

Abstract
Summary
Nonribosomally synthesized peptides (NRPs) are natural products with widespread applications in medicine and biotechnology. Many algorithms have been developed to predict the substrate specificities of nonribosomal peptide synthetase adenylation (A) domains from DNA sequences, which enables prioritization and dereplication, and integration with other data types in discovery efforts. However, insufficient training data and a lack of clarity regarding prediction quality have impeded optimal use. Here, we introduce prediCAT, a new phylogenetics-inspired algorithm, which quantitatively estimates the degree of predictability of each A-domain. We then systematically benchmarked all algorithms on a newly gathered, independent test set of 434 A-domain sequences, showing that active-site-motif-based algorithms outperform whole-domain-based methods. Subsequently, we developed SANDPUMA, a powerful ensemble algorithm, based on newly trained versions of all high-performing algorithms, which significantly outperforms individual methods. Finally, we deployed SANDPUMA in a systematic investigation of 7635 Actinobacteria genomes, suggesting that NRP chemical diversity is much higher than previously estimated. SANDPUMA has been integrated into the widely used antiSMASH biosynthetic gene cluster analysis pipeline and is also available as an open-source, standalone tool.
Availability and implementation
SANDPUMA is freely available at https://bitbucket.org/chevrm/sandpuma and as a docker image at https://hub.docker.com/r/chevrm/sandpuma/ under the GNU Public License 3 (GPL3).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Microbial nonribosomal peptide synthetases (NRPSs) are an important source of complex natural molecules of high therapeutic and biotechnological value. These large, modular protein-systems are found in a variety of microbes and produce structurally and functionally diverse specialized peptide metabolites used as antibiotics, anticancers, immunosuppressants, food additives and crop protection agents. For instance, the NRP vancomycin is used in the clinic as a first-line treatment for methicillin-resistant Staphylococcus aureus and other complex, life-threatening infections (Liu et al., 2011). In their natural setting, NRPs often have potent bioactivity and mediate diverse ecological interactions. For example, the NRP dentigerumycin is produced by the fungus-growing ant symbiont Pseudonocardia, which selectively inhibits the opportunistic pathogen Escovopsis (Oh et al., 2009).
Unlike in ribosomally mediated elongation, the order, identity and processing of peptides incorporated by an NRPS are dictated by its assembly-line module and domain structure (Fischbach and Walsh, 2006; Walsh, 2015). Typically, an NRPS module is comprised of at least three domains that together elongate a growing peptide chain by one amino acid (Walsh, 2015). These core domains include a peptidyl carrier protein (PCP) that tethers the substrate to the NRPS, an adenylation (A) domain that confers specificity for an amino acid substrate, and a condensation (C) domain that catalyzes the formation of peptide bonds between the amino acid substrate and the growing peptide chain (Walsh, 2015).
The advent of genome sequencing has fueled the discovery of thousands of NRPS biosynthetic gene clusters (BGCs). Many NRPS BGCs have more than ten modules and over 500 unique NRPS substrates have been described (Caboche et al., 2010). These features underscore the tremendous combinatorial potential and structural diversity within NRP biosynthesis. Methods utilizing amino acid motif-based analyses (Bachmann and Ravel, 2009; Knudsen et al., 2015; Röttig et al., 2011; Stachelhaus et al., 1999), profile Hidden Markov Models (pHMMs; Khayatt et al., 2013; Minowa et al., 2007; Prieto et al., 2012), latent semantic indexing (Baranašić et al., 2014) and Support Vector Machines (SVMs; Rausch et al., 2005; Röttig et al., 2011) have been developed to computationally assign an A-domain’s substrate specificity directly from its genomic sequence. Additionally, predictions for all individual A-domains in a BGC have been combined to perform predictions of either core or full NRP chemical structures (Li et al., 2009; Medema et al., 2011; Skinnider et al., 2015) enabling streamlined discovery and systematic prioritization efforts. Moreover, they have been used together with mass spectrometric data to dereplicate molecules (Ibrahim et al., 2012; Mohimani et al., 2014) and assign them to their most probable BGCs (Medema et al., 2014a).
Three problems currently prevent the optimal use of substrate prediction algorithms for NRPS A-domains: (i) no estimations are available for the reliability of individual predictions, (ii) the accuracy of available algorithms on novel data is unclear and (iii) training sets have not seen major updates in years. Here, we address all three of these issues. To evaluate the predictability of A-domains in an evolutionary context, we designed a phylogenetically driven algorithm, prediCAT, that calculates a confidence score for each A-domain based on comparative metrics against A-domains of known specificity. Moreover, it allows for more confident predictions in cases of recent evolutionary events. Then, we compiled a large set of experimentally validated substrate specificities from the Minimum Information about a Biosynthetic Gene Cluster (Medema et al., 2015; MIBiG) database and scientific literature, which was used as a test set for accurate benchmarking of all available algorithms. Furthermore, the newly identified set of A-domains was combined with previous training data to retrain all high-performing classes of algorithms, which were then combined into a single ensemble method, SANDPUMA, to optimize both precision and recall. This method was designed for automatic retraining to ensure its training data remains comprehensive as more NRPS BGCs are experimentally characterized in MIBiG. Finally, to demonstrate the opportunities of the high-quality predictions made available by SANDPUMA, the algorithm was applied to 7635 publicly available Actinobacteria genomes to systematically assess NRPS chemical diversity within this taxonomic group. The analysis of 83 589 A-domains in these genomes and the identification of 6049 NRPS BGCs with at least 3 A-domains. These revealed 458 distinct NRP superfamilies, paving the way for high-quality genome-based prioritization of NRP structural diversity.
2 Materials and methods
2.1 Individual algorithms
2.1.1 prediCAT monophyly
Individual query domains were aligned to the respective training set via MAFFT v7.123b (Katoh and Standley, 2013) with a gap open penalty of 5. Leading and trailing overhangs were identified in the query and trimmed. A new multiple sequence alignment (MSA) was created via MAFFT with the default gap open penalty (1.53) and a tree was generated from this MSA by FastTree v2.1.3 (Price et al., 2010). Internal nodes were assigned specificities based on their leaves’ annotations and each leaf node was assigned a grouping based on monophyletic specificities. Branch lengths were calculated between query and training-set leaves. Queries of branch distance less than 0.005 were assigned the specificity of the closest neighbor leaf. For queries of branch distance greater than 0.005, specificity was assigned based on the bounded, monophyletic group to which it belonged (see Fig. 1C). If no such group could be identified, no prediction was made (see Fig. 1E).
Fig. 1.
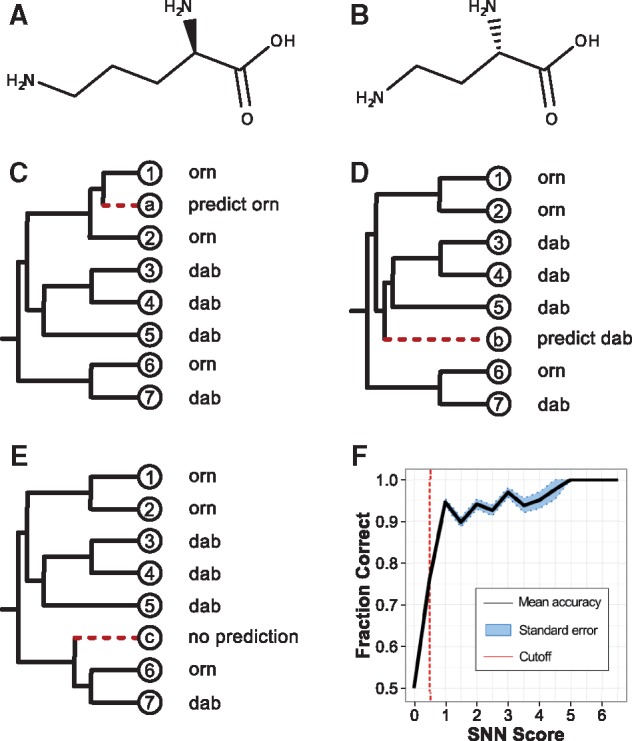
Overview of prediCAT. Structural similarities of (A) ornithine (orn) and (B) 2,4-diaminobutyric acid (dab). Examples of (C) monophyly prediCAT classification, (D) nearest neighbor (NN) prediCAT classification and (E) an ambiguous tree for which no prediCAT call is made. (F) Accuracy of prediCAT across SNN scores (bins of size 0.5)
2.1.2 prediCAT scaled nearest neighbor scores
Trees generated from prediCAT were used to calculate Scaled Nearest Neighbor (SNN) scores for each query. The branch distance between two known reference sequences was used to normalize the branch length of the query sequence to its nearest neighbor. We arrived empirically at a nearest neighbor distance cutoff (ce) of 2.5 from cross-validation results, as distances above 2.5 failed to give reliable results (see Supplementary Fig. S1). For distances less than this cutoff, the distance was transformed to be on a scale from 0 (distance 2.5) to 1 (distance 0). This scoring was repeated for the next n neighbors that both shared substrate specificity with the nearest neighbor and had normalized branch lengths less than the 2.5 distance cutoff. These were summed to give a final scaled nearest neighbor (SNN) score. Together this gives the following equation:
| (1) |
An SNN threshold of 0.5 was used for standalone and ensemble methods (see Fig. 1F).
2.1.3 Support vector machines
Support Vector Machines (SVMs) were used to predict substrate specificity as previously described (Röttig et al., 2011). Each SVM model maximizes the margin of a separating hyperplane between sequence representations specific to one given substrate or cluster versus representations that are specific to others. Sequence representations were comprised of 34 active site residues situated within 8 Ångström (Å) of the A-domain binding pocket (Röttig et al., 2011). Training sequences and queries are aligned to these A-domain loci using a pHMM (NRPS A-domain AMP-binding; PFAM ID PF00501.21). Determined sequence loci were then extracted as signatures and encoded with a numerical feature representation.
We retrained all NRPSpredictor2 models for sequences that predict single acid substrate specificity. For this we extended the training data from the original NRPSpredictor2 with our training sequences. If a combination of extracted signature with experimental substrate specificity occurred multiple times in the training data, we used this combination only once so to not overestimate benchmark performance. We used the radial basis function kernel as kernel function for all retrained models. Optimized model parameters were the penalty parameter, C, and the kernel width parameter, γ. For each specific training set, we determined the optimal parameters with a nested cross-validation to choose between inductive and transductive SVM approaches.
2.1.4 Active site motifs
Stachelhaus et al. have previously described specificity-conferring primary amino acid loci within A-domains which are putative constituents of the binding pocket (Stachelhaus et al., 1999). Of these 10 loci, the lysine residue at alignment position 517 was found to be invariant and thus excluded from further analysis. The remaining 9 loci were extracted from query sequences and assigned a specificity. Queries were aligned to four A-domains from the Stachelhaus study (GrsA, SrfAB-2, GrsB3 and CssA9; Stachelhaus et al., 1999) by MAFFT with a gap open penalty of 3.40. MSAs were automatically checked for alignment quality and passing active site motif (ASM) sequence signatures were assigned based on known specificities. Subsequent ASM searches looked first for motif matches of 9 (exact), 8, or 7 residues, whichever was highest.
2.1.5 Profile hidden Markov models
Profile Hidden Markov Models (pHMMs) were generated per methods described in Khayatt et al. (2013). Briefly, training sequences were aligned by default MAFFT. Sequences were iteratively trimmed and realigned to eliminate leading and trailing gaps (to a minimum of 360 positions). A tree was created from the final MSA by ClustalW (Larkin et al., 2007) and monophyletic groupings of shared substrate specificities were identified. pHMMs of each monophyletic group were created (and subsequently searched) with HMMER3 (Eddy, 2011). Performance of pHMMs was checked against the published Khayatt pHMMs and returned near identical results (192 correct versus 191 correct for Khayatt and this study respectively).
2.2 Benchmarking existing methods
prediCAT and pHMMs were trained as previously described on the A-domain dataset described in Khayatt et al. (2013). Manually curated and MIBiG sequences were used to benchmark prediCAT (both monophyly and SNN ≥ 0.5) and pHMM methods against existing Bachmann-Ravel (Bachmann and Ravel, 2009), Minowa (Minowa et al., 2007), NRPSpredictor2 (both Stachelhaus and SVM; Röttig et al., 2011), NRPSsp (Prieto et al., 2012) and SEQL-NRPS (Knudsen et al., 2015) algorithms in both accuracy and coverage. For sequences with shared coverage, methods were compared pairwise and used to calculate significant differences in sequence predictions by a McNemar’s test.
2.3 Full dataset cross-validation performance
The full dataset was randomized and broken into ∼10% subsets (either 93 or 92 A-domain sequences). For each subset, prediCAT, SVM, ASM and pHMM algorithms were trained on the remaining 90% of the data. Method accuracy was assessed by querying the subset against these models. 100 query sets and training sets were assessed (10 randomizations each with 10 subsets) totaling 9280 individual queries for each method to robustly estimate the performance of each method (see Supplementary Fig. S2).
ASM, SVM, prediCAT (monophyly and SNN ≥ 0.5) and pHMM methods were subjected to ten cross-validation resampling analyses, as described above. Precision was calculated as tp/(tp + fp) and recall was calculated as tp/(tp + fn) where tp, fp and fn are the number of true positives, false positives and false negatives, respectively. F-scores were calculated as the harmonic mean of precision and recall. Corresponding taxonomic data was gathered from NCBI and taxon-specific performance was quantified.
2.4 SANDPUMA ensemble method
A decision tree incorporating percent identity to the best match in the training set and results from each algorithm (ASM, SVM, prediCAT monophyly, prediCAT SNN ≥ 0.5 and pHMM) was used to calculate an ensemble specificity call through the supervised machine learning package scikitlearn (Pedregosa et al., 2012). Maximum depths of 20, 30, 40 and 50 nodes and minimum leaf supports of 2, 5, 10, 15 and 20 were tested (see Supplementary Fig. S3) and maximum depth of 40 and minimum leaf support of 10 was selected to minimize overfitting and maximize accuracy. SANDPUMA was then trained with all permutations of three out of four individual methods (of ASM, SVM, prediCAT and pHMM) to assess the contribution of each individual method to accuracy (see Supplementary Fig. S4). The maximum depth of these subsetted methods was scaled to 30 (0.75*40) so to avoid overfitting in this comparison. Accuracy of individual decision paths within the full ensemble algorithm was quantified (see Supplementary Fig. S5) and paths less than 50% accurate were deemed unreliable and removed. A second cross-validation was used to benchmark the individual and SANDPUMA methods. Accuracy and coverage were compared to the constituent methods and specificity precision and recall were calculated.
2.5 NRPS categorization of Actinobacteria
Open reading frames (ORFs) were predicted with prodigal v2.60 (Hyatt et al., 2010) in closed end mode from 7635 Actinobacteria genomes downloaded from NCBI. A-domains were identified through scanning against PF00501.21, CoA-ligase domain and peptidyl carrier protein pHMMs (described in antiSMASH; Blin et al., 2017) with HMMER3 (Eddy, 2011). Hits with higher bitscores for PF00501.21 than CoA-ligase which also had a PCP in the same ORF were called true A-domains and used in further analysis.
To construct a reference species tree, HMMER3 was used to search the Actinobacteria genomes using TIGRFAM HMMs of 92 genes conserved across bacteria. Hits were concatenated and used to build a multi-locus protein alignment with MAFFT. These were converted to nucleotide alignments and a phylogeny was created with RAxML 8.1.24 (Stamatakis, 2014) under a GTRGAMMA substitution model with 100 bootstraps. Deinococcus geopthermalis DSM 11300 was used as an outgroup.
SANDPUMA was employed to make substrate specificity predictions. Groupings of A-domains within 10 kb of each other were marked as putative BGCs. Pairwise percent identity comparisons were made using DIAMOND (Buchfink et al., 2015) and a sequence similarity matrix was created. Distance between clusters was calculated a combination of the Jaccard index (Lin et al., 2006) and Domain Duplicate Score (DDS) of substrate specificity groups of A domains (not of Pfam domains, as previously described by Cimermancic et al., 2014), weighted at 0.667 and 0.333 respectively. Distances of less than 0.1 were used to construct an NRPS BGC superfamily network, which was visualized in networkx (https://networkx.github.io/) and Cytoscape (http://www.cytoscape.org/).
Alignments from the reference species tree were used to segregate genomes into taxonomic bins of 97% nucleotide identity. Bins were chosen at random (with replacement) and a genome from within this bin was chosen at random (without replacement). Rarefaction curves (and extrapolation to 15 000 genomes) was performed on samplings of 3000, 4000, 5000, 6000, 7000 and 7635 by EstimateS (Colwell et al., 2012) with 100 randomizations and 750 knots.
3 Results
3.1 A new algorithm to assess substrate predictability and recent evolutionary events
While various computational methods for predicting NRPS A-domain substrates exist, no algorithms report a quantitative level of confidence associated with their predictions. Furthermore, since current methods rely on limited training data, they often struggle to accurately predict substrates of A-domains that have undergone recent evolutionary shifts in specificity. Duplication, divergence, recombination events, (Crüsemann et al., 2013; Diminic et al., 2014; Medema et al., 2014a; Rounge et al., 2008) and mutations in or around the active site (Cruz-Morales et al., 2016; Stachelhaus et al., 1999) are the major evolutionary mechanisms by which substrate switching and expansion can occur. Confidently predicting the effect of mutations on substrate specificity relies on the breadth of data used for training and the model’s ability to accurately reflect biology.
To address these challenges, we developed prediCAT (Predictions through Comparative A-domain Trees) which leverages comparative genomics to predict substrate specificity. In prediCAT, phylogenetic reconstructions of A-domain evolutionary histories are used to predict substrates in one of two ways: (i) query A-domains which fall within monophyletic clades of shared substrate specificities are assigned the specificity of that clade, and (ii) if this does not apply, a query is assigned the specificity of its nearest-neighbor. A scaled nearest-neighbor (SNN) scoring metric was developed (see Fig. 1) to assess and report the confidence of prediCAT predictions. The SNN score is a summation of the normalized, reversed branch lengths between a query and the nearest neighbors that share substrate specificities (i.e. the SNN score increases as more neighboring training sequences of short branch length share the nearest-neighbor’s specificity; see methods for details). An analysis of the relationship between SNN scores and prediction accuracies showed that query A domains with scores below 0.5 show low predictability, with a steep rise at a value around 0.5 (see Fig. 1F). Based on this result, we implemented an SNN cutoff of equal to or greater than 0.5. Queries with SNN scores lower than this threshold will not be given a prediction. We observed 84.1% accuracy after introduction of the SNN score cutoff compared to 50.7% without it. 40% of input sequences were no longer given a prediction. Moreover, the SNN score was implemented to quantitatively estimate the probability that a given query domain is predicted correctly, based on the average results of other queries at the same SNN score window (see Supplementary Table S1). All in all, the prediCAT-SNN algorithm thus provides a means for high-precision substrate specificity prediction (by design opting for a lower recall), while the SNN metric accurately quantifies the predictability of any A domain sequence.
3.2 A large set of new A-domains with known specificities
To accurately assess and compare the various versions of the prediCAT algorithm (monophyly assignment only, nearest-neighbor forced assignment, or with SNN score cutoff; see Methods) and all previously designed algorithms for A-domain substrate specificity prediction, we set out to gather a large set of A-domains that are confidently linked to substrates by experimental evidence yet have not been included in previous training sets. In this study, sufficient experimental evidence was defined as structure-based inferences, confirmatory activity assays, and/or other genetic validations of structure. To add A-domain sequences to the set previously described by Khayatt et al. (2013), we used two sources: experimentally supported A-domains from MIBiG NRPS BGCs (published after Khayatt, 2013) and an additional set of manually curated, experimentally verified domains from recent scientific literature. The set was checked for redundancy (100% protein identity), after which a total dataset of 928 unique A-domain sequences was established, made up by 494 domains from the Khayatt dataset and 434 newly added ones (see Fig. 2; for the full dataset with accession numbers, see Supplementary Table S2). The new data constituted an increase of 139 A-domains from Actinobacteria and 162 from Proteobacteria (increases of 104% and 141% respectively). Together, the newly added A-domains covered 116 unique substrate specificities and increased the total number of available sequences by 87%.
Fig. 2.

Dataset (A) source, (B) phylum distribution of the total dataset and (C) phylum distribution of the train and test datasets
3.3 Systematic benchmarking shows key differences in accuracy between algorithms
The availability of this large new set of A domains that were all experimentally supported and not part of previous training sets provided a unique opportunity to systematically assess and compare the accuracies of all prediction methods. The range of methods tested employed either prediCAT, ASM, pHMM, SVM, or greedy coordinate-descent algorithms (see Supplementary Table S3); to compare them, all algorithms were run on each of the 434 A domains, and the results were compared to the experimental data from literature to estimate their accuracy.
We hypothesized that some algorithms might perform relatively better for query sequences with high percent identity (PID) to their nearest neighbor in the training set, while others might perform better for sequences distantly related to all sequences in the training set. Indeed, the PrediCAT SNN + monophyly method, which emphasizes the identification of recent evolutionary events, was more accurate than all other algorithms for query sequences of high percent identity to the training set, while the NRPSPredictor2 SVM, which uses general physicochemical properties of amino acids close to the active sites, performed better at low PID (see Fig. 3A). PrediCAT SNN and monophyly methods did suffer from poor coverage (they often gave no prediction), especially at low PID, and were unable to make a confident prediction for many query sequences (see Fig. 3B). A prediCAT nearest neighbor prediction with no SNN cutoff (prediCAT monophyly + NN) greatly improved coverage at the cost of accuracy. In pairwise comparisons of shared coverage, NRPSPredictor2 ASM, NRPSPredictor2 SVM, prediCAT SNN and prediCAT monophyly were the best performing methods (see Fig. 3C). The most significant discrepancies in sequence-to-sequence prediction exist between NRPSPredictor2 ASM/SVM and pHMM-based methods, with pHMMs vastly underperforming against other methods (see Fig. 3C, Supplementary Table S4).
Fig. 3.
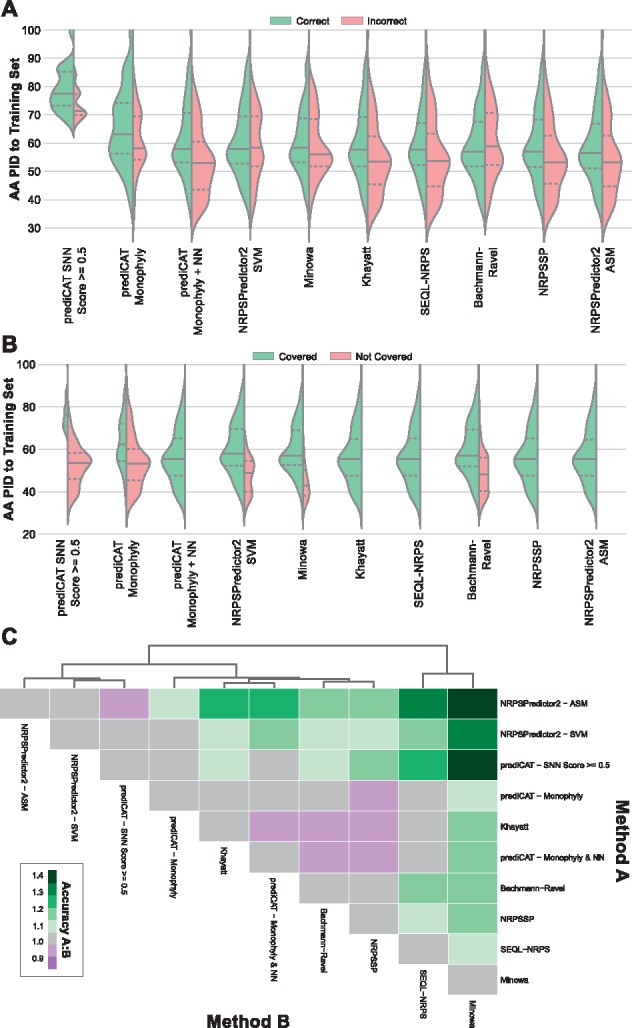
Comparison of current NRPS prediction algorithms. (A) Accuracy and (B) coverage distributions of NRPS prediction methods at different sequence percent identity to the training set. Solid and dashed lines indicate means and quartiles, respectively. Training data for prediCAT methods from Khayatt dataset. All other algorithms were used as released. Test data was comprised of A-domains from MIBiG (post-Khayatt) and manual sources. PID was calculated as protein sequence identity of test queries to the Khayatt dataset. (C) Pairwise accuracy ratios of shared coverage. Ratio calculated as (fraction correct method A) / (fraction correct method B)
3.4 SANDPUMA: an ensemble method that outperforms individual algorithms
The subclassification in PID classes during benchmarking made clear that the various algorithms may have complementary strengths, rather than a single algorithm outperforming all others in all cases. Hence, we hypothesized that an ensemble method might outperform individual methods. Therefore, we developed SANDPUMA (Specificity of AdenylatioN Domain Prediction Using Multiple Algorithms), which combined versions of the prediCAT SNN, prediCAT monophyly, ASM, pHMM and SVM methods that were all retrained on all 928 experimentally supported A domains.
A cross-validation of all 928 domains (see Fig. 4 and Supplementary Fig. S6) served as training data for ensemble substrate predictions. ASM, SVM, prediCAT monophyly and prediCAT SNN methods shared considerable coverage, with 91.4% of queries covered by two or more algorithms and only 5.6% of queries not covered by any method (see Supplementary Fig. S6A). As in the earlier analysis, both prediCAT methods performed well at over 80% accuracy, but had considerable drops in coverage under 80% PID (see Supplementary Fig. S6B and C). On average, ASMs performed best against similar sequences (>60% PID) while prediCAT Monophyly returned the most accurate predictions at lower PIDs (<60%) (see Supplementary Fig. S6B).
Fig. 4.
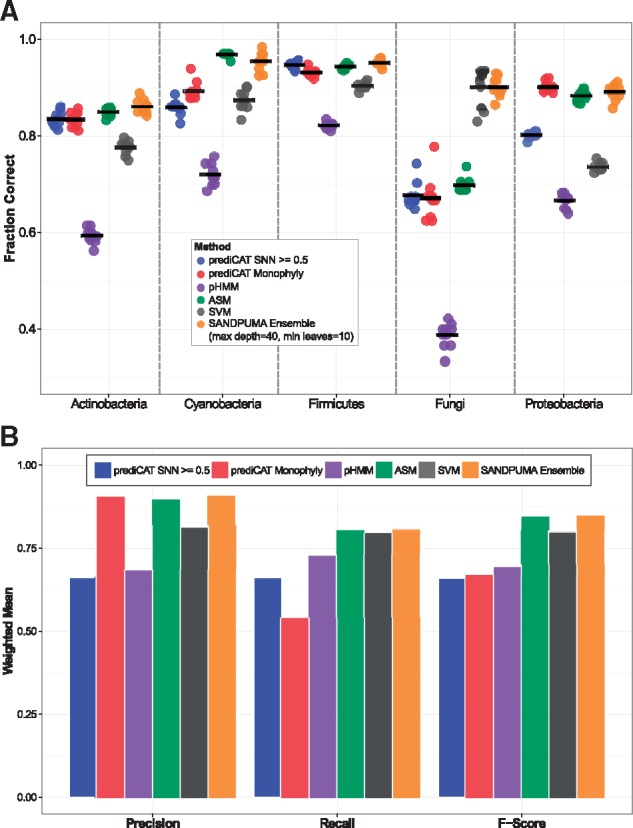
Cross-validation. Ten independent random shuffles were performed to segregate the full dataset into tenths (92 or 93 sequences). Each 1/10 was used to test methods trained on the remaining 9/10. (A) Accuracy of methods across different taxonomic groups. (B) Means are weighted by frequency in the cross-validation set. Weighted means of precision, recall and F-score are shown across individual and ensemble methods
SANDPUMA, a decision tree schema, was built to predict specificities based on the protein percent identify (PID) to the training A-domain set and the predictions of the individual ASM, SVM, prediCAT SNN, prediCAT monophyly and pHMM methods. A maximum tree depth of 40 nodes and a minimum leaf support of 10 samples was chosen to maximize accuracy while minimizing the potential of overfitting (see Supplementary Fig. S3). Accuracy assessment of each decision tree path based on cross-validation was used to identify and exclude unreliable paths from the algorithm (see Supplementary Fig. S5), as we reasoned that it would be preferable not to give a prediction if the probability of accurate assignment would be low. A second cross-validation was performed to benchmark the decision tree method, performing individual and ensemble predictions with newly randomized training data. As above, using trusted decision tree paths from the first cross-validation resulted in high accuracy and coverage predictions (see Supplementary Fig. S7). Importantly, the observation that an independent cross-validation does not significantly impact performance provides confidence that our predictive model is robust and accuracy is fairly estimated.
Importantly, low coverage issues that some individual high-precision methods exhibited were no longer present in the SANDPUMA ensemble implementation (see Supplementary Fig. S6C). Also, SANDPUMA exhibits high accuracies across many taxonomic groups and has greater precision, recall and F-scores than its constitutive methods across taxonomic groups (see Fig. 4). The expanded dataset and new method implementation resulted in improved F-scores for many amino acids (see Supplementary Table S5) and showed improvements over all individual methods (see Supplementary Table S6), while expanding the number of amino acid specificities for which predictions are available (see Supplementary Table S7).
3.5 NRPS diversity of Actinobacteria
As NRPS BGCs are a large and important biosynthetic class of secondary metabolites, we sought to showcase the scalability of SANDPUMA’s NRPS classification by describing the diversity of NRPS biosynthesis across all publicly available Actinobacteria genomes. Actinobacteria are prolific producers of secondary metabolites with diverse biological activities. Many of these compounds have been used in medical and biotechnological contexts, and studying their ability to mediate community dynamics has deepened our understanding of microbial interactions (Lewin et al., 2016).
83 589 A-domains of NRPS BGCs were analyzed in 7635 Actinobacteria genomes from 69 genera (see Supplementary Fig. S8) and clustered by Jaccard and Domain Duplicate Score (DDS) similarity metrics into superfamilies. Rather than using Pfam domains as basic units (Cimermancic et al., 2014), A-domain classes were used (according to methods previously published in Nguyen et al., 2016) for superfamily clustering. From the 6049 NRPS BGCs with at least three A-domains, 458 superfamilies were identified, 71 of which were comprised of five or more BGC examples. Interestingly, 273 superfamilies had only a single BGC representative. Furthermore, correction for NRP BGCs at contig breaks suggests these estimates are not biased by gene cluster fragmentation (see Supplementary Fig. S9). We see that many cluster superfamilies are genus-specific or of low genus diversity while others are dispersed across many genera within Actinobacteria (see Fig. 5A). A rarefaction analysis and extrapolation from a random sampling of taxonomic bins (97% pairwise identity across 92 conserved genes) suggests current estimates of NRP superfamily diversity are undercounted and there are many more chemical scaffolds to be found (see Fig. 5B).
Fig. 5.
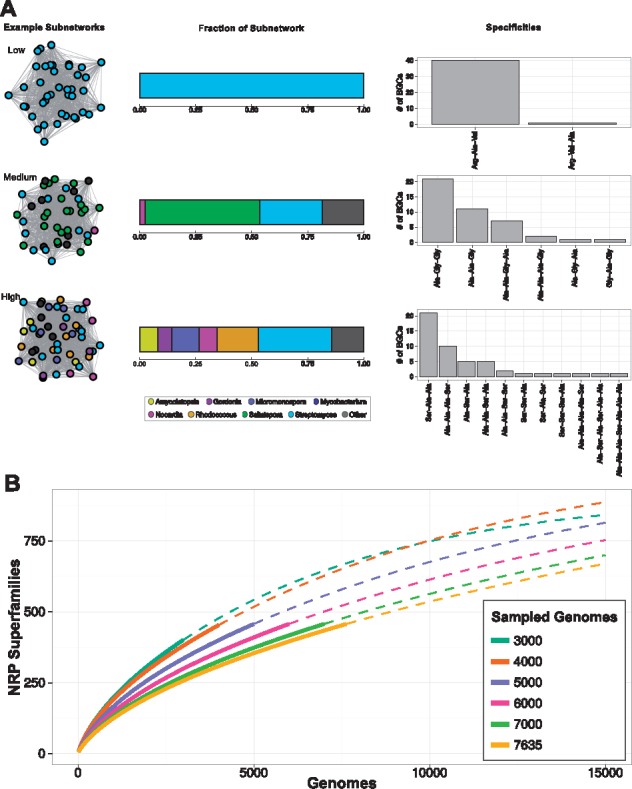
NRP biosynthesis across Actinobacteria. (A) Representative low, medium and high diversity subgraphs colored by genus with their respective compositions and A-domain specificities. (B) Rarefaction curve of NRP Superfamilies. Solid lines denote the sampled data and dashed lines denote values that are extrapolated. The full genome set (with no random sampling) is shown in yellow (Color version of this figure is available at Bioinformatics online.)
4 Discussion
The addition of experimentally characterized datasets from the literature and MIBiG provided a unique opportunity to perform a fair and comprehensive comparison of the wide range of published algorithms for A-domain substrate specificity prediction. Multiple conclusions can be drawn from the results: (i) in pairwise comparisons of the performance of existing methods on recently published experimental data (see Fig. 3C), NRPSPredictor2 and prediCAT SNN methods performed best and hierarchically cluster together; (ii) Minowa and SEQL-NRPS methods also cluster together, representing the group in which observed performance was poorest; (iii) various methods based on pHMMs (Khayatt, NRPSsp and Minowa) do not cluster closely in terms of performance. Minowa represents the pHMM method with the oldest training data (2007), and the significant differences in performance to NRPSsp (2012) and Khayatt (2013) (see Supplementary Table S4) suggest the importance of comprehensive training data in whole-domain predictive methods, especially when integrated as structure prediction models in widely used BGC analysis suites such as antiSMASH (Blin et al., 2017) and PRISM (Skinnider et al., 2015).
The integrated SANDPUMA ensemble algorithm, which combines results from ASM, SVM, pHMM and prediCAT methods into a single prediction, is significantly more accurate than individual methods (see Table 1). Importantly, SANDPUMA’s improved accuracy likely reflects the strengths of the individual algorithms in certain taxonomic groups (see Fig. 4A). For example, while the SVM method is strong for fungi and weak for proteobacteria, SANDPUMA performs well on sequences from both taxa. The ability to perform well across taxa is especially important as genomic information for an increasing number of species grows exponentially and NRP biosynthetic discovery increases from non-traditional sources such as protists (O’Neill et al., 2016) and even metazoans (Shou et al., 2016). Similarly, NRPSs from certain important ecosystems such as the human microbiome (Donia et al., 2014) are poorly covered by current training data. To better cover uncharted taxonomic and biosynthetic areas of NRP diversity in the future, projects for large-scale and systematic experimental characterization of unknown A-domains would be highly beneficial. In such an approach, uncharacterized A-domains would be selected to maximize taxonomic and functional coverage, be codon-optimized and synthesized for expression in a suitable heterologous host, and be profiled in detail using ATP-PPi exchange assays. Besides characterization of A-domains of unknown function, it would also be highly beneficial to perform similar high-throughput profiling of A-domains whose substrate specificity has been inferred from the final natural product structure, as the amino acid observed in the final structure is not always identical to the amino acid selected by the A-domain (Challis et al., 2000). The amino acid can be subject to post-assembly-line modifications by tailoring enzymes which obscure the true substrates for these domains (Challis et al., 2000). With currently available data, it is difficult to reliably distinguish recent evolution of A-domain substrate specificities from recent evolutionary changes in post-assembly-line modifications in such cases.
Table 1.
Comparison of SANDPUMA to individual methods
| Individual method | Shared cross-validation queries | Individual accuracy | SANDPUMA accuracy | McNemar P-value |
|---|---|---|---|---|
| ASM | 7255 | 0.905 | 0.922 | 6.59E−07 |
| SVM | 7201 | 0.824 | 0.908 | 1.95E−90 |
| prediCAT Monophyly | 4846 | 0.899 | 0.945 | 4.25E−36 |
| prediCAT SNN ≥ 0.5 | 5116 | 0.879 | 0.939 | 1.27E−52 |
| pHMMs | 7935 | 0.727 | 0.899 | 2.03E−246 |
The goal of a supervised machine learning decision tree is to create a predictive model from multivariate inputs by inferring decision rules from the data features. In SANDPUMA, these features are a query A-domain’s percent identity to the training set and its classifications by each individual algorithm. In contrast to unsupervised machine learning approaches, SML frameworks build models from user-defined training data only. This distinction is especially important in SANDPUMA, as only structurally supported or experimentally validated A-domain substrate specificities are used to fit the model, which eliminates the incorporation of unverified classifications into the predictive model. Overfitting remains a concern for large tree networks with many decision nodes, and we have addressed this by constraining the model to a maximum tree depth (40) and a minimum number of leaves that support a given decision path (10) (see Supplementary Fig. S3). The accuracy achieved by SANDPUMA leverages the strengths of ASM, SVM, prediCAT and pHMMs and is reliant on all these methods (see Supplementary Fig. S4). This is especially true of A-domain specificities with few examples in the training data (see Supplementary Fig. S4B). Furthermore, the ensemble SANDPUMA outperforms the best individual methods for shared queries (those queries for which predictions are returned by both SANDPUMA and the individual method; see Table 1).
In cross-validation, unreliable decision paths (<50% accurate) are followed for only 13.7% of the total data (see Supplementary Fig. S5), so their removal further improved overall accuracy of SANDPUMA while only slightly decreasing coverage (see Supplementary Fig. S7). We feel that, in general, high precision is more important than high recall, and that algorithms should not be forced to output predictions in cases where no confident prediction can be made. The accuracy of the followed decision tree path is reported to the end user to help assess the confidence of a given prediction. The decision tree architecture of SANDPUMA allows for an elegant way of incorporating this into its classifications. Furthermore, the reporting of both prediCAT SNN score and SANDPUMA decision tree path accuracy allows for users to estimate both how closely and consistently a query clades with training data and how accurately current algorithms are able to make predictions for the specificity finally assigned by SANDPUMA.
Accurate A-domain substrate predictions are critical in fueling high-throughput NRP discovery and in describing nature’s chemical ecology. Actinobacteria represent a major source of bioactive small molecules and BGCs encoding NRP biosynthesis are found in most major Actinobacteria genera (see Supplementary Fig. S8). Almost 60% of the NRP superfamilies identified within publicly available Actinobacteria genomes were comprised of only a single example which argues that despite the exponential growth of sequencing data, genomic NRP BGC discovery efforts are nowhere near saturation. Our metrics for superfamily diversity remain high when analyzing only NRP BGCs that are more than 10 kb from a contig break (see Supplementary Fig. S9), suggesting these estimates reflect the true natural diversity. The low genus-level diversity observed in many NRP superfamilies (see Fig. 5A) further suggests many novel NRP superfamilies have yet to be uncovered. Rarefaction analysis and extrapolation of NRP superfamilies across genomes (see Fig. 5B) supports this hypothesis. Moreover, it highlights the implicit taxonomic biases in available genomes that impact such analyses. With random sampling from taxonomic bins to correct for these biases, as fewer genomes are sampled the rarefaction curve exhibits a much steeper slope (and thus is more difficult to accurately extrapolate). Together, these findings suggest much NRP chemical diversity remains to be discovered, especially in sparsely sampled genera, and previous reports (Doroghazi et al., 2014) of NRP chemical diversity are likely underestimated.
With the increasing frequency of metagenomic analyses and large-scale genome studies, SANDPUMA provides a robust tool for guiding prioritization and dereplication of NRPs within large datasets. The ability to prioritize novel scaffolds and analogs within superfamilies of interest greatly increases the power of genomic natural product discovery efforts. Furthermore, studies elucidating the evolutionary mechanisms through which A-domain specificities evolve have the potential to inform rational engineering and strategies. These advances rely on accurate predictions. SANDPUMA provides a powerful framework that optimizes current capabilities for adenylation domain substrate specificity prediction, and (through its connection to MIBiG) is designed to be easily extendable with new data from both low- and high-throughput experimental sources in the future. Therefore, it provides a key step in the optimization of approaches to connect genomic and metabolomic data (Doroghazi et al., 2014; Medema et al., 2014b; Mohimani et al., 2014; Wang et al., 2016), which will be key drivers of future efforts in genome-based natural product discovery.
SANDPUMA is offered as open-source software and has been integrated into the antiSMASH (Blin et al., 2017) biosynthetic gene cluster analysis platform to provide both flexibility and general applicability for expert and non-expert users alike.
Supplementary Material
Acknowledgements
The authors thank Xiaowen Lu for inspiring BGC distance calculations, Aldo Gonzalez-Ortiz for data retrieval and Heidi Horn for constructive comments and suggestions.
Funding
M.G.C. was supported by National Institutes of Health National Research Service Award T32 GM008505. M.H.M. was supported by VENI grant 863.15.002 from The Netherlands Organization for Scientific Research (NWO) M.G.C. and C.R.C were supported by National Institutes of Health U19 Al109673.
Conflict of Interest: none declared.
References
- Bachmann B.O., Ravel J. (2009) Chapter 8. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods in enzymology, 458, 181–217. [DOI] [PubMed] [Google Scholar]
- Baranašić D. et al. (2014) Predicting substrate specificity of adenylation domains of nonribosomal peptide synthetases and other protein properties by latent semantic indexing. J. Ind. Microbiol. Biotechnol., 41, 461–467. [DOI] [PubMed] [Google Scholar]
- Blin K. et al. (2017) antiSMASH 4.0––improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res., 1854, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B. et al. (2015) Fast and sensitive protein alignment using DIAMOND. Nat. Methods, 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Caboche S. et al. (2010) Diversity of monomers in nonribosomal peptides: towards the prediction of origin and biological activity. J. Bacteriol., 192, 5143–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis G.L. et al. (2000) Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem. Biol., 7, 211–224. [DOI] [PubMed] [Google Scholar]
- Cimermancic P. et al. (2014) Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell, 158, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell R.K. et al. (2012) Models and estimators linking individual-based and sample-based rarefaction, extrapolation and comparison of assemblages. J. Plant Ecol., 5, 3–21. [Google Scholar]
- Crüsemann M. et al. (2013) Evolution-guided engineering of nonribosomal peptide synthetase adenylation domains. Chem. Sci., 4, 1041. [Google Scholar]
- Cruz-Morales P. et al. (2016) Phylogenomic analysis of natural products biosynthetic gene clusters allows discovery of arseno-organic metabolites in model streptomycetes. Genome Biol. Evol., 8, 1906–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diminic J. et al. (2014) Evolutionary concepts in natural products discovery: what actinomycetes have taught us. J. Ind. Microbiol. Biotechnol., 41, 211–217. [DOI] [PubMed] [Google Scholar]
- Donia M.S. et al. (2014) A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell, 158, 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroghazi J.R. et al. (2014) A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat. Chem. Biol., 10, 963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S.R. (2011) Accelerated profile HMM searches. PLoS Comput. Biol., 7, e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach M., Walsh C. (2006) Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem. Rev., 5, 3468–3496. [DOI] [PubMed] [Google Scholar]
- Hyatt D. et al. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim A. et al. (2012) Dereplicating nonribosomal peptides using an informatic search algorithm for natural products (iSNAP) discovery. Proc Natl Acad Sci USA, 109, 19196–19201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. (2013) MAFFT Multiple Sequence Alignment Software Version 7: improvements in performance and usability. Mol. Biol. Evol., 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayatt B.I. et al. (2013) Classification of the adenylation and acyl-transferase activity of NRPS and PKS systems using ensembles of substrate specific hidden Markov models. PloS One, 8, e62136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen M. et al. (2015) Computational discovery of specificity-conferring sites in non-ribosomal peptide synthetases. Bioinformatics, btv600. [DOI] [PubMed] [Google Scholar]
- Larkin M.A. et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics, 23, 2947–2948. [DOI] [PubMed] [Google Scholar]
- Lewin G.R. et al. (2016) Evolution and ecology of actinobacteria and their bioenergy applications. Annu. Rev. Microbiol., 70, 235–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.H. et al. (2009) Automated genome mining for natural products. BMC Bioinformatics., 10, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K. et al. (2006) An initial strategy for comparing proteins at the domain architecture level. Bioinformatics, 22, 2081–2086. [DOI] [PubMed] [Google Scholar]
- Liu C. et al. (2011) Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin. Infect. Dis., 52, 285–292. [DOI] [PubMed] [Google Scholar]
- Medema M.H. et al. (2011) AntiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res., 39 (Suppl. 2), 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema M.H. et al. (2014a) A systematic computational analysis of biosynthetic gene cluster evolution: lessons for engineering biosynthesis. PLoS Comput. Biol., 10, e1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema M.H. et al. (2014b) Pep2Path: automated mass spectrometry-guided genome mining of peptidic natural products. PLoS Comput. Biol., 10, e1003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema M.H. et al. (2015) Minimum information about a biosynthetic gene cluster. Nat. Chem. Biol., 11, 625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowa Y. et al. (2007) Comprehensive analysis of distinctive polyketide and nonribosomal peptide structural motifs encoded in microbial genomes. J. Mol. Biol., 368, 1500–1517. [DOI] [PubMed] [Google Scholar]
- Mohimani H. et al. (2014) NRPquest: coupling mass spectrometry and genome mining for nonribosomal peptide discovery. J. Nat. Prod., 77, 1902–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D.D. et al. (2016) Indexing the Pseudomonas specialized metabolome enabled the discovery of poaeamide B and the bananamides. Nat. Microbiol., 2, 16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill E.C. et al. (2016) Gene Discovery for Synthetic Biology In: Methods in Enzymology. Vol. 576, 1st edn. Elsevier Inc. pp. 99–120. [DOI] [PubMed]
- Oh D.C. et al. (2009) Dentigerumycin: a bacterial mediator of an ant-fungus symbiosis. Nat. Chem. Biol., 5, 391–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F. et al. (2012) Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res., 12, 2825–2830. [Google Scholar]
- Price M.N. et al. (2010) FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS ONE, 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto C. et al. (2012) NRPSSP: Non-ribosomal peptide synthase substrate predictor. Bioinformatics, 28, 426–427. [DOI] [PubMed] [Google Scholar]
- Rausch C. et al. (2005) Specificity prediction of adenylation domains in nonribosomal peptide synthetases (NRPS) using transductive support vector machines (TSVMs). Nucleic Acids Res., 33, 5799–5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röttig M. et al. (2011) NRPSpredictor2 – a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res., 39, W362–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rounge T.B. et al. (2008) Recombination and selectional forces in cyanopeptolin NRPS operons from highly similar, but geographically remote Planktothrix strains. BMC Microbiol., 8, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou Q. et al. (2016) A hybrid polyketide–nonribosomal peptide in nematodes that promotes larval survival. Nat. Chem. Biol., 12, 770–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinnider M.A. et al. (2015) Genomes to natural products PRediction Informatics for Secondary Metabolomes (PRISM). Nucleic Acids Res., 9140, gkv1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachelhaus T. et al. (1999) The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol., 6, 493–505. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. (2014) RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C.T. (2015) Insights into the chemical logic and enzymatic machinery of NRPS assembly lines. Nat. Prod. Rep., 00, 1–9. [DOI] [PubMed] [Google Scholar]
- Wang M. et al. (2016) Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol., 34, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.