

Abstract
Motivation
The Human Reference Epigenome Map, generated by the Roadmap Epigenomics Consortium, contains thousands of genome-wide epigenomic datasets that describe epigenomes of a variety of different human tissue and cell types. This map has allowed investigators to obtain a much deeper and more comprehensive view of our regulatory genome, e.g. defining regulatory elements including all promoters and enhancers for a given tissue or cell type. An outstanding task is to combine and compare different epigenomes in order to identify regions with epigenomic features specific to certain types of tissues or cells, e.g. lineage-specific regulatory elements. Currently available tools do not directly address this question. This need motivated us to develop a tool that allows investigators to easily identify regions with epigenetic features unique to specific epigenomes that they choose, making detection of common regulatory elements and/or cell type-specific regulatory elements an interactive and dynamic experience.
Results
An online tool EpiCompare was developed to assist investigators in exploring the specificity of epigenomic features across selected tissue and cell types. Investigators can design their test by choosing different combinations of epigenomes, and choosing different classification algorithms provided by our tool. EpiCompare will then identify regions with specified epigenomic features, and provide a quality assessment of the predictions. Investigators can interact with EpiCompare by investigating Roadmap Epigenomics data, or uploading their own data for comparison. We demonstrate that by using specific combinations of epigenomes we can detect developmental lineage-specific enhancers. Finally, prediction results can be readily visualized and further explored in the WashU Epigenome Browser.
Availability and implementation
EpiCompare is freely available on the web at http://epigenome.wustl.edu/EpiCompare/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The Roadmap Epigenomics Consortium generated a reference catalogue of human epigenomes across a variety of tissue and cell types (Roadmap Epigenomics et al., 2015). Using this resource, investigators can compare the epigenomes of different tissue and cell types and identify regulatory elements such as enhancers, promoters, and regions occupied by epigenetic features that are unique to a specific tissue or cell type, as well as those that are shared by multiple tissue and cell types.
One common application utilizing the Human Reference Epigenome is the identification of tissue or cell type-specific enhancers. Enhancers are cis-regulatory elements playing essential roles in regulating the spatial and temporal pattern of gene expression (Blackwood and Kadonaga, 1998). Many enhancers function in a tissue or cell type-specific manner (Ernst et al., 2011; Heintzman et al., 2009; Shen et al., 2012). Disruption of enhancer functions can often lead to diseases (Sakabe et al., 2012). Many studies revealed that enhancers significantly overlap with disease-causal variants and such variants are often enriched in enhancers specific to cell types that are implicated in the specific diseases (Claussnitzer et al., 2015; Ernst et al., 2011; Farh et al., 2015; Hoffman et al., 2013; Roadmap Epigenomics et al., 2015; Song and Chen, 2015; Zhou et al., 2015). Hence, a comprehensive list of tissue or cell type-specific enhancers could have significant clinical impact.
The identification of tissue-specific histone marks including H3K27ac and H3K4me1 can help identify tissue or cell type-specific enhancers. Enhancers are epigenetically defined by the presence of H3K4me1 and the absence of H3K4me3 (Heintzman et al., 2007). H3K27me3 is a repression histone mark that is associated with polycomb complex (Barski et al., 2007). The combination of H3K4me1 and H3K27me3 marks poised enhancers, which silence developmental genes in embryonic stem cells (ESCs) and keep them poised for activation in differentiating cells (Creyghton et al., 2010). H3K27ac is a mark of active enhancers and promoters and distinguishes active enhancers from poised enhancers. Combination of H3K4me1 and H3K27ac modifications is used to identify active enhancers (Prescott et al., 2015). Therefore, combination of different histone marks can be used to predict tissue or cell type-specific poised/active enhancers.
Given datasets of multiple histone modifications for a specific cell type, several tools, including ChromHMM (Ernst and Kellis, 2010; Ernst and Kellis, 2012), RFECS (Rajagopal et al., 2013), and Segway (Hoffman et al., 2012), can define chromatin states across the cell’s epigenome and/or define regulatory elements such as enhancers. While the above tools are designed for a single sample, tools like hiHMM (Sohn et al., 2015) and TreeHMM (Biesinger et al., 2013) can define chromatin states in multiple cell types or multiple species simultaneously. But these tools cannot be readily applied to detect tissue or cell type-specific enhancers. Several efforts have been devoted to define tissue or cell type-specific enhancers. For example, the FANTOM5 Consortium identified active enhancers for a large number of human tissue and cell types by using bidirectional capped RNA data (Andersson et al., 2014). They called differentially expressed enhancers across all tissue and cell types, using Kruskal–Wallis rank sum tests. To define tissue differentially expressed enhancers, for example, for the brain, they further performed pair-wise, post-hoc tests, and required the enhancers to be differentially expressed between brain tissues and at least one non-brain tissue. A limitation of this approach is that such differentially expressed enhancers are often expressed in multiple tissue and cell types and are not specific to a single tissue or cell type. Furthermore, since the enhancers are marked by active transcription, poised enhancers are likely to be missed. Indeed, the active enhancers identified by FANTOM5 had 231 fold more bidirectional capped RNA reads than polycomb-repressed enhancers (Andersson et al., 2014).
The Roadmap Epigenomics Project used a tool called HoneyBadger2 to define tissue or cell type-specific enhancers using k-means clustering. Regions that were clustered together share similar epigenetic profiles across a variety of tissue and cell types. A given cluster may have a pattern such that the enhancer signals are predominantly present in certain tissues, but not in other tissues. Such regions were defined as tissue-specific enhancers. However, this approach is based on unsupervised learning, and as such, clusters are not directly assigned to a specific tissue. Other groups characterized the cell-type specificity of enhancers in human and mouse using clustering methods (Ernst et al., 2011; Heintzman et al., 2009; Shen et al., 2012; Won et al., 2013), but did not provide tools to define cell-type specificity. Tools like MultiGPS (Mahony et al., 2014) and dPCA (Ji et al., 2013) were designed to compare Chip-seq data between two conditions but not readily adaptable to compare enhancers or histone modifications between groups of tissue and cell types. Another tool, ChromDiff (Yen and Kellis, 2015) compared chromatin states across different group of samples. For each given region, ChromDiff calculated the percent coverage for each chromatin state in each sample and corrected them based on sample metadata. Then it tested for difference of corrected values between two groups of samples for each chromatin state using statistical test such as Mann–Whitney–Wilcoxon test and identified significant regions with specific chromatin states. The tool can be applied to identify tissue or cell type-specific enhancers if ChromHMM models are defined, but can be difficult to use by experimental biologists due to the lack of a user-friendly interface.
To address these needs, we have developed an online tool EpiCompare to help investigators to analyze the Roadmap Epigenomics data. Investigators can easily identify regions with epigenomic features specific to combinations of tissue or cell types. Several classification methods are provided, including the clustering method used by the Roadmap Epigenomics Project (Roadmap Epigenomics et al., 2015). Investigators can compare enhancers, promoters, and specific histone marks using any combination of tissue and cell types, using Roadmap data and/or their own data. Investigators can test a variety of hypotheses by designing specific combinations of epigenome comparisons, and EpiCompare provides a quality assessment of the predictions. The predicted regions can be readily visualized and further explored within the WashU Epigenome Browser. EpiCompare makes Roadmap reference epigenomes more easily usable by experimental biologists in order to enhance their research.
2 Materials and methods
2.1 Datasets
The Roadmap Epigenomics Consortium uses the ChromHMM tool to generate chromatin states for different tissue and cell types. The type and number of chromatin states depends on the histone modification data provided. The 15-state ChromHMM model integrates histone modifications H3K4me1, H3K4me3, H3K9me3, H3K27me3, and H3K36me3, while the 18-state ChromHMM model integrates the five marks in the 15-state model plus H3K27ac (Roadmap Epigenomics et al., 2015). From the Roadmap Epigenomics Project, we obtained 15-state and 18-state ChromHMM models, and processed peak data [obtained from MACS (Zhang et al., 2008)] for H3K27ac, H3K4me1, H3K4me3 and H3K27me3 marks for all tissue and cell types. Chromatin states are predicted for each 200 base pair (bp) window. The 15-state ChromHMM model defines enhancers as state numbers 6, 7, 12, corresponding to genic enhancers, enhancers, and bivalent enhancers, respectively. The 18-state ChromHMM model defines enhancers as state numbers 7, 8, 9, 10, 11, 15, corresponding to genic enhancer 1, genic enhancer 2, active enhancer 1, active enhancer 2, weak enhancer, and bivalent enhancer, respectively. Further, for all processed peak data, the coordinates are mapped to 200bp windows by requiring at least 50bp overlapping. Only peaks with q-value less than 0.01 are considered. Each feature above—the enhancer state or epigenomic modification peak—is converted into binary presence or absence of the feature in each 200 bp window, denoted by 1 or 0. A table is generated for each feature by summarizing the presence or absence of the feature in all samples across windows where at least one sample has the feature.
2.2 Classification methods
EpiCompare contains three methods for identifying regions with epigenomic features specific to combinations of tissue or cell types (Supplementary Fig. S1). All methods require the definition of foreground samples and background samples by users. Foreground samples are the group of samples for which we identify specific regions. Background samples are the group of samples against which we compare foreground samples. The principle of all methods is, to define regions with features specific in foreground samples, the features should be enriched in the foreground samples but depleted in the background samples.
The first method implements a frequency cutoff. For each region (in this case each 200bp genomic window), the percentages of samples having the feature in the foreground samples and background samples are calculated. If the percentage of samples having the feature in the foreground samples is greater than or equal to the defined minimal foreground cutoff (default is 80%) and the percentage of samples having the feature in the background samples is less than or equal to the defined maximal background cutoff (default is 20%), then the region is defined as a positive region. These positive regions are further ranked by the difference between the percentage of samples having the feature in the foreground samples and background samples so users can prioritize top-ranked regions.
The second method implements Fisher’s exact test. For each 200 bp window, a contingency table composed of the number of samples with or without the feature in foreground samples and background samples is calculated. Fisher’s exact test is used to examine whether the percentage of features in the foreground samples is significantly greater than in the background samples. The p-value is corrected by multiple hypothesis testing using the Benjamini–Hochberg procedure, and regions with q-value less than a cutoff (default is 0.01) are identified and ranked by their q-values. The statistical power of the test depends on the number of foreground samples and background samples and having more samples can provide more statistical power to identify more significant q-values (See Supplementary Note S6). Therefore, when the number of foreground samples or background samples is small, investigators can use q-value as a ranking measure and obtain the top candidates by setting a higher q-value threshold. We also evaluated the false positive rate of Fisher’s exact test (See Supplementary Note S7).
The third method implements k-means clustering based on a Jaccard-index distance, similar to the clustering method used in HoneyBadger2 (Roadmap Epigenomics et al., 2015). First, k-means clustering is performed on regions in the binary data table for each feature. R package flexclust is used for clustering (Leisch, 2006). We determined the optimal cluster number by the elbow method and the silhouette method (Kodinariya and Makwana, 2013) (See Supplementary Note S8). The optimal cluster number for all features is close and around 140, so we provide the optimal cluster number for all features to be 140. In addition to the default number, we provide several other options (i.e. cluster number 90, 200, and 250) to give users flexibility. Next, the percentage of regions having the feature is calculated for each cluster and defined as a feature density table (number of clusters times number of samples). Finally, a cluster specific for a tissue/cell type should have higher feature density in that tissue/cell type than in the background samples. Specifically, to identify clusters specific for foreground samples, we select clusters satisfying the following two conditions: first, the median of feature densities of foreground samples in a cluster is greater than or equal to a threshold (default is 0.4); second, it should also be greater than or equal to the highest feature density in the background samples of that same cluster (this threshold can be set to any percentile of feature densities in the background samples).
3 Results
3.1 Performance comparison
To identify regions with epigenomic features specific to combinations of tissue or cell types, we applied three different methods: frequency cutoff, Fisher’s exact test, and k-means clustering, as described in Methods. The most important parameters for all the methods are choices of foreground samples and background samples (see Methods). The main assumption we make is that the epigenomic features we focus on are enriched in foreground samples but depleted in background samples. Identified regions were tested using the following validation methods: GREAT analysis, enrichment for DNase I hypersensitive sites (DHS) and H3K27ac peaks, and the tissue enrichment index, contribution measure (CTM) (see Supplementary Note S1). CTM measures how much a sample or a group of samples contributes to the total amount of signal (e.g. read density for H3K27ac) combined by all samples in a region (Pan et al., 2013). To further evaluate the performance directly, we randomly picked 20 identified regions and visualized them in WashU Epigenome Brower with chromatin states and histone modification tracks. We used adult brain tissues as foreground samples and evaluated the efficacy of the three methods in identifying adult brain-specific enhancers using enhancers defined by 15-state ChromHMM model. Seven adult brain samples were available from the Roadmap Epigenomics Project. We compared them to 91 other samples with available H3K27ac data. Since the clustering method does not provide ranks, we obtained a list of adult brain-specific enhancers using the clustering method with default settings. We then picked an equal number of regions in ascending order of ranks using the frequency cutoff and Fisher’s exact test methods.
First, we examined the overlap of enhancers found by three methods (Supplementary Fig. S2). Out of 188 076 identified adult brain-specific enhancers (i.e. 200 bp windows), 148 170 overlapped between the frequency cutoff and Fisher’s exact test; 133 370 overlapped between k-means clustering and Fisher’s exact test; and 144 182 overlapped between frequency cutoff and k-means clustering. 123 746 were shared across all three methods.
Next, we tested our predicted brain-specific enhancers using the three validation methods. Using the GREAT (McLean et al., 2010), we found that adult brain-specific enhancers identified by each of three methods were strongly associated with brain functions such as myelination, regulation of action potential and regulation of synaptic plasticity (Fig. 1(a); Supplementary Fig. S3). The brain-specific enhancers predicted by all three methods also had much higher enrichment for H3K27ac peaks in brain tissues compared to other tissues (Fig. 1(b) and Supplementary Fig. S4). Overall, the enrichment in brain tissues was higher for the frequency cutoff and Fisher’s exact test methods than for the clustering method. The brain-specific enhancers predicted by all three methods also had much higher CTM index in brain tissues than in other tissues for H3K27ac-based CTM distribution (Fig. 1(c); Supplementary Fig. S5), underscoring the brain specificity of the enhancer histone modification in the identified regions. The brain tissue CTM distributions for regions identified by the three methods almost superimposed each other (Supplementary Fig. S5). A visualization of randomly picked 20 brain-specific enhancers identified from Fisher’s exact test showed most regions had much stronger H3K4me1/H3K27ac peaks in the foreground samples than the background samples (Supplementary Fig. S6). In summary, the validation results confirmed that our methods can effectively identify tissue-specific enhancers. Similarly, the same methods can be applied to identify other epigenomic modifications that are tissue or cell-type specific.
Fig. 1.
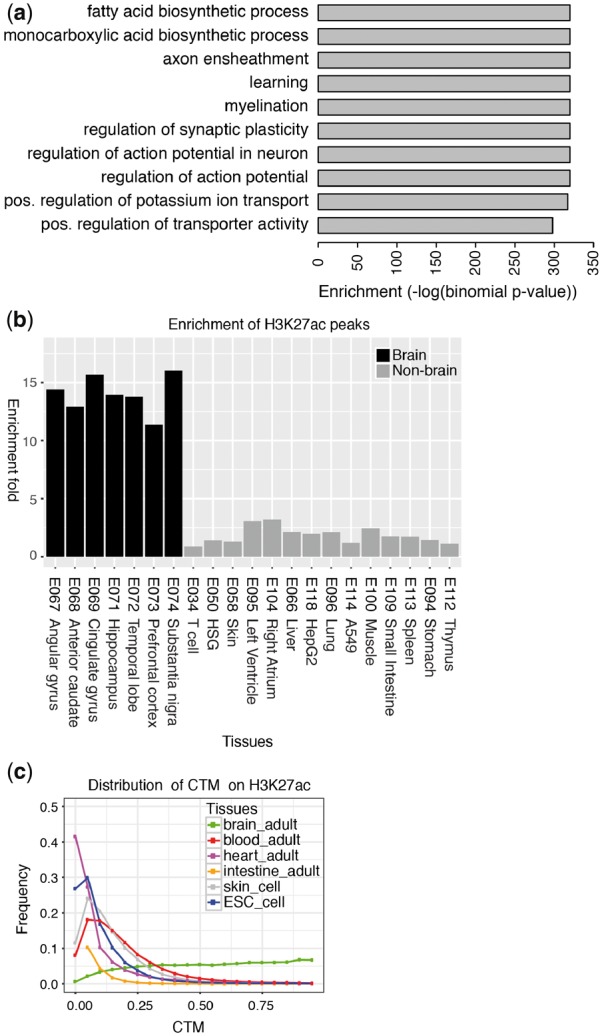
Validation of predicted brain-specific enhancers by Fisher’s exact test method. (a) Enriched GO terms and their binomial p-values based on GREAT. The top 10 GO terms are displayed here. (b) Enrichment of H3K27ac peaks in brain tissues and non-brain tissues for predicted adult brain-specific enhancers by Fisher’s exact test. (c) The distribution of tissue enrichment index CTM based on H3K27ac expression data for predicted adult brain-specific enhancers by Fisher’s exact test
Since FANTOM5 defined active enhancers for a variety of tissue and cell types by their differential expression patterns, we compared brain-specific enhancers identified by Fisher’s exact test on enhancers defined by 15-state ChromHMM model and the FANTOM5. First, we examined the overlap between these two methods. The FANTOM5 enhancers were not binned on 200bp windows, so we mapped them onto 200bp windows. 89 of 208 804 regions by ChromHMM-based method overlapped with 1578 binned FANTOM5 brain enhancers (hypergeometric test, p = 10−26). The overlap was small because only 11% of H3K4me1/H3K27ac loci overlapped the FANTOM5 enhancers (Andersson et al., 2014), and enhancers defined by ChromHMM included active enhancers (characterized by H3K4me1/H3K27ac loci), poised enhancers (characterized by H3K4me1/H3K27me3 loci), and other types of enhancers (single H3K4me1 mark or single H3K27ac mark). Although the overlap with the FANTOM5 brain enhancers was small, it was highly significant. In contrast, 35 regions by ChromHMM-based method overlapped with 3409 binned FANTOM5 blood enhancers (hypergeometric test, P = 0.98), suggesting the overlap was specific to brain. Second, we plotted enrichment of H3K27ac for the shared regions, as well as regions unique to each method (Supplementary Fig. S7). Finally, we randomly picked 20 regions that were unique to each method, and visualized them on the WashU Epigenome Browser in gene set view (Supplementary Figs S8 and S9). Interestingly, we found that many FANTOM5-defined brain-specific enhancers are defined as promoters by using Roadmap Epigenomics data, with clear and strong promoter histone mark support (i.e. H3K4me3). Moreover, these regions also have high H3K27ac in the background samples.
Using similar analysis as above, we compared identifying brain-specific enhancers using Fisher’s exact test and ChromDiff (See Supplemental Note S9). We found enhancers identified from Fisher’s exact test and ChromDiff largely overlapped (80%). Enhancers that were unique to Fisher’s exact test had much stronger enrichment of H3K27ac in brain samples than ChromDiff but also had higher enrichment in the background samples. Therefore anecdotally EpiCompare seems to have better sensitivity, while ChromDiff seems to exhibit better specificity, at a comparable statistical cutoff. ChromDiff is a command line only program, while EpiCompare provides a much more user-friendly interface and includes access to WashU Epigenome Browser, allowing biologists to better explore their result.
The k-means clustering method in our tool is similar to the clustering method used in HoneyBadger2 tool with the exception that enhancers defined by the 15-state ChromHMM model in HoneyBadger2 were further filtered by DHS before used for clustering. To demonstrate that our clustering method is comparable to HoneyBadger2, we compared adult brain-specific enhancers identified by the two approaches. We used 250 clusters as a close approximation of 246 clusters in HoneyBadger2 tool. We identified 158 110 regions with our approach and 86 019 regions with HoneyBadger2. For the comparison, we randomly picked 86 019 regions from the total regions identified by our approach. By comparing the enrichment of H3K27ac peaks in the foreground samples and background samples between our clustering method and HoneyBadger2, we found that both methods had similar enrichment in the foreground samples (t-test, P = 0.87) and also in the background samples (t-test, P = 0.98) (Supplementary Fig. S10a). When we examined the CTM distribution of H3K27ac, we found that the brain tissue CTM distributions for regions identified by the two methods almost superimposed each other (Supplementary Fig. S10b). Thus our clustering method is comparable to the clustering method in HoneyBadger2 tool.
After demonstrating that our methods can identify tissue-specific enhancers, we determined the impact of sample size on performance: i.e. the impact of the number of foreground samples and the number of background samples (see Supplementary Note S2). First, to examine how the number of foreground samples affects the performance, we predicted adult brain-specific enhancers by using different number of foreground samples while fixing background samples. To assess performance, we computed the average enrichment of H3K27ac peaks in the seven adult brain samples and also in selected background samples because we expect that tissue-specific enhancers should have higher enrichment in the foreground samples and lower enrichment in the background samples. We found that with increasing foreground samples, the performance of all three methods increased. This is illustrated by increasing H3K27ac enrichment in the foreground samples, and relatively stable depletion in the background sample (Fig. 2a).
Fig. 2.

The effect of sample size on the performance of adult brain specific-enhancer predictions. (a) How the number of foreground samples influences the performance with fixed background samples. (b) How the number of background samples influences the performance with fixed foreground samples
To examine how the number of background samples affects the performance, we predicted adult brain-specific enhancers by using different number of background samples while fixing foreground samples. The enrichment of H3K27ac in the foreground samples seemed to be quite stable across a range of numbers of background samples used (Fig. 2b). However, depletion of H3K27ac in background samples seemed to be quite sensitive to the number of background samples used. A larger number of background samples did improve the specificity effectively, underscoring the importance of having a comprehensive collection of epigenomes, such as those made available by the Roadmap Epigenomics project.
Finally, we demonstrate that our simple but versatile framework allows investigators to design any combination of epigenome comparison to identify specific epigenomic features associated with specific biological entities. For example, by combining samples that share the same developmental origin, one might be able to identify specific regulatory mechanisms for this developmental lineage. This is particularly useful when samples representing cells in early development are difficult to obtain. Here we set out to define endoderm-specific enhancers by comparing nine adult tissues derived from the endoderm to other background tissues (see Supplementary Note S3). The enhancers were defined using 18-state ChromHMM model. We identified 13 728 regions using frequency cutoff method, 46 859 regions using Fisher’s exact test method, and 29 386 regions using k-means clustering method with 140 clusters. We picked top 13 728 from Fisher’s exact test for the following analysis. The predicted regions exhibited much stronger enrichment of DHS in endoderm-derived tissues than in other tissues (Fig. 3a;Supplementary Fig. S11). Moreover, when subjected to analysis by the GREAT tool, these regions were strongly associated with biological processes related to epithelial cell functions (Fig. 3b), a well-known derivative function common for endoderm-derived tissues (Zorn and Wells, 2009). A visualization of randomly picked 20 endoderm-specific enhancers identified from Fisher’s exact test showed most regions had much stronger H3K4me1/H3K27ac peaks in the foreground samples than the background samples (Supplementary Fig. S12).
Fig. 3.

Identification of endoderm-specific enhancers by Fisher’s exact test method. (a) Enrichment of DHS for endoderm-specific enhancers identified by Fisher’s exact test. (b) Enriched GO terms and their binomial p-values based on GREAT. Top 10 terms are displayed. (c) Enrichment of TF binding motifs in endoderm-specific enhancers by Fisher’s exact test. Top 15 TFs are displayed. (d) The putative gene regulatory networks for endoderm tissues based on identified enhancers. (e) The expression profiles of epigenetic marks for enhancers in the network in endoderm tissues and non-endoderm tissues, ESCs and ESC-derived endoderm cells. (f) A browser example of merged endoderm-specific enhancers. Blue is endoderm tissues, brown is non-endoderm tissues, and red is ESCs and ESC-derived endoderm cells
To further explore the functions of these endoderm-specific enhancers, we identified potential regulatory transcription factors (TFs) interacting with these regions by HOMER (Heinz et al., 2010). The top enriched TFs are all important for endoderm specification, including FoxA family TFs (FoxA1, FoxA2), GATA family TFs (Gata4), HNF1, HNF4a, and others (Fig. 3c). FoxA family and GATA family TFs are key players in the transcriptional regulatory network of the endoderm (Zorn and Wells, 2009). FoxA1 and FoxA2 are pioneer TFs that remodel chromatin environment and facilitate recruitment of other TFs (Cirillo et al., 2002). FoxA1 and FoxA2 are homologous and required for the development of endoderm tissues such as liver, lung, intestine and pancreas (Gao et al., 2008; Gosalia et al., 2015; Lee et al., 2005; Wan et al., 2005). Like FoxA1 and FoxA2, HNF1 and HNF4a play key regulatory roles in liver, pancreas, and intestine development (DeLaForest et al., 2011; Pontoglio, 2000; Yang et al., 2016). Moreover, the foregut markers PDX1 and the hindgut marker CDX2 were also highly enriched (P = 10−5 for PDX1 and CDX2 motifs) (Spence et al., 2011). To further support the function of the top enriched TFs in endoderm tissues, many of them were highly expressed in endoderm tissues comparing to non-endoderm tissues, ESCs and ESC-derived multipotent endoderm cells (Roadmap Epigenomics et al., 2015) (Supplementary Fig. S13).
Using top enriched TFs that were also highly expressed in adult endoderm tissues compared to non-endoderm adult tissues, we identified 4 upstream TF candidates - FoxA1, FoxA2, HNF1b, HNF4a, and were able to build a transcriptional regulatory network for them and shared target genes by linking enhancers with TF binding sites to nearest genes (Fig. 3d;Supplementary Table S1) using previously described methods (Lee et al., 2015). The reconstructed network recapitulated many important gene regulation relationships in endoderm development and differentiation. For example, the FoxA family TFs cooperate with HNF1b and HNF4a to regulate intestinal epithelial cell function (Yang et al., 2016). FoxA2, HNF1b, and HNF4a were shown to bind to a large number of target regions in intestinal epithelial cell line (Yang et al., 2016). The 72 shared target genes for the 4 TFs were enriched for signaling pathways required for cell proliferation and differentiation including WNT, BMP, VEGF and Hippo signaling (Supplementary Table S2) (Kamburov et al., 2009). The median expression level of these genes was significantly higher in endoderm tissues than that in non-endoderm tissues (t-test, P = 5e−5) (Supplementary Fig. S14). To further confirm that the network was activated in endoderm tissues, we examined the profile of epigenetic marks (DNase I, H3K27ac and DNA methylation) on all enhancers in this network across different tissues, including adult endoderm tissues, fetal endoderm tissues, endoderm cells, non-endoderm tissues and ESCs. These enhancers showed strong expression of DNase I and H3K27ac mark and low DNA methylation only in adult endoderm tissues and fetal endoderm tissues (Fig. 3e). Figure 3f gave an example of merged endoderm-specific enhancers. The enhancers had strong DHS and H3K27ac peaks and low DNA methylation level in both adult and fetal endoderm tissues but not others. The evidence suggests that this regulatory cascade is active in fetal and adult endoderm tissues, but not in ESC-derived endoderm cells which presumably have not committed to a special endoderm cell type and also not in non-endoderm tissues.
3.2 Web server
The tool EpiCompare is freely available online. It was written in R using the Shiny framework and hosted by open source shiny server (Chang, 2015). The home page includes a simple and intuitive user interface for the selection of foreground samples and background samples from a list of human tissue and cell types available from the Roadmap Epigenomics Consortium (Supplementary Fig. S15). Options for selecting different classification methods and parameters are also provided. It also provides the option of uploading user’s data for analysis. The results page provides analysis results, including H3K27ac enrichment and tissue enrichment index using H3K27ac expression data. Results are presented as a table of identified regions, and can be downloaded for further analysis. Each region is linked to the WashU Epigenome Browser (Zhou et al., 2011) where users can visualize, explore, and compare their epigenomic patterns in different tissue/cell types. The help page gives a tutorial on how to use EpiCompare.
4 Discussion
We have developed an online tool EpiCompare to help investigators to analyze the Roadmap Epigenomics data. The presented data showed that the tool can easily identify regulatory elements such as enhancers, promoters, and regions occupied by epigenetic features that are unique to a specific tissue or cell type, as well as those that are shared by multiple tissue and cell types. Our tool is designed specifically for biologists in such a way that no programming or data processing capacity is required to perform genome-wide analysis. We demonstrated that our tool could identify endoderm-specific enhancers and analysis on these enhancers revealed the regulatory network common to all endoderm tissues.
In identifying regions with epigenomic features specific to combinations of tissue or cell types, EpiCompare has several advantages over existing methodologies reported in the FANTOM5, Roadmap, and others. First, investigators can compare enhancers, promoters, and specific histone marks using any combination of tissue and cell types depending on their needs. This enables the identification of specific epigenomic features associated with specific biological entities, such as lineage-specific enhancers. Second, the tool is user-friendly so that an experimental biologist with little or no programming experience can easily use. Investigators can test a variety of hypotheses by designing specific combinations of epigenome comparisons using Roadmap data and/or their own data, and EpiCompare provides a quality assessment of the predictions. The predicted regions can be readily visualized and further explored using the WashU Epigenome Browser.
EpiCompare has some limitations. First, the regulatory elements used in this tool are defined based on the ChromHMM model. Although considered the state-of-the-art, ChromHMM model still has limited sensitivity and specificity, especially for identifying enhancers (Song and Chen, 2015). The performance of predicting tissue or cell type-specific enhancers is clearly dependent on the performance of ChromHMM. Second, EpiCompare is based on comparison of binary data including chromatin states and histone mark peaks. It could potentially miss regions with quantitatively different signal between samples. For example, it could not distinguish a weak enhancer from a strong enhancer if both had signals over the threshold. It could also not distinguish two quantitatively different weak enhancers which were below the threshold. These cases are false negatives for EpiCompare. The comparison of binary data can also lead to false positives if two samples had very similar signal at one region, with one above the threshold and the other below the threshold. Third, we implemented three very simple statistical models, and potentially could oversimplify the problem of identifying tissue or cell type-specific features. Frequency cutoff method uses simple cutoffs, and Fisher’s exact test assumes the occurrence of features as hypergeometric distribution while k-means clustering method assumes certain number of clusters in the data and groups them based on similarity. All of them assume the independence of samples, but biological samples are clearly not independent from each other. The statistical models also do not consider the distribution of each feature along the genome of each sample. However, we are encouraged by the strong performance of these simple models, and anticipate that development of more sophisticated models will surely improve the accuracy of feature identification.
Supplementary Material
Acknowledgements
The authors thank Dr. Bo Zhang and all members in Ting Wang’s lab for suggestions on the manuscript and the website.
Funding
This work was supported by the National Institutes of Health [R01HG007354, R01HG007175, R01ES024992, U01CA200060, U24ES026699, U01HG009391]; and the American Cancer Society Research Scholar Grant [RSG-14-049-01-DMC].
Conflict of Interest: none declared.
References
- Andersson R. et al. (2014) An atlas of active enhancers across human cell types and tissues. Nature, 507, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A. et al. (2007) High-resolution profiling of histone methylations in the human genome. Cell, 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Blackwood E.M., Kadonaga J.T. (1998) Going the distance: a current view of enhancer action. Science, 281, 60–63. [DOI] [PubMed] [Google Scholar]
- Biesinger J. et al. (2013) Discovering and mapping chromatin states using a tree hidden Markov model. BMC Bioinformatics, 14 (Suppl. 5), S4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W. et al. (2015) shiny: Web Application Framework for R. R package version 0.12.1. [Google Scholar]
- Cirillo L.A. et al. (2002) Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell, 9, 279–289. [DOI] [PubMed] [Google Scholar]
- Claussnitzer M. et al. (2015) FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N Engl J Med, 373, 895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton M.P. et al. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA, 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLaForest A. et al. (2011) HNF4A is essential for specification of hepatic progenitors from human pluripotent stem cells. Development, 138, 4143–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. (2010) Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol., 28, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. (2012) ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods, 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J. et al. (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature, 473, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh K.K. et al. (2015) Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature, 518, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N. et al. (2008) Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev., 22, 3435–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosalia N. et al. (2015) FOXA2 regulates a network of genes involved in critical functions of human intestinal epithelial cells. Physiol. Genomics, 47, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman N.D. et al. (2009) Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature, 459, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman N.D. et al. (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet., 39, 311–318. [DOI] [PubMed] [Google Scholar]
- Heinz S. et al. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman M.M. et al. (2012) Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods, 9, 473–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman M.M. et al. (2013) Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res., 41, 827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H. et al. (2013) Differential principal component analysis of ChIP-seq. Proc. Natl. Acad. Sci. USA, 110, 6789–6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamburov A. et al. (2009) ConsensusPathDB–a database for integrating human functional interaction networks. Nucleic Acids Res., 37, D623–D628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodinariya T., Makwana P. (2013) Review on determining number of cluster in k-means clustering. Int. J. Adv. Res. Comput. Sci. Manag. Stud., 1, 90–95. [Google Scholar]
- Lee C.S. et al. (2005) The initiation of liver development is dependent on Foxa transcription factors. Nature, 435, 944–947. [DOI] [PubMed] [Google Scholar]
- Lee H.J. et al. (2015) Developmental enhancers revealed by extensive DNA methylome maps of zebrafish early embryos. Nat. Commun., 6, 6315.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leisch F. et al. (2006) A Toolbox for K-Centroids Cluster Analysis. Computational Statistics and Data Analysis, 51, 526–544. [Google Scholar]
- Mahony S. et al. (2014) An integrated model of multiple-condition ChIP-Seq data reveals predeterminants of Cdx2 binding. PLoS Comput. Biol., 10, e1003501.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean C.Y. et al. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol., 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.B. et al. (2013) PaGenBase: a pattern gene database for the global and dynamic understanding of gene function. PLoS One, 8, e80747.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontoglio M. (2000) Hepatocyte nuclear factor 1, a transcription factor at the crossroads of glucose homeostasis. J. Am. Soc. Nephrol., 11 (Suppl. 16), S140–S143. [PubMed] [Google Scholar]
- Prescott S.L. et al. (2015) Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell, 163, 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal N. et al. (2013) RFECS: a random-forest based algorithm for enhancer identification from chromatin state. PLoS Comput. Biol., 9, e1002968.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics C. et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature, 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe N.J. et al. (2012) Transcriptional enhancers in development and disease. Genome Biol., 13, 238.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y. et al. (2012) A map of the cis-regulatory sequences in the mouse genome. Nature, 488, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn K.A. et al. (2015) hiHMM: Bayesian non-parametric joint inference of chromatin state maps. Bioinformatics, 31, 2066–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J., Chen K.C. (2015) Spectacle: fast chromatin state annotation using spectral learning. Genome Biol., 16, 33.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence J.R. et al. (2011) Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature, 470, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H. et al. (2005) Compensatory roles of Foxa1 and Foxa2 during lung morphogenesis. J. Biol. Chem., 280, 13809–13816. [DOI] [PubMed] [Google Scholar]
- Won K.J. et al. (2013) Comparative annotation of functional regions in the human genome using epigenomic data. Nucleic Acids Res., 41, 4423–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R. et al. (2016) Hepatocyte nuclear factor 1 coordinates multiple processes in a model of intestinal epithelial cell function. Biochim. Biophys. Acta, 1859, 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen A., Kellis M. (2015) Systematic chromatin state comparison of epigenomes associated with diverse properties including sex and tissue type. Nat. Commun., 6, 7973.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol., 9, R137.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. et al. (2011) The Human Epigenome Browser at Washington University. Nat. Methods, 8, 989–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. et al. (2015) Epigenomic annotation of genetic variants using the Roadmap Epigenome Browser. Nat. Biotechnol., 33, 345–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn A.M., Wells J.M. (2009) Vertebrate endoderm development and organ formation. Annu. Rev. Cell Dev. Biol., 25, 221–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.