

Abstract
Summary
Next-generation sequencing has been widely applied to understand the complexity of non-coding RNAs (ncRNAs) in the last decades. Here, we present CPSS 2.0, an updated version of CPSS 1.0 for small RNA sequencing data analysis, with the following improvements: (i) a substantial increase of supported species from 10 to 48; (ii) improved strategies applied to detect ncRNAs; (iii) more ncRNAs can be detected and profiled, such as lncRNA and circRNA; (iv) identification of differentially expressed ncRNAs among multiple samples; (v) enhanced visualization interface containing graphs and charts in detailed analysis results. The new version of CPSS is an efficient bioinformatics tool for users in non-coding RNA research.
Availability and implementation
CPSS 2.0 is implemented in PHP + Perl + R and can be freely accessed at http://114.214.166.79/cpss2.0/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Development of large-scale sequencing technology has yielded extensive small RNA (sRNA) sequencing data. Many small RNA analysis tools have been developed accordingly, such as Chimira (Vitsios and Enright, 2015), WapRNA (Zhao et al., 2011), Oasis (Capece et al., 2015), sRNAtoobox (Rueda et al., 2015), mirTool 2.0 (Wu et al., 2013), MAGI (Kim et al., 2014), ISRNA (Luo et al., 2014) and our published work, CPSS 1.0 (Zhang et al., 2012).
With increasing evidence showing that small RNAs function in the context of complex regulatory network (Bracken et al., 2016), a systematic interpretation platform of sRNA data is still in great demand. However, current tools provide simple small RNA profiling rather than a systematic analysis. Their main limitations are as followed: (i) Their analysis cannot provide comprehensiveness and profoundness at the same time. For instance, Chimira is specified in detecting miRNA modification. Although many tools such as sRNAtoolbox and Oasis are equipped with multiple modules, they fail to integrate each other. Users should conduct unnecessary intermediate submission. (ii) Most of existing methods are short of graph presentations of the results. In some cases, even they provide plenty of graphs, lack of clear illustration and appropriate layout does not help to improve their popularity. (iii) Owing to the fixed analysis report, users are not able to modulate parameters after the completion of analysis.
To meet the urgent demand, we updated CPSS 1.0 to CPSS 2.0, including the following improvements: (i) Within a single submission, CPSS 2.0 is able to deliver analysis report from ncRNA quantification to miRNA target prediction and annotation of single and multiple datasets. With lncRNA and circRNA added to the system, CPSS 2.0 assembles the most abundant ncRNA modules. The number of supported species is also substantially increased from 10 to 48. All databases and software integrated in CPSS 2.0 are updated to the latest version. (ii) CPSS 2.0 classifies all results into two main categories ‘General Results’ and ‘Functional Analysis’. Each has several subcategories, presenting results in graphs and charts, which is very helpful for users with an intuitive understanding of statistic data. (iii) On each detailed result page, CPSS 2.0 provides search function for user to search specific terms or values. On GO, Pathway and Protein domain detailed pages, user could modify default parameters, P-value and enrichment fold. Taken together, CPSS 2.0 is the most comprehensive webserver so far among all available tools. Detailed comparison in specific modules we deemed essential or important is provided in the Supplementary table S1. We believe that CPSS 2.0 could assist users in a comprehensive and effective manner.
2 Workflow
The overall workflow of CPSS 2.0 is shown in Supplementary Figure S1. Users can submit input data in FASTA format or FASTA files compressed in *.tar.gz format. After genome alignment with Bowtie (Langmead et al., 2009), CPSS 2.0 first matches genome mapped reads with several reference sequences using Bowtie or Blast in the following order: precursor miRNA, mature, piRNA, circRNA, lncRNA, Rfam. repeats and mRNA, and then classifies them into known miRNAs, known piRNAs, mRNAs, repeat-associated RNA, circRNA, lncRNA and other types of ncRNAs (sRNA, tRNA, snRNAs and snoRNA). Expression of these classified RNAs are normalized based on the absolute counts of mapped reads (Normalized counts are displayed by Reads per Million, RPM). Sequences that are mapped to the reference genome but cannot be assigned to any of the above categories (defined as unclassified sequences) are used to predict novel miRNAs by Mireap (https://sourceforge.net/projects/mireap/). Secondary structure of predicted miRNAs are drawn by RNAfold (Lorenz et al., 2011). CPSS 2.0 implements miRanda (John et al., 2004) to identify known and novel miRNA targets, performing on the most abundant known/novel miRNA for single sample and for all the differentially expressed miRNAs among multiple samples or between two experimental groups. To further explore the potential biological function of predicted miRNA targets, CPSS 2.0 annotates them with Gene Ontology (GO), pathway, protein-domain information and extracts enriched annotation terms. Genes included in the enriched terms are matched with STRING (Szklarczyk et al., 2014) database to retrieve protein-protein interaction (PPI) information. Visualization of the PPI network is implemented by vis.js library (http://visjs.org/).
Currently, CPSS 2.0 is compatible with 48 reference genomes across vertebrates, insects, deuterostomes, nematodes and plants. It is ready-to-use for most users with pre-set analysis parameters. For users with advanced needs, parameters for each analysis step can be modified. CPSS 2.0 is able to complete group analysis of 10 samples with default parameters within 3 hours. For single or paired samples, its duration is even shorter. Detailed user instruction as well as materials and methods are provided in Supplementary information.
3 Main additions
3.1 Workflow modify
Due to decreasing sequencing cost, researchers are able to conduct multiple samples sequencing within a single dataset to deliver a more solid scientific conclusion. However, CPSS 1.0 is only able to analyze small RNA sequencing data of single or paired samples, and does not support more than two samples. CPSS 2.0 meets the demand to handle multiple samples dataset. Differently expressed ncRNAs between groups or among samples are retrieved while processing multiple samples. For ncRNAs, whose expressions satisfied the giving P-value and fold change criteria are marked as statistical significance. All significantly expressed miRNAs are selected for further functional analysis, including target prediction, GO, pathway, protein domain and PPI annotation. In order to better understand the underlying biological processes, enrichment analysis is also performed on the annotation terms to identify significantly enriched targets.
As CPSS 2.0 mainly focuses on ncRNA detection, quantification and function analysis of predicted miRNA targets, the detection of miRNA modification and editing are removed from current workflow (users could use our DeAnniso (Zhang et al., 2016) for their interests in the detection and annotation of miRNA isoform). Modules for the detection and quantification of circRNA and lncRNA are provided as addition categories for ncRNA classification.
3.2 Software and database update
CPSS 1.0 can only analyze small RNA sequencing data from 10 species of animals, which is not compatible with biology research in highly specific area. CPSS 2.0 integrates 38 more species, including 17 plants (such as Populus trichocarpa, Prunus persica and Zea mays), 31 animals (such as Danio rerio, Drosophila melanogaster and Caenorhabditis elegans). 11 of them are mammals such as Mus musculus and Bos Taurus. And 5 of the 11 mammal species are primates including Homo sapiens, Gorilla gorilla and Macaca mulatta. Reference sequences of all species integrated in CPSS 2.0 for reads alignment are updated to the latest version. Databases used for function annotation of predicted miRNA targets are also updated to the latest version. Detailed information is shown in Supplementary Table S2.
CPSS 2.0 removed some redundant softwares implemented in the workflow of CPSS 1.0. CPSS 2.0 employs Bowtie to map sequencing reads to reference genome and part of ncRNA databases. Unnecessary miRNA target prediction tools are also removed. These deleted tools are published as database and cannot be updated frequently to cover the newly discovered miRNAs recorded from sequencing data. CPSS 2.0 implements miRanda for target prediction due to its wide acceptance and high efficiency.
3.3 Interaction and visualization strengthen
CPSS 2.0 has a brand-new user-friendly interface (Fig. 1). Take group analysis as an example, users can select file and assign samples into two groups by optimized parameters of each analyze step or simply staying with default parameters. After clicking the ‘Submit’ button, the job will be uploaded and a progress bar will reflect the real-time status of this submitted job. When an analysis step is completed, the progress bar will be refreshed automatically and the general results of this step will be displayed as well. Moreover, if users click the green characters under the ‘completed green logo’ on the progress bar, they will be directed to the results of that part. By click the ‘HERE’ button at each result section, the detailed results will be shown in a new page. Each detailed result pages includes a search function to find specific terms or values. For GO, pathway and protein domain analysis, users can optimize parameters and rerun analysis at each detailed result page. If email address is provided, once the job is finished, a reminder will be sent by email. Users can also retrieve the analysis results from the stored jobs with a unique ID generated randomly by the server for each job.
Fig. 1.
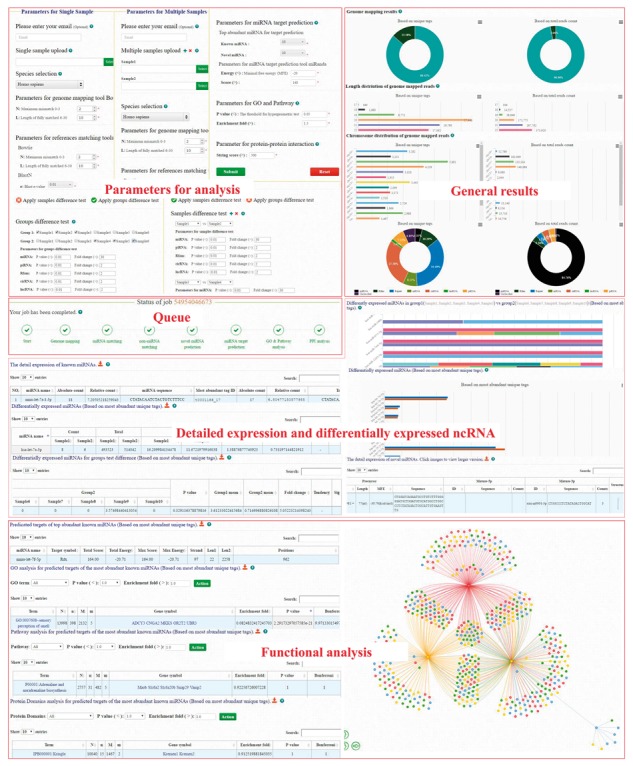
Parameter and summary result of CPSS 2.0
Supplementary Material
Acknowledgements
The authors would like to thank all the CPSS users for their helpful suggestions.
Author contribution
Y.Z. conceived and designed the project. C.W. and J.G. constructed the pipeline. R.B. developed the web interface. C.W. and H.Z. wrote the paper. X.J., Q.Z., Y.Z. and Q.S. revised the manuscript. Y.Z. and Q.S. supervised the project.
Funding
This work was supported by the National Key Research and Developmental Program of China (2016YFC1000605), National Basic Research Program of China (2013CB945502 and 2014CB943101), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB19000000), the National Natural Science Foundation of China (31371519, 31501202, 31630050, 31301227, 31501199, 31571555 and 61571414), the Natural Science Foundation of China - Israel Science Foundation (31461143013-1183/14), Major Program of Development Foundation of Hefei Centre for Physical Science and Technology (2014FXZY003), the Fundamental Research Funds for the Central Universities (WK2340000069).
Conflict of Interest: none declared.
References
- Bracken C.P. et al. (2016) A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet., 17, 719–732. [DOI] [PubMed] [Google Scholar]
- Capece V. et al. (2015) Oasis: online analysis of small RNA deep sequencing data. Bioinformatics, 31, 2205–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B. et al. (2004) Human microRNA targets. PLoS Biol., 2, e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. et al. (2014) MAGI: a Node. js web service for fast microRNA-Seq analysis in a GPU infrastructure. Bioinformatics, 30, 2826–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. et al. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol., 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R. et al. (2011) ViennaRNA Package 2.0. Algorithms Mol. Biol., 6, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G.Z. et al. (2014) ISRNA: an integrative online toolkit for short reads from high-throughput sequencing data. Bioinformatics, 30, 434–436. [DOI] [PubMed] [Google Scholar]
- Rueda A. et al. (2015) sRNAtoolbox: an integrated collection of small RNA research tools. Nucleic Acids Res., 43, W467–W473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D. et al. (2014) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res., gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitsios D.M., Enright A.J. (2015) Chimira: analysis of small RNA sequencing data and microRNA modifications. Bioinformatics, btv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J. et al. (2013) mirTools 2.0 for non-coding RNA discovery, profiling, and functional annotation based on high-throughput sequencing. RNA Biol., 10, 1087–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2012) CPSS: a computational platform for the analysis of small RNA deep sequencing data. Bioinformatics, 28, 1925–1927. [DOI] [PubMed] [Google Scholar]
- Zhang Y. et al. (2016) DeAnnIso: a tool for online detection and annotation of isomiRs from small RNA sequencing data. Nucleic Acids Res., gkw427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W. et al. (2011) wapRNA: a web-based application for the processing of RNA sequences. Bioinformatics, 27, 3076–3077. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.