCycler 480 SYBR Green master mix on a Roche LightCycler 480 platform using primers listed in Supplemental Table S2. Transcript abundance was determined by the ΔΔCt method and normalized to the geometric mean of Y45F10D.4, cdc-42, ama-1, and pmp-3 mRNAs. Relative values were determined by normalizing to wild type in each individual trial. Outliers were determined by the ROUT method with Q = 1% and excluded (Motulsky and Brown 2006).
CRISPR/Cas9 genome editing
Protocols for recombinant Cas9 purification and design and synthesis of guide RNAs and homology-directed repair (HDR) templates are described in the Supplemental Material. For each targeting event, 8 mg/mL recombinant Cas9 was complexed for 15 min at 37°C with an equal volume of a 2:2(:2):1:1 mixture of 4 µg/µL target sgRNA 1, 4 µg/µL target sgRNA 2 (0.5 µg/µL HDR template DNA; efa-6 only), 4 µg/µL dpy-10 sgRNA, and 40 µM dpy-10 HDR DNA oligonucleotide to induce the Roller phenotype (Paix et al. 2015). Cas9 complexes were then injected into the distal gonads of 20 wild-type young adults. After 3–4 d, Rol F1s were isolated and screened by PCR (primers in Supplemental Table S3) for the desired genome modifications. All mutations were confirmed by Sanger sequencing (Supplemental Table S4).
Bursting assay
For each sample, eight gravid adults were placed on a seeded plate of nematode growth medium (NGM) to lay eggs for 90 min, after which adults were removed and the eggs laid were counted. Plates were incubated for 5 d at 20°C followed by counting of healthy, burst, or dead adults on the plate. The difference between total number of adults and the original egg count was recorded as number of progeny that did not reach adulthood.
Brood size assay
For each trial, one healthy L4 larva was placed on a seeded NGM plate and allowed to develop for 5 d at 20°C. Hatched larvae were picked off plates and counted before they reached reproductive age.
Accession numbers
RNA-seq data and reanalyses of published small RNA data were deposited in the NCBI Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo) under superseries accession number GSE89890.
Supplementary Material
Acknowledgments
We thank D. Nix, T. Mosbruger, C. Stubben, the Huntsman Cancer Institute Bioinformatics Shared Resource, and the University of Utah Center for High-Performance Computing for bioinformatics guidance and resources. We are grateful to M. Krause and members of the Bass and Cazalla laboratories for discussion, advice, and reviews of this manuscript. This work was supported by funding to B.L.B. from the National Institute of General Medical Sciences (RO1GM044073) and the National Institute on Aging (8DP1AG044162). D.P.R. was supported by a National Institutes of Health Developmental Biology Training Grant (5T32HD07491).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.310672.117.
References
- Aoki K, Moriguchi H, Yoshioka T, Okawa K, Tabara H. 2007. In vitro analyses of the production and activity of secondary small interfering RNAs in C. elegans. EMBO J 26: 5007–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe A, Belicard T, Le Pen J, Sarkies P, Frezal L, Lehrbach NJ, Felix MA, Miska EA. 2013. A deletion polymorphism in the Caenorhabditis elegans RIG-I homolog disables viral RNA dicing and antiviral immunity. Elife 2: e00994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Ruby JG, Claycomb JM, Chiang R, Fahlgren N, Kasschau KD, Chaves DA, Gu W, Vasale JJ, Duan S, et al. 2008. PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Mol Cell 31: 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billi AC, Fischer SE, Kim JK. 2014. Endogenous RNAi pathways in C. elegans. WormBook (ed. The C. elegans Research Community). WormBook 10.1895/wormbook.1.170.1, http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blango MG, Bass BL. 2016. Identification of the long, edited dsRNAome of LPS-stimulated immune cells. Genome Res 26: 852–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld AL, Jose AM. 2016. Reproducible features of small RNAs in C. elegans reveal NU RNAs and provide insights into 22G RNAs and 26G RNAs. RNA 22: 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Franz CJ, Jiang H, Jiang Y, Wang D. 2017. An evolutionarily conserved transcriptional response to viral infection in Caenorhabditis nematodes. BMC Genomics 18: 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Lee J, Carroll D, Kim JS, Lee J. 2013. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics 195: 1177–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish-Bowden A. 1985. Nomenclature for incompletely specified bases in nucleic acid sequences: recommendations 1984. Nucleic Acids Res 13: 3021–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deffit SN, Hundley HA. 2016. To edit or not to edit: regulation of ADAR editing specificity and efficiency. Wiley Interdiscip Rev RNA 7: 113–127. [DOI] [PubMed] [Google Scholar]
- Deffit SN, Yee BA, Manning AC, Rajendren S, Vadlamani P, Wheeler EC, Domissy A, Washburn MC, Yeo GW, Hundley HA. 2017. The C. elegans neural editome reveals an ADAR target mRNA required for proper chemotaxis. Elife 6: e28625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake M, Furuta T, Suen KM, Gonzalez G, Liu B, Kalia A, Ladbury JE, Fire AZ, Skeath JB, Arur S. 2014. A requirement for ERK-dependent Dicer phosphorylation in coordinating oocyte-to-embryo transition in C. elegans. Dev Cell 31: 614–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecsedi M, Rausch M, Grosshans H. 2015. The let-7 microRNA directs vulval development through a single target. Dev Cell 32: 335–344. [DOI] [PubMed] [Google Scholar]
- Emmons SW, Klass MR, Hirsh D. 1979. Analysis of the constancy of DNA sequences during development and evolution of the nematode Caenorhabditis elegans. Proc Natl Acad Sci 76: 1333–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Ramaswami G, Li JB, Samuel CE. 2016. Editing of cellular self-RNAs by adenosine deaminase ADAR1 suppresses innate immune stress responses. J Biol Chem 291: 6158–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein B, Agranat-Tamir L, Light D, Ben-Naim Zgayer O, Fishman A, Lamm AT. 2017. A-to-I RNA editing promotes developmental stage-specific gene and lncRNA expression. Genome Res 27: 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang S, Bochner AF, Pavelec DM, Burkhart KB, Harding S, Lachowiec J, Kennedy S. 2008. An Argonaute transports siRNAs from the cytoplasm to the nucleus. Science 321: 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhang R, Wang J, Ding SW, Lu R. 2013. Homologous RIG-I-like helicase proteins direct RNAi-mediated antiviral immunity in C. elegans by distinct mechanisms. Proc Natl Acad Sci 110: 16085–16090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Muller AM, Higuchi M, Seeburg PH. 2004. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem 279: 4894–4902. [DOI] [PubMed] [Google Scholar]
- Hundley HA, Bass BL. 2010. ADAR editing in double-stranded UTRs and other noncoding RNA sequences. Trends Biochem Sci 35: 377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundley HA, Krauchuk AA, Bass BL. 2008. C. elegans and H. sapiens mRNAs with edited 3′ UTRs are present on polysomes. RNA 14: 2050–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Ewbank JJ. 2015. Signaling in the innate immune response. WormBook (ed. The C. elegans Research Community). WormBook, 10.1895/wormbook.1.83.2, http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight SW, Bass BL. 2002. The role of RNA editing by ADARs in RNAi. Mol Cell 10: 809–817. [DOI] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, et al. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham NR, Zuker M. 2008. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol 453: 3–31. [DOI] [PubMed] [Google Scholar]
- Motulsky HJ, Brown RE. 2006. Detecting outliers when fitting data with nonlinear regression—a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 7: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. 2016. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 17: 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell MA, Mannion NM, Keegan LP. 2015. The epitranscriptome and innate immunity. PLoS Genet 11: e1005687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Rasoloson D, Seydoux G. 2015. High efficiency, homology-directed genome editing in C. elegans using CRISPR/Cas9 ribonucleoprotein complexes. Genetics 201: 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak J, Fire A. 2007. Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science 315: 241–244. [DOI] [PubMed] [Google Scholar]
- Parry DH, Xu J, Ruvkun G. 2007. A whole-genome RNAi Screen for C. elegans miRNA pathway genes. Curr Biol 17: 2013–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. 2015. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43: 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CM, Montgomery TA, Breen PC, Ruvkun G. 2012. MUT-16 promotes formation of perinuclear mutator foci required for RNA silencing in the C. elegans germline. Genes Dev 26: 1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Menet JS, Rosbash M. 2012. Nascent-seq indicates widespread cotranscriptional RNA editing in Drosophila. Mol Cell 47: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Jan C, Player C, Axtell MJ, Lee W, Nusbaum C, Ge H, Bartel DP. 2006. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell 127: 1193–1207. [DOI] [PubMed] [Google Scholar]
- Sarkies P, Ashe A, Le Pen J, McKie MA, Miska EA. 2013. Competition between virus-derived and endogenous small RNAs regulates gene expression in Caenorhabditis elegans. Genome Res 23: 1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden ADJ, Smith CWJ. 2001. RNAi is antagonized by A → I hyper-editing. EMBO Rep 2: 1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiani P, Montano M, Puca A, Solovieff N, Kojima T, Wang MC, Melista E, Meltzer M, Fischer SE, Andersen S, et al. 2009. RNA editing genes associated with extreme old age in humans and with lifespan in C. elegans. PLoS One 4: e8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijen T, Steiner FA, Thijssen KL, Plasterk RHA. 2007. Secondary siRNAs result from unprimed RNA synthesis and form a distinct class. Science 315: 244–247. [DOI] [PubMed] [Google Scholar]
- Simmer F, Tijsterman M, Parrish S, Koushika SP, Nonet ML, Fire A, Ahringer J, Plasterk RHA. 2002. Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr Biol 12: 1317–1319. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thivierge C, Makil N, Flamand M, Vasale JJ, Mello CC, Wohlschlegel J, Conte D Jr, Duchaine TF. 2012. Tudor domain ERI-5 tethers an RNA-dependent RNA polymerase to DCR-1 to potentiate endo-RNAi. Nat Struct Mol Biol 19: 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonkin L, Bass BL. 2003. Mutations in RNAi rescue aberrant chemotaxis of ADAR mutants. Science 302: 1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonkin LA, Saccomanno L, Morse DP, Brodigan T, Krause M, Bass BL. 2002. RNA editing by ADARs is important for normal behavior in Caenorhabditis elegans. EMBO J 21: 6025–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasale JJ, Gu W, Thivierge C, Batista PJ, Claycomb JM, Youngman EM, Duchaine TF, Mello CC, Conte D Jr. 2010. Sequential rounds of RNA-dependent RNA transcription drive endogenous small-RNA biogenesis in the ERGO-1/Argonaute pathway. Proc Natl Acad Sci 107: 3582–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitali P, Scadden AD. 2010. Double-stranded RNAs containing multiple IU pairs are sufficient to suppress interferon induction and apoptosis. Nat Struct Mol Biol 17: 1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley CR, Li JB. 2017. Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol 18: 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf MB, Johnson WE, Bass BL. 2011. Improved annotation of C. elegans microRNAs by deep sequencing reveals structures associated with processing by Drosha and Dicer. RNA 17: 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf MB, Shepherd BA, Johnson WE, Bass BL. 2012. Effects of ADARs on small RNA processing pathways in C. elegans. Genome Res 22: 1488–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MC, Kakaradov B, Sundararaman B, Wheeler E, Hoon S, Yeo GW, Hundley HA. 2014. The dsRBP and inactive editor ADR-1 utilizes dsRNA binding to regulate A-to-I RNA editing across the C. elegans transcriptome. Cell Rep 6: 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker NC, Maity TS, Ye X, Aruscavage PJ, Krauchuk AA, Liu Q, Bass BL. 2011. Dicer's helicase domain discriminates dsRNA termini to promote an altered reaction mode. Mol Cell 41: 589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whipple JM, Youssef OA, Aruscavage PJ, Nix DA, Hong C, Johnson WE, Bass BL. 2015. Genome-wide profiling of the C. elegans dsRNAome. RNA 21: 786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Lamm AT, Fire AZ. 2011. Competition between ADAR and RNAi pathways for an extensive class of RNA targets. Nat Struct Mol Biol 18: 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Vallandingham J, Shiu P, Li H, Hunter CP, Mak HY. 2014. The DEAD box helicase RDE-12 promotes amplification of RNAi in cytoplasmic foci in C. elegans. Curr Biol 24: 832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yigit E, Batista PJ, Bei Y, Pang KM, Chen CC, Tolia NH, Joshua-Tor L, Mitani S, Simard MJ, Mello CC. 2006. Analysis of the C. elegans Argonaute family reveals that distinct Argonautes act sequentially during RNAi. Cell 127: 747–757. [DOI] [PubMed] [Google Scholar]
- Zhao HQ, Zhang P, Gao H, He X, Dou Y, Huang AY, Liu XM, Ye AY, Dong MQ, Wei L. 2015. Profiling the RNA editomes of wild-type C. elegans and ADAR mutants. Genome Res 25: 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang JJ, Banse SA, Hunter CP. 2013. The nuclear argonaute NRDE-3 contributes to transitive RNAi in Caenorhabditis elegans. Genetics 194: 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.