

Abstract
Summary
Rapid RNA synthesis of comprehensive single mutant libraries and targeted multiple mutant libraries is enabling new multidimensional chemical approaches to solve RNA structures. PCR assembly of DNA templates and in vitro transcription allow synthesis and purification of hundreds of RNA mutants in a cost-effective manner, with sharing of primers across constructs allowing significant reductions in expense. However, these protocols require organization of primer locations across numerous 96 well plates and guidance for pipetting, non-trivial tasks for which informatics and visualization tools can prevent costly errors. We report here an online tool to accelerate synthesis of large libraries of desired mutants through design and efficient organization of primers. The underlying program and graphical interface have been experimentally tested in our laboratory for RNA domains with lengths up to 300 nucleotides and libraries encompassing up to 960 variants. In addition to the freely available Primerize-2D server, the primer design code is available as a stand-alone Python package for broader applications.
Availability and Implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
In cells and viruses, RNA molecules modulate translation, genome silencing and editing, as well as other crucial biological processes (Khalil and Collins, 2010; Qi and Arkin, 2014). Studies of non-coding RNA molecules can now leverage systematic mutagenesis and compensatory mutations to infer accurate structures of RNA domains. The mutate-and-map (M2) approach provides rich base-pairing information by assessing which nucleotides are ‘released’ upon making single mutations at every other nucleotide; and mutate-map-rescue (M2R) tests base pairs by detecting rescue of the RNA structure by predicted compensatory mutations (Tian and Das, 2016). For these studies, RNA synthesis can be achieved at low cost by in vitro transcription from DNA templates harboring different mutations. For templates under 300 bps, PCR-based assembly of mixtures of short primers is time- and cost-effective (Thachuk and Condon, 2007; Tian et al., 2015) compared to plasmid-based techniques that require bacterial cloning (Firnberg and Ostermeier, 2012); multi-channel pipetting can further reduce time and expense. Nonetheless, such mutagenesis libraries often involve hundreds of constructs, requiring careful layout of primer sequences to avoid errors in pipetting. With the increasing demand for validating or falsifying structural models, there is a need for straightforward tools to automate the design and organization of these primer sets. For high-throughput RNA mutagenesis studies so far, design of primer plates has relied on unpublished scripts that required MATLAB setup. Here, we describe the Primerize-2D server, which supports on-line design of primers and their organization into 96-well plates for M2 and M2R studies.
2 Results
A previous application of Primerize supported automated primer design for single construct assembly (Tian et al., 2015). The new Primerize-2D server supports M2 plates, which enable separate synthesis of RNA single mutants for ‘two-dimensional’ datasets (Kladwang et al., 2011); and M2R sets that allow testing of base pairs in secondary structures through double mutations (Tian et al., 2014) (Supplementary Fig. S1). Both application pages provide one-click demos of the 158-nt Tetrahymena group I intron P4-P6 domain example. To submit a job, the DNA template sequence (and at least one secondary structure for M2R, which can be obtained from RNA structure prediction algorithms or prior M2 experiments) is required. The user can specify a particular set of primers from a previous single assembly design (manually or channeled from shortcut buttons); otherwise a solution from Primerize will be computed and used. For M2R designs, when several secondary structures are provided, the DIFF mode is activated, which compares base pairs across all structures and targets pairs that discriminate between structures. This DIFF mode is useful when multiple secondary structure hypotheses are under investigation. Additionally, advanced options are available, including sequence numbering offset (to allow the mutation positions to match conventional numbering), position range over which mutations should be designed, and mutation library (by default, nucleotides are mutated to their complement; additional choices are available).
After job submission, the web page reports the assigned JOB_ID and run status. A typical job takes approximately 2 seconds. Once finished, the output is automatically displayed on the same page (Fig. 1 and Supplementary Fig. S2). First, the web page displays a text representation of input sequence and target structures aligned, with numbering and chosen mutation range highlighted. This is helpful for double-checking if the advanced options are correct. For M2R designs, the targeted base pairs as well as mode are marked. Next, the page displays a graphical schematic of the 96-well primer plates to be ordered for DNA assembly. For each primer, constructs that share the same primer as the wild-type (WT) construct are colored differently. For each well, a tooltip displays details on mouse hover. Last, the PCR assembly scheme shows how the primers map into the full-length template sequence, as in conventional assembly of a single template.
Fig. 1.
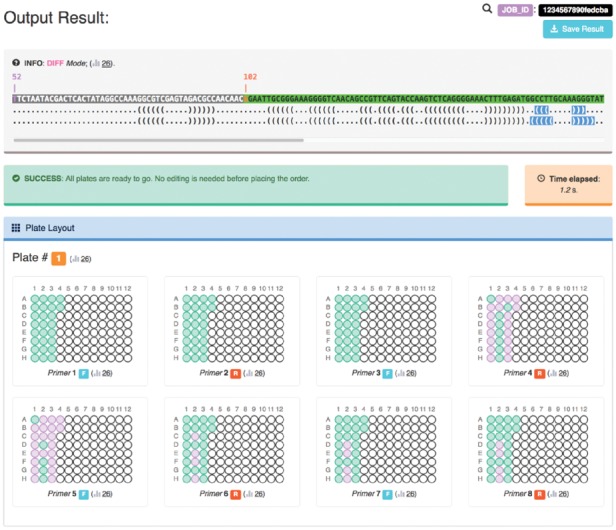
Primerize-2D server output interface for M2R demo
All Primerize results displayed on the web page are available for download, and Excel workbooks with plate spreadsheets are provided for 96-well plate ordering from vendors such as Integrated DNA Technologies (Coralville, IA). SVG images of each plate, construct list and assembly schemes are included for convenience. Job results are saved anonymously on the server for 9 months, and are only accessible by the corresponding JOB_ID. Additional tutorial pages for server usage, plate-format order guidance and experimental protocols are described to assist setup, as well as links to published studies and common workflows that leverage multiple mutant libraries to model structures of new RNAs.
The Primerize server, previously built with CherryPy (The CherryPy team), was reconfigured in the more versatile Django framework (Django Software Foundation), for web service and job management. For reliability and long-term support, our server is hosted at AWS (Amazon Web Services). All connections are under HTTPS protocol for enhanced security. The front-end provides a modern responsive interface with cross-browser compatibility, thanks to jQuery (The jQuery Foundation), Bootstrap (The Bootstrap team) and D3.js (Mike Bostock).
To facilitate wider use of Primerize, we migrated our previous MATLAB scripts to Python. The resulting Python package is available as a stand-alone distribution, and it is the same one used by the Primerize-2D server. The Primerize package can be optionally configured with Numba (Continuum Analytics) for loop optimization. Online documentation and examples are available as part of RiboKit (https://daslab.github.io/Primerize/). The source code can be downloaded freely at the Primerize server.
3 Summary
We have developed an easy-to-use tool for primer design of single assembly, mutate-and-map plates and mutation/rescue sets needed for RNA structural dissection through multidimensional chemical mapping workflows (Tian and Das, 2016). The server is built with fast calculation, customizable parameters, anonymized access and long-term support. The algorithm source code is released as a stand-alone Python package to facilitate broader usage. We hope the Primerize-2D server will contribute to RNA bioscience by facilitating structure inference through multidimensional chemical mapping.
Supplementary Material
Acknowledgements
The authors thank W. Kladwang, C. Geniesse and other members of Das laboratory for extensive testing of the web server and code package.
Funding
This work was supported by the National Institutes of Health (R01GM102519 to R.D.); a Stanford Graduate Fellowship to S.T.; and a Burroughs-Wellcome Foundation Career Award at the Scientific Interface to R.D.
Conflict of Interest: none declared.
References
- Firnberg E., Ostermeier M. (2012) PFunkel: Efficient, expansive, user-defined mutagenesis. PLoS ONE, 7, e52031.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil A.S., Collins J.J. (2010) Synthetic biology: applications come of age. Nat. Rev. Genet., 11, 367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladwang W. et al. (2011) A two-dimensional mutate-and-map strategy for non-coding RNA structure. Nat. Chem., 3, 954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L.S., Arkin A.P. (2014) A versatile framework for microbial engineering using synthetic non-coding RNAs. Nat. Rev. Microbiol., 12, 341–354. [DOI] [PubMed] [Google Scholar]
- Thachuk C., Condon A. (2007). On the design of oligos for gene synthesis. In: Proceedings of the 7th IEEE International Conference on Bioinformatics and Bioengineering, 2007 pp. 123–130.
- Tian S. et al. (2014) High-throughput mutate-map-rescue evaluates SHAPE-directed RNA structure and uncovers excited states. RNA, 20, 1815–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S., Das R. (2016) RNA structure through multidimensional chemical mapping. Q. Rev. Biophys., 49, 1–30. [DOI] [PubMed] [Google Scholar]
- Tian S. et al. (2015) Primerize: Automated primer assembly for transcribing non-coding RNA domains. Nucleic Acids Res., 43, W522–W526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.