

Abstract
A growing body of evidence shows that gene expression in multicellular organisms is controlled by the combinatorial function of multiple transcription factors. This indicates that not the individual transcription factors or signaling molecules, but the combination of expressed regulatory molecules, the regulatory state, should be viewed as the functional unit in gene regulation. Here, I discuss the concept of the regulatory state and its proposed role in the genome-wide control of gene expression. Recent analyses of regulatory gene expression in sea urchin embryos have been instrumental for solving the genomic control of cell fate specification in this system. Some of the approaches that were used to determine the expression of regulatory states during sea urchin embryogenesis are reviewed. Significant developmental changes in regulatory state expression leading to the distinct specification of cell fates are regulated by gene regulatory network circuits. How these regulatory state transitions are encoded in the genome is illuminated using the sea urchin endoderm–mesoderms cell fate decision circuit as an example. These observations highlight the importance of considering developmental gene regulation, and the function of individual transcription factors, in the context of regulatory states.
Keywords: regulatory state, gene regulatory networks, sea urchin development, gene regulation
Introduction
Echinoderms have over the past decades served as a premier system for analysis of developmental gene regulatory networks (GRNs), and the insights deriving from this field have broad implications on many other branches of biology, including functional genomics [1, 2]. The premise of the GRN concept is that the system of regulatory interactions between regulatory genes, encoding transcription factors and signaling molecules, controls the developmental expression of all genes in the genome in every part of an organism and throughout its formation and subsequent lifetime. To experimentally address the complexity of information encoded in developmental GRNs is therefore not a simple task. Focusing on the regulatory states, the active states of a developmental GRN, proves to be an important step in illuminating the underlying GRN. Regulatory states are defined by the set of functionally active transcription factors expressed together within a nucleus at levels high enough to occupy relevant DNA-binding sites and execute regulatory functions. Furthermore, the specific functional status of transcription factors regulated downstream of signaling pathways has to be taken into account. However, regulatory states are not just the product of developmental GRNs, they also represent important functional units in the control of gene expression. Thus, while individual transcription factors and signaling pathways may function in multiple developmental processes, context-specific gene regulation is achieved through the combinatorial function of transcription factors constituting a specific regulatory state.
Several observations highlight the importance of regulatory states in the control of developmental gene expression. First, countless cis-regulatory experiments show that the activation of cis-regulatory modules (CRMs) in multicellular animals requires the combinatorial function of multiple qualitatively distinct transcription factors, indicating that individual transcription factors, even when present at high levels, are not sufficient to activate specific gene expression [3–6]. Second, for most genes for which developmental regulation has been analyzed, specific CRMs that account for the specific gene expression pattern can be identified, demonstrating that spatial and temporal expression of genes during development is predominantly regulated at the transcriptional level [1]. In addition, once transcripts are expressed, further layers of regulation by posttranscriptional and posttranslational processes will affect the levels and function of expressed molecules. Third, many transcription factors are expressed in multiple developmental contexts, where they predominantly regulate the expression of context-specific sets of target genes, as shown, for example, for Pax6 in the mouse eye, brain and pancreas [7], which supports the idea that transcription factor binding as well as function is often determined by the context-specific regulatory state. The fourth type of evidence derives from developmental control mechanisms. Thus, when monitoring the mechanisms underlying the spatial specification of cell fate domains, such as, for example, the definition of the limb bud in a specific position of the embryo [8], novel expression of one or several transcription factors is usually a key step. These transcription factors are neither expressed during the developmental phase preceding the definition of the cell fate domain nor are they expressed in cells surrounding the domain. Thus, in the vast majority of cases, developmental functions depend on a qualitative change in the combination of transcription factors expressed together. Altogether, these and many other observations show that gene regulation is a function of regulatory states and not just of individual transcription factors.
Figure 1 summarizes the role of regulatory states in developmental gene regulation. The expression of regulatory states is directly controlled by upstream developmental GRNs in time and space. In turn, the set of transcription factors constituting the regulatory state controls the expression of all genes in the genome. In the following, I will first consider the control of gene expression downstream of a given regulatory state, before discussing approaches to identify regulatory state expression that have been successfully applied in sea urchin embryos, and finally turn to the developmental control of regulatory state expression in the differential specification of cell fates.
Figure 1.
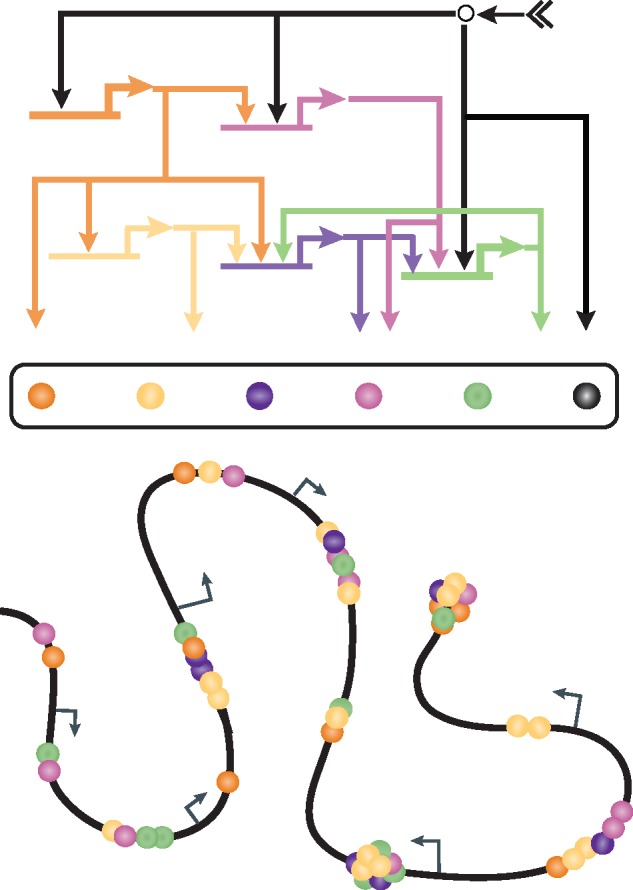
Regulatory states in developmental gene regulation. Scheme showing the expression of a cell fate-specific regulatory state (center) as controlled by the circuitry of a developmental GRN (top). The regulatory state consists of transcription factors co-expressed in a given nucleus, some of which are regulated by developmental signaling interactions (black). While the expression of all genes depends on the regulatory state, the specific combination of transcription factors regulating individual genes may vary (bottom). (A colour version of this figure is available online at: https://academic.oup.com/bfg)
Function of regulatory states in control of gene expression
Countless analyses demonstrate the expression of cell fate-specific combinations of transcription factors in different developmental contexts, as recently reviewed [1]. They confirm that although individual transcription factors may be expressed in multiple developmental contexts, regulatory states are expressed specifically in given embryonic domains at given developmental times. In most cases, cell fate-specific gene expression is therefore controlled by qualitatively different regulatory states, and not just by subtle quantitative differences in the relative abundance of transcription factors constituting a regulatory state. The importance of combinatorial gene regulation is perhaps best illustrated by considering the control of gene expression downstream of signaling pathways. Specific signal response transcription factors control the expression of a large set of genes in response to a given signaling interaction [1, 9, 10]. Yet, although the same signaling interactions are deployed repeatedly throughout development, the set of target genes controlled by these signal response transcription factors is specific to a given developmental context. Thus, other transcription factors within the specific regulatory states contribute to the selection of target genes and to the regulation of their expression. Regulatory states are therefore not just random collections of co-expressed individual transcription factors, but in respect to the genes expressed in any given nucleus, the regulatory state has to be considered as a regulatory unit. When addressing the function of any given transcription factor, it will be in respect to the regulatory context for which these functions are described.
This raises the question of why individual transcription factors are not sufficient to control gene expression. A possible explanation for the requirement of combinatorial gene regulation is that transcription factors controlling a given CRM are not equivalent in their function. Although perhaps some of the transcriptional activators might be able to substitute for one another, some regulatory functions can only be executed by specifically dedicated transcription factors. The best described examples are pioneer factors that have the capacity to bind DNA even when wrapped around histones [11]. Thus, pioneer factors are thought to play a particular role in the initial identification of cis-regulatory sequences. Binding of pioneer factors is a requirement for other transcription factors to have access to the regulatory sequences and contribute to the control of gene expression. But pioneer factors are only part of the equation. Pioneer factors can be expressed broadly such as FoxA in the endoderm or Zelda in the early Drosophila embryo, and nevertheless contribute to the regulation of genes that are expressed exclusively in only a subset of cells [4, 12–14]. For example, FoxA binding to the regulatory sequences of the liver gene Alb1 will only lead to gene expression in the liver, while this gene remains silent in other foregut endoderm cells, despite the binding of FoxA to the Alb1 locus [12]. Other transcription factors that control the expression of Alb1 function together with FoxA to ensure liver-specific gene expression. Although our current understanding of specific transcription factor functions is limited, it is conceivable that other types of functional specialization exist among those transcription factors bound together to a specific CRM, accounting for the requirement for combinatorial function of different regulatory factors. For instance, some transcription factors are responsible for the looping of a CRM to its basal transcriptional promoter to initiate gene transcription [15, 16]. Other transcription factors recruit enzymes that enable various modifications of specific amino acid residues within associated histones. For example, in skeletal muscle cells, G9a, a lysine methyltransferase, is recruited to the myogenin promoter by the transcription factor Sharp-1, and contributes to the repression of myogenin ([17]; for a recent discussion see [1]). The recruitment of broadly expressed cofactors, including histone modification enzymes, to the regulatory sequences of genes that are expressed in a cell fate-specific manner is an important function of transcription factors, and may affect the ability of additional transcription factors to bind to regulatory sequences.
Division of labor among transcription factors is also indicated by the observation that although a combination of factors is required for the control of gene expression, not all factors necessarily do so by direct sequence-specific binding to DNA. A recent study demonstrates that transcription factors required for expression of genes in response to estrogen stimulation are recruited to the regulatory sequences of their target genes by binding to a transcription factor recognizing the CRMs [18]. Although both Gata3 and the retinoic acid receptor (RAR) are required for activation of gene expression, neither of them appeared to bind to CRMs through their cognate binding sites, but by binding to the estrogen receptor-α. These results indicate that the precise regulatory function of each transcription factor within the context of a given CRM may range from the initial sequence-specific identification of the CRM, the opening of the chromatin by recruitment of cofactors, the recruitment of additional transcription factors, to the looping of the CRM to the basal transcription start site, and most likely includes additional functions. In the absence of any of these functions, gene expression will not be supported. The function of the regulatory state therefore lies in reading regulatory DNA sequences, and thus in determining which genes will be expressed in a given nucleus. However, to initiate gene transcription, the function of additional cofactors and histone modification enzymes is required, and they are recruited to CRMs by sequence-specific transcription factors.
Although the regulatory state should be considered as a functional unit in the sense that it determines the set of expressed genes, not all genes expressed downstream of a given regulatory state are controlled in the same manner, as indicated in Figure 1. Thus, comparing the regulation of differentiation genes expressed within a given cell type shows that only a subset of the regulatory state will be used to drive the expression of each gene. Even for those CRMs active within the same regulatory context, the precise grammar can be different among these CRMs, in respect to the type of transcription factors contributing to gene regulation, and in respect to the number and position of binding sites for each regulatory factor. For example, the expression of crystallin genes in the mouse lens epithelium is driven by Pax6, c-Maf, Six3, Sox2, RAR/RXR and other regulatory factors, but the precise combination of these factors binding to the enhancers and promoters of each crystallin gene varies [1, 19]. Similarly, genes expressed preferentially in sea urchin skeletogenic cells are differentially affected by the perturbation of two upstream transcription factors, Ets1 and Alx1 [20]. Some genes are affected by perturbation of Ets1, some by perturbation of Alx1, some by both perturbations and some were not affected at all, indicating regulatory inputs other than Ets1 and Alx1. Thus, even when exposed to the same regulatory state, individual genes are not controlled by exactly the same set of transcription factors, but different subsets of transcription factors within the same regulatory state will control the expression of individual genes within a nucleus. However, only the complete regulatory state will lead to the expression of the entire set of genes.
Analysis of developmental regulatory state expression
To reveal the mechanisms controlling developmental gene expression, identification of regulatory states is therefore essential. Once the sea urchin genome sequence became available, the echinoderm community collaborated in a large-scale effort to compile the spatial and temporal expression profiles of various functional classes of molecules, most prominently transcription factors and molecules involved in various signaling pathways, during early embryogenesis [21–31]. On the basis of these gene expression patterns, lists of candidate genes were assembled for each embryonic domain indicating the potential players of the GRN underlying specification of the domain. This systematic analysis of regulatory gene expression patterns was a crucial factor in the success of subsequent GRN analyses. Instead of working with just a small representation of expressed transcription factors, where the presence of additional layers of indirect regulatory interactions can never be ruled out, having a genome-wide survey of transcription factor expression allows for a comprehensive analysis of all regulatory functions of a regulatory state. As a result, developmental GRNs have been experimentally analyzed for the early cell fates of almost the entire embryo of the purple sea urchin Strongylocentrotus purpuratus [32–40]. These GRNs demonstrate how genomic programs operate the specification of diverse cell fates during early embryogenesis and they shed light on the function of regulatory factors in diverse developmental contexts.
The systematic analysis of developmental regulatory states provides an important starting point for the analysis of developmental GRNs. The initial analysis of regulatory gene expression in sea urchin embryos was based on the quantitative analysis of expression levels for all known regulatory genes in the sea urchin genome, identifying those regulatory genes expressed at levels that potentially support regulatory function. A previous computational simulation showed that in this embryo, developing at 15°C, about 10 transcripts per cell are sufficient to produce several hundred molecules of transcription factor molecules within a few hours, sufficient to activate downstream gene expression [41]. Assuming that in the early embryo, each cell fate domain consists of at least 16 cells, the whole embryo should show at least 160 copies of regulatory gene mRNA to produce functional levels of transcription factors [23]. Thus, regulatory genes expressed at levels of at least 150–300 transcripts/embryo were included in the spatial analysis of gene expression. The most important data set showing the developmental expression of regulatory genes derives from whole-mount in situ hybridization experiments carried out at multiple stages of embryogenesis [42]. Although such data are not quantitative, they show at once the expression of an individual regulatory gene in all cells of the embryo.
A crucial step for using large sets of spatial gene expression data in GRN analysis is the transformation of qualitative descriptions of expression patterns to digitalized Boolean annotations. Thus, the spatial distribution of regulatory gene transcripts throughout the entire embryo is annotated as Boolean ON/OFF states of gene expression [32, 38]. Gene expression is considered ON where transcripts are detectable by in situ hybridization and thus possibly sufficiently prevalent to indicate regulatory function, or OFF where transcripts are not detectable. As a result of this annotation, the gene expression status is provided for all cells of the embryo, irrespective of the gene expression pattern, thus discriminating between genes not being expressed and genes for which no data are available. In the context of GRNs, absence of a transcription factor in a given cell fate domain can be just as important as its presence elsewhere, particularly in the case of transcription factors that function as repressors. The systematic digital annotation of regulatory gene expression patterns in echinoderms has generated an invaluable resource that allows for comparative analyses of large collections of spatial expression data, which would not be possible based on qualitative image data alone. For every regulatory gene that is part of a developmental GRN in sea urchin embryos, these data are publicly accessible through the BioTapestry GRN models, as can be seen, for example, at http://grns.biotapestry.org/SpEndomes/ [43–45]. The Boolean annotation of regulatory gene expression also allows the direct analysis of regulatory state expression, making it possible to rapidly identify within a large set of spatial expression data those regulatory genes expressed in a given embryonic domain at a given time in development. Whole-mount in situ hybridization analyses have thus in echinoderms served an invaluable purpose to relatively quickly assess expression of individual genes throughout the embryo and at different developmental times. Despite the relative low throughput of this approach, the early definition of standards for the echinoderm community and the consistent annotation of gene expression patterns have over the years led to the accumulation of a precious data set. These expression data were initially mostly restricted to early development of the sea urchin embryo but is currently being extended to include the entire first 72 h of sea urchin development ([46, 47]; Valencia, J. & I.S.P, unpublished data).
In addition to transcription factors, intercellular signaling interactions are important components of developmental GRNs. The control of gene expression downstream of signaling interactions is executed by dedicated signal response transcription factors whose regulatory function is determined by the signaling pathway [10]. A complete description of the regulatory state therefore not only includes expression of transcription factors but also information about presence or absence of signaling interactions. For example, the function of Tcf, the transcription factor controlled by Wnt signaling, is decided by association with a cofactor. In the presence of Wnt signaling, Tcf binds to nuclearized β-catenin, a co-activator of gene expression, while in the absence of Wnt signaling and nuclear β-catenin, the co-repressor Groucho binds to Tcf and leads to repression of Tcf target genes [48]. System-level analyses of the spatial expression of signaling ligands and receptors and the functional assessment of given signaling interactions have shown the contribution of diverse signaling interactions to the developmental specification of cell fates in the sea urchin embryo [49–51].
Spatial expression data provide critical information for GRN analysis, as they allow direct comparison of expression of either individual regulatory genes or entire regulatory states across the entire embryo or at least several cellular domains neighboring the gene expression domain. In addition, determining the quantitative levels of regulatory gene expression is also relevant, perhaps not so much for understanding the differential specification of cell fates as for understanding the kinetics of the developmental process. Thus, the rate of transcript accumulation will determine the temporal delay between onset of regulatory gene transcription and onset of target gene expression, and therefore the kinetic behavior of the GRN [41, 52]. Quantitative data of developmental gene expression have been obtained for early sea urchin embryogenesis [53–55]. Even though these quantitative data are often generated with whole embryos, and thus only reveal the total transcript level per embryo, when combined with spatial expression data that reveal the number of cells in which gene expression occurs, an estimate for transcript levels per cell can be obtained. Furthermore, transcript levels have also been determined for isolated cell types such as skeletogenic cells and pigment cells [20, 56], and eventually, single-cell transcriptomes will expand our current understanding of regulatory states in each embryonic domain.
Control of developmental regulatory state expression
During development, regulatory state expression changes both in time and in space to define the body plan and progressively specify its constituent cell fates. Thus, the identification of regulatory mechanisms for given developmental functions often starts with characterizing the change in regulatory state associated with the developmental process. A Boolean computational model of the endomesoderm GRN was able to reproduce the expression of regulatory states during sea urchin development, showing that developmental changes in regulatory state expression are indeed controlled by the underlying GRNs [57].
An example for how the developmental expression of regulatory states is controlled by GRN circuitry is shown in Figure 2. This circuit is a component of the sea urchin endomesoderm GRN that encodes the distinct specification of mesodermal and endodermal cell fates [37, 38, 58]. Both cell fates are specified in the descendants of a common cell lineage, the veg2 cells. Of all the regulatory genes involved in endodermal and mesodermal specification, only few are expressed during earliest developmental stages. Analyzing the spatial expression of these regulatory genes shows that endomesoderm progenitors express an ‘endomesodermal’ regulatory state, composed of transcription factors later associated with either endodermal (such as Hox11/13b and FoxA) or mesodermal (such as Gcm) specification [37]. However, distinct populations of cells can be identified a few hours later, expressing either endodermal or mesodermal regulatory states (Figure 2). The reason for mesodermal genes being expressed first in the endomesoderm precursors and later in the mesodermal progenitors is their regulation by Delta/Notch signaling, induced by the expression of Delta ligand in adjacent skeletogenic mesoderm cells [59]. In turn, endodermal genes are controlled by Tcf/β-catenin that is present initially in all veg2 descendants because of maternal anisotropies [37, 60, 61]. Thus, in early endomesoderm progenitors, both Tcf/β-catenin and SuH/NICD are available, leading to the co-expression of regulatory genes later specific to either endoderm or mesoderm. Interestingly, gcm and hox11/13b show similar kinetics in expression levels during early specification of endomesoderm progenitors, despite being controlled by different regulatory mechanisms.
Figure 2.

Control of regulatory state expression in the endoderm/mesoderm cell fate decision. (A) Early in development, endodermal and mesodermal regulatory genes are co-expressed in endomesoderm progenitor cells (12 h), but expression eventually resolves into two different spatial domains (18 h) giving rise to endoderm expressing foxA and mesoderm expressing gcm (60 h). (B) Temporal expression profiles of endodermal and mesodermal regulatory genes show that gcm (mesoderm) and hox11/13b (endoderm) are the earliest genes expressed in endomesoderm cells. Despite being controlled by different regulatory mechanisms, both genes are initially expressed with similar accumulation rates. (C) Developmental GRN controlling endoderm/mesoderm cell fate decision showing that gcm is controlled by Delta/Notch signaling and is upstream of a positive feedback circuit active after 18 h. Spatial expression of the endoderm GRN is controlled by Tcf/β-catenin. Both endoderm and mesoderm GRNs are active in endomesoderm progenitors without cross-interaction, and are turned off in cells of the alternative cell fate. Regulatory states are shown in top-right corner. (A colour version of this figure is available online at: https://academic.oup.com/bfg)
Once the endomesoderm progenitors divide, about half of the daughter cells become physically separated from the Delta signaling source, terminating mesodermal gene expression. These cells continue to express endodermal regulatory genes and will give rise to foregut and midgut endoderm. Those daughter cells that continue to receive Delta/Notch signaling initially continue to express an endomesodermal regulatory state. However, within a couple of hours, these cells turn off endodermal regulatory genes by a Delta/Notch signaling-dependent mechanism that leads to the clearance of nuclear β-catenin and the conversion of Tcf into a repressor by binding to Groucho [38]. Eventually, Delta/Notch signaling turns off also in mesodermal cells, and expression of mesodermal regulatory genes is maintained by a positive feedback circuit between gcm, gatae and six1/2 [58]. The dramatic change in regulatory state expression during the endoderm/mesoderm cell fate decision in the sea urchin embryo is therefore the result of two independent GRNs, activated together in endomesoderm progenitors, and the subsequent inactivation of either the mesodermal or endodermal GRNs in endodermal or mesodermal progenitors, respectively [38]. Even when both GRNs are co-expressed, no interaction between the two cell fate GRNs is observed. Thus, the mixed endomesodermal regulatory state is only the result of the combined activity of two GRNs, indicating that regulatory states themselves can be just as modular as their underlying GRNs. Ultimately, changes in the geometry of the embryo, differential properties of signaling molecules and developmental time all contribute to the differential readout of the regulatory genome in progenitors versus descendants and endoderm versus mesoderm.
What this small circuit for separation of endoderm and mesoderm in the sea urchin embryo demonstrates, is that ultimately the DNA sequences responsible for given developmental functions such as the endoderm–mesoderm cell fate decision may be just a few binding sites, a tiny fraction of the regulatory genome, active for just a short time and thus basically undetectable based on genome-scale approaches without prior knowledge of the responsible transcription factors and the precise developmental timing [62]. However, where the developmental expression of regulatory states and the changes in regulatory states associated with a developmental function are identified, finding the underlying control mechanism within the regulatory genome becomes feasible. In the sea urchin, the consistent analysis and annotation of regulatory gene expression profiles, and the regulatory states that were deduced from these data, have provided a significant contribution to the analysis of GRNs. Similar data sets are available also for other animals, although often presented at the level of single genes and not in the form of regulatory states. However, the approaches used for identification of developmental regulatory states in the sea urchin embryo are to a large extent applicable to other systems as well and should enhance the identification of developmental mechanisms and their origin within the regulatory genome.
Key Points
Regulatory states are the set of co-expressed transcription factors.
Regulatory states determine the combinatorial control of gene expression.
Extensive analysis of developmental regulatory state expression enables the identification of GRNs.
Developmental changes in regulatory states determine the differential specification of cell fates and are controlled by developmental GRNs.
Acknowledgments
I wish to thank Deanna Thomas for her work in preparing the figures and Jonathan Valencia for providing the image showing gcm expression in the late embryo. I am deeply grateful to Eric Davidson for countless discussions over the years that contributed to the thoughts expressed here.
Funding
This work was supported by National Institutes of Health Grant HD-037105.
Biography
Isabelle S. Peter is a research professor at the California Institute of Technology. Her laboratory studies gene regulatory networks in development and evolution, with particular focus on sea urchins.
References
- 1. Peter IS, Davidson EH.. Genomic Control Process, Development and Evolution. San Diego: Academic Press/Elsevier, 2015. [Google Scholar]
- 2. Peter IS, Davidson EH.. Chapter 13: Implications of developmental gene regulatory networks inside and outside developmental biology In: Paul MW. (ed). Current Topics in Developmental Biology, Vol. 117 San Diego: Academic Press/Elsevier, 2016, 237–51. [DOI] [PubMed] [Google Scholar]
- 3. Thanos D, Maniatis T.. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 1995;83:1091–100. [DOI] [PubMed] [Google Scholar]
- 4. Xu Z, Chen H, Ling J, et al. Impacts of the ubiquitous factor Zelda on Bicoid-dependent DNA binding and transcription in Drosophila. Genes Dev 2014;28:608–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Swanson CI, Evans NC, Barolo S.. Structural rules and complex regulatory circuitry constrain expression of a Notch- and EGFR-regulated eye enhancer. Dev Cell 2010;18:359–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yuh CH, Bolouri H, Davidson EH.. Genomic cis-regulatory logic: experimental and computational analysis of a sea urchin gene. Science 1998;279:1896–902. [DOI] [PubMed] [Google Scholar]
- 7. Xie Q, Yang Y, Huang J, et al. Pax6 interactions with chromatin and identification of its novel direct target genes in lens and forebrain. PLoS One 2013;8:e54507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duboc V, Logan MP.. Regulation of limb bud initiation and limb-type morphology. Dev Dyn 2011;240:1017–27. [DOI] [PubMed] [Google Scholar]
- 9. Schuijers J, Mokry M, Hatzis P, et al. Wnt-induced transcriptional activation is exclusively mediated by TCF/LEF. EMBO J 2014;33:146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barolo S, Posakony JW.. Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling. Genes Dev 2002;16:1167–81. [DOI] [PubMed] [Google Scholar]
- 11. Zaret KS, Carroll JS.. Pioneer transcription factors: establishing competence for gene expression. Genes Dev 2011;25:2227–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zaret KS, Watts J, Xu J, et al. Pioneer factors, genetic competence, and inductive signaling: programming liver and pancreas progenitors from the endoderm. Cold Spring Harb Symp Quant Biol 2008;73:119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liberman LM, Stathopoulos A.. Design flexibility in cis-regulatory control of gene expression: synthetic and comparative evidence. Dev Biol 2009;327:578–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pearson JC, Watson JD, Crews ST.. Drosophila melanogaster Zelda and Single-minded collaborate to regulate an evolutionarily dynamic CNS midline cell enhancer. Dev Biol 2012;366:420–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ren X, Siegel R, Kim U, et al. Direct interactions of OCA-B and TFII-I regulate immunoglobulin heavy-chain gene transcription by facilitating enhancer-promoter communication. Mol Cell 2011;42:342–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo Y, Monahan K, Wu H, et al. CTCF/cohesin-mediated DNA looping is required for protocadherin α promoter choice. Proc Natl Acad Sci USA 2012;109:21081–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ling BM, Gopinadhan S, Kok WK, et al. G9a mediates Sharp-1-dependent inhibition of skeletal muscle differentiation. Mol Biol Cell 2012;23:4778–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu Z, Merkurjev D, Yang F, et al. Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell 2014;159:358–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cvekl A, Duncan MK.. Genetic and epigenetic mechanisms of gene regulation during lens development. Prog Retin Eye Res 2007;26:555–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rafiq K, Shashikant T, McManus CJ, et al. Genome-wide analysis of the skeletogenic gene regulatory network of sea urchins. Development 2014;141:2542. [DOI] [PubMed] [Google Scholar]
- 21. Howard-Ashby M, Materna SC, Brown CT, et al. Identification and characterization of homeobox transcription factor genes in Strongylocentrotus purpuratus, and their expression in embryonic development. Dev Biol 2006;300:74–89. [DOI] [PubMed] [Google Scholar]
- 22. Howard-Ashby M, Materna SC, Brown CT, et al. Gene families encoding transcription factors expressed in early development of Strongylocentrotus purpuratus. Dev Biol 2006;300:90–107. [DOI] [PubMed] [Google Scholar]
- 23. Howard-Ashby M, Materna SC, Brown CT, et al. High regulatory gene use in sea urchin embryogenesis: implications for bilaterian development and evolution. Dev Biol 2006;300:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Materna SC, Howard-Ashby M, Gray RF, et al. The C2H2 zinc finger genes of Strongylocentrotus purpuratus and their expression in embryonic development. Dev Biol 2006;300:108–20. [DOI] [PubMed] [Google Scholar]
- 25. Sodergren E, Weinstock GM, Davidson EH, et al. The genome of the sea urchin Strongylocentrotus purpuratus. Science 2006;314:941–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tu Q, Brown CT, Davidson EH, et al. Sea urchin Forkhead gene family: phylogeny and embryonic expression. Dev Biol 2006;300:49–62. [DOI] [PubMed] [Google Scholar]
- 27. Arnone MI, Rizzo F, Annunciata R, et al. Genetic organization and embryonic expression of the ParaHox genes in the sea urchin S. purpuratus: insights into the relationship between clustering and colinearity. Dev Biol 2006;300:63–73. [DOI] [PubMed] [Google Scholar]
- 28. Rizzo F, Fernandez-Serra M, Squarzoni P, et al. Identification and developmental expression of the ets gene family in the sea urchin (Strongylocentrotus purpuratus). Dev Biol 2006;300:35–48. [DOI] [PubMed] [Google Scholar]
- 29. Croce JC, Wu SY, Byrum C, et al. A genome-wide survey of the evolutionarily conserved Wnt pathways in the sea urchin Strongylocentrotus purpuratus. Dev Biol 2006;300:121–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lapraz F, Rottinger E, Duboc V, et al. RTK and TGF-beta signaling pathways genes in the sea urchin genome. Dev Biol 2006;300:132–52. [DOI] [PubMed] [Google Scholar]
- 31. Walton KD, Croce JC, Glenn TD, et al. Genomics and expression profiles of the Hedgehog and Notch signaling pathways in sea urchin development. Dev Biol 2006;300:153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li E, Cui M, Peter IS, et al. Encoding regulatory state boundaries in the pregastrular oral ectoderm of the sea urchin embryo. Proc Natl Acad Sci USA 2014;111:E906–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li E, Materna SC, Davidson EH.. Direct and indirect control of oral ectoderm regulatory gene expression by Nodal signaling in the sea urchin embryo. Dev Biol 2012;369:377–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Materna SC, Ransick A, Li E, et al. Diversification of oral and aboral mesodermal regulatory states in pregastrular sea urchin embryos. Dev Biol 2013;375:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oliveri P, Carrick DM, Davidson EH.. A regulatory gene network that directs micromere specification in the sea urchin embryo. Dev Biol 2002;246:209–28. [DOI] [PubMed] [Google Scholar]
- 36. Oliveri P, Tu Q, Davidson EH.. Global regulatory logic for specification of an embryonic cell lineage. Proc Natl Acad Sci USA 2008;105:5955–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peter IS, Davidson EH.. The endoderm gene regulatory network in sea urchin embryos up to mid-blastula stage. Dev Biol 2010;340:188–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peter IS, Davidson EH.. A gene regulatory network controlling the embryonic specification of endoderm. Nature 2011;474:635–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Davidson EH, Rast JP, Oliveri P, et al. A genomic regulatory network for development. Science 2002;295:1669–78. [DOI] [PubMed] [Google Scholar]
- 40. Davidson EH, Rast JP, Oliveri P, et al. A provisional regulatory gene network for specification of endomesoderm in the sea urchin embryo. Dev Biol 2002;246:162–90. [DOI] [PubMed] [Google Scholar]
- 41. Bolouri H, Davidson EH.. Transcriptional regulatory cascades in development: initial rates, not steady state, determine network kinetics. Proc Natl Acad Sci USA 2003;100:9371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ransick A. Detection of mRNA by in situ hybridization and RT-PCR. Dev Biol 2004;74:601–20. [DOI] [PubMed] [Google Scholar]
- 43. Longabaugh WJ. BioTapestry: a tool to visualize the dynamic properties of gene regulatory networks. Methods Mol Biol 2012;786:359–94. [DOI] [PubMed] [Google Scholar]
- 44. Longabaugh WJ, Davidson EH, Bolouri H.. Computational representation of developmental genetic regulatory networks. Dev Biol 2005;283:1–16. [DOI] [PubMed] [Google Scholar]
- 45. Longabaugh WJ, Davidson EH, Bolouri H.. Visualization, documentation, analysis, and communication of large-scale gene regulatory networks. Biochim Biophys Acta 2009;1789:363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Annunciata R, Perillo M, Andrikou C, et al. Pattern and process during sea urchin gut morphogenesis: the regulatory landscape. Genesis 2014;52:251–68. [DOI] [PubMed] [Google Scholar]
- 47. Andrikou C, Pai CY, Su YH, et al. Logics and properties of a genetic regulatory program that drives embryonic muscle development in an echinoderm. Elife 2015;4:e07343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Range RC, Venuti JM, McClay DR.. LvGroucho and nuclear beta-catenin functionally compete for Tcf binding to influence activation of the endomesoderm gene regulatory network in the sea urchin embryo. Dev Biol 2005;279:252–67. [DOI] [PubMed] [Google Scholar]
- 49. Cui M, Siriwion N, Li E, et al. Specific functions of the Wnt signaling system in gene regulatory networks throughout the early sea urchin embryo. Proc Natl Acad Sci USA 2014;111:E5029–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Materna SC, Davidson EH.. A comprehensive analysis of Delta signaling in pre-gastrular sea urchin embryos. Dev Biol 2012;364:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Luo YJ, Su YH.. Opposing nodal and BMP signals regulate left-right asymmetry in the sea urchin larva. PLoS Biol 2012;10:e1001402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ben-Tabou de-Leon S, Davidson EH.. Modeling the dynamics of transcriptional gene regulatory networks for animal development. Dev Biol 2009;325:317–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tu Q, Cameron RA, Davidson EH.. Quantitative developmental transcriptomes of the sea urchin Strongylocentrotus purpuratus. Dev Biol 2014;385:160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Materna SC, Nam J, Davidson EH.. High accuracy, high-resolution prevalence measurement for the majority of locally expressed regulatory genes in early sea urchin development. Gene Expr Patterns 2010;10:177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gildor T, Malik A, Sher N, et al. Quantitative developmental transcriptomes of the Mediterranean sea urchin Paracentrotus lividus. Mar Genomics 2016;25:89–94. [DOI] [PubMed] [Google Scholar]
- 56. Barsi JC, Tu Q, Calestani C, et al. Genome-wide assessment of differential effector gene use in embryogenesis. Development 2015;142:3892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Peter IS, Faure E, Davidson EH.. Feature Article: predictive computation of genomic logic processing functions in embryonic development. Proc Natl Acad Sci USA 2012;109:16434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ransick A, Davidson EH.. Cis-regulatory logic driving glial cells missing: self-sustaining circuitry in later embryogenesis. Dev Biol 2012;364:259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ransick A, Davidson EH.. cis-regulatory processing of Notch signaling input to the sea urchin glial cells missing gene during mesoderm specification. Dev Biol 2006;297:587–602. [DOI] [PubMed] [Google Scholar]
- 60. Logan CY, Miller JR, Ferkowicz MJ, et al. Nuclear beta-catenin is required to specify vegetal cell fates in the sea urchin embryo. Development 1999;126:345–57. [DOI] [PubMed] [Google Scholar]
- 61. Ben-Tabou de-Leon SB, Davidson EH.. Information processing at the foxa node of the sea urchin endomesoderm specification network. Proc Natl Acad Sci USA 2010;107:10103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Peter IS. A view on systems biology beyond scale and method In: Green S. (ed). Philosophy of Systems Biology: Perspectives from Scientists and Philosophers. Cham: Springer International Publishing, 2017, 237–45. [Google Scholar]