

Abstract
Motivation
The size and complexity of modern large-scale genome variation studies demand novel approaches for exploring and sharing the data. In order to unlock the potential of these data for a broad audience of scientists with various areas of expertise, a unified exploration framework is required that is accessible, coherent and user-friendly.
Results
Panoptes is an open-source software framework for collaborative visual exploration of large-scale genome variation data and associated metadata in a web browser. It relies on technology choices that allow it to operate in near real-time on very large datasets. It can be used to browse rich, hybrid content in a coherent way, and offers interactive visual analytics approaches to assist the exploration. We illustrate its application using genome variation data of Anopheles gambiae, Plasmodium falciparum and Plasmodium vivax.
Availability and implementation
Freely available at https://github.com/cggh/panoptes, under the GNU Affero General Public License.
1 Introduction
Two important trends are reshaping the genome variation data landscape: the advent of massively parallel sequencing methods, and the formation of large consortia, consisting of many international collaborators contributing to a single data repository with rich metadata. As a consequence, the creation and analysis of these data has reached a new level, both in terms of size and complexity.
Unlocking the full potential of such complex data often requires interactive, visual exploration by skilled experts, complementary to conventional computational analyses (Keim et al., 2006). However, current standard visualization tools focus only on specific aspects of the data, such as genotype viewers, genome browsers, phylogeny viewers, or geographic mapping tools (examples: Huson et al., 2007; Manske and Kwiatkowski, 2009; Robinson et al., 2011; Wang et al., 2012). There is a distinct need for a browsing framework that integrates all relevant aspects in a single, coherent environment, serving as a one-stop shop for the various components of a dataset including derived results such as statistical calculations. Only in this way can a researcher obtain rapid responses to a wide variety of questions, with user journeys based on a rich set of navigational paths between the different components. This need has already been identified in other areas of research that face similar problems, such as functional genomics (Chelaru et al., 2014).
Knowledge creation in large collaborative consortia is often complicated by being a distributed process, with collaborators from around the globe interacting with a dataset and exchanging findings. A data exploration framework should facilitate this process by offering appropriate tools to easily share results, and by focusing on ease of access. Project collaborators with different areas of expertise can contribute valuable, complementary insights, but they may not have the technical background required to deal with traditional bioinformatics data distribution channels, such as an FTP site with VCF files. Those with the technical background will often want to see an overview before going further. To lower the barrier for exploring the data, a user-friendly web application is a prerequisite, avoiding the need for client side software installation, data download and computational power, and enabling sharing of specific data views via hyperlinks.
Another key requirement is fast handling of very large datasets. A rich navigational context only works well if it responds almost instantaneously, encouraging the user to drill deeper into the data. Given the size of current genome variation datasets, achieving this in a web browser requires some tailored technological solutions.
Here we describe Panoptes, an open source framework for collaborative exploration of large-scale genome variation datasets. It focuses on supporting genetic data in the context of rich metadata, such as geographic and time information for samples. We elaborate on some technology choices made to address the previously mentioned challenges, and illustrate its usage for the study of genome variation in the human malaria parasites Plasmodium falciparum and Plasmodium vivax, and the malaria mosquito Anopheles gambiae, using data from various MalariaGEN projects (MalariaGEN, 2008). As an example of scale the A. gambiae dataset comprises 765 samples from 9 populations with genotype calls at over 95 million unfiltered variant sites.
2 Materials and methods
2.1 Architecture
A key objective of Panoptes is the ability to browse a rich set of metadata in conjunction with the genome variation data, and create useful navigational paths between all aspects of the dataset. We addressed this by creating a two-level architecture.
The first level consists of a set of generic features that can be applied to any data table, regardless of the specific nature of its content. These include a table view, graphical query builder, interactive plotting tools and various search and selection mechanisms. A user has to learn these tools only once, and can apply them on any type of data in a deployment, such as lists of variants, samples, genomic regions or sampling sites.
On a second level, specific types of data can be augmented with more specialized functionality, through the declaration of additional semantics. For example, a data table can be declared to contain items with genomic positions (such as a table of single nucleotide polymorphisms), causing the software to activate more specific features, such as visualization of properties as tracks on the genome browser. Other specific declarations include genotype calls, genomic regions, samples, geographical coordinates and dates.
Panoptes gains much of its power from this deep integration of a broad suite of interactive, generic functionality applicable to any data table, and specialized functionality for genome variation data. This combination guarantees flexibility in the way genome variation data and associated metadata can be explored, irrespective of particularities of specific data deployments. Figure 1 displays a typical data setup, and highlights how both the generic functionality and genome specific features interact with the data. Note that the framework is designed to serve pre-computed data, and the current design does not allow the user to initiate new calculations.
Fig. 1.
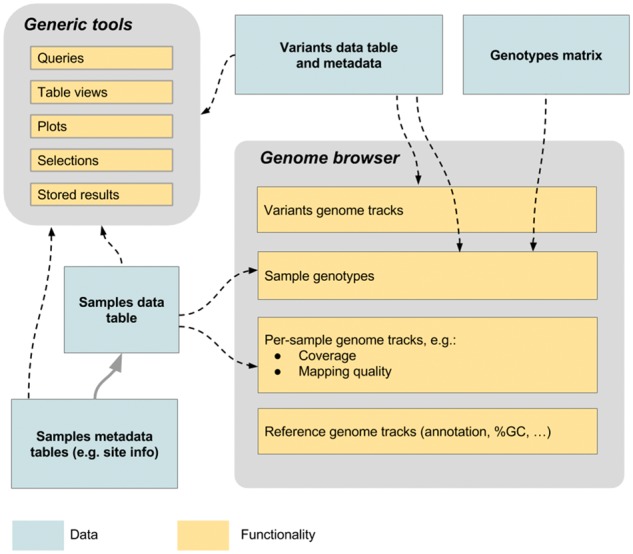
Diagram showing a typical data setup. A typical configuration in Panoptes for genome variation data. Data can be used as input for a range of generic tools, as well as specific functionality such as the genome browser. Not indicated are the interactions between the features. For example, the query tool can be used to filter the variants being showed on the genome browser
2.2 Generic functionality
2.2.1 Query builder & table browser
Filtering of datasets, based on certain properties, is an essential operation for interactive analysis. Examples include: filtering variants according to quality metrics, finding variants that have a high contrast in allele frequencies over geographical regions, or finding samples that were collected in a specific region. Panoptes contains an interactive, intuitive query builder that integrates with all data visualizations (Fig. 2). The most elementary exploration of a data table is through a paged table browser (example: http://tinyurl.com/jhk3zug). Combined with the interactive query builder and the ability to sort the items according to a property, this view provides a powerful way to reveal data items that fulfil certain criteria.
Fig. 2.

Graphical query builder. The query is presented as a vertical flow of data from the top (unfiltered data) to the bottom (filtered set), with each query statement represented as a block encountered in the flow. Statements that are combined with a logical AND operation (i.e. all have to be fulfilled) are displayed as a vertical stack. Statements that are combined with a logical OR operation (i.e. at least one has to be fulfilled) are presented as a horizontal bifurcation in the graph. The result table is visible in the background
2.2.2 Interactive plot tools
Although there are many excellent plot libraries such as R (http://www.r-project.org/), embedding a core set of plotting functionality within Panoptes was instrumental to achieve the goals of the software. Only in this way are interactive, rich user journeys through the data possible. For example, on a scatter plot representing principal components analysis results for samples, a user can click on an outlier point to obtain full information about that sample, and can subsequently navigate to a genome viewer displaying coverage and/or genotypes for this sample in a genomic region of choice. The plotting tools are deeply integrated with various interactive data selection mechanisms in the software. For example, a selection range on a histogram is automatically translated into a query for the corresponding value interval. Two plot types, a tree view and a geographical map plot, are specifically tailored for sample data. (example tree: http://tinyurl.com/gv9j5ya; map: http://tinyurl.com/zsnetay)
2.3 Genome specific functionality
A cornerstone of the Panoptes framework is the genome browser. Its main purpose is somewhat different from several well-established web tools, in the sense that it focuses on visualising rich genome variation data rather than genome annotation information. Settings declarations in the deployment directives define a set of tracks to be displayed on the browser, each one taken from data tables that are declared as containing genomic positions or regions. Numerical properties are visualized as graphs, whereas categorical properties are shown as colour coded markers or stacked bars. Typical use cases include visualization of variant properties such as average call quality, coverage or heterozygosity (example: http://tinyurl.com/zvp86jg).
A key genome browser track visualizes genotypes for a set of samples over a set of variants (Fig. 3). This track is defined using three components of the dataset: (i) a data table containing the samples and samples metadata, (ii) a data table containing the variant positions and their metadata and (iii) a data matrix containing the genotype calls. The calls matrix can have different properties such as the actual genotype call, coverage or confidence, and these properties can be visualized on the matrix via colour coding, transparency, or cell height (example: http://tinyurl.com/gtoodf2). A particularly powerful feature is the ability to use the interactive query builder to restrict the set of genotype calls currently displayed, for example to show only variants that pass certain filters, or to show only samples from a specific population. The user can sort samples in the track according to any sample metadata property, such as population or phenotype, or according to the genotype calls for one or more selected variants.
Fig. 3.

Genotypes visualization. Each row represents a sample, and each column indicates a variant on the genome. Cell colours indicate zygosity of a variant, and heights indicate sequencing coverage (Color version of this figure is available at Bioinformatics online.)
2.4 Visual analytics
Modern genome variation datasets can easily include thousands of samples characterized over tens of millions of variant positions, and are often accompanied by rich metadata. Samples can have geographic information, population information, time stamps, phenotypes and derived information such as sequencing and alignment metrics and PCA values. Variants can have different types of metadata such as quality and confidence metrics, coverage data, genetic effect information, allele frequencies over sample populations, and statistical calculations such as population genetic metrics and GWAS signals. Individual calls can also have several quality metrics attached. This can lead to complex, hybrid and high-dimensional datasets, making it hard to find interesting signals.
One way to address this data deluge is using visual analytics, which has been recognized as a valuable tool for knowledge creation in situations involving large and complex datasets (Keim et al., 2006). In this approach, knowledge is created through a rapid interaction cycle between visualizations and a human operator using her/his intelligence, creativity and experience to interpret the visualizations, and actively steer the cycles. In Panoptes, we include some visual analytics concepts to assist users in exploring large genome variation datasets.
A key concept from this approach, implemented in Panoptes, is the interaction cycle between the queries and data views. A view is always based on a query, which can be assembled using the visual query builder. Subsequently, in the view, a user can visually select a region (e.g. a rectangular or lasso selection region on a scatter plot), and turn this into a refinement of the current query. With the click of a button, this refined query can then be used as a starting point for another visualization, based on other properties of the data. This cyclic process can be used to drill down and identify regions in the multidimensional data space displaying a specific behaviour. A possible use case of this process is the identification of classes of outliers.
Another visual analytics concept embedded in Panoptes is linking and brushing (Keim, 2002). For any data type, a user can interactively select specific items using one of the visualizations. These selected items automatically become highlighted on all other visualizations that are currently active (Fig. 4, example: http://tinyurl.com/zbqwyle). This tool greatly assists in establishing a visual correspondence between complementary representations of a set of items.
Fig. 4.

Brushing and linking on multiple visualizations. Three alternative views of samples: a geographical map indicating origins of samples (top), a scatter plot showing sample PCA coordinates (bottom left), and a neighbour-joining tree (bottom right). A lasso tool can be used to select points in any of the three plots, and the selected set of samples is automatically highlighted in all plots (black circles around points in bottom plots and black arcs in top plot correspond to the same set of selected samples)
2.5 Collaborative exploration
Large scientific projects often involve many collaborators, spread across the world. Tools that simplify sharing data and insights are instrumental to streamline the communication in such projects. A number of features in Panoptes are designed to address this challenge.
First, the choice of a web app greatly simplifies collaboration and data sharing (Sagotsky et al., 2008). Collaborators do not need to download and install any software or data, and the hardware requirements are minimal: it runs on any up-to-date browser on virtually any platform, including mobile devices such as tablets. Optionally, Panoptes can use single sign-on authentication to restrict access to registered users only.
In addition, a user can instantly create a permanent link to any view created in the software, and share it with collaborators. Opening this link automatically re-creates the full application state the original user was looking at, including all settings, views, plots and active queries. In contrast to sharing a static representation (such as screenshots), a collaborator receiving this link can immediately take advantage of the Panoptes framework to start further exploration of the stored app snapshot, potentially re-sharing a follow-up result. The hyperlinks to application views provided as examples in this paper are based on this mechanism.
Users can attach notes to any item in the dataset, and create discussion threads that are accessible for full-text search. Users can also create permanently stored lists of selected data items, called subsets. These subsets can be shared with collaborators, and potentially modified by other users. A use case is the joint creation and maintenance of lists of interesting items that need further attention, such as variants that are candidate biomarkers.
2.6 Solutions for handling large amounts of data
Responsiveness of the application, with latencies below 500 ms, is considered a key ingredient for efficient visual exploration, because it encourages the user to drill deeper and explore more options (Liu and Heer, 2014; Piringer et al., 2009). Panoptes is designed to maintain a near real-time responsiveness, regardless of the size of the dataset served. Here, we describe some technology choices made to achieve this goal in a web browser, a platform that was traditionally considered to be ill suited for very responsive applications.
2.6.1 Single page application
Panoptes is designed as a Single Page Application (SPA), a web application architecture capable of providing a fluid user experience similar to a desktop application (Hales, 2012). The HTML page and JavaScript code are transferred to the client when the application starts, together with the set of essential data needed to construct the user interface. Any data required to build views are dynamically fetched from the server using AJAX requests and an efficient binary and compressed encoding scheme. Compared to the traditional multi-page navigation web architecture, the SPA model dramatically reduces the amount of visible server round trips, and never disrupts the visual appearance of the client user interface. Since all views are rendered client-side, many user interactions such as zooming and panning are instantaneous, as they do not require a server request at all.
2.6.2 HTML5 canvas
Panoptes relies on rich graphical visualizations, dynamically rendered in the client web browser, and often containing tens of thousands of components. HTML5 offers two choices for client-side rendering of graphics: the vector-based SVG element, and the raster-based Canvas element. Many standard graphics libraries, such as D3 (http://d3js.org/), are designed primarily for use with SVG. However, the SVG element relies on manipulations of the DOM tree, and does not scale well when more than 10 000 components need to be manipulated individually (Gong et al., 2012). Using the Canvas element, on most modern hardware, it is possible to achieve acceptable drawing speed for graphics in a web browser containing as many as 100 000 elements. For its visualizations, Panoptes relies on drawing functions that render to Canvas elements upon redraw request events (e.g. because of zooming or panning), using the standard HTML5 Canvas API.
2.6.3 Multi-resolution filtering
An area where a fluent, responsive UI is particularly important to a good user experience is the genome browser (Skinner et al., 2009). A single chromosome may represent a vast dynamic range of feature scale lengths, ranging from events happening at a single nucleotide to trends over tens of millions of base pairs. Phenomena at different scale lengths often correlate or confound each other, and the ability to rapidly and smoothly change the viewport zoom level and position greatly helps understanding these effects by giving context and a sense of coherency.
To achieve this in Panoptes, we adopted a multi-resolution filtering approach similar to (Robinson et al., 2011) in a web browser environment. During data import, binning and filtering is proactively calculated over the entire genome, in intervals with size 2n, for increasing integer n. Whenever a running Panoptes instance requires visualization of this data over a given genomic range, data at the appropriate filter level to achieve an optimal visual representation are fetched via a fast binary and compressed web transfer protocol. In this way, only a few thousand data points need to be transferred to the client, enabling almost instantaneous rendering, regardless the size of the genome or the zoom level. In addition, the Panoptes client pre-emptively fetches extra data for the left and right flank outside the visible viewport, enabling a very smooth zooming and panning experience. On a desktop computer equipped with a mouse, the user can use the scroll wheel to instantly zoom in and out, and drag the viewable area to pan along the genome without experiencing any rendering delay. On tablet devices, zooming and panning is achieved by pinch and swipe actions.
For numerical values, filtering over binned intervals includes by default calculating the average, minimum and maximum value. Panoptes represents the min–max range in each bin as a shaded background, and the average as a solid line on top (Fig. 5, example: http://tinyurl.com/j8ugf3x). Having visual information about minima and maxima at every zoom level gives a sense of the variation of the signal, and is crucial for situations where the extreme values of the signal are informative, such as spikes of significance in GWAS data. When sufficiently zoomed in, individual, clickable data points are displayed on top of the filtered trend information. For categorical values, frequency histograms are calculated over the binned intervals (example: http://tinyurl.com/jsxb5q2).
Fig. 5.

Different levels of zooming in the genome browser. Genome browser showing three numerical tracks. Left: full chromosome overview. Middle: 1 Mbp viewport. Right: zoom level at individual variants. The dark green line indicates the average value for each track, and the light green area shows the minimum and maximum value over the binned range. Individual, clickable variant data points are visible in the rightmost zoom level. Zooming and panning happens without any visible rendering delay (Color version of this figure is available at Bioinformatics online.)
2.6.4 Subsampling
Maintaining near real-time responsiveness when plotting and interacting with millions of data points is not possible with the current state of web technology. However, in case of large datasets, usually not all data points are needed, as plots are often used to spot global trends in the data structure. Therefore, we implemented a subsampling strategy in Panoptes. The user can dynamically change the size of the random subset used for plotting and querying, and other slices of random subsets can be visualized using a paging function (example: http://tinyurl.com/jzb29jd). Small subsets on the order of 50 000 data points can be used to visually explore the data structure interactively, maintaining a near real-time response time, whereas the subset size can be increased at any time at the cost of responsiveness, for example to spot rare outliers in a dataset.
3 Results
Panoptes was developed in the framework of the Malaria Genomic Epidemiology Network (MalariaGEN), a worldwide community of researchers studying the biology and epidemiology of malaria through the collection and analysis of genome variation data of human, parasite and mosquito populations (MalariaGEN, 2008). Sharing these data in an accessible way with the partners of MalariaGEN, and communicating them to the general scientific community, is an important and non-trivial challenge of the consortium, and since its inception the MalariaGEN resource centre has been developing innovative web tools to assist in this (Manske and Kwiatkowski, 2009; Preston et al., 2012). Panoptes was created as an extension of a web-based genotype exploration tool developed earlier (Miles et al., 2016), and has been successfully used for exploration of several large datasets created by the MalariaGEN network, both internally and publicly. Here, we describe three public datasets that are empowered by the Panoptes framework, using the current stable release (https://github.com/cggh/panoptes/releases/tag/Pn1.6.2). Note that these datasets are the results of work performed by three different research consortia. As a consequence, each set was produced with a different bioinformatics processing pipeline, and contains different types of information, representing the different priorities of the three projects. These datasets are included as an illustration of the Panoptes framework, and a full description of the various aspects of them is beyond the scope of this paper. For each dataset we include references where important additional context information can be found.
3.1 Anopheles gambiae dataset
The Anopheles gambiae 1000 Genomes Project (Ag1000G – https://www.malariagen.net/projects/ag1000g) is a global collaboration using whole genome deep sequencing to provide a high-resolution view of genetic variation in natural populations of Anopheles gambiae, the principal vector of Plasmodium falciparum malaria in Africa. Genome variation data is shared publicly on the web via Panoptes (https://www.malariagen.net/apps/ag1000g) (Ag1000G, 2016).
The current dataset comprises a variants table detailing approximately 95 million single nucleotide polymorphisms across 80 columns including regional allele frequencies, filters, quality scores and other metrics. The 765 samples used to discover the variants are detailed across 48 columns including geographic, contributor details and PCA coordinates. Diploid call, depth and quality are included for all 76 billion genotypes. A neighbour joining tree for the samples is included for each 2Mbase window across the genome. A movie highlighting some of the features of Panoptes using this dataset can be found at https://www.youtube.com/watch?v=LWCbi8t9Zug.
The stored view http://tinyurl.com/k4xbo3h provides an example of how visual exploration in Panoptes can reveal useful insights. It displays tracks on the genome browser containing levels of heterozygosity over the entire 3R chromosome arm for all Kenian samples. These tracks contain long runs of homozygosity (ROH), indicating a recent bottleneck in this population (see Ag1000G, 2016 for more details). For comparison, the stored view http://tinyurl.com/m6m2qbm provides the same view for the samples originating from Uganda, lacking such ROHs.
3.2 Plasmodium falciparum dataset
Pf3k (https://www.malariagen.net/projects/pf3k) is an international collaboration using whole genome deep sequencing to provide a high-resolution view of natural variation in the malaria parasite Plasmodium falciparum. Release 3 of the pilot data is shared publicly via Panoptes (https://www.malariagen.net/apps/pf3k/). The current dataset contains genotype information on 944270 variants over 2512 samples.
3.3 Plasmodium vivax dataset
The P.vivax Genome Variation project (https://www.malariagen.net/projects/p-vivax-genome-variation) aims to study and understand genome diversity in Plasmodium vivax, a parasite that causes malaria and, because it can remain dormant in the liver for years, is particularly hard to eliminate using conventional malaria control measures. Genome variation is shared publicly using Panoptes (https://www.malariagen.net/apps/pvgv). The current dataset contains information on 303616 variants in 228 samples (Pearson et al., 2016).
4 Discussion
The field of whole genome sequencing is rapidly maturing, and in the future genome variation data may become a commodity. Increasingly, value will be created by linking genetic information to rich complementary sample data, such as high quality, deep phenotypes. The success of these projects will be determined to a large extent by the ability to jointly interpret the genetic and phenotypic information. Because of the sheer complexity of such data and the huge variety of questions that can be asked, part of this process will require interactive data exploration and data mining. Visual analytics likely will prove a valuable tool to help address the challenge of distilling knowledge from such vast and complex datasets.
In Panoptes, we embraced one approach to this by combining a layer that captures generic interactive visual data exploration patterns, with a layer containing functionality specific for genome variation data. The deep integration of both aspects in a single, coherent framework allows a user to quickly obtain answers to questions that would be difficult and time consuming to address using a variety of standalone software tools. Exploring additional visual analytics solutions to tackle this challenge likely will become an active area of research and methods development in the coming years.
Another challenge stems from the rapidly increasing data sizes involved in this type of projects. Using a single server machine, Panoptes can cope with current data size challenges by relying on some technical solutions to handle large data volumes in near real-time. True scalability can be achieved by adopting a distributed parallel algorithm server architecture, as pioneered by MapReduce (Dean and Ghemawat, 2004). Another interesting path to pursue is using a column-oriented database such as MonetDB as a fast engine to serve real-time queries on genome variation data (Idreos et al., 2012).
In its present form, Panoptes is designed as an experimental lab where various kinds of data views and interactions are presented in a relatively unstructured way. In future versions, we intend to enhance the usability of the framework by having the ability to further stratify the data views and create pre-defined simple and commonly used navigational paths. A planned 2.0 release will focus on achieving these goals while preserving the flexibility and power of the framework. User feedback on the datasets empowered by Panoptes will serve as a guideline for these improvements.
The members of the A.gambiae 1000 Genomes Consortium, the MalariaGEN P.falciparum Community Project and the MalariaGEN P.vivax Community Project provided user feedback and assistance with software testing.
Acknowledgements
Jacob Almagro Garcia, Rachel Giacomantonio, Dushyanth Jyothi, Magnus Manske and James Stalker contributed valuable feedback during the development of the software.
Funding
The Centre for Genomics and Global Health is jointly funded by the UK Medical Research Council (MRC) and the UK Department for International Development (DFID) under the MRC/DFID Concordat agreement and is also part of the EDCTP2 programme supported by the European Union (G0600718; MR/M006212/1). This work was also supported by the Wellcome Trust with funding to the Wellcome Trust Sanger Institute (098051), the Wellcome Trust Centre for Human Genetics (090532/Z/09/Z) and to the Resource Centre for Genomic Epidemiology of Malaria (090770).
Conflict of Interest: none declared.
References
- Chelaru F. et al. (2014) Epiviz: interactive visual analytics for functional genomics data. Nat. Methods, 11, 938–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean J., Ghemawat S. (2004) MapReduce: Simplified Data Processing on Large Clusters. In: Proceedings of the 6th conference on operating systems design and implementation.
- Gong X. et al. (2012) Web visualization of distributed network measurement system based on HTML5. In: Proceedings of IEEE CCIS2012.
- Hales W. (2012) HTML5 and JavaScript Web Apps. O’Reilly Media.
- Huson D.H. et al. (2007) Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinformatics, 8, 460.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idreos S. et al. (2012) MonetDB: two decades of research in column-oriented database architectures IEEE Data Eng. Bull., 35, 40–45. [Google Scholar]
- Keim D.A. (2002) Information visualization and visual data mining. IEEE Trans. Visual. Comput. Graph.,7, 1–8. [Google Scholar]
- Keim D.A. et al. (2006) Challenges in Visual Data Analysis. In: Tenth International Conference on Information Visualization.
- Liu Z., Heer H. (2014) The effects of interactive latency on exploratory visual analysis. IEEE Trans. Visual. Comput. Graph. InfoVis 2014, 20, 2122–2131. [DOI] [PubMed] [Google Scholar]
- Malaria Genomic Epidemiology Network (2008) A global network for investigating the genomic epidemiology of malaria. Nature, 456, 732–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manske H.M., Kwiatkowski D. (2009) LookSeq: a browser-based viewer for deep sequencing data. Genome Res., 19, 2125–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles A. et al. (2016) Indels, structural variation, and recombination drive genomic diversity in Plasmodium falciparum. Genome Res., 26, 1288–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson R.D. et al. (2016) Genomic analysis of local variation and recent evolution in Plasmodium vivax. Nat. Genet., 48, 959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piringer H. et al. (2009) A multi-threading architecture to support interactive visual exploration. IEEE Trans. Visual. Comput. Graph., 15, 1113–1120. [DOI] [PubMed] [Google Scholar]
- Preston M.D. et al. (2012) VarB: a variation browsing and analysis tool for variants derived from next–generation sequencing data. Bioinformatics, 28, 2983–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.T. et al. (2011) Integrative genomics viewer. Nat. Biotechnol., 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagotsky J.A. et al. (2008) Life Sciences and the web: a new era for collaboration. Syst. Biol., 4, 201.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.E. et al. (2009) JBrowse: A next–generation genome browser. Genome Res., 19, 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Anopheles gambiae 1000 Genomes Consortium (2016) Natural diversity of the malaria vector Anopheles gambiae. BioRxiv, 096289. [Google Scholar]
- Wang J. et al. (2012) A brief introduction to web-based genome browsers. Brief. Bioinf., 14, 131–143. [DOI] [PubMed] [Google Scholar]